

Abstract
Exome sequencing of parent-offspring trios is a popular strategy for identifying causative genetic variants in children with rare diseases. This method owes its strength to the leveraging of inheritance information, which facilitates de novo variant calling, inference of compound heterozygosity, and the identification of inheritance anomalies. Uniparental disomy describes the inheritance of a homologous chromosome pair from only one parent. This aberration is important to detect in genetic disease studies because it can result in imprinting disorders and recessive diseases. We have developed a software tool to detect uniparental disomy from child–mother–father genotype data that uses a binomial test to identify chromosomes with a significant burden of uniparentally inherited genotypes. This tool is the first to read VCF-formatted genotypes, to perform integrated copy number filtering, and to use a statistical test inherently robust for use in platforms of varying genotyping density and noise characteristics. Simulations demonstrated superior accuracy compared with previously developed approaches. We implemented the method on 1057 trios from the Deciphering Developmental Disorders project, a trio-based rare disease study, and detected six validated events, a significant enrichment compared with the population prevalence of UPD (1 in 3500), suggesting that most of these events are pathogenic. One of these events represents a known imprinting disorder, and exome analyses have identified rare homozygous candidate variants, mainly in the isodisomic regions of UPD chromosomes, which, among other variants, provide targets for further genetic and functional evaluation.
Uniparental disomy (UPD) is a defect of inheritance in which both chromosomal homologs, or segments of homologs, in an individual’s genome originate from a single parent. Initially hypothesized by Engel (Engel 1980), and subsequently implicated in disease (Spence et al. 1988), instances of all but three of the 44 possible uniparental autosomal pairs have been reported (http://upd-tl.com/upd.html), with a population prevalence estimated to be approximately one in 3500 live births (Robinson 2000). The UPD chromosome can be characterized in four ways: (1) extent: affecting the whole chromosome (complete) or a portion of the chromosome (segmental); (2) zygosity: affecting all cells (constitutive) or a proportion of cells (mosaic); (3) by homolog segregation: whether the centromeric regions are identical (isodisomy) or represent both grandparental homologs (heterodisomy); and (4) by parental-origin: maternal or paternal. The origin of UPD often entails meiotic nondisjunction followed by a mitotic rescue event, but the possibility of crossing-over of homologs and missegregation of translocated chromosomes and other complex events are possible (Kotzot 2008).
UPD has three important disease associations: first, by disrupting the inheritance of essential parent-specific epigenetic modifications, causing imprinting disorders (Yamazawa et al. 2010); second, by converting deleterious alleles bequeathed from a heterozygous parent to a homozygous state, causing recessive disease (Zlotogora 2004); and third, by its relationship to incomplete trisomy rescue, resulting in residual trisomy mosaicism (Kotzot 2008). UPD is known to be a contributor to rare genetic diseases and its identification is an important part of the search for disease-causing variations. Recent clinical research involving high-throughput genome-wide SNP genotyping analysis of probands with intellectual disability has identified long regions of homozygosity (ROH) that were resolved into UPD events (Bruno et al. 2011; Papenhausen et al. 2011; Wiszniewska et al. 2013). In addition, SNP data facilitates the detection of mosaic events by detecting minor allele fractions (B-allele frequencies) with systematic departures from diploid genotypes that are not associated with apparent copy number changes (Pique-Regi et al. 2010; Van Loo et al. 2010; Jacobs et al. 2012). However, there are two drawbacks to using probands alone in UPD detection: First, UPD cannot be detected directly, but requires either experimental (microsatellite) validation or inference based on homozygosity contained to only a single chromosome; and second, this approach is blind to constitutive heterodisomic regions, as they do not produce homozygous genotypes or split in the B allele pattern.
Alternatively, UPD can be detected directly from genotypes in a proband and his or her parents, a parent–offspring trio, by searching for an enrichment of genotypes that are only compatible with uniparental inheritance. Important advantages of this approach include the discrimination of inherited ROH regions from isodisomic regions, greater resolution of UPD detection, and detection of heterodisomy. There are two previously described software tools available for detecting UPD from trio data: SNPtrio, a webtool published in 2007 that accepts as input Illumina BeadStudio or Affymetrix CNAT SNP data and uses a test to identify statistically unlikely runs of contiguous UPD-informative genotypes (Ting et al. 2007); and UPDtool, which detects groupings of non-Mendelian errors from tab-separated-value (TSV) custom genotype files and uses absolute cutoffs to select putative UPD regions and classify into UPD types (Schroeder et al. 2013). However, these tools share similar drawbacks, in that they do not avoid copy number deleted regions in the proband (a frequent source of false segmental isodisomy), require inputs limited to SNP genotyping software outputs or custom TSV files, and use statistical approaches inherently sensitive to platform genotyping density and quality. In addition, no previous tool has, to our knowledge, been systematically tested using whole-exome or whole-genome sequencing data.
The method described here, UPDio, accepts variant call format (VCF) (Danecek et al. 2011) formatted trio genotypes and compares the allelic composition of proband genotypes with parental genotypes. Unlike the previously developed methods that identify contiguous runs or groups of UPD-genotypes, this method aggregates UPD signatures on a whole-chromosomal basis, with subsequent inspection to refine the extent of the UPD. The per-chromosome binomial test can detect UPD events accurately from genotyping platforms of variable density, such as whole-exome data, SNP data, and whole-genome sequence data, without extensive platform-specific parameter manipulation. This method avoids copy number regions via the filtering of common and sample-specific copy number variable regions, regions that often result in false-positive UPD calls, thus increasing statistical power. Simulations of SNP and exome data at the default P-value threshold demonstrated high accuracy at detecting whole-chromosomal UPD and segmental UPD above 1 Mb for SNP data and 10 Mb for exome data. We applied this method for UPD detection on 1057 unique trios in a rare disease study, the Deciphering Developmental Disorders (Firth and Wright 2011) (DDD) project. We identified six individuals with a uniparentally inherited chromosome—of which one is associated with a known imprinting disorder—and carried out candidate variant selection from the exome data for these individuals.
Results
We developed an approach to identify pathogenic UPD events that is comprised of three steps: (1) genotype preparation, (2) UPD detection, and (3) candidate variant selection (Fig. 1). Genotype preparation begins with preprocessing the genotype data from SNP chip or exome sequencing data. Data preprocessing is critical and includes two steps: (1) removal of low-quality genotypes, (2) removal of genotyped sites within CNVs, since heterozygous deletions can masquerade as uniparental isodisomy. For exome data in which only nonreference genotypes are recorded (e.g., from single sample calling), an additional preprocessing step was used to introduce homozygous reference genotypes for common SNPs that are well covered and for which a nonreference genotype was not detected (Methods). Note that for multi-sample VCFs generated by multi-sample calling, this homozygous reference imputation step is not required.
Figure 1.

Study workflow. The study consisted of three main steps: data preparation, UPD detection, and candidate variant analysis. In the data preparation stage, we collected informative genotypes seen in all members of each trio. Either a multi-sample trio VCF or three single-sample VCFs can be used as input; the latter requires the annotation of homozygous reference genotypes, not usually encoded in single-sample VCF files. In the UPD detection stage, we selected trios containing a proband chromosome with an enrichment of UPD-informative genotypes. Exomes (five) available for samples with a detected UPD event were selected for the candidate workup analysis, in which we attempted to find rare protein-altering variants that may manifest in the proband’s phenotypes.
After preprocessing, the proband genotypes diagnostic for uniparental or biparental inheritance are counted on each chromosome. Uniparental genotypes can be quantitatively distinguished from one another by the relative proportions of the two different classes of genotype configurations that are diagnostic for uniparental inheritance (Table 1), or qualitatively by visualization. One class of uniparental genotype configuration is specifically informative for isodisomy (UI [uniparental–isodisomic]), and the other class does not distinguish heterodisomy from isodisomy (UA [uniparental–ambiguous]). Heterodisomic events contain only UA genotypes and lack UI genotypes, while isodisomic events contain mixtures of UA and UI genotypes.
Table 1.
Informative genotypes for UPD analyses
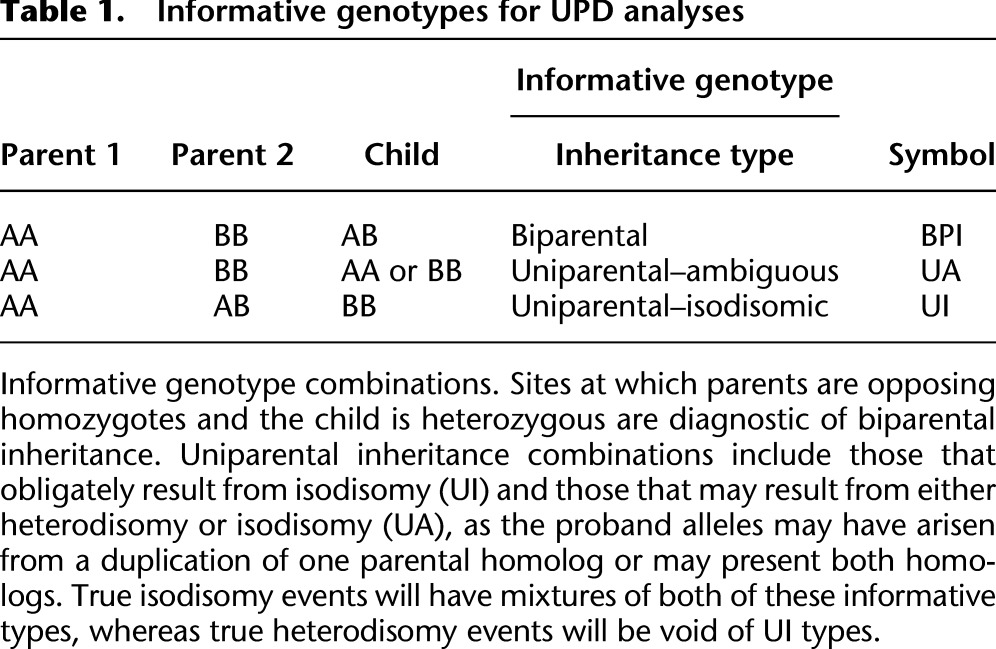
Simulations
We performed simulations to assess the accuracy of UPD calling in UPDio (see Methods). The sensitivity of UPD detection was measured at a range of sizes (1, 2, 5, 10, and 20 Mb) to test detection rates of segmental UPD and chromosome-wide, to test detection of complete UPD. Simulations were performed for heterodisomy and isodisomy from data generated by exome and SNP genotyping platforms.
The method is more sensitive for detecting isodisomy than heterodisomy, as expected given the greater number of informative sites for the former. Also, the method is more sensitive at a given size using SNP chip data than using exome data (Fig. 2), primarily due to both the greater density of genotyped sites, with a possible minor contribution from the likely higher genotype accuracy in SNP chips. At Bonferroni-adjusted significance threshold (light-blue line, P = 0.000568), we observed in SNP chip data nearly perfect sensitivity for detecting either class of UPD event (heterodisomy or isodisomy) at 5 Mb. At 2 Mb, 98% of isodisomy and 91% of heterodisomy could be detected. Sensitivity of isodisomy detection from exome data was 99% for isodisomy and 75% for heterodisomy at 10 Mb.
Figure 2.
Sensitivity of UPD detection simulations. Simulations to assess sensitivity of UPD detections at different sizes, from different data sources. (iUPD) Isodisomy; (hUPD) heterodisomy.
We defined specificity as the proportion of tested non-UPD trios that lacked maternal UPD calls. At the Bonferroni-adjusted P-value of 0.000568, specificity was 99% for exome data and 100% for SNP data. The cause of the single false-positive UPD event was found to be due to a slight excess of genotype errors resulting in an event called with a marginally significant P-value (0.00044).
Given that a size threshold for suspecting UPD in clinical molecular diagnostics is typically near 10 Mb (Conlin et al. 2010), the successful detection of UPD of this size is of practical utility. Indeed, even 2-Mb isodisomic events were detected accurately from SNP-chip data, a result likely due to low genotyping error rates and relatively uniform genotyping density; although at this size, the accuracy of detection of heterodisomy from SNP-chip data, and isodisomy and heterodisomy from exome data, was appreciably lower.
Comparing UPD detection software tools
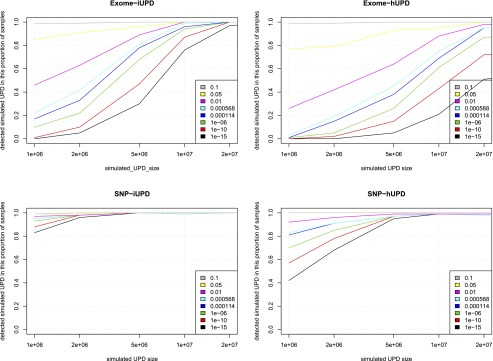
We assessed the strengths and limitations of three trio-based UPD detection tools: SNPtrio, UPDtool, and UPDio (Table 2). There are substantial differences in the interface, statistical methods, calibrations, and outputs of these three tools. One notable difference is the input format requirements. UPDtool requires the construction of custom tab-separated-value genotype files, while SNPtrio processes SNP-genotyping software output files, and UPDio reads VCF files, which is a platform-independent standard file format for genotype data. The underlying statistical methods vary as well. UPDio is the only tool that integrates CNV filtering during genotype parsing, which occurs before statistical analysis. In terms of calling confidence, UPDio and SNPtrio provide a P-value output measurement, while UPDtool does not provide a confidence score for its UPD detections. For threshold calibration, the webtool SNPtrio accepts a parameter “minimum number of SNPs in an event region”; UPDtool has a list of seven adjustable parameters (min_mes, window size, min_mes_fraction, min_hetero, min_iso, min_mes_paternal and max_mes_paternal); and lastly, UPDio allows for user control of the P-value threshold as a single parameter. Neither SNPtrio nor UPDtool parameters are recalibrated dynamically based on input data; instead, they are tuned for platforms resembling the density and noise characteristics of high-density SNP trios. In contrast, UPDio calculates a per-chromosome proportion-based statistic, which is innately normalized for input data of different global density and noise characteristics.
Table 2.
Software comparisons
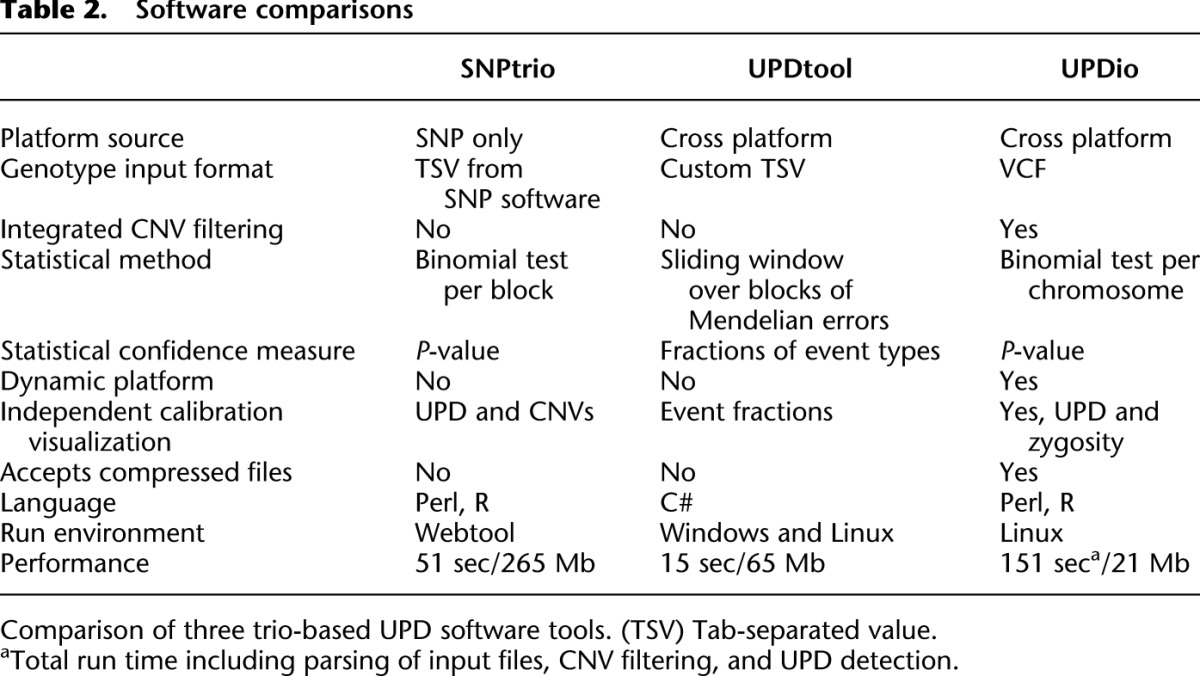
Simulations assessed the comparative accuracy of three trio-based UPD detection tools: SNPtrio, UPDtool, and UPDio. All three platforms were run using default parameters (Methods: Simulation Comparison), on the same simulated data sets (reformatted to accommodate each tool’s input requirements). Sensitivity results were tabulated as the proportion of tested samples with maternal UPD detection on the chromosome containing the simulated event (Fig. 3). Specificity was calculated as the proportion of samples not containing maternal UPD events in samples without obvious UPD events (Fig. 4).
Figure 3.
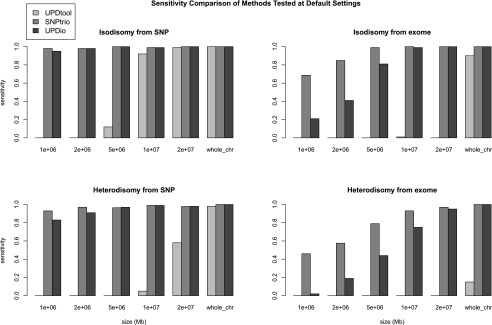
Sensitivity comparisons. Simulations were performed to measure the sensitivity of detecting introduced UPD events from SNP and exome data, ranging in size from 1 Mb to chromosomal.
Figure 4.

Specificity comparisons. Simulations on normal SNP and exome samples were compared to measure the proportion of samples without UPD detections.
Simulation results demonstrated that SNPtrio was the least specific algorithm (31% for SNP data and ∼0% for exome data), and UPDtool was the least sensitive tool, only capable of detecting the very largest UPD events. Unsurprisingly, specificity and sensitivity were inversely related. UPDtool was 100% specific, and made no false UPD assignments in normal samples from either SNP or exome data. UPDio was nearly as specific as UPDtool. SNPtrio was the most sensitive, which was most evident in the detection of smaller heterodisomic events from exome data. UPDio was only very slightly less sensitive than SNPtrio for events 10 Mb and greater in size in exome data and for events 1 Mb and greater in size in SNP data.
We produced receiver operator characteristic (ROC) curves to evaluate the calling performance of UPDio at various P-value thresholds (Fig. 5). The UPDio (“dio”) curves demonstrate excellent classification of UPD events from SNP platform at 5 Mb and 10 Mb. The classification of UPD events from exome data is noticeably weaker, especially for detection of heterodisomy at a size of 5 Mb. The Bonferroni corrected P-value of 0.000568 represents a good balance of sensitivity and specificity for both data types and both classes of UPD event. Thus, we decided to use this P-value as a default parameter for UPD calling in UPDio.
Figure 5.
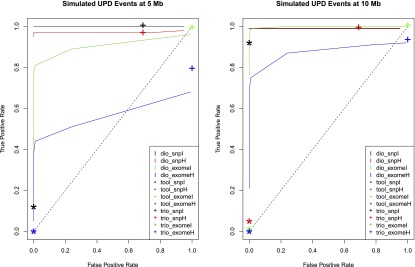
Receiver operator characteristic curve comparing UPD detection accuracy. Accuracy of UPD detection at different simulated UPD sizes. (dio) UPDio; (tool) UPDtool; (trio) SNPtrio.
For the two ROC curves we plotted the classification performance of UPDtool (“tool”) and SNPtrio (“trio”) for the calculated sensitivity and specificity of these programs at their default parameter settings. While most SNPtrio classifications demonstrated high true-positive rates, these came at the expense of very high false-positive rates that would require substantial additional downstream manual filtering such that large-scale application is inherently limited. On the other hand, UPDtool performance was characterized by low true-positive rates, near zero for most event types and platforms, with the notable exception of isodisomy from SNP data at a size of 10 Mb. In contrast, UPDio, using the default P-value threshold, detected a substantially higher ratio of true to false events compared with the other programs under all conditions. These differences are likely to be accentuated when implementing these tools for whole-genome sequence data sets. While SNPtrio and UPDtool are tuned for SNP data and to our knowledge not tested on whole-genome data, we obtained HapMap child–mother–father trio (NA12878, NA12891, NA12892) and CNV data (Mills et al. 2011) and implemented UPDio using a default Bonferroni-corrected P-value threshold. Whole-genome analysis counted an average of 278 informative genotypes per Mb, 20× greater density than our SNP platform, required 9 min and 27 Mb of memory and detected no UPD events beyond marginal significance.
UPD detections in the Deciphering Developmental Disorders (DDD) project
We performed UPD detection on 1057 unique parent-offspring trios in the DDD project. The majority (915) of these trios were analyzed by both SNP and exome data, with slightly more trios available from SNP data (1035) compared with exome data (937). We applied a P-value of 0.000568 as a statistical threshold (see section “Genotype Segregation and Statistical Analysis” in Methods) for identifying putative UPD events for further investigation.
We observed that the putative UPD events had calculated P-values that were clearly bimodal in distribution (Fig. 6), and this finding was consistent in results from both SNP and exome trio data. We observed that the extremely significant events were, in all cases, authentic UPD detections and these were selected for further analysis. The marginally significant events were consistently spurious UPD detections.
Figure 6.

DDD UPD P-value distributions. Distribution of the –log10 P-values for UPD detections from different data sources, with or without CNV data. Presence of sample-specific CNV data increases the proportion of extremely significant events and decreases the proportion of marginally significant events. P-value minimum truncated to 1 × 10−100.
We investigated the less-significant group of detections and observed differences between the two platforms regarding the number and underlying cause of these spurious events. The SNP data had 133 such events while the exome data had 70 such events. The underlying cause of these false detections in the SNP data usually (∼80% of the time) was due to misattribution of undetected (and thus unfiltered) CNV regions as isodisomy. This was especially true for the most significant events of this category; for example, a 1-Mb deletion (which escaped detection by aCGH due to low-quality array data) resulted in false signals of high significance (UI_P at 1 × 10−31 and UA_P of 1 × 10−22). In contrast, the underlying cause in the exome data in most (∼85%) cases was due to stochastic fluctuations of genotyping errors. The different underlying causes and number of the marginally significant events likely reflect underlying platform differences, namely that the SNP platform has far greater genotyping density, especially in noncoding regions, thus is more prone to detecting hemizygous genotypes within small deletions than the exome data, while the exome data (from single sample calling) has a slightly higher genotyping error rate, and is therefore more susceptible to the random aggregation of genotyping errors.
Large UPD events have substantial numbers of both UI and UA events. Consequently, binomial tests assessing the enrichment of both event types often redundantly detect these large UPD events by both signatures. We developed a visualization tool to illustrate the distribution of informative sites along each chromosome in a trio to clarify the type and extent of these events, which may include both isodisomy and heterodisomic regions (Fig. 7). In addition, the method provides additional output files to specify all informative genotype events comprising the UPD region.
Figure 7.

UPD example plot. A plot of QC-passing proband genotypes on each autosome. The position and color reflect zygosity (homozygous, heterozygous) and informative state (biparental inheritance, maternal isodisomy, maternal heterodisomy or isodisomy, paternal isodisomy, paternal heterodisomy or isodisomy). The figure displays each chromosome ideogram. Each chromosome has an x-axis (chromosome position) and y-axis (zygosity, and informative event type). In this case, the UPD event for chromosome 2 is depicted with a mixture of dark-green points (maternal isodisomy) and light-green points (maternal isodisomy or maternal heterodisomy). The zygosity row demonstrates homozygosity along the entirety of the chromosome, reflecting the complete isodisomy.
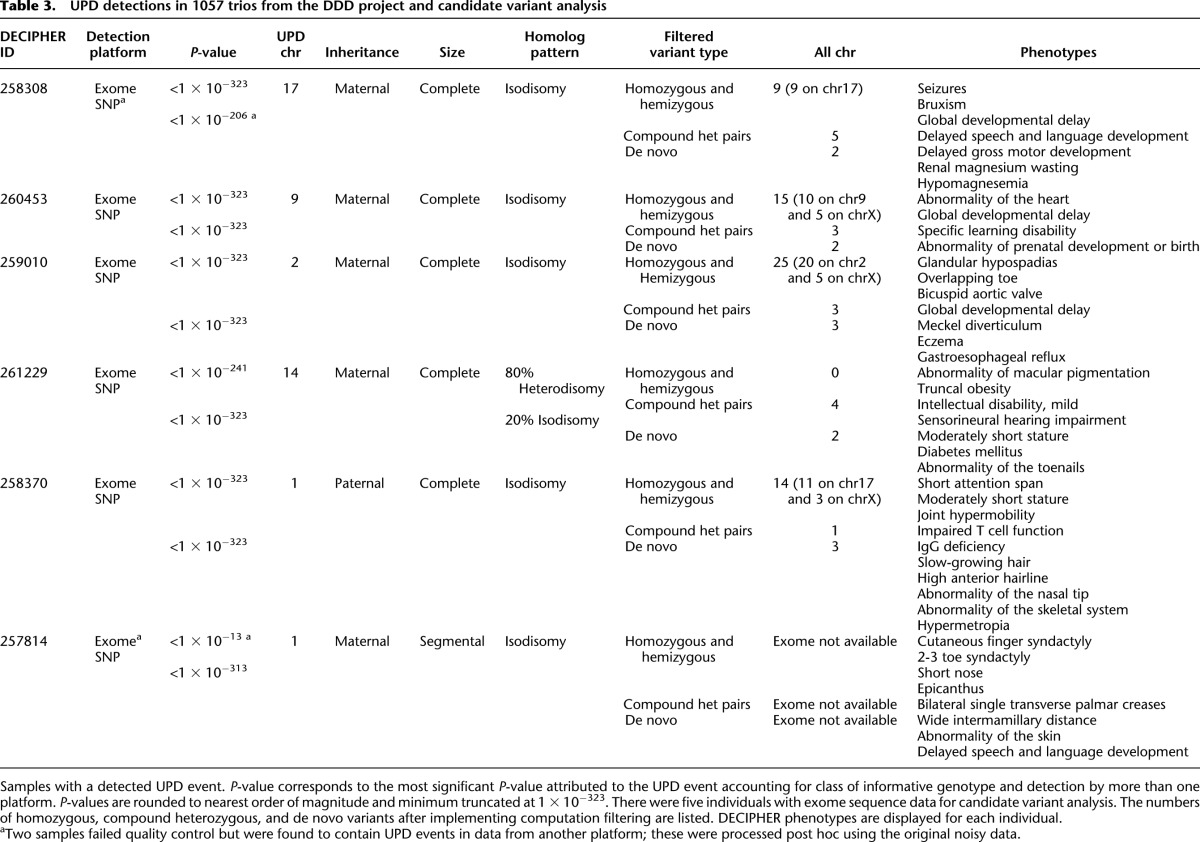
There were 10 extremely significant event types in SNP data and also in exome data, which reflected the redundant detection of five UI and UA events in both cases. Of the five UPD events, four of five in both sets were the identical event seen by both platforms. One UPD event was detected solely from SNP data, and a different event detected solely from exome data. In these two latter cases, data were not initially available from both platforms, but subsequent genotyping led to successful detection in both cases. Thus, in total, there were six unique probands with UPD events, and all events were detected using SNP data and exome sequence data. The six events comprised a variety of UPD events. Most (five of six) were maternal, most (five of six) involved the entire chromosome, and most (five of six) reflected isodisomy (Table 3).
Table 3.
UPD detections in 1057 trios from the DDD project and candidate variant analysis
Investigating UPD frequency
Compared with the widely quoted birth prevalence of UPD (1/3500) (Robinson 2000) the proportion of UPD events detected in the trio analyses (6/1057) described above is significantly higher (binomial test P-value 8.036 × 10−7). The UPD rate at birth in the general population has been estimated on extrapolation from clinically relevant UPD events at a single locus, and thus is potentially susceptible to variation among chromosomes in UPD rate. To generate an empirical estimate of the population prevalence of all classes of UPD would require dense genome-wide genotypes for tens of thousands of parent–offspring trios sampled randomly from the population; such data are not currently available. However, it is possible to estimate the rate of uniparental isodisomy from dense genome-wide genotypes on unrelated individuals since isodisomy manifests with an easily detectable signature: a long region of homozygosity. Other processes, such as consanguinity (Li et al. 2006) or cryptic relatedness (Astle and Balding 2009) similarly generate long regions of homozygosity, but are distinguishable from isodisomy because these other processes often involve multiple chromosomes and are rarely longer than 20 Mb (Li et al. 2006).
We used 16,881 samples from the Wellcome Trust Case Control Consortium (WTCCC) data set to develop an empirical estimate of the rate of complete uniparental isodisomy by observing the number of samples containing a single chromosome burden of large regions of homozygosity. First, we used PLINK (Purcell et al. 2007) to identify large (>10 Mb) tracts of homozygosity for each sample, and retained samples with a large homozygous region or regions confined to a single chromosome. There were many samples (N = 103), which satisfied this criterion. Of these, only a single sample appeared to have whole-chromosomal isodisomy, but a further five samples had significant homozygosity that extended over at least half of the chromosome (Supplemental Figs. 1–6). These five samples comprised four telomeric events on chromosomes 4, 21, 22, 22, and one on chromosome 4 with two large interstitial regions of homozygosity. As the homozygosity of these events covered the majority of the chromosome, and represent the only major tract of homozygosity in these genomes, we believe these events likely reflect mixtures of isodisomy and heterodisomy and are unlikely to reflect inherited homozygosity. Under the conservative assumption that all these chromosomes reflect complete uniparental disomy of a chromosome in these individuals, this represents a frequency of six uniparental disomy events in 16,881 individuals, which is not significantly different from the reported frequency of one in 3500 (binomial test P-value 0.4934). Notably, by enforcing the same criteria to define a UPD event (the majority of the chromosome homozygous and large homozygosity confined to a single chromosome), the five UPD detections in the DDD project (removing the one 10 Mb segmental event from the six in total) is still a significant enrichment compared with the population estimate (binomial test, P-value 1.751 × 10−5) and also a significant enrichment compared with the WTCCC data (Fisher exact test, P-value 0.0002598).
We also attempted to use the WTCCC data to investigate the prevalence of segmental UPD, however, despite stringent filtering of subchromosomal segments of homozygosity, we could not recapitulate the expected pattern of terminal segmental UPD events (http://upd-tl.com/upd.html; Supplemental Fig. 7). Therefore, we believed that most of the regions of segmental homozygosity in the WTCCC were not reflective of segmental UPD events and we considered that estimating population of prevalence of segmental UPD events from this data set would not be appropriate. Analyses of segmental UPD, which are typically mosaic (Rodriguez-Santiago et al. 2010), are better suited to algorithms that interrogate the B allele frequency, rather than genotype data.
Identifying plausibly pathogenic genetic variation in the DDD trios with UPD events
The segmental UPD on chromosome 1 is associated with a flanking de novo 12-Mb duplication/triplication event that is the likely pathogenic variant. Only one of the other five UPD events detected in the DDD trios is associated with a known imprinting disorder—the maternal UPD of chr14 known to cause Temple Syndrome (OMIM *605636 and #176270)—however, not all of the clinical features of this individual have a reported association with this syndrome. Thus, for this patient and the remaining four patients with UPD, it is of interest to identify other genetic variations that might account for the observed clinical features; so the exome data for these patients were examined to identify genetic variants potentially underlying the observed developmental disorders (see section “Identifying Candidate Variants” in Methods; Supplemental Table 1). We found that the UPD chromosome accounts for the vast majority of the rare homozygous variants predicted to impact upon protein function in each of the studied exomes (Table 3), however, we also broadened the search to other genetic models compatible with sporadic presentation in these families. Although each patient harbored rare, functional variants in known Mendelian disease genes, none of the specific variants we observed had previously been classified as being pathogenic. Therefore, experimental follow-up would be required to definitively implicate these novel variants with disease causation. Nevertheless, the exome analysis provided a rich source of plausible candidate variants for a follow-up investigation.
The patient with maternal UPD of chromosome 14 is a 15-yr-old girl. Temple Syndrome (maternal UPD14) accounts for most of the child’s phenotypes, including truncal obesity (weight 99th centile), moderately short stature (height first centile), and mild intellectual disability (Temple et al. 1991), while the diabetes mellitus phenotype is likely attributed to the metabolic consequences of the disorder (BMI 38; class II obesity). In addition, the child has sensorineural hearing impairment, a condition that is not a reported characteristic of the syndrome. Of the variants that remained after filtering, four were present in known Mendelian disease genes (DRD2, TTN, PLAU, TECTA). The gene TECTA encodes an extracellular matrix protein (tectorin alpha) of the tectorial membrane, the surface of the sensory epithelium of the cochlea (Balciuniene et al. 1999), and is a well known cause of autosomal dominant (MIM:601543) and autosomal recessive (MIM:603629) hearing loss. This proband had novel compound heterozygous variants, with a missense substitution inherited from the mother and a stop gained mutation inherited from the father. Neither parent has a documented hearing disability, suggesting that the compound heterozygosity has resulted in the recessive form of hearing loss in the child. Recently, a hearing-impaired proband with normal-hearing parents was found to contain compound heterozygous variants (missense and splicing mutation leading to truncated protein) in the TECTA gene, which was indicated to be pathogenic through in vitro functional characterization (Sagong et al. 2012).
The patient with UPD of chromosome 9 is a 15-yr-old male patient with developmental delay and intellectual disability, recruited following noninformative aCGH CNV analysis. His family history was notable for having several second-degree family members with similar phenotypes. The child also has a congenital heart defect. As the clinical features were relatively common among children with congenital disorders, it was more challenging to use phenotypic matching to identify specific genetic candidates in this patient. The child has four rare functional variants in Mendelian disease genes (MLLT3, LAMC3, HNRNPU, SLC6A8). The HNRNPU gene is a known intellectual disability gene, and the de novo variant is well supported by sequencing data (11 of 22 sequence reads in proband and absent in well-covered parents), although sequencing by capillary sequencing is ongoing. In addition, the child has a hemizygous missense variant in SLC6A8 and defects in this gene are known to cause X-linked intellectual disability through creatinine transport malfunction.
The patient with maternal UPD of chromosome 2 is a 7-yr-old male patient, with a complex phenotype profile including global developmental delay, glandular hypospadias, overriding toe and bicuspid aortic valve. Recently, a female child, also with UPD-maternal of chromosome 2 and complex phenotype, distinct from our patient, had been exome sequenced and many (18) candidate variants were identified on the UPD chromosome, none definitively pathogenic (Carmichael et al. 2012). None of that girl’s phenotypes are coincident with our patient, suggesting that an imprinting disease is not the likely cause of the diseases in these children. In our patient, the entire burden of rare homozygous coding variants (20 variants) lay on the UPD chromosome. We identified six variants in Mendelian disease genes (EIF2AK3, AGXT, PASK, L1CAM, CUL4B, GLA). Two of these variants are interesting with respect to the global developmental delay in this child. The child has a hemizygous variant in the GLA gene causing an amino acid change, D313Y, which has been exculpated as a cause of Fabry disease (Niemann et al. 2013), but recently associated with severely decreased GLA enzyme activity in plasma and the formation of white matter lesions in males (Lenders et al. 2013). Also potentially interesting is the rare, missense hemizygous (X chromosomal) variant in CUL4B, a gene well recognized to cause mental retardation and hypogonadism (Isidor et al. 2010), although our patient has a less severe phenotype than is typically associated with a null mutation in CUL4B.
The patient with UPD of chromosome 17 had delayed developmental milestones, growth retardation, microcephaly, and suffers from seizures intractable to medical intervention. She was found to have decreased serum magnesium and renal magnesium wasting but genetic testing for diseases of renal hypomagnesium wasting (TRPM6 and SCN1A gene testing) was normal. Her seizures did not resolve after intravenous magnesium infusion and resulting restoration of blood magnesium to normal range, suggesting that hypomagnesaemia alone is not the cause of her seizures. The child has three variants in Mendelian disease genes (FKBP10, SCN4A, CCDC40). The missense SNV in SCN4A is interesting as it is very rare (0.0012 minor allele frequency), inherited from a heterozygous parent, but made homozygous on the UPD chromosome, and located in a gene that encodes a subunit of a voltage-gated sodium channel. This sodium channel is implicated in a diversity of neuromuscular disorders, such as periodic paralysis and myotonia congenita, diseases that mimic seizure disorders (Stephenson et al. 2004; Uldall et al. 2006). While channelopathies often follow a dominant mode of inheritance (Koch et al. 1993), recessive modes have been seen as well (Trip et al. 2008), and several channel proteins are known to underlie severe seizure disorders, such as KCNQ2 (Ohtahara syndrome) (Yamatogi and Ohtahara 2002) and paralogs of SCN4A, such as SCN1A (Wolff et al. 2006), SCN2A (Kearney et al. 2001), and SCN9A (Singh et al. 2009). In addition, the child has compound heterozygous missense mutations in UNC13C, a gene known to result in an inability to learn complex motor tasks in mouse knockouts (Augustin et al. 2001).
The male patient with complete paternal UPD of chromosome 1 had diverse clinical features, including skeletal defects, immunological defects (IgG deficiency, impaired T-cell function), as well as phenotypes shared with the mother—short attention span and short stature. There were five variants in known Mendelian disease genes (GJA8, ADCK3, LMNA, EFHC2, KAL1), none of which is associated with phenotypes consistent with those in the proband. The majority of rare, homozygous variants were present on the UPD chromosome (11 on chr1 and three on chrX).
Discussion
The search for disease-causing variants is a central task of modern genomics. Advances in sequencing technology have greatly improved sequencing throughput (Mardis 2008), yet the identification of disease-causative variants is complicated by genetic diversity among individuals of our species (The 1000 Genomes Project Consortium 2010). As a result, study designs have evolved to narrow the field of candidate variants. Trio-based family designs (Evangelou et al. 2006) can substantially narrow the field of candidate high-penetrance variants by leveraging inheritance information in variant calling. In addition to the power for family-based studies to readily identify de novo variants and compound heterozygosity, and reduce population stratification in association analysis, they can be used to identify uniparental disomy.
Methods to detect UPD have evolved with our ability to interrogate the genome. Trio-based UPD calling has advantages compared with proband-based calling as the former can identify uniparentally inherited genotypes directly and is sensitive to heterodisomy, while the latter relies on detection of large (typically larger than 10 Mb) regions of homozygosity confined to a single chromosome and is blind to heterodisomy. On the other hand, this strategy of using informative genotypes as a signal for uniparental disomy can be polluted by hemizygous or erroneous genotypes that mimic uniparental signatures. UPDio has unique advantages compared with existing trio-based UPD detection programs for mitigating the effect of genotype errors and heterozygous deletions. First, genotype errors have the potential to hypersegment UPD calling in SNPtrio and UPDtool, tools that detect runs or blocks of UPD, but have little effect on disrupting the per chromosome rate of informative genotypes, the metric used by UPDio. Second, SNPtrio and UPDtool are vulnerable to false isodisomy created by hemizygous regions in the proband, while UPDio has an integrated CNV filter to avoid common CNV and user-specified sample-specific CNV regions before the binomial test is applied. Since deletions generate genotypic signatures identical to isodisomy, this step is essential to prevent the unintentional ascription of deletions as UPD. With the availability of tools to detect CNVs from SNP genotyping and exome data (Love et al. 2011; Fromer et al. 2012; Li et al. 2012; P Vijayarangakannan, T Fitzgerald, C Joyce, S McCarthy, ME Hurles, pers. comm.), our software tool enables users to remove these erroneous signatures from UPD analyses using data from a single platform, by providing sample-specific CNVs in BED (Quinlan and Hall 2010) or VCF format. In addition, the statistical test applied in UPDio intrinsically adjusts for differences in platform genotyping density, which varies in orders of magnitude between exome data, SNP data, and whole-genome data. Also, only UPDio outputs a measure of statistical confidence, a P-value that can be calibrated by the user to achieve the desired sensitivity and specificity. Only UPDio can read single-sample and multi-sample VCF files, the modern genotype file standard, and thus can be more easily assimilated as a module into existing pipelines. While UPDtool was the fastest method of the three tested, UPDio performs additional processing to cleanse poor-quality genotypes and avoid copy number regions; nevertheless, it completes UPD calling on high-density SNP trio data in under 3 min, and is the least memory intensive of the three methods for detecting UPD events. In fact, memory efficient iterator functions enabled UPDio to process a whole-genome trio using less memory than either of the competing programs used to process a SNP trio.
We compared the relative accuracy of the three trio-based UPD calling software using each tool’s default parameter settings on the same set of simulated data. We found marked differences in the sensitivity and specificity of these three software tools. The practical utility of SNPtrio is greatly hampered by its lack of specificity, whereas UPDtool exhibited very low sensitivity, was only capable of detecting the very largest of simulated UPD events, and would miss most small UPD events. In contrast, using default parameters, UPDio was sensitive and specific for simulated UPD events at 1 Mb from SNP data and 10 Mb from exome data, with broadly equivalent sensitivity to SNPtrio. There are several factors that likely account for these dramatic differences in calling accuracy. Probably the most important factor is due to the need to finely calibrate SNPtrio and UPDtool, which use statistical approaches that are more vulnerable than is UPDio to platform-differences in genotype density and genotype error rates. Unfortunately, unlike UPDio, SNPtrio and UPDtool do not offer a convenient user-adjustable threshold of statistical threshold.
The sensitivity and resolution of UPD detection is inherently determined by the density, distribution, and accuracy of genotyped sites. In our study, the sensitivity for detecting smaller UPD events was lower for trios in exome data primarily because the number of informative sites genotyped was (∼10×) fewer, although other factors, such as less even distribution and slightly higher genotyping error rate may have been contributory. Exome genotyping accuracy could potentially be improved by multi-sample calling, which would include explicit homozygous reference genotypes for all variants detected in one or more samples, and UPDio is capable of processing multi-sample VCF files. However, the technical noise (median of zero apparently uniparental genotypes per chromosome in SNP data, and a median of one apparently uniparental genotype per chromosome in exome data) is exceptionally low, allowing for detection sensitivity on either platform to be 100% for whole-chromosomal UPD events and sensitive for most simulated segmental events at the 1-Mb level in SNP data and the 10-Mb size for exome data. This size is clinically relevant as non-trio-based studies of UPD typically only investigate potential UPD when regions of homozygosity exceed 10 Mb (Conlin et al. 2010).
Smaller UPD events, such as those affecting 1 Mb in size, are challenging to detect due to a paucity of informative genotypes. For example, the SNP chip data contain on average only 14 informative genotypes per megabase window. Still, with high-quality genotypes, the occurrence by chance of 14 contiguous UPD-characteristic genotypes is a very unlikely event, and the previously developed contiguous runs of informative genotypes method may be marginally more sensitive than the proposed method at detecting events at this size. However, the contiguous runs method is also more likely to be sensitive to small runs of UPD-mimicing genotypes occurring by chance across the whole genome, lowering specificity. Moreover, smaller UPD events are less likely to be pathogenic and are much more likely to be mosaic (Kotzot 2008), implying that alternative UPD detection approaches, based on B-allele frequency of proband genotypes, would be more appropriate for segmental UPD events.
We implemented UPD detection on 1057 unique trios and identified four probands with whole-chromosomal isodisomy, one with whole-chromosomal heterodisomy, and one proband with segmental uniparental isodisomy of 10 Mb. Using UPDio, all six UPD events were easily called from both platforms yielding highly significant P-values in both SNP and exome data. Given this finding and the simulation results, this suggests that exome-based trio designs are appropriate to detect UPD, without the requirement to run SNP chips specifically for this purpose. Also interesting is the conspicuous lack of detection of complete heterodisomy events in this data set, which are invisible to proband-based inquiry but detectable using this trio-based analysis. This suggests that the prevalence of complete heterodisomy is indeed rare, although larger cohorts of analyzed trio data will prove useful to substantiate this conjecture. Heterodisomic events, by virtue of not homozygosing rare functional variants, can only be pathogenic through imprinting disorders.
The proportion of UPD events detected in the DDD project to date represents a nearly 25× enrichment for UPD as compared with what had been expected from prior population prevalence estimates, and is highly significant (the P-value compared with established estimates of one in 3500 is 9 × 10−7 [binomial test] and compared with a proportion of six of 16,881 in the WTCCC is 2 × 10−5 [Fisher exact test]). There are several explanations that could cause the high rate seen in our study: (1) a high false-positive rate in UPD detection in DDD, (2) the estimation of UPD prevalence in the population is an underestimate and the DDD study has higher prevalence of benign UPD by chance alone, (3) some of the UPD events are disease causing. There is over-whelming statistical evidence of UPD in all six cases from two independent platforms, suggesting that (1) is not the explanation. To address the question of whether UPD prevalence in the population has been underestimated we attempted to empirically estimate the rate of UPD using SNP genotyping data on unrelated individuals from the Wellcome Trust Case Control Consortium. There are limitations to this approach, mainly that it is indirect (we are only able to identify UPD by observing single-chromosome large runs of homozygosity, not directly from the inheritance patterns of individual genotypes), and confounded by other causes of large runs of homozygosity, such as identity by descent, identity by state, or loss of heterozygosity. Notwithstanding these limitations, we found that previous prevalence estimates about uniparental disomy in the human population are compatible with our observations. Therefore, the suggestion that some individuals with UPD in our study may have UPD-related disorders warrants further investigation.
Thus, we interrogated the probands harboring these UPD events for UPD-related diseases. In only one of these six individuals does the UPD region encompass a known imprinting disorder region (Temple Syndrome, UPD14 Maternal). This child’s phenotypes are consistent with the known manifestation of Temple Syndrome; however, the child also has hearing impairment, which may be explained by compound heterozygous variants in TECTA found during exome variant analysis. We identified possible disease-causing variants for the other probands who had UPD events and for whom exome sequence data were available. Similar to the offspring of consanguineous unions, UPD chromosomes provided a rich source of candidate recessive variants, since isodisomy can promote parental heterozygosity to homozygosity. We note that the complete isodisomy of chromosome 2 in one proband reflects homozygosity of ∼8% of the genome, which is a similar proportion to that expected among offspring of first-cousin marriages (1/16, ∼6%).
Candidate recessive variants were identified for each proband, although none is definitively pathogenic, these candidate genes deserve further investigation. We note that the specific ascertainment of patients in this study, whom are only recruited once clinical genetics services have failed to obtain a diagnosis, may bias against the discovery of UPD events that result in a well-recognized imprinting or recessive disorders for which routine diagnostic assays are available.
As sequencing technologies continue to increase the cost-effectiveness of genome-wide sequencing data, our ability to interrogate UPD will improve. The tool we developed can scale to interrogation of whole-genome genotype data, as files are read line-by-line without storing large data hashes, thus making efficient use of memory. Although we note that UPD detection is ultimately fundamentally limited to a resolution on the scale of tens of kilobases by the density of informative genotype configurations in the parents. Given the broad range of recessive and imprinted phenotypes associated with UPD, its detection should be a part of the genetic analysis for disease studies more broadly, as it is a small, but important piece of the puzzle of pathogenic genomic variation.
Methods
Genotype segregation and statistical analysis
A site genotyped in parents and proband is considered “informative” if it is diagnostic for uniparental or biparental inheritance (Table 1). Some genotype configurations supporting UPD are definitive for isodisomy (uniparental–isodisomic, UI), while others could reflect isodisomy or heterodisomy (uniparental–ambiguous, UA). These configurations can be further classified by maternal or paternal inheritance, reflecting a total of four uniparentally inherited signatures: UI_M, UI_P, UA_M, UA_P. Genotype configurations may also be supportive only of eudisomy, i.e., normal biparental inheritance (BPI).
The number of informative genotypes arising from maternal or paternal origin were counted for each chromosome and assessed for statistical significance. A binomial test was used to compare the proportion of genotypes supporting each of the four types of UPD on each chromosome to the genome-wide average proportion for that UPD type. Those chromosomes harboring an enrichment of UPD-type proportions are “called” as UPD if they were statistically unlikely. As a threshold of statistical significance we adjusted an initial 0.05 alpha using a Bonferroni correction to account for 88 tests (four different types of UPD event possible on each of 22 autosomes), yielding a P-value cutoff of 0.000568, which we demonstrated through simulations (see section “Simulations”) was a sensitive and specific detection calibration.
Deciphering Developmental Disorders sample recruitment
The Deciphering Developmental Disorders (DDD) project is a parent–offspring trio study. Its main aim is to identify the disease-causing variants in 12,000 children with undiagnosed severe developmental conditions. These children are referred to a clinical geneticist at one of 24 regional genetics services in the UK and Ireland where recruitment includes recording of detailed clinical information though the DECIPHER database (Firth et al. 2009) and collection of samples for DNA analyses. Proband DNA and parental DNA are genotyped genome-wide using SNP-chips and/or exome sequencing, and copy-number profiled in the proband using array Comparative Genomic Hybridization (aCGH). A total of 1057 unique trios were analyzed in this study, for which all probands had aCGH CNV data available and the vast majority had genome-wide genotype data available both from SNP chips and exome sequencing.
Exome processing
Exome sequencing genotypes were available for 937 (of 1057; 89% of) trios. Exome capture was performed with the Agilent SureSelect v.3 50-Mb baits and augmented with 5 Mb of custom regulatory sequences. Sequencing was performed with the Illumina HiSeq 2000 platform to greater than 50× mean coverage using paired-end 75-bp read-length sequence reads. Alignment to the genome reference GRCh37, version hs37d, used BWA (Li and Durbin 2009) version 0.5.9. Picard tools version 1.46 (McKenna et al. 2010) was used to mark duplicates, and GATK version 1.1-20 was used to realign indels and recalibrate indel quality scores. Single sample variant calling used SAMtools (Li et al. 2009) and GATK for single nucleotide variants (SNVs), and SAMtools and Dindel (Albers et al. 2011) for insertion–deletions (indels). Quality control filters (genotype quality <30.0, homopolymer runs >5, variant quality by depth <5.0, read depth <4 or >1200, strand bias >10.0) were applied. Only biallelic, autosomal SNVs and indels passing all filters were considered for analysis.
The genotype-calling pipeline we used is based on single-sample calling and creates single-sample VCF files, which do not contain positions that are homozygous for the reference base. To include these homozygous positions (required for deducting inheritance patterns), we made the assumption that common polymorphisms in well-covered exome-targeted regions were homozygous for the reference allele if no alternate allele was genotyped at that position. Accordingly, we explicitly annotated homozygous-reference genotypes to positions in our VCF files if the position was contained within the inner 80% of highly covered (30 median average sequence read depth) exome-targeted regions and the minor allele frequency (The 1000 Genomes Project Consortium 2010) of the variant was between 0.05 and 0.95. We ensured that this procedure maintained high genotyping calling accuracy by demonstrating high genotype dosage correlation (r = 0.9958, Pearson correlation, two-sided P-value < 2.2 × 10−16) among 1,369,049 QC-passed sites from 50 samples genotyped by SNP and exome platforms. Among the 937 trios analyzed by exome, the per-trio average of genotype positions in which all members of the trio were jointly genotyped was 54,394 positions, of which 3619, on average, were informative. Thus, the average density of informative exome sites per megabase was 1.2 (3619 * 1 × 106/3 × 109). We measured the noise floor of genotyping errors and calculated the median number of the four categories of uniparental informative event types, which was consistently one per chromosome. During UPD detection from SNP data, we detected a proband with a UPD event for which no exome data had been generated; exome analysis was performed for this trio post hoc to enable confirmatory validation of this event from exome data.
SNP processing
Genome-wide SNP array genotypes were available for 1041 trios. The SNP typing platform used was a custom genotyping chip, using a backbone of 733,059 HumanOmniExpress-12v1_A-b37 positions and the addition of 94,840 selected positions. Genotyping was performed using Illuminus (Teo et al. 2007), recorded in PLINK format, and converted to VCF format using plinkseq version 0.08. A set of 695,829 autosomal SNPs was used in this analysis. Samples were rejected on the basis of a high proportion of missing genotypes, but not due to outlying levels of heterozygosity rate, to prevent exclusion of samples that may contain UPD chromosomes. Among the 1041 trios available, 1035 SNP trios passed sample QC and were analyzed in this study. After UPD detection was performed in exome data, it was determined that one of these QC-failed samples in the SNP data was the father of a proband with a UPD event; this trio was processed post hoc to enable confirmatory validation of the UPD event in the SNP data. An average of 439,205 variant sites were genotyped per trio, of which 42,490 on average, were informative. Thus, the average density of informative SNP genotypes across one megabase was 14.2 (42,490 * 1 × 106/3 × 109). The median number of the four categories of uniparental informative event types was consistently zero per chromosome.
Avoiding positions in copy number variant regions
The diploid human genome can vary locally in copy number, through deletions and duplications of chromosomal segments. The majority of genotype callers, including those used in this study, are ignorant to changes in copy number, i.e., they assume diploidy, and interpret hemizygosity as diploid homozygosity. For the purpose of detecting uniparental disomy, this can be problematic because single-copy loci, which reflect (uniparental) monosomy by definition, could be spuriously identified as uniparental disomy. Therefore, we incorporated into the software tool a copy number filter that avoids genotyped sites present in or near (within 10 kb) deletions common in the population or present in the sample (using user-specified CNV data encoded in VCF or tab-separated-value format).
The list of common deletions was acquired by selecting copy number variable regions of greater than one percent frequency from a composite of multiple studies (Barnes et al. 2008; Conrad et al. 2010). Sample-specific CNV data were generated using a custom, exome-focused, 2,000,000 probe Agilent aCGH array. CNVs were called using CNsolidate, an in-house algorithm that integrates 12 change-point detection algorithms (T Fitzpatrick, P Vijayarangakannan, N Carter, M Hurles, in prep.). CNV data were available for all 1057 probands studied.
Simulation testing
We generated a variety of data sets to evaluate the detection accuracy of UPDio. In addition, we compared the detection accuracy of UPDio with two other trio-based UPD detection methods.
To prepare sensitivity evaluations we simulated the presence of a UPD event by introducing genotypes consistent with uniparental maternal inheritance into a proband VCF. Then, we implemented the three methods using each tool’s default parameters to detect maternal UPD events in a trio consisting of the original parents and the modified proband. For simulating heterodisomy, proband genotypes were substituted for both alleles of maternal genotypes in the selected regions. For simulating isodisomy, proband genotypes were substituted for homozygosity of one of the maternal alleles, chosen at random. We simulated complete UPD as well as segmental UPD at various sizes: 1, 2, 5, 10, and 20 Mb. Simulated regions of the required length were randomly placed across autosomes and selected unless the region overhung the edge of the chromosome or >25% of its length overlapped known UCSC-defined “gap” regions. For each permutation of UPD size, class, and platform, we simulated 100 trio data sets. Sensitivity was defined as the proportion of these trios with detection of the simulated maternal event by the algorithm.
For assessing specificity, we selected empirical genotype SNP and exome data from trios in which the probands had no obvious UPD events at Bonferroni-corrected P-values, nor contained any large (longer than 10 Mb) regions of homozygosity. By doing so, we reasoned that only genotyping errors and rare undetected CNVs would lead to false UPD detections. Specificity was then defined as the proportion of trios lacking any maternal UPD.
We used the procedure described above to calculate UPDio sensitivity and specificity at various P-value stringencies to construct receiver operator characteristic (ROC; true positive vs. 1—false positive rate) curves. In addition, we calculated the sensitivity and specificity of all three methods using default parameters. For UPDio, we used a Bonferroni-corrected P-value threshold. For UPDtool, we used the following defaults settings: min_mes (300), window size (10 kb), min_mes_fraction (1%), min_hetero (90%), min_iso (85%), min_mes_paternal (80%), and max_mes_paternal (20%). Although SNPtrio is supported as a webtool, the investigators kindly provided us with the source code, which we adapted to run locally. The webtool outputs and plots all events, regardless of P-value significance, and we likewise did not impose a threshold when running this tool.
Identifying candidate variants
High-quality exome data were available for five of six probands with detected UPD events and these samples were analyzed for several sources of candidate variants: inherited single nucleotide substitutions and indels; inherited compound heterozygotes; de novo substitutions and indels; and copy number variants. Homozygous substitions and indels and heterozygous de novo variants were analyzed if they were rare (below 0.5% frequency in 1000 Genomes [The 1000 Genomes Project Consortium 2010] and below 1.0% internal DDD-frequency), and in a functional or loss-of functional Variant Effect Predictor (VEP) version 2.6 category (splice donor variant, splice acceptor variant, stop gained, frameshift variant, stop lost, initiator codon variant, inframe insertion, inframe deletion, missense variant). The subset of CNVs that were on the UPD chromosome, were homozygous deletions, at least 5 kb, and overlapped at least one gene, were selected for scrutinized investigation. De novo variants were detected by DeNovoGear (Ramu et al. 2013), subjected to stringent algorithmic filtering and experimental validation. We used an in-house algorithmic approach to leverage inheritance information from parents to improve curation of proband genotypes and to detect compound heterozygous variants. Since our pipeline uses inheritance information to stringently curate variant detection, regions inherited from a single parent produce genotypes that fail these filters; therefore, variant detection on the UPD chromosome required tailored retention. A panel of pathogenicity scores was obtained for filtered variants, including Genomic Evolutionary Rate Profiling (GERP) (Cooper et al. 2005), Polymorphism Phenotyping (PolyPhen) (Ramensky et al. 2002), Sorting Intolerant From Tolerant (SIFT) (Ng and Henikoff 2003) scores, and Haploinsufficiency Score (Huang et al. 2010). All variants in the above categories were cataloged (see Supplemental Table 1). Additionally, variants were prioritized if they resided in genes associated with Mendelian disease; these genes were identified as being included in an internal database of congenital disease genes, “Developmental Disease Genes 2 Phenotype” (DDG2P), or present as “DM” variants (not “DM?” variants) in the Human Genome Mutation Database (HGMD) (Stenson et al. 2014). Variants were considered candidate variants if mutations in these genes were known to result in phenotypes consistent with the patient’s phenotype profile.
Prevalence data
The Wellcome Trust Case Control Consortium (WTCCC1; Acknowledgments) is a group of research centers in the UK that studies the genetic basis for common diseases. The WTCCC1 was a study composed of 14,000 individuals having one of seven diseases, and an additional 3000 individuals in control groups. The samples were genotyped using the Affymetrix 500k chip. We utilized a “missing genotype” quality-control metric by removing samples with >10% missing genotypes. Since isodisomy is expected to affect the average rate of genomic heterozygosity, we did not filter samples on abnormal rates of heterozygosity. A total of 16,881 individuals were included for analysis. We used PLINK (v1.07) (Purcell et al. 2007) to calculate runs of homozygosity that contained at least 50 homozygous positions and spanned at least 500 kb in size. We subsequently used custom Perl scripts to select samples with large (larger that 10 Mb) stretches of homozygosity and identify those samples containing large regions of homozygosity affecting only one chromosome.
Computational performance
The UPD calling method uses iterators to scan VCFs line by line, resulting in a low memory footprint (30 Mb of RAM per trio) regardless of genotyping density. The calling speed is reasonably quick (3 min for a SNP trio), and scales linearly with number of probes. Each trio can be run independently; therefore, the number of trios that can be analyzed simultaneously is only limited by the capacity of the data center used to drive the tool. The UPD code was written mainly in Perl v5.10.0. All required Perl modules are available on CPAN. A plotting tool is included that allows the visual display of aberrant genotypes and zygosity of the proband. Plotting requires the module ‘quantsmooth’ (Eilers and de Menezes 2005) available on CRAN.
Software availability
Software for UPD detection in trios, UPDio, is freely available at https://github.com/findingdan/UPDio. Instructions and preprocessing scripts are included to enable users to prepare VCF input files from custom exome capture designs.
Acknowledgments
We thank the DDD patients and their families for participating in this study. Many DDD project members provided constructive research ideas. Helen Firth, David Fitzpatrick, Karen Temple, and Wendy Jones provided valuable advice on clinical phenotypes. Jeff Barrett provided critical, constructive feedback. Saeed Al Turki and Parthiban Vijayarangakannan assisted in the analytical framework. Petr Danchek helped with SNP allelic codings, and Larry Singh helped with statistics development. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund, award HICF-1009-003. This study makes use of data generated by the UK10K Consortium, www.UK10K.org, Wellcome Trust award WT098051. This study also makes use of data generated by the Wellcome Trust Case Control Consortium, www.wtccc.org.uk, Wellcome Trust award 076113. D.A.K. is supported by a Wellcome Trust Sanger Institute Research Studentship.
Author contributions: D.A.K. and M.E.H. designed the method and wrote the manuscript. T.W.F. prepared the aCGH CNV data. R.M. provided coding insight and oversight. N.C., J.C.-S., D.J., S.M., F.S., and P.V. recruited and provided phenotypes for the six patients with UPD events.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.160465.113.
References
- The 1000 Genomes Project Consortium 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers CA, Lunter G, MacArthur DG, McVean G, Ouwehand WH, Durbin R 2011. Dindel: Accurate indel calls from short-read data. Genome Res 21: 961–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astle W, Balding DJ 2009. Population structure and cryptic relatedness in genetic association studies. Stat Sci 24: 451–471 [Google Scholar]
- Augustin I, Korte S, Rickmann M, Kretzschmar HA, Sudhof TC, Herms JW, Brose N 2001. The cerebellum-specific Munc13 isoform Munc13-3 regulates cerebellar synaptic transmission and motor learning in mice. J Neurosci 21: 10–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balciuniene J, Dahl N, Jalonen P, Verhoeven K, Van Camp G, Borg E, Pettersson U, Jazin EE 1999. α-tectorin involvement in hearing disabilities: One gene–two phenotypes. Hum Genet 105: 211–216 [DOI] [PubMed] [Google Scholar]
- Barnes C, Plagnol V, Fitzgerald T, Redon R, Marchini J, Clayton D, Hurles ME 2008. A robust statistical method for case-control association testing with copy number variation. Nat Genet 40: 1245–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno DL, White SM, Ganesamoorthy D, Burgess T, Butler K, Corrie S, Francis D, Hills L, Prabhakara K, Ngo C, et al. 2011. Pathogenic aberrations revealed exclusively by single nucleotide polymorphism (SNP) genotyping data in 5000 samples tested by molecular karyotyping. J Med Genet 48: 831–839 [DOI] [PubMed] [Google Scholar]
- Carmichael H, Shen Y, Nguyen T, Hirschhorn J, Dauber A 2012. Whole exome sequencing in a patient with uniparental disomy of chromosome 2 and a complex phenotype. Clin Genet 84: 213–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlin LK, Thiel BD, Bonnemann CG, Medne L, Ernst LM, Zackai EH, Deardorff MA, Krantz ID, Hakonarson H, Spinner NB 2010. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum Mol Genet 19: 1263–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, et al. 2010. Origins and functional impact of copy number variation in the human genome. Nature 464: 704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A 2005. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 15: 901–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. 2011. The variant call format and VCFtools. Bioinformatics 27: 2156–2158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers PH, de Menezes RX 2005. Quantile smoothing of array CGH data. Bioinformatics 21: 1146–1153 [DOI] [PubMed] [Google Scholar]
- Engel E 1980. A new genetic concept: Uniparental disomy and its potential effect, isodisomy. Am J Med Genet 6: 137–143 [DOI] [PubMed] [Google Scholar]
- Evangelou E, Trikalinos TA, Salanti G, Ioannidis JP 2006. Family-based versus unrelated case-control designs for genetic associations. PLoS Genet 2: e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth HV, Wright CF 2011. The Deciphering Developmental Disorders (DDD) study. Dev Med Child Neurol 53: 702–703 [DOI] [PubMed] [Google Scholar]
- Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM, Carter NP 2009. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources. Am J Hum Genet 84: 524–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, Moran JL, Chambert K, Banks E, Bergen SE, Ruderfer DM, Handsaker RE, McCarroll SA, O’Donovan MC, Owen MJ, et al. 2012. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am J Hum Genet 91: 597–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N, Lee I, Marcotte EM, Hurles ME 2010. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet 6: e1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isidor B, Pichon O, Baron S, David A, Le Caignec C 2010. Deletion of the CUL4B gene in a boy with mental retardation, minor facial anomalies, short stature, hypogonadism, and ataxia. Am J Med Genet A 152A: 175–180 [DOI] [PubMed] [Google Scholar]
- Jacobs KB, Yeager M, Zhou W, Wacholder S, Wang Z, Rodriguez-Santiago B, Hutchinson A, Deng X, Liu C, Horner MJ, et al. 2012. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet 44: 651–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney JA, Plummer NW, Smith MR, Kapur J, Cummins TR, Waxman SG, Goldin AL, Meisler MH 2001. A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience 102: 307–317 [DOI] [PubMed] [Google Scholar]
- Koch MC, Ricker K, Otto M, Wolf F, Zoll B, Lorenz C, Steinmeyer K, Jentsch TJ 1993. Evidence for genetic homogeneity in autosomal recessive generalised myotonia (Becker). J Med Genet 30: 914–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotzot D 2008. Complex and segmental uniparental disomy updated. J Med Genet 45: 545–556 [DOI] [PubMed] [Google Scholar]
- Lenders M, Duning T, Schelleckes M, Schmitz B, Stander S, Rolfs A, Brand SM, Brand E 2013. Multifocal white matter lesions associated with the D313Y mutation of the α-galactosidase A gene. PLoS ONE 8: e55565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LH, Ho SF, Chen CH, Wei CY, Wong WC, Li LY, Hung SI, Chung WH, Pan WH, Lee MT, et al. 2006. Long contiguous stretches of homozygosity in the human genome. Hum Mutat 27: 1115–1121 [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Lupat R, Amarasinghe KC, Thompson ER, Doyle MA, Ryland GL, Tothill RW, Halgamuge SK, Campbell IG, Gorringe KL 2012. CONTRA: Copy number analysis for targeted resequencing. Bioinformatics 28: 1307–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Mysickova A, Sun R, Kalscheuer V, Vingron M, Haas SA 2011. Modeling read counts for CNV detection in exome sequencing data. Stat Appl Genet Mol Biol 10: 1–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER 2008. The impact of next-generation sequencing technology on genetics. Trends Genet 24: 133–141 [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, et al. 2011. Mapping copy number variation by population-scale genome sequencing. Nature 470: 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S 2003. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31: 3812–3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann M, Rolfs A, Giese A, Mascher H, Breunig F, Ertl G, Wanner C, Weidemann F 2013. Lyso-Gb3 indicates that the alpha-galactosidase A mutation D313Y is not clinically relevant for Fabry disease. JIMD Rep 7: 99–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papenhausen P, Schwartz S, Risheg H, Keitges E, Gadi I, Burnside RD, Jaswaney V, Pappas J, Pasion R, Friedman K, et al. 2011. UPD detection using homozygosity profiling with a SNP genotyping microarray. Am J Med Genet A 155A: 757–768 [DOI] [PubMed] [Google Scholar]
- Pique-Regi R, Caceres A, Gonzalez JR 2010. R-Gada: A fast and flexible pipeline for copy number analysis in association studies. BMC Bioinformatics 11: 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. 2007. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM 2010. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S 2002. Human non-synonymous SNPs: Server and survey. Nucleic Acids Res 30: 3894–3900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramu A, Noordam MJ, Schwartz RS, Wuster A, Hurles ME, Cartwright RA, Conrad DF 2013. DeNovoGear: de novo indel and point mutation discovery and phasing. Nat Methods 10: 985–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson WP 2000. Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays 22: 452–459 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Santiago B, Malats N, Rothman N, Armengol L, Garcia-Closas M, Kogevinas M, Villa O, Hutchinson A, Earl J, Marenne G, et al. 2010. Mosaic uniparental disomies and aneuploidies as large structural variants of the human genome. Am J Hum Genet 87: 129–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagong B, Park HJ, Lee KY, Kim UK 2012. Identification and functional characterization of novel compound heterozygotic mutations in the TECTA gene. Gene 492: 239–243 [DOI] [PubMed] [Google Scholar]
- Schroeder C, Sturm M, Dufke A, Mau-Holzmann U, Eggermann T, Poths S, Riess O, Bonin M 2013. UPDtool: A tool for detection of iso- and heterodisomy in parent-child trios using SNP microarrays. Bioinformatics 29: 1562–1564 [DOI] [PubMed] [Google Scholar]
- Singh NA, Pappas C, Dahle EJ, Claes LR, Pruess TH, De Jonghe P, Thompson J, Dixon M, Gurnett C, Peiffer A, et al. 2009. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet 5: e1000649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence JE, Perciaccante RG, Greig GM, Willard HF, Ledbetter DH, Hejtmancik JF, Pollack MS, O’Brien WE, Beaudet AL 1988. Uniparental disomy as a mechanism for human genetic disease. Am J Hum Genet 42: 217–226 [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Shaw K, Phillips AD, Cooper DN 2014. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet 133: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temple IK, Cockwell A, Hassold T, Pettay D, Jacobs P 1991. Maternal uniparental disomy for chromosome-14. J Med Genet 28: 511–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo YY, Inouye M, Small KS, Gwilliam R, Deloukas P, Kwiatkowski DP, Clark TG 2007. A genotype calling algorithm for the Illumina BeadArray platform. Bioinformatics 23: 2741–2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JC, Roberson ED, Miller ND, Lysholm-Bernacchi A, Stephan DA, Capone GT, Ruczinski I, Thomas GH, Pevsner J 2007. Visualization of uniparental inheritance, Mendelian inconsistencies, deletions, and parent of origin effects in single nucleotide polymorphism trio data with SNPtrio. Hum Mutat 28: 1225–1235 [DOI] [PubMed] [Google Scholar]
- Trip J, Drost G, Verbove DJ, van der Kooi AJ, Kuks JB, Notermans NC, Verschuuren JJ, de Visser M, van Engelen BG, Faber CG, et al. 2008. In tandem analysis of CLCN1 and SCN4A greatly enhances mutation detection in families with non-dystrophic myotonia. Eur J Hum Genet 16: 921–929 [DOI] [PubMed] [Google Scholar]
- Uldall P, Alving J, Hansen LK, Kibaek M, Buchholt J 2006. The misdiagnosis of epilepsy in children admitted to a tertiary epilepsy centre with paroxysmal events. Arch Dis Child 91: 219–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Loo P, Nordgard SH, Lingjaerde OC, Russnes HG, Rye IH, Sun W, Weigman VJ, Marynen P, Zetterberg A, Naume B, et al. 2010. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci 107: 16910–16915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson J, Whitehouse W, Zuberi S 2004. Paroxysmal nonepileptic disorders: Differential diagnosis of epilepsy. In Epilepsy in Children, 2nd ed. (ed. Wallace SJ, Farrell K), pp. 4–20. CRC Press, Boca Raton, FL [Google Scholar]
- Wiszniewska J, Bi W, Shaw C, Stankiewicz P, Kang SH, Pursley AN, Lalani S, Hixson P, Gambin T, Tsai CH, et al. 2013. Combined array CGH plus SNP genome analyses in a single assay for optimized clinical testing. Eur J Hum Genet 22: 79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff M, Casse-Perrot C, Dravet C 2006. Severe myoclonic epilepsy of infants (Dravet syndrome): Natural history and neuropsychological findings. Epilepsia (Suppl 2) 47: 45–48 [DOI] [PubMed] [Google Scholar]
- Yamatogi Y, Ohtahara S 2002. Early-infantile epileptic encephalopathy with suppression-bursts, Ohtahara syndrome; its overview referring to our 16 cases. Brain Dev 24: 13–23 [DOI] [PubMed] [Google Scholar]
- Yamazawa K, Ogata T, Ferguson-Smith AC 2010. Uniparental disomy and human disease: An overview. Am J Med Genet C Semin Med Genet 154C: 329–334 [DOI] [PubMed] [Google Scholar]
- Zlotogora J 2004. Parents of children with autosomal recessive diseases are not always carriers of the respective mutant alleles. Hum Genet 114: 521–526 [DOI] [PubMed] [Google Scholar]