

Abstract
Genetic code expansion for synthesis of proteins containing noncanonical amino acids is a rapidly growing field in synthetic biology. Creating optimal orthogonal translation systems will require re-engineering central components of the protein synthesis machinery on the basis of a solid mechanistic biochemical understanding of the synthetic process.
The genetic code was thought to be immutable. At the time the genetic code table was defined, its evolution was most readily explained by Crick’s frozen accident theory1. Central to the theory was the idea that stability of the proteome and the accuracy of protein synthesis were essential for cell viability. These ideas emerged alongside a nascent molecular biology that developed before genome sequencing and proteomics, when much of what we now know of life’s microbial and molecular biodiversity was still undiscovered and unknown. Since the elucidation of the genetic code in the 1960s, the most exciting and surprising findings are those related to exceptions to the standard code and the discovery of diversity far greater than was anticipated in the mechanisms of aminoacyl-tRNA formation and protein synthesis.
The first indication that the genetic code might not be static came with the finding in the late 1970s that the UGA stop codon was reassigned to encode tryptophan in yeast mitochondria. Today, there are ~20 known variations to the standard genetic code2. Although many code variants occur in organelles, free-living and human-associated microbes are also known to have unique genetic codes3.
Selenocysteine (Sec), the twenty-first genetically encoded amino acid, was the next big surprise4 when it was uncovered in the 1980s that UGA is recoded to direct Sec incorporation into proteins in many species, including humans4. Accurate selenoprotein synthesis is essential for human health and development. Another stop codon (UAG) genetically encodes pyrrolysine (Pyl), which is essential in trimethylamine metabolism in archaeal methanogens5, and some organisms (for example, Desulfitobacterium) apparently encode 22 amino acids. One anaerobic bacterium was recently shown to dynamically alter the number of amino acids it encodes in a carbon source–dependent manner6. Ambiguous decoding in yeast7 and ‘open’ (unused) codons in some organisms (for example, in Mycoplasma, discussed in ref. 8) were other dogma-breaking findings that reshaped our view of protein synthesis and the genetic code.
Given these and other new findings, many of the basic assumptions underlying the presumed immutability of the genetic code are now known to be false or incomplete. Cells tolerate9 and can even derive selective advantage7 from ambiguous decoding, cells can encode more than 20 amino acids, and codon reassignment and recoding is possible. Recent developments10 demonstrate not only that the genetic code can evolve but also that rewiring translation to genetically encode more (possibly many more) than 20 amino acids, primarily by recoding UAG, is both feasible and desirable. Expanding the genetic code has emerged as a definitive goal of synthetic biology, and successes in establishing ~100 distinct noncanonical amino acid (ncAA) orthogonal translation systems (OTSs) have enabled facile in vivo production of proteins with hardwired post-translational modifications, photocaged labile amino acids or site-specific fluorescent labels10, suggesting that further expansion is possible.
Despite these successes, certain amino acids, including biomedically relevant methylated and some phosphorylated amino acids, are refractory to traditional genetic code expansion techniques. There seem to be limitations in the natural protein synthesis machinery that must be better understood to further rewire the genetic code. Since the 1960s, the field of protein synthesis has developed a detailed mechanistic and structural understanding of aminoacyl-tRNA formation and elongation factor interaction with tRNA and the ribosome as well as a structural understanding of mechanisms involved in codon reading, protein synthesis fidelity and quality control. This field is a source of inspiration and methodology that will aid further efforts to expand the genetic code. In this Commentary, we wish to highlight insights in the field of protein synthesis that could be used to further the design and evolution of very efficient and specific OTSs as well as identify experimental challenges that should be embraced by scientists in both protein synthesis and synthetic biology to advance this topic.
Biological parts
Expanding the genetic code beyond the 20 canonical amino acids requires (i) an ‘open’ codon (described below) to encode (ii) an ncAA that can permeate the cell, (iii) an aminoacyl-tRNA synthetase (AARS) capable of efficiently ligating a desired ncAA (iv) a tRNA that can decode the ‘open’ codon and (v) compatible elongation factors and ribosomes. Developing an efficient OTS thus demands optimization of all of the above components (Fig. 1)
Figure 1. Engineering efficient OT Ss.
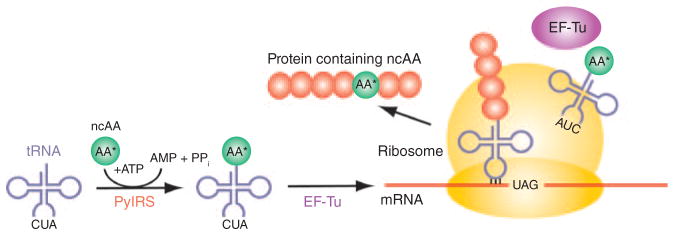
Protein synthesis requires amino acid (AA)-tRNAs as building blocks for ribosomal protein synthesis. An amino acid is ligated to the tRNA by a dedicated AARS. The product (AA-tRNA) is then delivered by an elongation factor (for example, EF-Tu) to the ribosome, where the anticodon of the tRNA matches the triplet codon of the mRNA. Protein synthesis using an expanded genetic code is shown with an ncAA incorporated by PylRS, tRNAPyl and EF-Tu at the UAG codon. For efficient ncAA incorporation, all three components of the above OTS should be optimized. First, an efficient and specific AARS needs to be developed for each ncAA (AA*). It will be necessary to combine the power of new methods to engineer and select mutant libraries. High-throughput screening of amino acid chemical libraries, biochemical analysis and structural determination are important factors in developing new OTSs. This cycle may need to be repeated several times to eventually produce AARS-ncAA pairs that match the efficiency and specificity of natural AARSs. Second, the synthetic tRNA carrying the desired ncAA must be orthogonal (that is, not recognized by endogenous AARSs) in each organism. Third, EF-Tu requires mutations to accommodate ncAAs with negative charges or bulky side chains. For certain ncAAs, even the ribosome may need to be engineered to improve ncAA incorporation in protein.
By definition, the AARS-tRNA orthogonal pair must not cross-react with endogenous AARS-tRNA pairs and are in this way ‘orthogonal’ to the translation machinery of the host cell. For genetic code expansion in Escherichia coli, the most successful orthogonal pairs are derived from archaea. Tyrosyl-tRNA synthetase from Methanocaldococcus jannaschii, pyrrolysyl-tRNA synthetase (PylRS) from Methanosarcina species and phosphoseryl-tRNA synthetase (SepRS) found in archaeal methanogens, are the main vehicles for code expansion. The tyrosyl-tRNA synthetase system has been used to install a diverse array of tyrosine derivatives, whereas PylRS and its engineered variants support translation with lysine, phenylalanine and Pyl derivatives, including click chemistry–reactive ncAAs10 (Fig. 2). Phosphoseryl-tRNA synthetase, which has a natural function in cysteine biosynthesis and Cys-tRNACys formation in archaea, was instrumental for expanding the genetic code of E. coli with phosphoserine11 and may have a similar role in expanding to other phosphoamino acids and analogs in the future.
Figure 2. Chemical diversity of amino acids in the standard and expanded genetic codes.

Current genetically encoded ncAAs are mainly derivatives of lysine and phenylalanine. Future efforts are needed to develop a wide variety of orthogonal systems to expand the genetic code with a much larger pool of ncAAs including diverse functional groups. The tree relates the canonical amino acids according to the JTT similarity matrix. Amino acid similarity reflects the substitution frequency of one amino acid for another in standardized sets of multiple sequence alignments.
Despite the development of numerous OTSs, many challenges remain. The most critical problem is the poor efficiency of engineered AARSs, which can partly be overcome by high cellular ncAA12. Compatibility of ncAA-tRNA with elongation factor Tu (EF-Tu) and the ribosome, ncAA permeability to the cell and stability in the cell are also important challenges. Translation with ncAAs is generally much less efficient than with standard amino acids, which results from the fact that ncAA aminoacylation efficiency is as low as aminoacylation with nearcognate amino acids (Table 1). For example, PylRS shows a similar catalytic efficiency in Pyl-tRNAPyl formation compared to typical AARS-tRNA pairs, but the engineered versions of the enzyme show a reduction in aminoacylation efficiency by a factor of ~1,000 (kcat/KM). AARSs are well known to show some activity with near-cognate amino acids, which leads to tRNAs charged with the ‘wrong’ amino acid and contributes to the overall mistranslation rate (~1 error in 10,000 amino acids). Furthermore, inefficient ncAA production by engineered AARS enzymes will allow successful competition by cellular amino acid–tRNAs with near-cognate anticodons13; this may substantially lower the yield of the desired ncAA-containing protein. Thus, optimization of expression of the orthogonal tRNA-AARS pair and high ncAA are prerequisites for high-yield expression and purity of the desired proteins. In addition, efficient UAG recoding requires removal of release factor (RF1 in E. coli) to allow efficient read-through by the orthogonal amino acid–tRNA14–16.
Table 1.
Catalytic efficiencies of natural and engineered AARSs with amino acids and ncAAs.
| Natural AARS (cognate amino acid) | AARS variant (near-cognate amino acid) | Loss of catalytic efficiency (x-fold)a | References |
|---|---|---|---|
| PylRS (Pyl) | PylRS variant (Ack)b | >7,000 | 26 |
| MetRS (Met) | MetRS variant (Anl)c | >800 | 27 |
| LeuRS (Leu) | LeuRS (Ile) | ~1,000 | 28 |
| PheRS (Phe) | PheRS (Tyr) | >5,000 | 29 |
| ThrRS (Thr) | ThrRS (Ser) | >500 | 30 |
Loss of catalytic efficiency is calculated as the ratio of the kcat/KM values of the AARS–amino acid pairs shown in the first and second columns.
N-acetyllysine.
Azidonorleucine.
A second problem is the primary focus, thus far, on engineering a small group of ‘parts’ out of those that are involved in the whole pathway of protein biosynthesis. Current directed evolution strategies rely on generating a library of random mutants at five or six sites in the substrate recognition pocket of the orthogonal AARS active site. Successive rounds of positive selection (antibiotic resistance) and negative selection (toxic protein production) are used to isolate orthogonal pair variants that specifically incorporate a desired ncAA and achieve a low background of endogenous amino acid insertion10.
Although these strategies have been successful at establishing site-directed insertion of ncAAs into proteins, ncAAs are not incorporated as efficiently as the standard 20 amino acids, in part owing to poor aminoacylation kinetics of engineered AARSs (Table 1). We believe that larger libraries of orthogonal pair mutants (involving more than six amino acids), generated by site-directed random mutagenesis in combination with error-prone PCR or using genome recombineering methods (for example, multiplex automated genome engineering (MAGE) or clustered regularly interspaced short palindromic repeats (CRISPR)-based approaches) in addition to new selection and screening schemes (for example, fluorescenceactivated cell sorting or directed evolution) will ultimately lead to more optimal orthogonal pairs (Fig. 1). Structure-guided design and rational active site mutagenesis have important roles in engineering enzymatic activity, and, in combination with directed evolution, these methods can successfully redesign active sites by, for example, converting a deaminase into a phosphatase17. These studies indicate that a diversity of approaches will be required to further engineer ncAA specificity and optimize tRNA acylation with ncAAs.
Optimization of OTS expression constructs, aminoacylation efficiency and engineering EF-Tu11 have also led to major improvements in the yield of ncAA-containing proteins. Even with these strategies, some ncAAs do not permeate the cell (and may require a dipeptide uptake approach), and others are chemically labile (including phosphorylated amino acids (phosphoAAs)) in the cellular environment. Amino acid import pathways and nonhydrolyzable or more chemically stable ncAA analogs must be considered. Moving beyond orthogonal pair design, ribosome mutagenesis and engineering biosynthetic pathways to produce ncAAs in vivo will be fascinating topics as synthetic biology develops.
Chemical parts
Compared to the 20 canonical amino acids, there exists in nature and in the laboratory a vast chemical diversity of amino acid side chains (Fig. 2). One thing that excites us about the future of synthetic biology will be technology that enables facile in vivo production of genetically encoded proteins with multiple hardwired post-translational modifications. In nature, there are over 150 known modified amino acids and an even greater diversity of side chain functional groups accessible to synthetic chemists (Fig. 2); the ability to harness this untapped chemical diversity would enable a diversity of new applications. Genetically encoded phosphoAAs, for example, will be invaluable in understanding the human kinome in health and disease and to ultimately develop therapeutic phosphoproteins. Photocage-protected phosphoAAs would provide time-resolved probing of cellular signaling pathways, and nonhydrolyzable analogs of phosphoAAs will allow production of a particular phosphoprotein (or of phosphoproteins) in desired phosphorylated states that are resistant to cellular phosphatases. Developing these systems will require exploration of more natural AARS systems that are suitable for a greater variety of ncAAs, for example, phosphoAAs, nonhydrolyzable analogs, photocaged amino acids and ncAAs for click chemistry (Fig. 2)10. Such ncAAs often carry charged or bulky side chains, which might be weakly recognized by the elongation factor11,18 and even the ribosome. Future studies of these translational factors are necessary to optimize the incorporation efficiencies of ncAAs.
As AARS enzymes are further engineered, scientists must be mindful of the fact that, though AARS enzymes were fashioned by evolution to have exquisite substrate specificity involving editing mechanisms that hydrolyze misacylated tRNA substrates19, evolution did not protect against non-natural substrate analogs that the cell might encounter. It has been well known that the E. coli wild-type translation machinery incorporates many non-natural amino acids into proteins20. For chemically and structurally similar amino acids, both naturally evolved and engineered AARSs, assumed to be specific for one amino acid or ncAA, often show polyspecificity by cross-reacting with multiple other ncAAs21,22. This fact may cause problems, especially when inserting multiple ncAAs into a single protein. Repeated rounds of further enzyme evolution (based on cycles of biochemical analysis followed by structure-inspired mutagenesis and selection of new AARS variants) are expected to decrease polyspecificity, leading to more specific orthogonal pairs.
Extending orthogonality
Successes in directed evolution experiments have enhanced ncAA incorporation in response to the three nonsense (that is, stop) codons and a few quadruplet codons10. Nonsense codons are also read by endogenous amino acid–tRNAs via near-cognate decoding13, and competition with peptide chain release factors is a considerable barrier to efficient decoding of stop codons with some ncAAs13. Quadruplet codons are poorly translated on the ribosome and face frame-shifting problems that result from erroneous decoding by tRNAs recognizing triplet codons. Although ribosome23 or tRNA engineering may overcome some of these challenges, there is a need to invest in projects that reassign (recode) sense codons to support incorporation of multiple ncAAs into a synthetic protein.
Given the degeneracy of the genetic code and the rules of codon recognition by tRNA anticodons, the notion emerged that between 30 and 40 sense codons suffice to encode an organism’s genetic information. Thus, a large number of codons (>20) should be potentially reassignable sense codons. Natural diversity provides some immediate entries into potential mechanisms to rewrite the genetic code2,3. For instance, the CUN (N denotes A, U, G and C) codons are reassigned from leucine to threonine in yeast mitochondria. Such recoding requires only a few mutations in the tRNA and AARS to form a new orthogonal pair (Fig. 3). Natural recoding events are rare, but we expect that with advances in genome sequencing technology (for example, high-coverage single-cell sequencing), combined with bioinformatics and biochemical studies, more recoding events and orthogonal AARS-tRNA pairs will be identified, particularly in organelles, bacteria and archaea with small genomes. These new AARS-tRNA pairs could serve as portable parts for genetic code expansion in microorganisms and even higher eukaryotes, although in each organism the orthogonality needs to be tested and perhaps reestablished10. Identifying a larger pool of natural orthogonal pairs would provide more routes to recode multiple sense codons in either natural or synthetic organisms. Such work will also reveal which sense codons are redundant (and potentially more ‘recodeable’) and which tRNAs are dispensable if specific sets of codons are absent. Post-transcriptional modifications are known to alter the decoding capacity and efficiency of aminoacyl-tRNAs; it is therefore essential to fully understand the modification patterns of tRNAs responsible for the codons of interest.
Figure 3. Reassignment of CUN codons in yeast mitochondria provides insight into sense codon recoding.
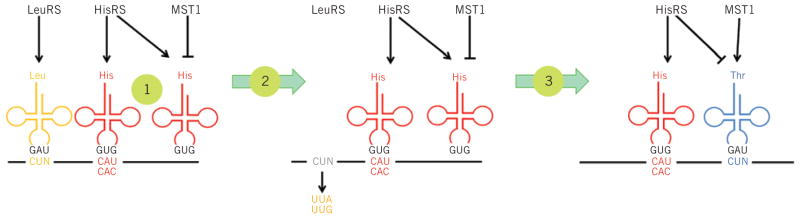
The ancestor mitochondria contain a tRNALeu with a UAG anticodon that pairs with CUN codons. Both the CUN codons and tRNALeu UAG were lost during evolution. A duplicated copy of tRNAHis then evolved to decode CUN. Although this new tRNA is no longer recognized by histidyl-tRNA synthetase (HisRS), it becomes a substrate for the coevolved threonyl-tRNA synthetase (MST1). With the emergence of the orthogonal MST1-tRNAThr UAG pair, CUN codons reappeared in the mitochondrial genome to complete the codon reassignment event25. This naturally evolved system could serve as a model for sense codon recoding.
Recoding sense codons in the laboratory faces several key challenges. Apart from finding natural species or creating synthetic organisms with open sense codons (that is, codons unused in the genome), a generic selection or screening method for sense codon reassignment needs to be developed to test the orthogonality of imported AARS-tRNA pairs and determine the efficiency of ncAA incorporation. Most AARSs recognize the cognate tRNA’s anticodon as the major identity element24. Thus, synthetic tRNAs intended to recode sense codons are likely to be recognized by endogenous AARSs and misacylated with canonical amino acids8. Further engineering of such synthetic tRNAs is needed to prevent cross-reaction with endogenous amino acids. For this reason, it is desirable to develop a completely orthogonal translation system that operates independently of the cell’s normal protein synthesis machinery.
Outlook
To reach the exciting goals we outline here regarding genetic code expansion and recoding, multiple components in the translational system should be optimized for efficiency and specificity. New selection methods for specific amino acids (such as those described in ref. 9) also need to be developed, and endogenous AARSs may need to be evolved together with new tRNAs to establish mutual orthogonality. Such efforts demand expertise from synthetic biologists in genome and metabolic engineering as well as continued contributions from the field of protein synthesis, which has so far uncovered the natural diversity of the genetic code2 and provided critical mechanistic insights. Together, researchers in these areas will enable future recoding efforts that should provide the basis for generating viable, engineered organisms with more diverse genetic codes and capabilities.
Acknowledgments
We are grateful to M. Englert, I. Heinemann, F. Isaacs and J. Rinehart for inspired discussions. Work in the authors’ laboratory was supported by the Division of Chemical Sciences, Geosciences and Biosciences, Office of Basic Energy Sciences of the US Department of Energy (DE-FG02-98ER20311); the National Institute of General Medical Sciences (GM22854); DARPA contracts N66001- 12-C-4020 and N66001-12-C-4211; and by the National Science Foundation (MCB-0950474).
Footnotes
Competing financial interests
The authors declare no competing financial interests.
Contributor Information
Patrick O’Donoghue, Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, Connecticut, USA.
Jiqiang Ling, Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, Connecticut, USA.
Yane-Shih Wang, Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, Connecticut, USA.
Dieter Söll, Email: dieter.soll@yale.edu, Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, Connecticut, USA. Department of Chemistry, Yale University, New Haven, Connecticut, USA.
References
- 1.Crick FHC. J Mol Biol. 1968;38:367–379. doi: 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- 2.Ambrogelly A, Palioura S, Söll D. Nat Chem Biol. 2007;3:29–35. doi: 10.1038/nchembio847. [DOI] [PubMed] [Google Scholar]
- 3.Campbell JH, et al. Proc Natl Acad Sci USA. 2013;110:5540–5545. doi: 10.1073/pnas.1303090110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Böck A, Stadtman TC. Biofactors. 1988;1:245–250. [PubMed] [Google Scholar]
- 5.Gaston MA, Jiang R, Krzycki JA. Curr Opin Microbiol. 2011;14:342–349. doi: 10.1016/j.mib.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prat L, et al. Proc Natl Acad Sci USA. 2012;109:21070–21075. doi: 10.1073/pnas.1218613110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bezerra AR, et al. Proc Natl Acad Sci USA. 2013;110:11079–11084. doi: 10.1073/pnas.1302094110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krishnakumar R, et al. Chem Bio Chem. 2013 http://dx.doi.org/10.1002/cbic.201300444.
- 9.Ruan B, et al. Proc Natl Acad Sci USA. 2008;105:16502–16507. doi: 10.1073/pnas.0809179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu CC, Schultz PG. Annu Rev Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- 11.Park HS, et al. Science. 2011;333:1151–1154. doi: 10.1126/science.1207203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiltschi B, Wenger W, Nehring S, Budisa N. Yeast. 2008;25:775–786. doi: 10.1002/yea.1632. [DOI] [PubMed] [Google Scholar]
- 13.O’Donoghue P, et al. FEBS Lett. 2012;586:3931–3937. doi: 10.1016/j.febslet.2012.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mukai T, et al. Nucleic Acids Res. 2010;38:8188–8195. doi: 10.1093/nar/gkq707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Isaacs FJ, et al. Science. 2011;333:348–353. doi: 10.1126/science.1205822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson DB, et al. Nat Chem Biol. 2011;7:779–786. doi: 10.1038/nchembio.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khare SD, et al. Nat Chem Biol. 2012;8:294–300. doi: 10.1038/nchembio.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ieong KW, Pavlov MY, Kwiatkowski M, Forster AC, Ehrenberg M. J Am Chem Soc. 2012;134:17955–17962. doi: 10.1021/ja3063524. [DOI] [PubMed] [Google Scholar]
- 19.Ling J, Reynolds N, Ibba M. Annu Rev Microbiol. 2009;63:61–78. doi: 10.1146/annurev.micro.091208.073210. [DOI] [PubMed] [Google Scholar]
- 20.Richmond MH. Bacteriol Rev. 1962;26:398–420. doi: 10.1128/br.26.4.398-420.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang YS, Fang X, Wallace AL, Wu B, Liu WR. J Am Chem Soc. 2012;134:2950–2953. doi: 10.1021/ja211972x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Young DD, et al. Biochemistry. 2011;50:1894–1900. doi: 10.1021/bi101929e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neumann H, Wang K, Davis L, Garcia-Alai M, Chin JW. Nature. 2010;464:441–444. doi: 10.1038/nature08817. [DOI] [PubMed] [Google Scholar]
- 24.Giegé R, Sissler M, Florentz C. Nucleic Acids Res. 1998;26:5017– 5035. doi: 10.1093/nar/26.22.5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Su D, et al. Nucleic Acids Res. 2011;39:4866–4874. doi: 10.1093/nar/gkr073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Umehara T, et al. FEBS Lett. 2012;586:729–733. doi: 10.1016/j.febslet.2012.01.029. [DOI] [PubMed] [Google Scholar]
- 27.Tanrikulu IC, Schmitt E, Mechulam Y, Goddard WA, III, Tirrell DA. Proc Natl Acad Sci USA. 2009;106:15285–15290. doi: 10.1073/pnas.0905735106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boniecki MT, Vu MT, Betha AK, Martinis SA. Proc Natl Acad Sci USA. 2008;105:19223–19228. doi: 10.1073/pnas.0809336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reynolds NM, et al. Proc Natl Acad Sci USA. 2010;107:4063–4068. doi: 10.1073/pnas.0909640107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ling J, Söll D. Proc Natl Acad Sci USA. 2010;107:4028–4033. doi: 10.1073/pnas.1000315107. [DOI] [PMC free article] [PubMed] [Google Scholar]