

Abstract
Obtaining high-quality sequence continuity of complex regions of recent segmental duplication remains one of the major challenges of finishing genome assemblies. In the human and mouse genomes, this was achieved by targeting large-insert clones using costly and laborious capillary-based sequencing approaches. Sanger shotgun sequencing of clone inserts, however, has now been largely abandoned, leaving most of these regions unresolved in newer genome assemblies generated primarily by next-generation sequencing hybrid approaches. Here we show that it is possible to resolve regions that are complex in a genome-wide context but simple in isolation for a fraction of the time and cost of traditional methods using long-read single molecule, real-time (SMRT) sequencing and assembly technology from Pacific Biosciences (PacBio). We sequenced and assembled BAC clones corresponding to a 1.3-Mbp complex region of chromosome 17q21.31, demonstrating 99.994% identity to Sanger assemblies of the same clones. We targeted 44 differences using Illumina sequencing and find that PacBio and Sanger assemblies share a comparable number of validated variants, albeit with different sequence context biases. Finally, we targeted a poorly assembled 766-kbp duplicated region of the chimpanzee genome and resolved the structure and organization for a fraction of the cost and time of traditional finishing approaches. Our data suggest a straightforward path for upgrading genomes to a higher quality finished state.
Complete high-quality sequence assembly remains a difficult problem for the de novo assembly of genomes (Alkan et al. 2011b; Church et al. 2011; Salzberg et al. 2012). Finishing of the human and mouse genomes involved selecting large-insert BAC clones and subjecting them to capillary-based shotgun sequence and assembly (English et al. 2012). Sanger-based assembly of large-insert clones has been typically a time-consuming and expensive operation requiring the infrastructure of large genome sequencing centers and specialists focused on particular problematic or repetitive regions (Zody et al. 2008; Dennis et al. 2012; Hughes et al. 2012). Such activities can significantly improve the quality of genomes, including the discovery of missing genes and gene families. A recent effort to upgrade the mouse genome assembly, for example, resulted in the correction or addition of 2185 genes, 61% of which corresponded to lineage-specific segmental duplications (Church et al. 2009). Within the human genome, there are >900 annotated genes mapping to large segmental duplications. About half of these map to particularly problematic regions of the genome where annotation and genetic variation are poorly understood (Sudmant et al. 2010). Such genes are typically missing or misassembled in working draft assemblies of genomes. These include genes such as the SRGAP2 family, which evolved specifically in the human lineage and is thought to be important in the development of the human brain (Charrier et al. 2012; Dennis et al. 2012). Other regions (e.g., 17q21.31 inversion) show incredible structural diversity, predispose specific populations to disease, and have been the target of remarkable selection in the human lineage (Stefansson et al. 2005; Zody et al. 2008; Steinberg et al. 2012). Such structurally complex regions were not resolved within the human reference sequence until large-insert clones were recovered and completely sequenced.
The widespread adoption of next-generation sequencing methods for de novo genome assemblies has complicated the assembly of repetitive sequences and their organization. Although we can generate much more sequence, the short sequence read data and inability to scaffold across repetitive structures translates into more gaps, missing data, and more incomplete reference assemblies (Alkan et al. 2011a; Salzberg et al. 2012). Due to budgetary constraints, traditional capillary-based sequencing capacity as well as genome finishing efforts have dwindled in sequencing centers leaving most of the complex regions of working draft genomes unresolved. Clone-based hierarchical approaches remain important for reducing the complexity of genomes, but even targeted sequencing of these clones using short-read data fails to completely resolve and assemble these regions due to the presence of highly identical repeat sequences common in mammalian genomes. Here, we tested the efficacy of a method developed for finishing microbial genomes (Chin et al. 2013) to a 1.3-Mbp complex region of human chromosome 17q21.31 previously sequenced and assembled using traditional Sanger-based approaches. We directly compared sequenced and assembled clones and validated differences to highlight advantages and limitations of the different technologies. We then applied the approach to a previously uncharacterized, highly duplicated region of the chimpanzee genome and show that we can rapidly resolve the structure and organization of the region using this approach.
Results
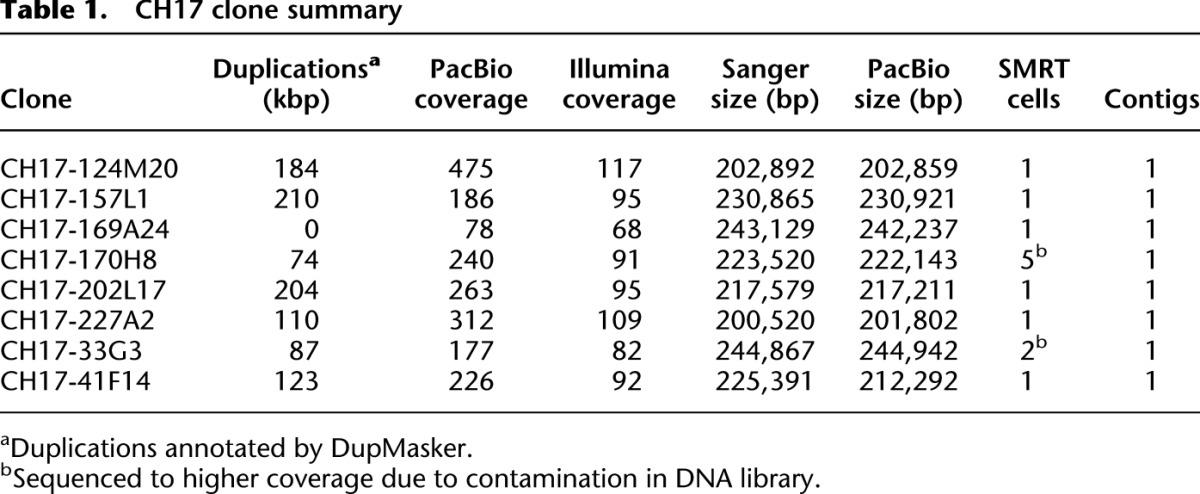
For the purpose of this study, we initially selected eight BAC clones from a hydatidiform mole sample corresponding to a complex 1.3-Mbp region of 17q21.31 (Fig. 1). The region was chosen because of its biomedical relevance and the difficulty it posed in the initial sequence and assembly of the human genome. Of the corresponding clone sequence, 55% consist of high-identity segmental duplications and the region is a site of large-scale structural polymorphisms that predisposes European and Mediterranean populations to recurrent microdeletions associated with the Koolen-DeVries syndrome (Zody et al. 2008; Steinberg et al. 2012). Although the targeted region is complex, we note that it does not contain any sequences that are recalcitrant to existing sequencing technologies. Its complexity lies in the presence of layers of common and low-copy repeat sequences, which complicates assembly at the whole-genome level and has typically required targeted clone-based approaches to resolve. We constructed 10-kbp insert sequence libraries and assembled PacBio sequence into consensus sequence contigs using the HGAP long-read assembler. Quiver was used to generate a final consensus with quality scores through the standard SMRT Analysis (v. 2.0.1) pipeline (Chin et al. 2013). In this study, the average subread length across all clones was 1.8 kbp (maximum length of 12.4 kbp) and sequence coverage ranged from 78- to 475-fold (average of 245-fold per clone). After vector trimming, we generated a single, linear contig sequence for each of the eight clones representing a total of 1,774,407 bp of “finished” sequence. We note that each of the eight clones assembled into a single contig (Table 1; Supplemental Table S1), with six clones assembled from a single SMRT Cell of data each.
Figure 1.
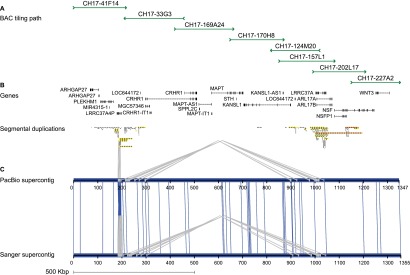
17q21.31 genomic target region. (A) Tiling path of eight large-insert BAC clones sequenced and assembled using both PacBio- and Sanger-based approaches. Clones were selected from a haploid complete hydatidiform mole source (CH17). (B) Gene annotation (RefSeq) and segmental duplication organization were obtained from GRCh37 using a custom liftover coordinate conversion tool that accounted for the difference in copy number between the mole haplotype and the reference. (C) Alignment of supercontigs built from the same eight clones using PacBio and Sanger assemblies. Sequence differences (vertical blue lines) and internal duplications (gray) are shown. The two supercontigs are 99.99% identical, excluding a collapsed higher-order repeat at the end of the PacBio assembly of CH17-41F14.
Table 1.
CH17 clone summary
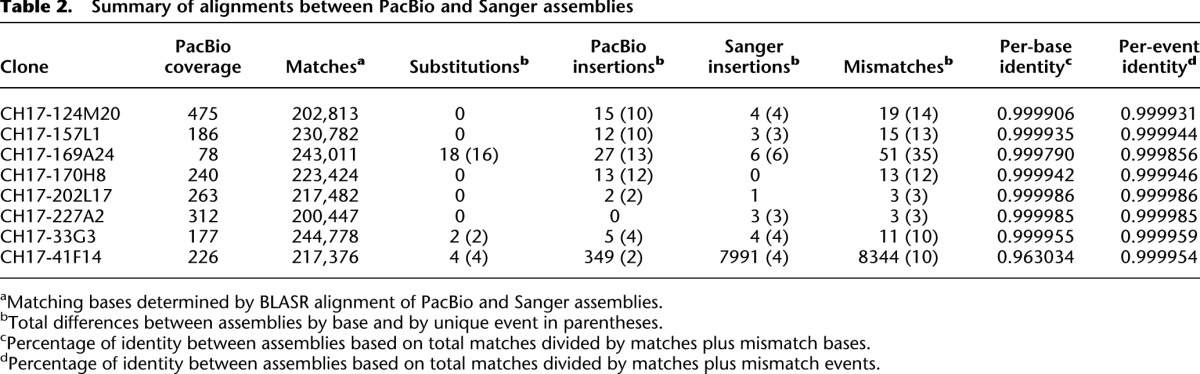
For each sequenced clone, we aligned the Sanger and PacBio HGAP assembled sequence contigs using BLASR (Chaisson and Tesler 2012) and identified all sequence differences <50 bp in length. A total of 125 sequence differences were identified in 1.77 Mbp of aligned sequence resulting in 99.994% sequence identity between the assemblies (Table 2). In this estimate, we count the total number of base pairs encompassing a given insertion/deletion (indel) event as opposed to counting an indel as a single difference. Relatively few sequence differences (24 or 19%) were sequence substitutions. For example, five of the aligned clones showed no sequence substitution difference between the two assemblies. The bulk of sequence differences, instead, corresponded to insertions (81% or 101 aligned base-pair differences). We note marked asymmetry between assemblies with 76 insertions found in the PacBio assemblies and only 25 insertions in the Sanger assemblies (Supplemental Fig. S1). Simple repeats contributed to 48% of the differences with 47 differences occurring within homopolymer runs (Supplemental Fig. S2) and 13 within dinucleotide repeats.
Table 2.
Summary of alignments between PacBio and Sanger assemblies
Overall, the assembled contigs showed remarkable similarity in length (1788 kbp Sanger vs. 1774 kbp PacBio). The difference in length was due primarily to one clone where there was evidence of larger structural differences between the assemblies (Fig. 2). The assembly of CH17-41F14 contained a 12-kbp complex higher-order repeat structure, which was expanded to 20 kbp in the Sanger assembly (Supplemental Fig. S3). Visualization of the corresponding read depth (Fig. 2) confirmed a PacBio misassembly—i.e., a symmetric increase of reads in the collapsed region of CH17-41F14 (Fig. 2). In addition to this PacBio misassembly, we discovered a 357-bp deletion in the Sanger assembly of CH17-41F14 (Supplemental Table S2), which was subsequently confirmed as bona fide in the PacBio assembly based on alignment of Illumina reads from the same clone to both assemblies (Supplemental Table S3). Interestingly, HGAP correctly assembled the clone CH17-227A2, which had been previously misassembled by an earlier long-read assembly algorithm, Allora, during our preliminary analysis (Supplemental Fig. S4).
Figure 2.
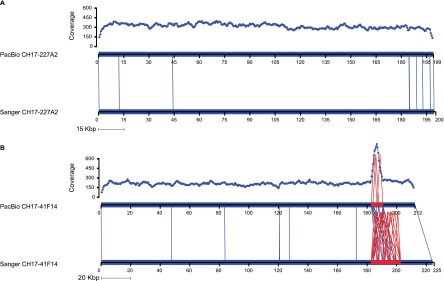
Concordant and discordant PacBio assemblies. (A) Alignment between PacBio (top) and Sanger (bottom) assemblies for CH17-227A2 using Miropeats (Parsons 1995) shows virtually no differences. Note the uniform sequence coverage between 200- and 300-fold. Mismatches/indels are indicated by vertical blue lines. (B) Alignment between PacBio and Sanger assemblies for clone CH17-41F14. A spike of increased sequence coverage across the internal repeat and the reduced complexity of the repeat compared with the Sanger assembly clearly define a collapse of a higher-order repeat from 20 to 12 kbp within the PacBio assembly. The uniformity of sequence coverage may be used as one indicator of potential misassembly.
To determine if the smaller sequence differences were errors in the PacBio or the Sanger assembly, we sequenced the same eight clones to high coverage (average 94-fold) using a Nextera Illumina sequencing pipeline (Adey et al. 2010). Short-read sequencing data were insufficient to assemble the complete insert, even in the case of clone CH17-170H8 where the longest exact repeat, 76 bases, is shorter than the read length (Supplemental Table S4). Local alignment of the Illumina assemblies allowed us to unambiguously validate 44 differences between the assemblies (Table 3; Supplemental Fig. S5). Illumina sequencing supported 31 PacBio and 13 Sanger differences. The majority of variants supported in PacBio assemblies (97%) clustered within complex repetitive regions. For example, a 372-bp region in the Sanger assembly of CH17-169A24 accounted for 24 unambiguous differences, suggesting that this region had been misassembled in the Sanger assembly (Supplemental Fig. S6). The remaining validated PacBio difference corresponded to a homopolymer repeat. Similarly, the validated differences within the Sanger assemblies shared common features. Five (38%) of the validated Sanger variants occurred within simple repeat sequences. For the four validated Sanger variants within homopolymers, one of the alternate PacBio variants added an extra base, one added two bases, and one removed a single base. The remaining eight variants validated within the Sanger assemblies were evenly split between complex indels and mismatches where the PacBio assembly had potentially misassembled segments with no coverage between regions of normal coverage. We manually inspected the capillary traces for five of the eight clones at 23 total mismatch positions between Sanger and PacBio assemblies. Of these mismatches, 20 previously ambiguous mismatches were validated for the Sanger assemblies and three mismatches previously validated for PacBio were also supported by the capillary traces.
Table 3.
Total mismatches between assemblies validated by Illumina reads
Since the average sequence coverage per clone was relatively high (245-fold), we performed two experiments to estimate the minimum coverage required to properly assemble clones into a single contig. In the first experiment, we randomly subsampled (100-fold coverage) and assembled ∼20 iterations per clone. We measured the success of these assemblies by their identity to the corresponding Sanger assembly, median number of assembled contigs, and total bases assembled per clone. The mean identity of assemblies ranged from 99.98% to 99.99%. Four of five clones had a median of one contig, while one clone, CH17-124M20, had a median contig count of two (Supplemental Fig. S11; Supplemental Table S5). The identity between single-contig subsampled assemblies and their Sanger counterparts was 0.01% lower than the original HGAP assemblies with all reads (Table 2; Supplemental Table S5). Interestingly, one assembly of the clone CH17-41F14 matched the length of the Sanger assembly by adding ∼8 kbp in the complex repetitive region that had collapsed in the original assembly (Supplemental Fig. S12). However, this subsampled assembly had lower overall identity with the Sanger sequence at 99.95% compared with the original assembly’s 99.99%. These results suggest that the previously recommended coverage of 100-fold for high-quality libraries is a minimum requirement for high-accuracy BAC assembly (Chin et al. 2013).
In the second experiment, we empirically assessed the efficacy of pooling BAC clones in individual SMRT Cells to determine if distinct assemblies of high quality could be produced. All pooled clones had been previously sequenced and assembled from single SMRT Cells with three out of four clones assembling into a single contig (Supplemental Table S6). We tested a pool of two clones (Pool #1: CH251-75B17 and CH277-30K2) and a second pool of three clones (Pool #2: CH251-75B17, CH251-182P19, and CH277-80C4). In each case, clones were isolated independently and DNA normalized prior to library construction. Four of the five pooled clones assembled into single contigs. The clone CH277-30K2, which had previously assembled into one contig, assembled into two from the pooled data. Interestingly, the clone CH277-80C4 assembled into one contig from the pool with an additional 13 kbp of sequence compared with its single SMRT Cell assembly of five contigs. The single and pooled assemblies for the remaining clones were structurally concordant and ranged in alignment identity between 99.85% and 99.99%.
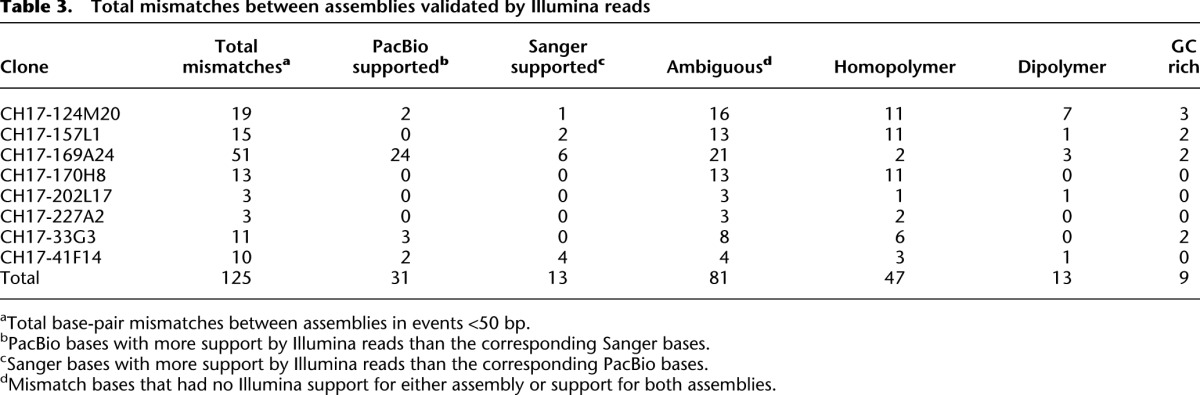
To demonstrate the utility of this approach for upgrading working draft assemblies, we identified five clones (CH251) corresponding to two complex segmental duplications within an orthologous region of the Smith-Magenis syndrome (SMS) in the chimpanzee (see Methods). We specifically selected this region because our previous analysis had shown it to be a site of complex lineage-specific duplications that had not been properly assembled in the chimpanzee genome (Sudmant et al. 2013). Moreover, the first chimpanzee analog of a genomic disorder had been identified within this region. The chimpanzee showed an SMS-like phenotype although the breakpoints of this rearrangement could not be reliably identified due to misassembly of the segmental duplications (Sudmant et al. 2013). Each chimpanzee BAC clone was sequenced and assembled as described above and each clone assembled into a single insert of the expected length. Two supercontigs were generated corresponding to 504 kbp of segmental duplication. This expanded to 766 kbp when including one orphan capillary clone sequence that had not yet been incorporated in the chimpanzee assembly (Supplemental Table S7). A comparison to the current chimpanzee genome assembly (panTro4) showed that 241 kbp of sequence was completely absent from the chimpanzee whole-genome assembly (Fig. 3). The remaining 525 kbp which showed homology with sequence in panTro4 was distributed to six contigs, most of which were not localized (i.e., assigned to the random bin on unmapped chromosome). Only one ∼44-kbp region was assigned correctly to a map location on chromosome 17. Alignment of supercontigs with the genome assembly revealed hundreds of small and large inconsistencies with an overall sequence identity of 94.69%.
Figure 3.
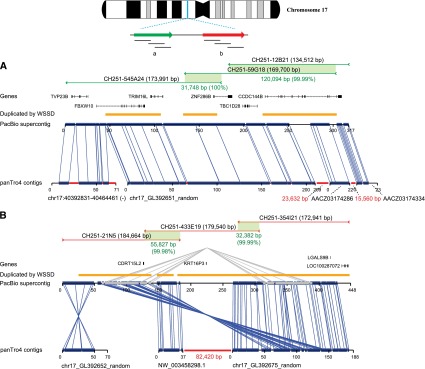
Upgrading a chimpanzee genomic region. Sequence and assembly of six large-insert clones (CH251) from two segmental duplication blocks (red and green) are aligned to their corresponding sequences from the 17p11.2 Smith-Magenis region of the chimpanzee reference assembly (panTro4). Clones were sequenced and assembled from the (A) distal and (B) proximal segmental duplication blocks. The PacBio assembly was compared with the corresponding working draft sequences from panTro4. The alignment identity of panTro4 contigs without gap sequence and the PacBio supercontigs is 94.69% over 525 kbp of aligned sequence. Thirty-one percent (241/766 kbp) of the chimpanzee sequence is missing within the working draft assembly. The average sequence identity for phred >30 bp from BAC end sequence (BES) mappings was 99.72% (16,174/16,220 high-quality bases) and 99.98% (156,955/156,991 high-quality bases) from fosmid end sequence (FES) mappings. Gaps in the panTro4 contigs are indicated in red. Gene annotations are shown based on a custom liftover from RefSeq annotations of GRCh37 in the corresponding regions of 17p11.2. The missing sequence corresponds to high-identity segmental duplications (orange bars represent segmental duplications predicted by whole-genome shotgun sequence detection or WSSD). The clone CH251-545A24 was previously sequenced with capillary sequencing (GenBank accession: AC183294).
To assess the accuracy of this new assembly, we mapped both chimpanzee BAC end and fosmid end sequences (BES and FES) to the assembled contigs (Fig. 4; Supplemental Fig. S7) restricting alignments to high-quality base pairs from the capillary traces (phred quality score >30). The mean identity of all BES alignments was 99.72% (16,174/16,220 high-quality bases). Twelve clones mapped concordantly (mean identity of 99.99%), five mapped discordantly with both ends (mean identity of 99.32%), and six mapped with one end only (mean identity of 99.03%). Alignment of concordant and discordant chimpanzee FES to CH251 supercontigs showed a mean alignment of 99.98% (156,955/156,991 high-quality bases). A total of 138 clones mapped concordantly (mean identity of 99.98%) while 17 mapped discordantly in pairs (mean identity of 99.98%) and 20 mapped with only one end (mean identity of 99.99%). We note that the assembled contigs are largely composed of high-identity duplications and many of the lower-identity discordant read pairs likely originate from paralogs or alternate structural haplotypes within the chimpanzee. Importantly, analysis of the fosmid insert size distribution based on mapped FES to the chimpanzee supercontig shows a tight insert size distribution (37 ± 3 kbp) revealing that 99% of the assembly was spanned correctly by fosmid end sequence pairs of high identity (Supplemental Fig. S8). These data confirm the order, orientation, and sequence accuracy of the clone-based assembly of this complex region of the chimpanzee genome (Supplemental Fig. S7).
Figure 4.
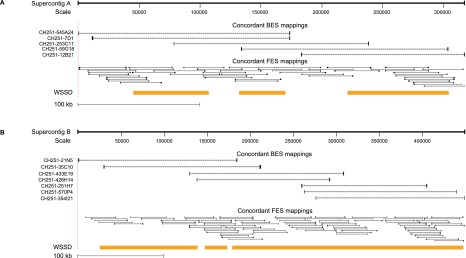
Support for chimpanzee supercontig architecture from clone end mappings. Concordant BES and FES alignments confirm order and orientation of (A) distal and (B) proximal chimpanzee supercontig assemblies. One-hundred-twenty-five paired-end sequences that map with >99.8% sequence identity are depicted. Both analyses support high-quality assembly of these complex regions of the chimpanzee genome.
Discussion
Our data suggest that SMRT sequencing of large-insert clones can significantly improve sequence assembly within complex repetitive regions of genomes, including segmental duplications. Clones assembled both with capillary-based and SMRT sequencing compared favorably in length and sequence accuracy (99.994%). The most common error within the assembled clones was the addition of a single base pair particularly in homopolymer runs, which is consistent with previous reports of potential artifacts of SMRT technology (Au et al. 2012; Okoniewski et al. 2013). High sequence coverage (>90-fold) and the single-base-pair error correction model afforded by Quiver were key to accurate assembly. In addition, the long reads were critical to traversing common repeats. It is instructive, for example, that sequence collapses were restricted to the largest and most identical tandem repeats within each clone. The HGAP assembler (Chin et al. 2013), which sub-selects the longest reads upon which to scaffold an assembly, readily resolved a 2-kbp artifactual duplication from our preliminary Allora assemblies but was unable to fully resolve a 20-kbp higher-order tandem repeat. In the case of the latter, it is interesting to note that the longest read generated for the clone CH17-41F14 (∼12.3 kbp) was shorter than the tandem duplication and that the largest repeat sequence generated in this assembly was ∼12 kbp. We predict that larger clone libraries and longer subread lengths will be required to resolve these most problematic regions. Automated gel electrophoresis systems such as BluePippin (Sage Science) may be particularly useful in this regard because they facilitate the preparation of larger insert libraries that can traverse larger repeats.
Despite these limitations, application of the PacBio sequencing approach confers significant advantages in terms of cost, labor, and throughput. Sequencing centers recently estimated that finishing a single BAC clone to high quality (QV > 45) using capillary-based approaches now costs between $4000 and $5000 per clone. Approximately 30–50 clones per month could be completely sequenced and assembled given a staff of three to four dedicated persons within The Genome Institute at Washington University. We estimate that with one PacBio RS sequencing machine, a single technician with part-time bioinformatics support can produce ∼100–120 clone assemblies per month with ∼85% being completely finished with an estimated error of one mismatch/10,000 using the HGAP/Quiver assembly approach. The cost per finished clone is estimated at approximately $625 (per SMRT Cell)—based on a survey of cost-center rates of five centers currently operating PacBio RS machines. Of course, the cost decreases and throughput increases multifactorially if BACs are pooled. We note that our benchmark pooling experiments were performed with a PacBio RS machine with 75,000 ZMWs (zero-mode waveguides). Current upgrades (PacBio RS II) double the number of productive ZMWs (n = 150,000) and increase movie times making larger BAC pooling schemes feasible. We caution that target regions frequently harbor internal large repeats and automated assembly benefits from both high coverage and the reduced complexity of the large-insert target. Downsampling and pooling experiments highlight the need for sufficient coverage (≥100×) and high-quality DNA libraries for each clone. Barcoding, which is now possible, may further improve pooling of multiple clones within a single SMRT Cell (http://www.pacificbiosciences.com/pdf/TN_Multiplexing_Targeted_Sequencing_Using_Barcodes.pdf).
One approach to improve existing working draft genome assemblies would be to leverage the extensive BES data for many mammalian genomes to select large-insert clones spanning gaps and repeats and mapping to collapsed regions of segmental duplication. All BAC clones mapping to a problematic region (as well as extending 50–100 kbp outside of it for anchoring purposes) could be selected and sequenced to high coverage in 96-well pools using a Nextera Illumina-based sequencing protocol (Adey et al. 2010). Although de novo assemblies of 150 bp Nextera reads tend to fragment within homopolymer and SINE/Alu repeats, the mapping positions of short reads from clones could be used to define an optimal tiling path of clones (∼10–20 clones per region). Once a tiling path of clones has been established for each region, clones could be sequenced in pools of two to three clones, assembled using HGAP/Quiver, and validated by mapping fosmid paired-end sequences to the final assemblies. Clones that failed to assemble into a single contig could be subjected to higher coverage using one clone prep per SMRT Cell. For genomes without fosmid end sequence data, gel extraction of DNA and sequencing using orthogonal chemistries would be an important development to enable validation of de novo assemblies.
It should be emphasized that this procedure is a targeted one rather than a genome-wide approach. Other strategies have been described to upgrade draft genome assemblies by leveraging long-read sequence data or long-range information provided from Hi-C sequence data (English et al. 2012; Burton et al. 2013). While these methods systematically improve chromosomal contiguity across the genome (as measured by N50 contig length), they fail to accurately assemble the most complex regions of segmental duplications (Burton et al. 2013). Regions targeted by our approach are frequently missing or grossly misassembled by whole-genome shotgun sequence assembly using either capillary or next-generation sequencing platforms (Alkan et al. 2011b), still requiring high-quality sequencing of large-insert clones to correctly resolve. Analysis of the mouse and human genomes suggests that these typically correspond to 300–500 regions (∼140–150 Mbp) per genome, including in some cases almost entire chromosomes, such as the Y chromosome (Hughes et al. 2012). The approach we have described provides a strategy to resolve these more structurally complex regions during the final stages of assembly, ensuring that the 1000–2000 genes mapping therein become incorporated within future mammalian genome assemblies (Alkan et al. 2011b; Church et al. 2011).
Methods
PacBio DNA preparation
BAC DNA from CHORI-17 (CH17) and CHORI-251(CH251) clone libraries (http://bacpac.chori.org) was isolated using a High Pure Plasmid Isolation Kit from Roche Applied Science per manufacturer instructions using 10 mL LB media with Chloramphenicol selective marker. We isolated 10 preps per BAC yielding ∼10 µg of starting material.
PacBio sequencing
Approximately 5 µg of BAC DNA was mechanically sheared to a size of ∼8 kbp, using the Hydroshear system and large assembly at a shearing speed of 9 for 20 cycles per manufacturer instructions. SMRTbell libraries were prepared for each sample by ligation of hairpin adaptors at both the ends (Travers et al. 2010), using PacBio DNA Template Prep Kit 2.0 (3–10 kbp) for SMRT sequencing with C2 chemistry on the PacBio RS according to manufacturer instructions. Libraries were purified using (0.45×) Agencourt AMPure beads to remove short sheared inserts <1.5 kbp. The sheared DNA template was characterized for size distribution using an Agilent Bioanalyzer 2100 along with a DNA 12000 chip, and the means from the fragment distribution were between 7 and 9 kbp, while the overall fragment inserts' distribution ranged from ∼2 to 13 kbp (Supplemental Fig. S9). Sequencing primers were annealed to the templates at a final concentration of 5 nM template DNA, and DNA polymerase enzyme C2 was complexed per manufacturer’s recommendation for small-scale libraries. DNA/Polymerase Binding Kit 2.0 (PacBio) was used for setting up enzyme template complexes and libraries were loaded on to the 75,000 ZMWs following instructions in the complex setup and loading calculator provided by the manufacturer. Sequencing Kit 2.0 (PacBio) was used for sequencing using a 45-min sequence capture protocol along with stage start to maximize subread length, on the PacBio RS. With the exception of accidental and intentional pooling, each SMRT Cell contained a single BAC. For pooling experiments, libraries were made individually following “Reduced Input 10 kbp Template Preparations” per manufacturer instructions. BACs were pooled with a finished library using roughly equimolar concentrations and sequenced in a standard diffusion run.
Clone sequence assembly
De novo assembly of BAC inserts was performed using the standard SMRT Analysis (v. 2.0.1) pipeline. Reads were masked for vector sequence (pBACGK1.1) and assembled with HGAP followed by consensus sequence calling with Quiver (Supplemental Fig. S10; Chin et al. 2013). HGAP creates a scaffold assembly using the longest reads (e.g., >7 kbp) as seeds to recruit additional subreads as a scaffold, while Quiver is a multi-read consensus algorithm that takes advantage of the full information from the raw pulse and base call information generated during SMRT sequencing. Final assembly was performed using a minimum read length of 500 bp and minimum read quality of 0.80 on a PC cluster (eight cores/10 GB of RAM) running RedHat 6 SE. We screened unsplit PacBio reads in FASTA format with cross_match using the recommended settings for contamination screening (–minmatch 10 –minscore 20 –screen). PacBio assemblies were reviewed for misassembly by visualizing read depth of PacBio reads in Parasight (http://eichlerlab.gs.washington.edu/jeff/parasight/index.html) using coverage summaries generated during the resequencing protocol. Sanger assemblies were obtained from NCBI by accession ID (Supplemental Table S8). De novo assembly of short-read data was performed with iCAS (ftp://ftp.sanger.ac.uk/pub/badger/aw7/icas_README).
Illumina sequencing of BAC clones
BAC DNA isolation and library preparation was performed as described by Steinberg et al. (2012).
Sequence alignment
We compared Sanger and PacBio assemblies for each clone using BLASR (Chaisson and Tesler 2012) (–maxLCPLength 16 –bestn 1 –m 0) and visualized these for larger structural rearrangements using Miropeats (Parsons 1995). Alignment identity was calculated from the total number of single-base-pair matches between assemblies divided by the total number of contiguous mismatch events, including substitutions, insertions, and deletions. From the BLASR alignments, we determined the coordinates for each mismatch in both assemblies to create a set of PacBio and Sanger variant pairs. We annotated a subset of these variants that qualified as components of homopolymers, dipolymers, or GC-rich regions based on the context of their adjacent bases. We identified the corresponding regions for the two chimpanzee supercontigs in panTro4 using NCBI’s default MEGABLAST settings and aligned the resulting sequences to the supercontigs with BLASR and Miropeats (s = 1000).
To validate the differences we observed between PacBio and Sanger assemblies, we mapped 76-bp Illumina reads from each BAC to both assemblies and chose the variant in each difference that was unambiguously supported by the short reads. Clone pools were sequenced to high coverage using the Nextera protocol described above and mapped with mrsFAST 2.4.0.4 in single-end mode with an edit distance of zero to ensure that only reads with perfect matches counted as support for variants. For a variant to be supported by the short reads, we required at least one read to span the variant and anchor in sequence that was neither homopolymer nor dipolymer. If one variant in a difference had short-read support and the alternate variant did not, the variant with support was considered validated. In the case where neither or both variants in a difference had support, the difference was considered ambiguous. We performed the same experiment with whole-genome sequence (WGS) from three high-coverage individuals (NA12891, NA18507, and NA18508). Differences between calls from BAC and WGS reads were attributed to potential cell-line variants.
Data access
All sequence assemblies from this study have been submitted to the NCBI GenBank (http://www.ncbi.nlm.nih.gov/genbank/) under accession nos. AC254814–AC254826. Accessions are linked to clone names in Supplemental Tables S7 and S8. Underlying raw sequence reads have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) under accession number SRP035602. See also BioProject (PRJNA236102; http://www.ncbi.nlm.nih.gov/bioproject/).
Competing interest statement
S.R., L.H., S.W.T., and J.K. are employees of Pacific Biosciences, Inc., a company that commercializes DNA sequencing technologies, and E.E.E. is on the scientific advisory boards (SABs) of DNAnexus, Inc. and was an SAB member of Pacific Biosciences, Inc. (2009–2013) and SynapDx Corp. (2011–2013).
Acknowledgments
We thank C. Campbell, D. Alexander, and A. Klammer for helpful conversations, K. Mohajeri and L. Harshman for assistance in sample preparation, and T. Brown for assistance in manuscript preparation. This work was supported, in part, by US National Institutes of Health (NIH) grants HG002385 and HG004120 to E.E.E. E.E.E. is an investigator of the Howard Hughes Medical Institute.
Author contributions: E.E.E. and S.W.T. designed experiments; F.A. prepared DNA; M.M. and S.R. prepared libraries and generated sequence data; P.H.S. and M.Y.D. identified clones for sequencing; J.H., L.H., M.C., T.A.G., and C.A. performed bioinformatics analyses; T.A.G. and R.K.W. performed targeted capillary sequencing of clones; J.H. and E.E.E. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.168450.113.
References
- Adey A, Morrison HG, Asan, Xun X, Kitzman JO, Turner EH, Stackhouse B, MacKenzie AP, Caruccio NC, Zhang X, et al. 2010. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol 11: R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C, Cardone MF, Catacchio CR, Antonacci F, O’Brien SJ, Ryder OA, Purgato S, Zoli M, Della Valle G, Eichler EE, et al. 2011a. Genome-wide characterization of centromeric satellites from multiple mammalian genomes. Genome Res 21: 137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan C, Sajjadian S, Eichler EE 2011b. Limitations of next-generation genome sequence assembly. Nat Methods 8: 61–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au KF, Underwood JG, Lee L, Wong WH 2012. Improving PacBio long read accuracy by short read alignment. PLoS ONE 7: e46679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton J, Adey A, Patwardhan RP, Qiu R, Kitzman J, Shendure J 2013. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol 31: 1119–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, Tesler G 2012. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): Application and theory. BMC Bioinformatics 13: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrier C, Joshi K, Coutinho-Budd J, Kim J-E, Lambert N, de Marchena J, Jin WL, Vanderhaeghen P, Ghosh A, Sassa T, et al. 2012. Inhibition of SRGAP2 function by its human-specific paralogs induces neoteny during spine maturation. Cell 149: 923–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, et al. 2013. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10: 563–569 [DOI] [PubMed] [Google Scholar]
- Church DM, Goodstadt L, Hillier LW, Zody MC, Goldstein S, She X, Bult CJ, Agarwala R, Cherry JL, DiCuccio M, et al. 2009. Lineage-specific biology revealed by a finished genome assembly of the mouse. PLoS Biol 7: e1000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church DM, Schneider VA, Graves T, Auger K, Cunningham F, Bouk N, Chen HC, Agarwala R, McLaren WM, Ritchie GR, et al. 2011. Modernizing reference genome assemblies. PLoS Biol 9: e1001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MY, Nuttle X, Sudmant PH, Antonacci F, Graves TA, Nefedov M, Rosenfeld JA, Sajjadian S, Malig M, Kotkiewicz H, et al. 2012. Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication. Cell 149: 912–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- English AC, Richards S, Han Y, Wang M, Vee V, Qu J, Qin X, Muzny DM, Reid JG, Worley KC, et al. 2012. Mind the gap: Upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 7: e47768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, Skaletsky H, Brown LG, Pyntikova T, Graves T, Fulton RS, Dugan S, Ding Y, Buhay CJ, Kremitzki C, et al. 2012. Strict evolutionary conservation followed rapid gene loss on human and rhesus Y chromosomes. Nature 483: 82–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okoniewski MJ, Meienberg J, Patrignani A, Szabelska A, Matyas G, Schlapbach R 2013. Precise breakpoint localization of large genomic deletions using PacBio and Illumina next-generation sequencers. Biotechniques 54: 98–100 [DOI] [PubMed] [Google Scholar]
- Parsons J 1995. Miropeats: Graphical DNA sequence comparisons. Comput Appl Biosci 11: 615–619 [DOI] [PubMed] [Google Scholar]
- Salzberg SL, Phillippy AM, Zimin A, Puiu D, Magoc T, Koren S, Treangen TJ, Schatz MC, Delcher AL, Roberts M, et al. 2012. GAGE: A critical evaluation of genome assemblies and assembly algorithms. Genome Res 22: 557–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Helgason A, Thorleifsson G, Steinthorsdottir V, Masson G, Barnard J, Baker A, Jonasdottir A, Ingason A, Gudnadottir VG, et al. 2005. A common inversion under selection in Europeans. Nat Genet 37: 129–137 [DOI] [PubMed] [Google Scholar]
- Steinberg KM, Antonacci F, Sudmant PH, Kidd JM, Campbell CD, Vives L, Malig M, Scheinfeldt L, Beggs W, Ibrahim M, et al. 2012. Structural diversity and African origin of the 17q21.31 inversion polymorphism. Nat Genet 44: 872–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A, Sampas N, Bruhn L, Shendure J, Eichler EE 2010. Diversity of human copy number variation and multicopy genes. Science 330: 641–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Huddleston J, Catacchio CR, Malig M, Hillier LW, Mohajeri K, Kondova I, Bontrop RE, Persengiev S, Antonacci F, et al. 2013. Evolution and diversity of copy number variation in the great ape lineage. Genome Res 23: 1373–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers KJ, Chin CS, Rank DR, Eid JS, Turner SW 2010. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic Acids Res 38: e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zody MC, Jiang Z, Fung HC, Antonacci F, Hillier LW, Cardone MF, Graves TA, Kidd JM, Cheng Z, Abouelleil A, et al. 2008. Evolutionary toggling of the MAPT 17q21.31 inversion region. Nat Genet 40: 1076–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]