

Abstract
Tumor angiogenesis is one of the hallmarks of tumor progression and is essential for invasiveness and metastasis. Myeloid inflammatory cells, such as immature myeloid precursor cells, also termed myeloid-derived suppressor cells (MDSCs), neutrophils, and monocytes/macrophages, are recruited to the tumor microenvironment by factors released by the malignant cells that are subsequently “educated” in situ to acquire a pro-invasive, pro-angiogenic, and immunosuppressive phenotype. The proximity of myeloid cells to endothelial cells (ECs) lining blood vessels suggests that they play an important role in the angiogenic response, possibly by secreting a network of cytokines/chemokines and inflammatory mediators, as well as via activation of ECs for proliferation and secretion of pro-angiogenic factors. Interleukin-1 (IL-1) is an “alarm,” upstream, pro-inflammatory cytokine that is generated primarily by myeloid cells. IL-1 initiates and propagates inflammation, mainly by inducing a local cytokine network and enhancing inflammatory cell infiltration to affected sites and by augmenting adhesion molecule expression on ECs and leukocytes. Pro-inflammatory mediators were recently shown to play an important role in tumor-mediated angiogenesis and blocking their function may suppress tumor progression. In this review, we summarize the interactions between IL-1 and other pro-angiogenic factors during normal and pathological conditions. In addition, the feasibility of IL-1 neutralization approaches for anti-cancer therapy is discussed.
Keywords: IL-1α, IL-1β, VEGF, VEGFR1, VEGFR2, angiogenesis, myeloid cells, inflammation
Introduction
Postnatal angiogenesis, first described by Folkman in the 70's, referred to a process by which mature endothelial cells (ECs) proliferate and sprout to create new blood vessels. Usually, quiescent ECs are inactive for months and even years; they proliferate only upon angiogenic activation due to various pathological conditions (Carmeliet and Jain, 2000; Carmeliet, 2003, 2005; Ribatti, 2009). Initially, the angiogenic process was considered to represent the outcome of straightforward interactions between pro-angiogenic factors and specific signaling receptors on the membranes of ECs (Folkman, 1995, 2001, 2003; Folkman and D'Amore, 1996). Although many molecules are involved in angiogenesis, VEGF stands out because it has a direct mitogenic effect on ECs and functions as a key regulator of both physiological and pathological angiogenesis (Thomas, 1996; Alitalo and Carmeliet, 2002; Shibuya, 2006a,b). VEGFA belongs to the VEGF family and through activation of two tyrosine kinase receptors, VEGFR1 (Flt-1) and VEGFR2 (FLK-1), plays a major role in the angiogenic response. The biological effects of VEGF are thought to be mediated mainly by VEGFR2, whereas VEGFR1 can transduce a weak, intracellular signal in ECs and was initially thought to be a decoy receptor, thus serving as a negative regulator of angiogenesis (Hiratsuka et al., 1998). At the same time, VEGFR1 is expressed on different myeloid cells, such as macrophages and is directly involved in the migration of these cells to inflammatory areas (Shibuya, 2006a,b). Recently, it was found that inflammation usually accompanies tumor development and the mechanism by which inflammation contributes to tumor-mediated angiogenesis has been the subject of intensive study.
Pro-inflammatory cytokines are potent factors in the induction and support of angiogenesis. Of special relevance to angiogenesis are the “alarm cytokines” IL-1 and TNFα that are generated by macrophages immediately after confrontation with inflammatory stimuli. These stimuli induce the expression of pro-inflammatory genes in diverse stromal/inflammatory cells, which ultimately results in a local cascade of cytokines and small effector molecules that initiate, propagate, and sustain inflammation (Dinarello, 1996; Apte and Voronov, 2002, 2008; Balkwill, 2002; Apte et al., 2006a,b; Balkwill and Mantovani, 2012). IL-1 and TNFα also increase the expression of adhesion molecules on both ECs and leukocytes, promoting leukocyte infiltration from the blood into inflamed tissues (Apte and Voronov, 2002, 2008; Apte et al., 2006a,b).
The direct relationship between inflammation and tumor-mediated angiogenesis has prompted the development of novel anti-tumor therapies based on the attenuation of inflammation.
In this review, the interaction of IL-1 molecules with a critical pro-angiogenic factor, VEGF, and the effects of IL-1 on ECs and myeloid cells during the angiogenic response are discussed.
The IL-1 molecules
The IL-1 family consists of agonistic and antagonistic molecules, as well as receptors. The two major agonistic proteins are IL-1α and IL-1β. The third important molecule is the IL-1 receptor antagonist (IL-1Ra), which is a physiological inhibitor of pre-formed IL-1; it binds to IL-1 receptors without transmitting an activation signal (reviewed in Dinarello, 1996; Stylianou and Saklatvala, 1998; Sims et al., 2001; Apte and Voronov, 2002, 2008; Arend, 2002; Dinarello, 2002; O'Neill, 2002; Braddock and Quinn, 2004; Apte et al., 2006a,b; Mantovani et al., 2008; Mantovani and Sica, 2010).
Many cell types produce and secrete IL-1α, IL-1β, and IL-1Ra upon activation with microbes, microbial products, cytokines, and other environmental stimuli, as well as products of damaged tissue (reviewed in Dinarello, 1996, 2006; Apte and Voronov, 2008; Voronov et al., 2013). IL-1α and IL-1β are synthesized as precursors of 31 kD that are further processed by proteases to their mature secreted 17 kD forms. IL-1 differs from most other cytokines by the lack of a signal sequence, thus not passing through the endoplasmic reticulum-Golgi pathway. The mechanisms of IL-1 secretion are not yet completely understood. IL-1Ra, which has a signal peptide, is secreted in the ER-Golgi exocytic pathway.
IL-1β is not present in homeostatic conditions; it is induced and secreted only upon inflammatory signals. The IL-1β precursor is biologically inactive until it is enzymatically cleaved into the mature secreted form by the IL-1β-converting enzyme (caspase-1) that is activated in the cytosol on the inflammasome platform (reviewed in Dinarello, 2009, 2011a; Eisenbarth and Flavell, 2009; Franchi et al., 2009; Martinon et al., 2009; Latz, 2010; Schroder and Tschopp, 2010). IL-1β, due to the fact that it is extensively secreted, has been considered to be the major pro-inflammatory molecule. At low local doses, IL-1β induces limited inflammatory responses, followed by activation of specific immune mechanisms, while at high doses, IL-1β induces broad inflammation accompanied by tissue damage and immune suppression (reviewed in Voronov et al., 2013).
IL-1α, in contrast, is present in homeostatic conditions in many cells. IL-1α is only rarely secreted by living cells and in most cases is undetectable in body fluids. Its protein is translated as a precursor (ProIL-1α), which is further processed by the Ca2+-dependent protease, calpain, into the mature 17 kD form and the 16 kD N-terminal cleavage product- the propiece of IL-1α, also known as the IL-1α N-terminal peptide (IL-1NTP). Intracellular ProIL-1α is present in many cells because they contain calpain inhibitors and are thus unable to process and secrete IL-1α (Afonina et al., 2011; Di Paolo and Shayakhmetov, 2013; Zheng et al., 2013). Recently, a novel mechanism to control IL-1α activity has been described by Zheng et al. (2013) and reviewed in Di Paolo and Shayakhmetov (2013). Thus, under normal conditions, IL-1α is synthesized as a p33 precursor that is sequestered in the cytosol by IL-1 receptor type 2 (IL-1R2), where it cannot be cleaved by proteases or activate IL-1 receptor type 1 (IL-1R1) signaling. However, under inflammatory signals, after inflammasome activation, IL-1R2 is cleaved by caspase-1 and ProIL-1α is further processed by calpain to the highly active p17 mature IL-1α form, which can be secreted from cells.
A biologically active membrane-associated form of IL-1α (23 kD) that is anchored to the membrane via a mannose-like receptor has been observed in activated cells that express the cytokine. However, it is not clear how IL-1α is inserted into the membrane.
IL-1α and IL-1β signal through the same IL-1Rs, which belong to the immunoglobulin (Ig) supergene family and are extensively expressed on many cell types. IL-1R1 (80 kD) is a signaling receptor, whereas IL-1R2 (68 kD) serves as a decoy target, acting to reduce excessive amounts of IL-1 (reviewed in Apte and Voronov, 2008; O'Neill, 2008; Dinarello, 2009, 2011b; Garlanda et al., 2009; Gabay et al., 2010; Sims and Smith, 2010; Voronov et al., 2013). Following the binding of IL-1 to IL-1R1, a second chain, i.e., the IL-1R acceptor protein (IL-1RAcP), is recruited. This heterodimeric complex triggers IL-1 signaling by activating the IL-1 receptor-associated kinase (IRAK) and ultimately leads to the activation of NF-κB and its target genes. On the contrary, IL-1R2 and the IL-1Ra do not form this heterodimeric complex with the IL-1RAcP and therefore do not recruit IRAK. While IL-1α and IL-1β signals through the same receptors, they differ dramatically in some biological functions (reviewed in Apte and Voronov, 2008; O'Neill, 2008; Dinarello, 2009, 2011b; Garlanda et al., 2009; Gabay et al., 2010; Sims and Smith, 2010; Voronov et al., 2013).
The differential effects of these molecules on angiogenesis are discussed in this review.
The effects of IL-1 on ECs and its crosstalk with pro-angiogenic molecules
ECs are the main cells that are involved in both the normal and pathological angiogenic response. Multiple studies have shown in vitro effects of recombinant IL-1 on parameters related to the physiology of ECs, including their activation, as evidenced by morphological changes, increased migration and proliferation and ultimately organization into tube-like structures (reviewed in Voronov et al., 2007, 2010a, 2013; Apte and Voronov, 2008). As IL-1 is a strong activator of ECs, it induces profound changes in gene expression and function that allow these cells to participate actively in inflammatory reactions, immunity, and blood vessel formation. For example, IL-1β induces morphological transformation in human dermal micro-vascular endothelial cells (HDMECs), accompanied by an increased growth rate, loss of contact inhibition, and an increase in the permeability of confluent EC monolayers (Bokhari et al., 2006). Nevertheless, it is not known whether the described effects of IL-1 on ECs are direct, due to its action, or indirect, due to IL-1-induced cytokines produced by ECs. It was found that in vitro, IL-1β increases expression of FGF2 in ECs through activation of NF-κB (Lee and Kay, 2012) and also induces expression of different chemokines, cytokines, direct angiogenic factors, and adhesion molecules on ECs (Breviario et al., 1990; Sica et al., 1990; Kang et al., 2006). In addition, IL-1β interaction with IL-1R1 on ECs induces migration of the cells and tube formation, mainly via activation of p38-mitogen-activated protein kinase (MAPK) and MAPK-activated protein kinase 2 (Jagielska et al., 2012). Moreover, IL-1β up-regulates expression of VEGF and its receptors on ECs (Berse et al., 1999) or aortic smooth muscle cells (Stavri et al., 1995; Maruyama et al., 1999; Nasu et al., 2006) and VEGF secretion from these cells was significantly inhibited by the addition of IL-1Ra. In vivo, IL-1 also plays a synergistic, pro-angiogenic role with some pro-angiogenic factors (Friesel and Maciag, 1999). For example, the synergistic effects of VEGF and IL-1 in the up-regulation of genes of growth factors and inflammatory cytokines in ECs have been described and a 60% overlapping of genes induced by each of these cytokines was observed (Schweighofer et al., 2009). VEGF/IL-1 induced genes comprise mainly a group of genes with NFAT, as well as NF-κB binding sites in their promoters; VEGF-A preferentially uses NFAT and IL-1 uses NF-κB to induce these genes. It was also observed that both VEGF and IL-1β increase the permeability of ECs via a Src-dependent pathway (Sheikpranbabu et al., 2009).
Using a Matrigel model, we found that both rIL-1β and rVEGF induce each other and are both essential to induce an angiogenic response. Thus, neutralization of either of these cytokines abrogated the angiogenic response. The importance of the interaction between IL-1β and VEGF was also demonstrated by the weak angiogenic response observed in Matrigel plugs loaded with rVEGF in mice deficient in IL-1β or its signaling receptor (Carmi et al., 2013). Inhibition of VEGFR1 signaling in Matrigel plugs supplemented with recombinant cytokines (IL-1β or VEGF) abrogated angiogenesis, probably by inhibiting recruitment of bone marrow-derived (BMD) myeloid cells. Thus, in multiple studies, synergy of IL-1β and VEGF has been described; however, our studies demonstrate the necessity of both factors in angiogenic responses. IL-1β is also an essential factor for endothelial precursor cells (EPCs) to mature into ECs and this IL-1β-induced effect was facilitated by the addition of VEGF. For example, when either EPCs or myeloid circulating angiogenic cells from lupus patients were co-cultured with IL-1β and VEGF, their capacity to proliferate and differentiate into mature ECs was synergistically increased (Thacker et al., 2010).
There are fewer studies on the role of IL-1α in angiogenesis. We found that IL-1α has less potent pro-angiogenic effects compared to IL-1β in its recombinant form (unpublished results). Nevertheless, in vivo studies on the effects of IL-1α and IL-1β on angiogenesis revealed that both agonistic molecules are important, but probably work through different pathways. Thus, IL-1α can stimulate a high angiogenic response by recruiting macrophages that are an abundant source of FGF (reviewed in Sano et al., 1990; Brogi et al., 1993; Dinarello, 1996; Rider et al., 2011, 2012), or other VEGF-expressing inflammatory cells (Salven et al., 2002). In addition, a dose-dependent effect of IL-1α in de novo synthesis of VEGF by human peripheral mononuclear cells (PBMCs) was shown. This effect was blocked by treatment with VEGFR2 antibodies, whereas neutralization of VEGFR1 induced only marginal effects. Both in vitro and in vivo, IL-1α can stimulate ECs to secrete IL-8 and during in vivo angiogenesis, the source of IL-1 can be from PBMCs or activated platelets (Kaplanski et al., 1994a,b). IL-1α is also released from ECs following stress signals, such as starvation or TNF activation (Berda-Haddad et al., 2011) and in addition, IL-1α activates ECs to express CXCL1, VCAM-1, and ICAM-1, thus promoting trans-endothelial-migration of inflammatory cells (Thornton et al., 2010).
IL-1 in inflammation-induced angiogenesis
In the last decade, the link between inflammation and angiogenesis has become recognized. Extensive angiogenesis was observed in chronic inflammation, for example in rheumatoid arthritis and inflammatory bowel disease (Folkman, 2001, 2003). In both acute and chronic inflammation, functional changes in the vasculature were found. In acute inflammation, these changes include an increase in permeability, extensive EC mitotic activity, and remodeling of capillaries. Upon chronic inflammation, vascular dilation and an increase in capillary density was observed. During inflammation, ECs actively recruit immune cells from the circulation into the underlying tissue, where they play a role in angiogenesis. An increase in growth factor and cytokine production, due to inflammation, also leads to proliferation of ECs.
The role of the IL-1 molecules in promoting inflammation-induced angiogenesis was studied by us, using the model of Matrigel plugs supplemented with supernatants of hypoxic macrophages, as such, or after activation with LPS as an inflammatory stimulus (Carmi et al., 2009). We showed that neutralization of IL-1 in supernatants of hypoxic macrophages, particularly IL-1β, completely abrogated cell infiltration and angiogenesis in Matrigel plugs, concomitant to dramatically reduced VEGF levels. Supernatants from macrophages of IL-1β knockout (KO) mice did not induce this inflammatory or angiogenic response. The importance of IL-1 signaling in the host was demonstrated by the dramatic reduction of inflammation-induced angiogenic responses in Matrigel plugs that contained supernatants derived from WT macrophages implanted in IL-1RI KO mice. Using the aortic sprouting assay, it was shown that IL-1 does not directly activate EC migration, proliferation, and organization into blood vessel-like structures, but rather activates infiltrating myeloid cells to produce a cascade of cytokines/chemokines, which further activate tissue resident ECs to produce direct pro-angiogenic factors, such as VEGF (Carmi et al., 2009). Similarly, Nakao et al., demonstrated that ectopic expression of IL-1β in the cornea induces increased angiogenesis, which is dependent on infiltration of COX-2-positive macrophages that activate the angiogenic process in a complex manner (Nakao et al., 2005). An example of an indirect effect of IL-1β on angiogenesis is the pro-angiogenic activity of osteopontin-treated monocytes on chick embryo chorioallantoic membranes that is completely abrogated by neutralization with anti-IL-1β antibodies (Naldini et al., 2006). In addition, systemic treatment with IL-1Ra prevented the formation of new blood vessels in corneas impregnated with VEGF or basic FGF (Coxon et al., 2002). In conclusion, IL-1, most probably the actively secreted form of IL-1β, functions directly or indirectly on myeloid infiltrating cells, as well as ECs, and thus regulates inflammation-induced angiogenic responses.
IL-1 molecules play a key role in the tumor microenvironment
The induction of angiogenesis is considered to be a hallmark of cancer and is important for tumor growth and dissemination (Folkman, 1995, 2001, 2003; Folkman and D'Amore, 1996). An initially developing tumor is dormant until it undergoes the angiogenic switch, as initially described by Folkman. During this switch, the balance between the pro-angiogenic and anti-angiogenic factors changes and angiogenic stimuli are predominant. One of the major mechanisms of the angiogenic switch is enhanced expression and secretion of angiogenic factors, mainly VEGF, by the malignant cells. However, the mechanisms of the angiogenic switch are not completely elucidated, as detectable tumors usually have undergone this switch and already have an established vascular network. As indicated, tumor vascularization occurs through “classical” angiogenesis, as well as via vasculogenesis, which involves cells recruited from the BM (Carmeliet, 2005; De Palma et al., 2005; Bertolini et al., 2006). Thus, circulating VEGFR2-positive cells were shown to be initiators of vasculogenesis; however, conflicting findings on their ability to incorporate into newly formed blood vessels at tumor sites have been reported (De Palma et al., 2003; Gothert et al., 2004; Peters et al., 2005; Purhonen et al., 2008; Carmi et al., 2013). EPCs were initially described by Asahara, who reported on their ability to repair angiogenesis in ischemic tissues (Asahara et al., 1999). It was found that IL-1β can mobilize EPCs in a VEGF-dependent manner by regulation of VEGF and VEGFR2 expression on ECs, thus supporting neovascularization (Amano et al., 2004).
In further studies, a population of hematopoietic cells expressing endothelial markers, such as CD31, VEGFR2, and Tie2, was described and termed myeloid angiogenic cells (MACs) (Medina et al., 2010). These cells express high levels of pro-angiogenic and macrophage type 2 markers (Medina et al., 2011) and their function at the tumor site is still disputed. In some models, MACs were shown to differentiate into mature ECs that incorporate into the vasculature (Kawamoto et al., 2001; Urbich et al., 2003); however, upon intravitreal injection, MACs enhanced vascular repair of ischemic retinopathy, but preserved their original phenotype and did not differentiate into mature ECs. In addition, BMD cells of myeloid origin were shown to be essential for tumor-mediated angiogenesis. The proximity of BMD cells, including MACs, to ECs lining blood vessels suggests their paracrine role, facilitating the EC response during angiogenesis. These cells probably do not incorporate into blood vessels but function as supporting cells (Ziegelhoeffer et al., 2004).
Various populations of BMD myeloid cells are found in the tumor microenvironment. These include circulating monocytes, Tie-2 expressing monocytes (TEMs), myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs) and neutrophils (reviewed in Murdoch et al., 2008; Wels et al., 2008; Coffelt et al., 2010; Ferrara, 2010a; Ruffell et al., 2012; Sica et al., 2012; Chambers et al., 2013; De Palma and Lewis, 2013; Favre et al., 2013). The precise relationship among the myeloid cell types is uncertain and it is not known whether they act in the tumor microenvironment in redundant or specific manners. In various tumor experimental systems, diverse myeloid cell populations dominate. Mantovani et al. suggested a unifying hypothesis that myeloid cells at tumor sites actually represent a continuum of cell populations that all share properties of M2-macrophages, which promote tumor-mediated angiogenesis and progression (Mantovani and Sica, 2010; Allavena and Mantovani, 2012). Myeloid cells perform paracrine functions in the tumor-mediated environment, secreting angiogenic factors, and providing the inflammatory milieu for the angiogenic response. For example, MDSCs and TAMs secrete VEGF and MMP9, which increases the bioavailability of VEGF sequestered in the extracellular matrix by its proteolytic activity (reviewed in Murdoch et al., 2008; Mantovani et al., 2009; Gabrilovich et al., 2012).
IL-1, as a major hematopoietic and pro-inflammatory cytokine has dramatic effects on the generation of myeloid cells in the BM and on their recruitment to tumor sites, as well as on the in situ phenotype/function of these cells in the tumor microenvironment (reviewed in Apte and Voronov, 2008; Voronov et al., 2013). For example, in vitro, addition of IL-1 to BMCs increases secretion of VEGF and the total number of BMCs expressing CD34 or Flk-1 (Qin et al., 2006). Indeed, the absence of IL-1β in the tumor microenvironment limited the recruitment of FGF1-producing mononuclear cells to tumor sites (Prudovsky et al., 2003). Thus, it was suggested that IL-1 can affect BMD differentiation or induce them to secrete pro-inflammatory or angiogenic factors (reviewed in Apte and Voronov, 2008; Voronov et al., 2013), which are also involved in the recruitment of additional myeloid cells to tumor sites. Indeed, the importance of the VEGFR1/VEGF-A axis in recruiting myeloid cells that express VEGFR1 to tumor sites has been noted, and in mice deficient in VEGFR1, decreased recruitment of macrophages and other myeloid cells into tumors was observed (Duyndam et al., 2002). In addition, depletion of VEGFR1 in the renal cell carcinoma model leads to inhibition of macrophage recruitment to tumors (Li et al., 2011).
In our studies, the role of microenvironment-derived IL-1 on the recruitment of VEGFR1-positive BMD cells to tumor sites was confirmed. We characterized a new auto-induction circuit, IL-1β/VEGF, which acts via interactions between BMDs, mainly VEGFR1+/IL-1R1+ immature myeloid cells (MDSCs) and to a lesser extent macrophages, and tissue-resident ECs (Carmi et al., 2013). Myeloid cells do not directly stimulate ECs for migration, proliferation, and subsequent blood vessel formation. However, they produce IL-1β and a network of other pro-inflammatory cytokines/molecules, such as Bv8, CCL2 and CCL3, etc., which subsequently activate resting tissue-resident ECs to produce VEGF, as well as other direct pro-angiogenic factors, such as PlGF and bFGF. Thus, IL-1β provides the inflammatory microenvironment for angiogenesis and tumor progression. We have shown that IL-1β inhibition stably reduces tumor growth, by limiting inflammation and inducing the maturation of MDSCs into M1 macrophages, which do not promote tumor invasiveness and can be cytotoxic/cytostatic for tumor cells. In addition, M1 macrophages also serve as antigen-presenting cells for inducing anti-tumor immunity. These results suggest that IL-1β, apart from its ability to affect recruitment of myeloid cells, controls their maturation or activation for a pro-tumorigenic phenotype (Song et al., 2005; Bunt et al., 2006; Tu et al., 2008; Carmi et al., 2013). For example, Hagenmann et al. showed that IL-1R signaling in macrophages is essential for M2 polarization induced by conditioned medium of tumor cells (Hagemann et al., 2008). Other studies also showed the involvement of IL-1β in myeloid cell differentiation. Thus, IL-1β inhibits the skewing of myeloid cells present in proangiogenic cultures from circulating angiogenic cells to mature dendritic cells (DCs) (Mohty et al., 2003; Denny et al., 2007). In addition, IL-1β can impair maturation in DCs treated with rapamycin (Turnquist et al., 2008).
Our experimental system of tumor cells encapsulated in Matrigel plugs allows the study of the early angiogenic response and even the angiogenic switch. In this case, VEGF produced by ECs possibly synergizes with direct angiogenic factors secreted by the malignant cells. Traditionally, it was thought that ECs respond to VEGF during the angiogenic response and produce very little or undetectable amounts of VEGF (Kerbel, 2008). In other studies, using large, established tumors that are densely infiltrated by “activated” myeloid cells, it was shown that VEGF mainly originates from such myeloid cells and it controls tumor angiogenesis (reviewed in Apte and Voronov, 2008; Murdoch et al., 2008; Coffelt et al., 2010; Ferrara, 2010a; Mantovani and Sica, 2010; Qian and Pollard, 2010; Ruffell et al., 2010; Allavena and Mantovani, 2012; Gabrilovich et al., 2012). In the early stages of tumor development, as assessed in our studies, myeloid cells are probably not yet sufficiently activated to produce VEGF. Thus, at different stages of tumor progression, VEGF is secreted by diverse microenvironmental cells and it acts together with angiogenic factors of tumor cell origin.
Results of our studies have shown that IL-1β is a major mediator in the tumor microenvironment, which plays a crucial role in the angiogenic response. IL-1β acts together with VEGF in mounting and maintaining tumor-mediated angiogenesis.
The differential effects of IL-1 molecules in tumor-induced angiogenesis and invasiveness
High levels of IL-1 molecules are found in experimental tumor models and in human malignancies and IL-1 has been implicated as a key factor in tumor progression. IL-1 exerts its proliferative and angiogenic effects in the tumor microenvironment, mainly via interaction with stromal, inflammatory, as well as the malignant cells, stimulating tumor cell proliferation and invasion through autocrine or paracrine loops. For example, IL-1β induces secretion of chemokines by both tumor and tumor-microenvironmental cells and thus promotes cancer invasiveness (Portier et al., 1993; Suswam et al., 2005; Apte et al., 2006a; Naldini et al., 2010). A few studies have documented constitutive IL-1β protein production in human and animal cancer cell lines, including sarcomas and ovarian cell carcinomas (Dinarello, 1996; Lewis et al., 2006). In patients, overexpression of IL-1β has been described in solid tumors, including breast, colon, lung, head and neck cancers, and melanomas. In some tumors, IL-1, as well as other pro-inflammatory cytokines, is induced by oncogenes that transform the cell, thus providing the microenvironment for invasiveness of the malignant cells. In other types of tumors, IL-1 is induced only in the invasiveness and metastasis phases. The switch-on of IL-1 genes in malignant cells is induced by genetic alterations and possibly also by microenvironment-derived signals. Indeed, high IL-1 concentrations within the tumor microenvironment have been reported in numerous studies in cancer patients and experimental models and are associated with a more virulent tumor phenotype (reviewed in Elaraj et al., 2006; Lewis et al., 2006).
In the tumor milieu, IL-1 induces expression of various metastatic mediators, such as matrix metalloproteinases (MMP), VEGF, IL-8, IL-6, TNFα, and TGFβ (reviewed in Dinarello, 1996; Apte and Voronov, 2002, 2008; Apte et al., 2006b; Lewis et al., 2006; Voronov et al., 2013). Previously, in a transgenic model of Myc-dependent carcinogenesis, IL-1β was characterized as the principal effector molecule in the onset of angiogenesis, via MMP-mediated sequestration of extracellular matrix-associated VEGF and its further ligation to its cognate receptor on ECs (Shchors et al., 2006).
Our studies throughout the years have assessed the role of the IL-1 molecules in different phases of the malignant process, such as carcinogenesis, tumor angiogenesis, and invasiveness (Song et al., 2003, 2005; Krelin et al., 2007). Using a transplantable fibrosarcoma cell line transfected with cDNAs of the active forms of IL-1β, i.e., the mature form of IL-1β or the mature form of IL-1β ligated to a signal sequence (ssIL-1β), in which IL-1β is actively secreted through the endoplasmic reticulum-Golgi pathway, we observed that invasiveness of the different tumor cell lines was directly proportional to the amount of IL-1β secreted by the malignant cells (Song et al., 2003, 2005). In this system, invasiveness correlated with increased angiogenesis and accumulation of MDSCs in the spleen and tumor, which leads to general anergy. Similar observations were described in other experimental systems using IL-1β-transfected tumor cells (Saijo et al., 2002; Nakao et al., 2005; Bunt et al., 2006). We have hypothesized that initially small amounts of tumor cell-derived IL-1β induce a local inflammatory response, which subsequently recruits and activates BMDs that further secrete IL-1β, as well as an entire cytokine network that promotes tumor-mediated angiogenesis and tumor progression.
In various tumor models, we found that IL-1β induces increased tumor growth, invasiveness, and angiogenesis, when compared to IL-1α (Song et al., 2003; Voronov et al., 2003, 2010b). However, in some tumor cells, active secretion of IL-1α is observed (Apte and Voronov, 2008; Voronov et al., 2013).
In some tumors, such as breast carcinoma or pancreatic cancer, a pro-angiogenic signature correlated with IL-1α signaling (Matsuo et al., 2009; Guo and Gonzalez-Perez, 2011; Zhou et al., 2011). In these studies, IL-1α was shown to be involved in the induction of VEGF/VEGFR2 in the tumor microenvironment, through a cascade involving leptin/Notch. Secreted IL-1α may promote invasiveness in a similar manner to secreted IL-1β in these tumors.
However, in most malignancies, IL-1 is produced not only by tumor cells, but by other cells, such as neutrophils, macrophages, and MDSCs, recruited to the tumor microenvironment (reviewed in Apte and Voronov, 2008; Witz, 2008; Balkwill and Mantovani, 2010; Demaria et al., 2010; Grivennikov et al., 2010; Hanahan and Weinberg, 2011; Voronov et al., 2013).
These recruited cells thereby promote inflammation in the tumor microenvironment and thus increase the angiogenic response, tumor cell invasiveness and metastasis (Goldberg and Schwertfeger, 2010; Naldini et al., 2010; Schmid et al., 2011; Carmi et al., 2013).
Using IL-1 KO mice, we demonstrated that microenvironment-derived IL-1β, and to a much lesser extent IL-1α, is responsible for in vivo tumor angiogenesis and invasiveness of B16 melanoma cells. Significantly decreased angiogenesis was observed in IL-1β deficient mice injected with either melanoma cells or melanoma cell-containing Matrigel plugs, when compared to wild-type mice. In IL-1α KO mice, tumor growth and angiogenesis in Matrigel plugs were observed to a lesser extent than in WT mice, but higher than those in IL-1β KO mice (Voronov et al., 2003). Addition of recombinant IL-1 into Matrigel plugs containing B16 cells in IL-1β KO mice partially restored the angiogenic response, while addition of IL-1Ra to B16-containing Matrigel plugs in wild-type mice inhibited the ingrowth of the blood vessel network into the plugs. In subsequent studies, the angiogenic potential of IL-1 was confirmed by Elaraj et al., who observed high expression of IL-1 mRNA in more than half of all tested metastatic human tumor specimens, including non-small-cell lung carcinoma, colorectal adenocarcinoma, and melanoma tumor samples. Supernatants from IL-1 expressing cell lines induced a significant increase in EC monolayer permeability, a hallmark event in early angiogenesis. Systemic treatment with recombinant IL-1Ra resulted in significant inhibition of xenograft growth and neovessel density of IL-1-producing tumor cell lines. Nevertheless, the addition of IL-1Ra did not have any effect in non-IL-1-producing tumor lines (Elaraj et al., 2006).
Anti-IL-1 therapy in anti-tumor approaches
Since tumor development is related to and dependent on the angiogenic response, anti-angiogenic therapy targeting blood vessel formation was used to suppress existing tumors and to prevent metastasis. Various studies used different anti-angiogenic molecules or agents to limit tumor angiogenesis. The main molecule used in these experiments was VEGF, which is the most abundant pro-angiogenic molecule. A number of preclinical studies demonstrated a significant inhibition of tumor growth in various types of cancer by blocking VEGF (Ferrara and Kerbel, 2005). VEGF has been approved by the FDA to be used in cancer patients to inhibit tumor growth. Anti-VEGF monoclonal antibodies (bevacizumab, also called Avastin) were tested in various metastatic cancers in humans, usually in combination with chemotherapy (Ferrara et al., 2005; Carmeliet and Jain, 2011). However, some patients are refractory to this treatment and tumor recurrence or even metastases were observed after initial tumor shrinkage. It is beyond the scope of this review to summarize the effects of anti-VEGF therapy in cancer patients. In animal models, it was also shown that termination or interruption of VEGF neutralization induces rapid vascular regrowth in tumors and limits tumor-suppressive effects of VEGF treatment (reviewed in Kubota, 2012; Bellou et al., 2013). The dramatic response of the tumor microenvironment to VEGF inhibition may be due to the key role VEGF plays in normal cell viability, including ECs, myeloid cells, and possibly other cell types involved in tissue homeostasis (Chung and Ferrara, 2011; Lazarus and Keshet, 2011; Luo et al., 2011; Potente et al., 2011). For example, blocking VEGF induces EC apoptosis, resulting in the loss of integrity of blood vessels, and disturbance of tissue homeostasis. Due to these effects, VEGF blockage damages not only tumor blood vessels, but also healthy vessels, thus occasionally resulting in severe complications, such as hemorrhagic or thrombotic events (Verheul and Pinedo, 2007).
Different mechanisms have been proposed to explain rebound angiogenesis following VEGF neutralization (Casanovas et al., 2005; Ferrara, 2010b; Chung and Ferrara, 2011; Carmi et al., 2013). For example, intrinsic mechanisms, such as the adaptation of malignant cells to low VEGF levels by selecting tumor cell variants that are less dependent on VEGF for their survival/proliferation or variants with enhanced expression/secretion of other pro-angiogenic factors, such as bFGF or PlGF have been described (Casanovas et al., 2005; Fischer et al., 2007; Kubota, 2012). Following VEGF neutralization, extrinsic mechanisms also contribute to tumor recurrence by altering microenvironment cells to express different or additional angiogenic factors, thus facilitating tumor recurrence. These include induced secretion of Bv8 by MDSCs and a subsequent VEGF-independent angiogenic response (Shojaei et al., 2007; Carmi et al., 2013) or expression of PDGF-C in cancer-associated fibroblasts (Bergers and Hanahan, 2008; Crawford et al., 2009). In our studies, we described a novel mechanism of rebound angiogenesis after VEGF inhibition in Matrigel plugs containing tumor cells (Carmi et al., 2013). This resulted in significant and consistently elevated expression of direct angiogenic factors, such as bFGF, PlGF, and PDGF, as well as VEGF in VEGFR1+ immature myeloid cells, mostly MDSCs. The re-programming of VEGFR1+ immature myeloid cells into active pro-angiogenic cells also involves increased expression of hypoxia-inducible factor 1α, a key transcription factor regulating angiogenesis (Cramer and Johnson, 2003; Semenza, 2011). In contrast, in “uninterrupted angiogenesis” (without anti-VEGF treatment), as described above, ECs, rather than myeloid cells, are the major cells that produce VEGF and other direct angiogenic factors (Carmi et al., 2013).
In recent years, myeloid, cell-driven angiogenesis was shown to contribute to refractoriness or resistance to VEGF inhibition. For example, as discussed above, MDSCs can secrete Bv8, which further upregulates granulocyte colony-stimulating factor that promotes tumor growth and angiogenesis in a VEGF-independent manner. Therefore, intervention in myeloid cell recruitment to tumor sites in order to modulate differentiation or neutralization of specific pro-inflammatory or pro-angiogenic factors secreted by these cells, are now widely discussed. For example, blocking CSF-1 was shown to be effective in suppressing tumor angiogenesis (Kubota, 2012). In our studies, we found that blocking a major, alarm, pro-inflammatory molecule derived from myeloid cells, i.e., IL-1β can lead to a significant amelioration of tumor angiogenesis (Voronov et al., 2003; Carmi et al., 2013). The inhibition of IL-1β does not affect the healthy vascular system or tissue homeostasis and is more suitable for anti-angiogenic therapy than anti-VEGF approaches. IL-1β neutralization largely inhibited infiltration of myeloid cells, which are obligatory for tumor-mediated angiogenesis. Furthermore, IL-1β neutralization induces maturation of immature myeloid cells into anti-invasive M1 macrophages.
We recently found that IL-1β neutralization inhibited pro-inflammatory genes, such as Bv8, CCL2 and CCL3 in myeloid cells, and did not significantly alter the expression of angiogenic factors. IL-1β inhibition also reduced in vivo tumor development in mice injected subcutaneously with tumor cells. This inhibition was long-lasting and without recurrence a contrast to that observed with anti-VEGF (Carmi et al., 2013).
These results have provided the rationale to use IL-1β neutralization as a treatment for tumor-mediated angiogenesis, and subsequent tumor progression.
Indeed, the positive role IL-1Ra plays in reducing the angiogenic response and in attenuating tumor progression has been demonstrated by us (Song et al., 2003; Voronov et al., 2003) and by others (Saijo et al., 2002; Nakao et al., 2005; Bunt et al., 2006; Schmid et al., 2011). IL-1 neutralization agents are available at the present time (Dinarello, 2010a,b; Dinarello and van der Meer, 2012). The IL-1Ra, also called Anakirna (Kineret; Amgen/Biovitrum) has been shown to be safe and effective in alleviating symptoms of rheumatoid arthritis and other auto-inflammatory diseases. Continuous delivery systems of recombinant IL-1Ra or cells over-expressing IL-1Ra, encapsulated within alginate-poly (L-lysine)-alginate (APA) microspheres, reduced the tumor burden and inhibited tumor-mediated angiogenesis when implanted into tumor-bearing mice (Bar et al., 2004; Lavi et al., 2007). Similarly, over-expression of the IL-1Ra in human melanoma cell lines expressing endogenous IL-1, inhibits tumor growth and metastasis in human melanoma xenografts in nude mice through its effects on the microenvironment (Weinreich et al., 2003; Elaraj et al., 2006). Altogether, these results provide pre-clinical support for the use of IL-1Ra in cancer therapy.
Recently, some novel IL-1 neutralization agents, such as humanized anti-IL-1β, an IL-1 trap, as well as other agents that indirectly inhibit IL-1β production, have been developed (Dinarello, 2011b). These agents are now ready for testing in cancer patients. Optimally, IL-1 neutralization should be most effective in patients with minimal residual disease (MRD), to prevent tumor-induced angiogenesis, recurrence, and metastasis.
Conclusions
Finally, this review demonstrated the important role of the IL-1 family molecules, especially IL-1β, in the tumor microenvironment. IL-1β represents a major upstream cytokine that controls the local pro-inflammatory cascade and thereby affects the balance between protective immunity and destructive inflammation. IL-1 modulates diverse cells in the tumor microenvironment and acts together with VEGF in mounting and maintaining tumor-mediated angiogenesis (Figure 1). Thus, its neutralization should reduce tumor progression and invasiveness via its affect on pro-tumorigenic cells, and on the tumor-induced angiogenic switch. Further characterization of the optimal conditions for IL-1 neutralization could lead to the application of anti-IL-1 approaches in cancer therapy.
Figure 1.
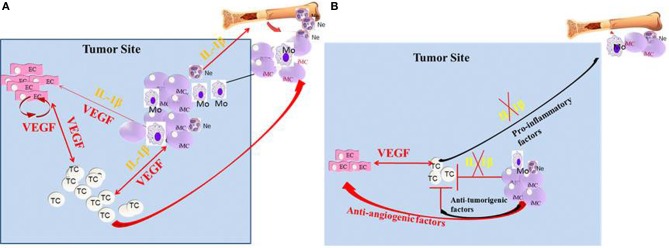
Crosstalk between IL-1β and VEGF in the tumor microenvironment. (A) In the tumor microenvironment, IL-1β can be expressed initially by the malignant cells. In turn, IL-1β can recruit myeloid cells from the BM and further activate them to secrete pro-inflammatory and pro-angiogenic molecules, such as VEGF and IL-1β. Furthermore, IL-1β keeps myeloid cells in their immature stage, where they are pro-invasive and immunosuppressive. IL-1β can also activate tissue-resident ECs to produce VEGF and other pro-angiogenic factors. The malignant cells can also be activated by IL-1β for increased invasiveness. VEGF in the tumor microenvironment activates ECs and myeloid cells and also has homeostatic effects on these cells. (B) In the absence of IL-1β, inflammation, as well as VEGF production is diminished, which results in reduced invasiveness. Lack of IL-1β in the tumor microenvironment also induces the maturation of MDSCs into anti-tumor M1 macrophages.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank their collaborators: Prof. Charles A. Dinarello, University of Colorado, Denver; Prof. Yochiro Iwakura, University of Tokyo, Tokyo, Japan; Prof. Hynda K. Kleinman, NIH, Bethesda, MD, USA, and Dr Monika Huszar, Kaplan Hospital, Rehovot, Israel. We would like to thank our technician, Mrs. Malka White for her devoted technical and editorial help, as well as to our students, whose work is discussed in this review. Ron N. Apte was supported by the following grants: FP7: Cancer and Inflammation (INFLA-CARE), Israel Ministry of Science (MOS) jointly with the Deutsches Krebsforschungscentrum (DKFZ), Heidelberg, Germany, the Israel Science Foundation funded by the Israel Academy of Sciences and Humanities, the United States-Israel Bi-national Foundation (BSF), the Israel Cancer Association and the Israel Ministry of Health Chief Scientist's Office. Prof. Ron N. Apte is an incumbent of the Irving Isaac Sklar Chair in Endocrinology and Cancer, Ben Gurion University of the Negev. Elena Voronov was supported by the FP7: Cancer and Inflammation and the Israel Science Foundation, Israel Cancer Association, the Israel Ministry of Health Chief Scientist's Office, the United States-Israel Bi-national Foundation (BSF), and the Israel Science Foundation funded by the Israel Academy of Sciences and Humanities.
Glossary
Abbreviations
- IL-1
Interleukin-1
- IL-1Ra
Interleukin-1 receptor antagonist
- VEGF
Vascular endothelial growth factor
- VEGFR
Vascular endothelial growth factor receptor
- TNF
Tumor necrosis factor
- TGFβ
Tumor growth factor beta
- PlGF
Placental growth factor
- bFGF
Basic Fibroblast growth factor.
References
- Afonina I. S., Tynan G. A., Logue S. E., Cullen S. P., Bots M., Luthi A. U., et al. (2011). Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1alpha. Mol. Cell 44, 265–278 10.1016/j.molcel.2011.07.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alitalo K., Carmeliet P. (2002). Molecular mechanisms of lymphangiogenesis in health and disease. Cancer Cell 1, 219–227 10.1016/S1535-6108(02)00051-X [DOI] [PubMed] [Google Scholar]
- Allavena P., Mantovani A. (2012). Immunology in the clinic review series; focus on cancer: tumour-associated macrophages: undisputed stars of the inflammatory tumour microenvironment. Clin. Exp. Immunol. 167, 195–205 10.1111/j.1365-2249.2011.04515.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano K., Okigaki M., Adachi Y., Fujiyama S., Mori Y., Kosaki A., et al. (2004). Mechanism for IL-1 beta-mediated neovascularization unmasked by IL-1 beta knock-out mice. J. Mol. Cell. Cardiol. 36, 469–480 10.1016/j.yjmcc.2004.01.006 [DOI] [PubMed] [Google Scholar]
- Apte R. N., Dotan S., Elkabets M., White M. R., Reich E., Carmi Y., et al. (2006b). The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 25, 387–408 10.1007/s10555-006-9004-4 [DOI] [PubMed] [Google Scholar]
- Apte R. N., Krelin Y., Song X., Dotan S., Recih E., Elkabets M., et al. (2006a). Effects of micro-environment- and malignant cell-derived interleukin-1 in carcinogenesis, tumour invasiveness and tumour-host interactions. Eur. J. Cancer 42, 751–759 10.1016/j.ejca.2006.01.010 [DOI] [PubMed] [Google Scholar]
- Apte R. N., Voronov E. (2002). Interleukin-1-a major pleiotropic cytokine in tumor-host interactions. Semin. Cancer Biol. 12, 277–290 10.1016/S1044-579X(02)00014-7 [DOI] [PubMed] [Google Scholar]
- Apte R. N., Voronov E. (2008). Is interleukin-1 a good or bad “guy” in tumor immunobiology and immunotherapy? Immunol. Rev. 222, 222–241 10.1111/j.1600-065X.2008.00615.x [DOI] [PubMed] [Google Scholar]
- Arend W. P. (2002). The balance between IL-1 and IL-1Ra in disease. Cytokine Growth Factor Rev. 13, 323–340 10.1016/S1359-6101(02)00020-5 [DOI] [PubMed] [Google Scholar]
- Asahara T., Masuda H., Takahashi T., Kalka C., Pastore C., Silver M., et al. (1999). Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 85, 221–228 10.1161/01.RES.85.3.221 [DOI] [PubMed] [Google Scholar]
- Balkwill F. (2002). Tumor necrosis factor or tumor promoting factor? Cytokine Growth Factor Rev. 13, 135–141 10.1016/S1359-6101(01)00020-X [DOI] [PubMed] [Google Scholar]
- Balkwill F., Mantovani A. (2010). Cancer and inflammation: implications for pharmacology and therapeutics. Clin. Pharmacol. Ther. 87, 401–406 10.1038/clpt.2009.312 [DOI] [PubMed] [Google Scholar]
- Balkwill F. R., Mantovani A. (2012). Cancer-related inflammation: common themes and therapeutic opportunities. Semin. Cancer Biol. 22, 33–40 10.1016/j.semcancer.2011.12.005 [DOI] [PubMed] [Google Scholar]
- Bar D., Apte R. N., Voronov E., Dinarello C. A., Cohen S. (2004). A continuous delivery system of IL-1 receptor antagonist reduces angiogenesis and inhibits tumor development. FASEB. J. 18, 161–163 10.1096/fj.03-0483fje [DOI] [PubMed] [Google Scholar]
- Bellou S., Pentheroudakis G., Murphy C., Fotsis T. (2013). Anti-angiogenesis in cancer therapy: hercules and hydra. Cancer Lett. 338, 219–228 10.1016/j.canlet.2013.05.015 [DOI] [PubMed] [Google Scholar]
- Berda-Haddad Y., Robert S., Salers P., Zekraoui L., Farnarier C., Dinarello C. A., et al. (2011). Sterile inflammation of endothelial cell-derived apoptotic bodies is mediated by interleukin-1alpha. Proc. Natl. Acad. Sci. U.S.A. 108, 20684–20689 10.1073/pnas.1116848108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G., Hanahan D. (2008). Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 8, 592–603 10.1038/nrc2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berse B., Hunt J. A., Diegel R. J., Morganelli P., Yeo K., Brown F., et al. (1999). Hypoxia augments cytokine (transforming growth factor-beta (TGF-beta) and IL-1)-induced vascular endothelial growth factor secretion by human synovial fibroblasts. Clin. Exp. Immunol. 115, 176–182 10.1046/j.1365-2249.1999.00775.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolini F., Shaked Y., Mancuso P., Kerbel R. S. (2006). The multifaceted circulating endothelial cell in cancer: towards marker and target identification. Nat. Rev. Cancer 6, 835–845 10.1038/nrc1971 [DOI] [PubMed] [Google Scholar]
- Bokhari S. M., Zhou L., Karasek M. A., Paturi S. G., Chaudhuri V. (2006). Regulation of skin microvasculature angiogenesis, cell migration, and permeability by a specific inhibitor of PKCalpha. J. Invest. Dermatol. 126, 460–467 10.1038/sj.jid.5700071 [DOI] [PubMed] [Google Scholar]
- Braddock M., Quinn A. (2004). Targeting IL-1 in inflammatory disease: new opportunities for therapeutic intervention. Nat. Rev. Drug Discov. 3, 330–339 10.1038/nrd1342 [DOI] [PubMed] [Google Scholar]
- Breviario F., Proserpio P., Bertocchi F., Lampugnani M. G., Mantovani A., Dejana E. (1990). Interleukin-1 stimulates prostacyclin production by cultured human endothelial cells by increasing arachidonic acid mobilization and conversion. Arteriosclerosis 10, 129–134 10.1161/01.ATV.10.1.129 [DOI] [PubMed] [Google Scholar]
- Brogi E., Winkles J. A., Underwood R., Clinton S. K., Alberts G. F., Libby P. (1993). Distinct patterns of expression of fibroblast growth factors and their receptors in human atheroma and nonatherosclerotic arteries. Association of acidic FGF with plaque microvessels and macrophages. J. Clin. Invest. 92, 2408–2418 10.1172/JCI116847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunt S. K., Sinha P., Clements V. K., Leips J., Ostrand-Rosenberg S. (2006). Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J. Immunol. 176, 284–290 [DOI] [PubMed] [Google Scholar]
- Carmeliet P. (2003). Angiogenesis in health and disease. Nat. Med. 9, 653–660 10.1038/nm0603-653 [DOI] [PubMed] [Google Scholar]
- Carmeliet P. (2005). Angiogenesis in life, disease and medicine. Nature 438, 932–936 10.1038/nature04478 [DOI] [PubMed] [Google Scholar]
- Carmeliet P., Jain R. K. (2000). Angiogenesis in cancer and other diseases. Nature 407, 249–257 10.1038/35025220 [DOI] [PubMed] [Google Scholar]
- Carmeliet P., Jain R. K. (2011). Molecular mechanisms and clinical applications of angiogenesis. Nature 473, 298–307 10.1038/nature10144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmi Y., Dotan S., Rider P., Kaplanov I., White M. R., Baron R., et al. (2013). The role of IL-1beta in the early tumor cell-induced angiogenic response. J. Immunol. 190, 3500–3509 10.4049/jimmunol.1202769 [DOI] [PubMed] [Google Scholar]
- Carmi Y., Voronov E., Dotan S., Lahat N., Rahat M. A., Fogel M., et al. (2009). The role of macrophage-derived IL-1 in induction and maintenance of angiogenesis. J. Immunol. 183, 4705–4714 10.4049/jimmunol.0901511 [DOI] [PubMed] [Google Scholar]
- Casanovas O., Hicklin D. J., Bergers G., Hanahan D. (2005). Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 8, 299–309 10.1016/j.ccr.2005.09.005 [DOI] [PubMed] [Google Scholar]
- Chambers S. E., O'Neill C. L., O'Doherty T. M., Medina R. J., Stitt A. W. (2013). The role of immune-related myeloid cells in angiogenesis. Immunobiology 218, 1370–1375 10.1016/j.imbio.2013.06.010 [DOI] [PubMed] [Google Scholar]
- Chung A. S., Ferrara N. (2011). Developmental and pathological angiogenesis. Annu. Rev. Cell Dev. Biol. 27, 563–584 10.1146/annurev-cellbio-092910-154002 [DOI] [PubMed] [Google Scholar]
- Coffelt S. B., Lewis C. E., Naldini L., Brown J. M., Ferrara N., De Palma M. (2010). Elusive identities and overlapping phenotypes of proangiogenic myeloid cells in tumors. Am. J. Pathol. 176, 1564–1576 10.2353/ajpath.2010.090786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coxon A., Bolon B., Estrada J., Kaufman S., Scully S., Rattan A., et al. (2002). Inhibition of interleukin-1 but not tumor necrosis factor suppresses neovascularization in rat models of corneal angiogenesis and adjuvant arthritis. Arthritis Rheum. 46, 2604–2612 10.1002/art.10546 [DOI] [PubMed] [Google Scholar]
- Cramer T., Johnson R. S. (2003). A novel role for the hypoxia inducible transcription factor HIF-1alpha: critical regulation of inflammatory cell function. Cell Cycle 2, 192–193 10.4161/cc.2.3.402 [DOI] [PubMed] [Google Scholar]
- Crawford Y., Kasman I., Yu L., Zhong C., Wu X., Modrusan Z., et al. (2009). PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell 15, 21–34 10.1016/j.ccr.2008.12.004 [DOI] [PubMed] [Google Scholar]
- Demaria S., Pikarsky E., Karin M., Coussens L. M., Chen Y. C., El-Omar E. M., et al. (2010). Cancer and inflammation: promise for biologic therapy. J. Immunother. 33, 335–351 10.1097/CJI.0b013e3181d32e74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny M. F., Thacker S., Mehta H., Somers E. C., Dodick T., Barrat F. J., et al. (2007). Interferon-alpha promotes abnormal vasculogenesis in lupus: a potential pathway for premature atherosclerosis. Blood 110, 2907–2915 10.1182/blood-2007-05-089086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Palma M., Lewis C. E. (2013). Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 23, 277–286 10.1016/j.ccr.2013.02.013 [DOI] [PubMed] [Google Scholar]
- De Palma M., Venneri M. A., Galli R., Sergi Sergi L., Politi L. S., Sampaolesi M, et al. (2005). Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 8, 211–226 10.1016/j.ccr.2005.08.002 [DOI] [PubMed] [Google Scholar]
- De Palma M., Venneri M. A., Naldini L. (2003). In vivo targeting of tumor endothelial cells by systemic delivery of lentiviral vectors. Hum. Gene Ther. 14, 1193–1206 10.1089/104303403322168028 [DOI] [PubMed] [Google Scholar]
- Dinarello C. A. (1996). Biologic basis for interleukin-1 in disease. Blood 87, 2095–2147 [PubMed] [Google Scholar]
- Dinarello C. A. (2002). The IL-1 family and inflammatory diseases. Clin. Exp. Rheumatol. 20, S1–13 [PubMed] [Google Scholar]
- Dinarello C. A. (2006). Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am. J. Clin. Nutr. 83, 447S-455S [DOI] [PubMed] [Google Scholar]
- Dinarello C. A. (2009). Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 27, 519–550 10.1146/annurev.immunol.021908.132612 [DOI] [PubMed] [Google Scholar]
- Dinarello C. A. (2010a). Anti-inflammatory agents: present and future. Cell 140, 935–950 10.1016/j.cell.2010.02.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello C. A. (2010b). Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev. 29, 317–329 10.1007/s10555-010-9229-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello C. A. (2011a). Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117, 3720–3732 10.1182/blood-2010-07-273417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello C. A. (2011b). A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur. J. Immunol. 41, 1203–1217 10.1002/eji.201141550 [DOI] [PubMed] [Google Scholar]
- Dinarello C. A. S. A., van der Meer J. W. (2012). Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 11, 633–652 10.1038/nrd3800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo N. C., Shayakhmetov D. M. (2013). Interleukin-1 receptor 2 keeps the lid on interleukin-1alpha. Immunity 38, 203–205 10.1016/j.immuni.2013.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyndam M. C., Hilhorst M. C., Schluper H. M., Verheul H. M., van Diest P. J., Kraal G., et al. (2002). Vascular endothelial growth factor-165 overexpression stimulates angiogenesis and induces cyst formation and macrophage infiltration in human ovarian cancer xenografts. Am. J. Pathol. 160, 537–548 10.1016/S0002-9440(10)64873-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenbarth S. C., Flavell R. A. (2009). Innate instruction of adaptive immunity revisited: the inflammasome. EMBO Mol. Med. 1, 92–98 10.1002/emmm.200900014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elaraj D. M., Weinreich D. M., Varghese S., Puhlmann M., Hewitt S. M., Carroll N. M., et al. (2006). The role of interleukin 1 in growth and metastasis of human cancer xenografts. Clin. Cancer Res. 12, 1088–1096 10.1158/1078-0432.CCR-05-1603 [DOI] [PubMed] [Google Scholar]
- Favre J., Terborg N., Horrevoets A. J. (2013). The diverse identity of angiogenic monocytes. Eur. J. Clin. Invest. 43, 100–107 10.1111/eci.12009 [DOI] [PubMed] [Google Scholar]
- Ferrara N. (2010a). Role of myeloid cells in vascular endothelial growth factor-independent tumor angiogenesis. Curr. Opin. Hematol. 17, 219–224 10.1097/MOH.0b013e3283386660 [DOI] [PubMed] [Google Scholar]
- Ferrara N. (2010b). Pathways mediating VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev. 21, 21–26 10.1016/j.cytogfr.2009.11.003 [DOI] [PubMed] [Google Scholar]
- Ferrara N., Hillan K. J., Novotny W. (2005). Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem. Biophys. Res. Commun. 333, 328–335 10.1016/j.bbrc.2005.05.132 [DOI] [PubMed] [Google Scholar]
- Ferrara N., Kerbel R. S. (2005). Angiogenesis as a therapeutic target. Nature 438, 967–974 10.1038/nature04483 [DOI] [PubMed] [Google Scholar]
- Fischer C., Jonckx B., Mazzone M., Zacchigna S., Loges S., Pattarini L., et al. (2007). Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell 131, 463–475 10.1016/j.cell.2007.08.038 [DOI] [PubMed] [Google Scholar]
- Folkman J. (1995). Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1, 27–31 10.1038/nm0195-27 [DOI] [PubMed] [Google Scholar]
- Folkman J. (2001). Angiogenesis-dependent diseases. Semin. Oncol. 28, 536–542 10.1016/S0093-7754(01)90021-1 [DOI] [PubMed] [Google Scholar]
- Folkman J. (2003). Fundamental concepts of the angiogenic process. Curr. Mol. Med. 3, 643–651 10.2174/1566524033479465 [DOI] [PubMed] [Google Scholar]
- Folkman J., D'Amore P. A. (1996). Blood vessel formation: what is its molecular basis? Cell 87, 1153–1155 10.1016/S0092-8674(00)81810-3 [DOI] [PubMed] [Google Scholar]
- Franchi L., Eigenbrod T., Munoz-Planillo R., Nunez G. (2009). The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 10, 241–247 10.1038/ni.1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesel R., Maciag T. (1999). Fibroblast growth factor prototype release and fibroblast growth factor receptor signaling. Thromb. Haemost. 82, 748–754 [PubMed] [Google Scholar]
- Gabay C., Lamacchia C., Palmer G. (2010). IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 6, 232–241 10.1038/nrrheum.2010.4 [DOI] [PubMed] [Google Scholar]
- Gabrilovich D. I., Ostrand-Rosenberg S., Bronte V. (2012). Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 12, 253–268 10.1038/nri3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlanda C., Anders H. J., Mantovani A. (2009). TIR8/SIGIRR: an IL-1R/TLR family member with regulatory functions in inflammation and T cell polarization. Trends Immunol. 30, 439–446 10.1016/j.it.2009.06.001 [DOI] [PubMed] [Google Scholar]
- Goldberg J. E., Schwertfeger K. L. (2010). Proinflammatory cytokines in breast cancer: mechanisms of action and potential targets for therapeutics. Curr. Drug Targets 11, 1133–1146 10.2174/138945010792006799 [DOI] [PubMed] [Google Scholar]
- Gothert J. R., Gustin S. E., van Eekelen J. A., Schmidt U., Hall M. A., Jane S. M., et al. (2004). Genetically tagging endothelial cells in vivo: bone marrow-derived cells do not contribute to tumor endothelium. Blood 104, 1769–1777 10.1182/blood-2003-11-3952 [DOI] [PubMed] [Google Scholar]
- Grivennikov S. I., Greten F. R., Karin M. (2010). Immunity, inflammation, and cancer. Cell 140, 883–899 10.1016/j.cell.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S., Gonzalez-Perez R. R. (2011). Notch, IL-1 and leptin crosstalk outcome (NILCO) is critical for leptin-induced proliferation, migration and VEGF/VEGFR-2 expression in breast cancer. PLoS ONE 6:e21467 10.1371/journal.pone.0021467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann T., Lawrence T., McNeish I., Charles K. A., Kulbe H., Thompson R. G., et al. (2008). “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J. Exp. Med. 205, 1261–1268 10.1084/jem.20080108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- Hiratsuka S., Minowa O., Kuno J., Noda T., Shibuya M. (1998). Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc. Natl. Acad. Sci. U.S.A. 95, 9349–9354 10.1073/pnas.95.16.9349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagielska J., Kapopara P. R., Salguero G., Scherr M., Schutt H., Grote K., et al. (2012). Interleukin-1 assembles a proangiogenic signaling module consisting of caveolin-1, tumor necrosis factor receptor-associated factor 6, p38-mitogen-activated protein kinase (MAPK), and MAPK-activated protein kinase 2 in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 32, 1280–1288 10.1161/ATVBAHA.111.243477 [DOI] [PubMed] [Google Scholar]
- Kang H. W., Torres D., Wald L., Weissleder R., Bogdanov A. A., Jr. (2006). Targeted imaging of human endothelial-specific marker in a model of adoptive cell transfer. Lab. Invest. 86, 599–609 10.1038/labinvest.3700421 [DOI] [PubMed] [Google Scholar]
- Kaplanski G., Farnarier C., Benoliel A. M., Foa C., Kaplanski S., Bongrand P. (1994b). A novel role for E- and P-selectins: shape control of endothelial cell monolayers. J. Cell Sci. 107(Pt 9), 2449–2457 [DOI] [PubMed] [Google Scholar]
- Kaplanski G., Farnarier C., Kaplanski S., Porat R., Shapiro L., Bongrand P., et al. (1994a). Interleukin-1 induces interleukin-8 secretion from endothelial cells by a juxtacrine mechanism. Blood 84, 4242–4248 [PubMed] [Google Scholar]
- Kawamoto A., Gwon H. C., Iwaguro H., Yamaguchi J. I., Uchida S., Masuda H., et al. (2001). Therapeutic potential of ex vivo expanded endothelial progenitor cells for myocardial ischemia. Circulation 103, 634–637 10.1161/01.CIR.103.5.634 [DOI] [PubMed] [Google Scholar]
- Kerbel R. S. (2008). Tumor angiogenesis. N. Engl. J. Med. 358, 2039–2049 10.1056/NEJMra0706596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krelin Y., Voronov E., Dotan S., Elkabets M., Reich E., Fogel M., et al. (2007). Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res. 67, 1062–1071 10.1158/0008-5472.CAN-06-2956 [DOI] [PubMed] [Google Scholar]
- Kubota Y. (2012). Tumor angiogenesis and anti-angiogenic therapy. Keio J. Med. 61, 47–56 10.2302/kjm.61.47 [DOI] [PubMed] [Google Scholar]
- Latz E. (2010). The inflammasomes: mechanisms of activation and function. Curr. Opin. Immunol. 22, 28–33 10.1016/j.coi.2009.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavi G., Voronov E., Dinarello C. A., Apte R. N., Cohen S. (2007). Sustained delivery of IL-1Ra from biodegradable microspheres reduces the number of murine B16 melanoma lung metastases. J. Control Release 123, 123–130 10.1016/j.jconrel.2007.07.015 [DOI] [PubMed] [Google Scholar]
- Lazarus A., Keshet E. (2011). Vascular endothelial growth factor and vascular homeostasis. Proc. Am. Thorac. Soc. 8, 508–511 10.1513/pats.201102-021MW [DOI] [PubMed] [Google Scholar]
- Lee J. G., Kay E. P. (2012). NF-kappaB is the transcription factor for FGF-2 that causes endothelial mesenchymal transformation in cornea. Invest. Ophthalmol. Vis. Sci. 53, 1530–1538 10.1167/iovs.11-9102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis A. M., Varghese S., Xu H., Alexander H. R. (2006). Interleukin-1 and cancer progression: the emerging role of interleukin-1 receptor antagonist as a novel therapeutic agent in cancer treatment. J. Transl. Med. 4, 48 10.1186/1479-5876-4-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Liu B., Dai Z., Tao Y. (2011). Knockdown of VEGF receptor-1 (VEGFR-1) impairs macrophage infiltration, angiogenesis and growth of clear cell renal cell carcinoma (CRCC). Cancer Biol. Ther. 12, 872–880 10.4161/cbt.12.10.17672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J., Xiong Y., Han X., Lu Y. (2011). VEGF non-angiogenic functions in adult organ homeostasis: therapeutic implications. J. Mol. Med. (Berl.) 89, 635–645 10.1007/s00109-011-0739-1 [DOI] [PubMed] [Google Scholar]
- Mantovani A., Romero P., Palucka A. K., Marincola F. M. (2008). Tumour immunity: effector response to tumour and role of the microenvironment. Lancet 371, 771–783 10.1016/S0140-6736(08)60241-X [DOI] [PubMed] [Google Scholar]
- Mantovani A., Sica A. (2010). Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr. Opin. Immunol. 22, 231–237 10.1016/j.coi.2010.01.009 [DOI] [PubMed] [Google Scholar]
- Mantovani A., Sica A., Allavena P., Garlanda C., Locati M. (2009). Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum. Immunol. 70, 325–330 10.1016/j.humimm.2009.02.008 [DOI] [PubMed] [Google Scholar]
- Martinon F., Mayor A., Tschopp J. (2009). The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27, 229–265 10.1146/annurev.immunol.021908.132715 [DOI] [PubMed] [Google Scholar]
- Maruyama K., Mori Y., Murasawa S., Masaki H., Takahashi N., Tsutusmi Y., et al. (1999). Interleukin-1 beta upregulates cardiac expression of vascular endothelial growth factor and its receptor KDR/flk-1 via activation of protein tyrosine kinases. J. Mol. Cell. Cardiol. 31, 607–617 10.1006/jmcc.1998.0895 [DOI] [PubMed] [Google Scholar]
- Matsuo Y., Sawai H., Ochi N., Yasuda A., Takahashi H., Funahashi H., et al. (2009). Interleukin-1alpha secreted by pancreatic cancer cells promotes angiogenesis and its therapeutic implications. J. Surg. Res. 153, 274–281 10.1016/j.jss.2008.04.040 [DOI] [PubMed] [Google Scholar]
- Medina R. J., O'Neill C. L., O'Doherty T. M., Knott H., Guduric-Fuchs J., Gardiner T. A., et al. (2011). Myeloid angiogenic cells act as alternative M2 macrophages and modulate angiogenesis through interleukin-8. Mol. Med. 17, 1045–1055 10.2119/molmed.2011.00129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina R. J., O'Neill C. L., Sweeney M., Guduric-Fuchs J., Gardiner T. A., Simpson D. A., et al. (2010). Molecular analysis of endothelial progenitor cell (EPC) subtypes reveals two distinct cell populations with different identities. BMC Med. Genomics 3:18 10.1186/1755-8794-3-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohty M., Vialle-Castellano A., Nunes J. A., Isnardon D., Olive D., Gaugler B. (2003). IFN-alpha skews monocyte differentiation into Toll-like receptor 7-expressing dendritic cells with potent functional activities. J. Immunol. 171, 3385–3393 [DOI] [PubMed] [Google Scholar]
- Murdoch C., Muthana M., Coffelt S. B., Lewis C. E. (2008). The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 8, 618–631 10.1038/nrc2444 [DOI] [PubMed] [Google Scholar]
- Nakao S., Kuwano T., Tsutsumi-Miyahara C., Ueda S., Kimura Y. N., Hamano S., et al. (2005). Infiltration of COX-2-expressing macrophages is a prerequisite for IL-1 beta-induced neovascularization and tumor growth. J. Clin. Invest. 115, 2979–2991 10.1172/JCI23298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini A., Filippi I., Miglietta D., Moschetta M., Giavazzi R., Carraro F. (2010). Interleukin-1beta regulates the migratory potential of MDAMB231 breast cancer cells through the hypoxia-inducible factor-1alpha. Eur. J. Cancer 46, 3400–3408 10.1016/j.ejca.2010.07.044 [DOI] [PubMed] [Google Scholar]
- Naldini A., Leali D., Pucci A., Morena E., Carraro F., Nico B., et al. (2006). Cutting edge: IL-1beta mediates the proangiogenic activity of osteopontin-activated human monocytes. J. Immunol. 177, 4267–4270 [DOI] [PubMed] [Google Scholar]
- Nasu K., Itoh H., Yuge A., Kawano Y., Yoshimatsu J., Narahara H. (2006). Interleukin-1beta regulates vascular endothelial growth factor and soluble fms-like tyrosine kinase-1 secretion by human oviductal epithelial cells and stromal fibroblasts. Gynecol. Endocrinol. 22, 495–500 10.1080/08916930600929487 [DOI] [PubMed] [Google Scholar]
- O'Neill L. A. (2002). Signal transduction pathways activated by the IL-1 receptor/toll-like receptor superfamily. Curr. Top. Microbiol. Immunol. 270, 47–61 10.1007/978-3-642-59430-4_3 [DOI] [PubMed] [Google Scholar]
- O'Neill L. A. (2008). The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol. Rev. 226, 10–18 10.1111/j.1600-065X.2008.00701.x [DOI] [PubMed] [Google Scholar]
- Peters B. A., Diaz L. A., Polyak K., Meszler L., Romans K., Guinan E. C., et al. (2005). Contribution of bone marrow-derived endothelial cells to human tumor vasculature. Nat. Med. 11, 261–262 10.1038/nm1200 [DOI] [PubMed] [Google Scholar]
- Portier M., Zhang X. G., Ursule E., Lees D., Jourdan M., Bataille R., et al. (1993). Cytokine gene expression in human multiple myeloma. Br. J. Haematol. 85, 514–520 10.1111/j.1365-2141.1993.tb03341.x [DOI] [PubMed] [Google Scholar]
- Potente M., Gerhardt H., Carmeliet P. (2011). Basic and therapeutic aspects of angiogenesis. Cell 146, 873–887 10.1016/j.cell.2011.08.039 [DOI] [PubMed] [Google Scholar]
- Prudovsky I., Mandinova A., Soldi R., Bagala C., Graziani I., Landriscina M., et al. (2003). The non-classical export routes: FGF1 and IL-1alpha point the way. J. Cell Sci. 116, 4871–4881 10.1242/jcs.00872 [DOI] [PubMed] [Google Scholar]
- Purhonen S., Palm J., Rossi D., Kaskenpaa N., Rajantie I., Yla-Herttuala S., et al. (2008). Bone marrow-derived circulating endothelial precursors do not contribute to vascular endothelium and are not needed for tumor growth. Proc. Natl. Acad. Sci. U.S.A. 105, 6620–6625 10.1073/pnas.0710516105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian B. Z., Pollard J. W. (2010). Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51 10.1016/j.cell.2010.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S. L., Li T. S., Takahashi M., Hamano K. (2006). In vitro assessment of the effect of interleukin-1beta on angiogenic potential of bone marrow cells. Circ. J. 70, 1195–1199 10.1253/circj.70.1195 [DOI] [PubMed] [Google Scholar]
- Ribatti D. (2009). The paracrine role of Tie-2-expressing monocytes in tumor angiogenesis. Stem Cells Dev. 18, 703–706 10.1089/scd.2008.0385 [DOI] [PubMed] [Google Scholar]
- Rider P., Carmi Y., Guttman O., Braiman A., Cohen I., Voronov E., et al. (2011). IL-1alpha and IL-1beta recruit different myeloid cells and promote different stages of sterile inflammation. J. Immunol. 187, 4835–4843 10.4049/jimmunol.1102048 [DOI] [PubMed] [Google Scholar]
- Rider P., Kaplanov I., Romzova M., Bernardis L., Braiman A., Voronov E., et al. (2012). The transcription of the alarmin cytokine interleukin-1 alpha is controlled by hypoxia inducible factors 1 and 2 alpha in hypoxic cells. Front. Immunol. 3:290 10.3389/fimmu.2012.00290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffell B., Affara N. I., Coussens L. M. (2012). Differential macrophage programming in the tumor microenvironment. Trends Immunol. 33, 119–126 10.1016/j.it.2011.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffell B., DeNardo D. G., Affara N. I., Coussens L. M. (2010). Lymphocytes in cancer development: polarization towards pro-tumor immunity. Cytokine Growth Factor Rev. 21, 3–10 10.1016/j.cytogfr.2009.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo Y., Tanaka M., Miki M., Usui K., Suzuki T., Maemondo M., et al. (2002). Proinflammatory cytokine IL-1 beta promotes tumor growth of Lewis lung carcinoma by induction of angiogenic factors: in vivo analysis of tumor-stromal interaction. J. Immunol. 169, 469–475 [DOI] [PubMed] [Google Scholar]
- Salven P., Hattori K., Heissig B., Rafii S. (2002). Interleukin-1alpha promotes angiogenesis in vivo via VEGFR-2 pathway by inducing inflammatory cell VEGF synthesis and secretion. FASEB J. 16, 1471–1473 [DOI] [PubMed] [Google Scholar]
- Sano H., Forough R., Maier J. A., Case J. P., Jackson A., Engleka K., et al. (1990). Detection of high levels of heparin binding growth factor-1 (acidic fibroblast growth factor) in inflammatory arthritic joints. J. Cell Biol. 110, 1417–1426 10.1083/jcb.110.4.1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M. C., Avraamides C. J., Foubert P., Shaked Y., Kang S. W., Kerbel R. S., et al. (2011). Combined blockade of integrin-alpha4beta1 plus cytokines SDF-1alpha or IL-1beta potently inhibits tumor inflammation and growth. Cancer Res. 71, 6965–6975 10.1158/0008-5472.CAN-11-0588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K., Tschopp J. (2010). The inflammasomes. Cell 140, 821–832 10.1016/j.cell.2010.01.040 [DOI] [PubMed] [Google Scholar]
- Schweighofer B., Testori J., Sturtzel C., Sattler S., Mayer H., Wagner O., et al. (2009). The VEGF-induced transcriptional response comprises gene clusters at the crossroad of angiogenesis and inflammation. Thromb. Haemost. 102, 544–554 10.1160/TH08-12-0830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza G. L. (2011). Hypoxia. Cross talk between oxygen sensing and the cell cycle machinery. Am. J. Physiol. Cell Physiol. 301, C550–C552 10.1152/ajpcell.00176.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shchors K., Shchors E., Rostker F., Lawlor E. R., Brown-Swigart L., Evan G. I. (2006). The Myc-dependent angiogenic switch in tumors is mediated by interleukin 1beta. Genes Dev. 20, 2527–2538 10.1101/gad.1455706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikpranbabu S., Kalishwaralal K., Venkataraman D., Eom S. H., Park J., Gurunathan S. (2009). Silver nanoparticles inhibit VEGF-and IL-1beta-induced vascular permeability via Src dependent pathway in porcine retinal endothelial cells. J. Nanobiotechnol. 7, 8 10.1186/1477-3155-7-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya M. (2006a). Differential roles of vascular endothelial growth factor receptor-1 and receptor-2 in angiogenesis. J. Biochem. Mol. Biol. 39, 469–478 10.5483/BMBRep.2006.39.5.469 [DOI] [PubMed] [Google Scholar]
- Shibuya M. (2006b). Vascular endothelial growth factor (VEGF)-Receptor2: its biological functions, major signaling pathway, and specific ligand VEGF-E. Endothelium 13, 63–69 10.1080/10623320600697955 [DOI] [PubMed] [Google Scholar]
- Shojaei F., Wu X., Zhong C., Yu L., Liang X. H., Yao J., et al. (2007). Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature 450, 825–831 10.1038/nature06348 [DOI] [PubMed] [Google Scholar]
- Sica A., Matsushima K., Van Damme J., Wang J. M., Polentarutti N., Dejana E., et al. (1990). IL-1 transcriptionally activates the neutrophil chemotactic factor/IL-8 gene in endothelial cells. Immunology 69, 548–553 [PMC free article] [PubMed] [Google Scholar]
- Sica A., Porta C., Morlacchi S., Banfi S., Strauss L., Rimoldi M., et al. (2012). Origin and functions of tumor-associated myeloid cells (TAMCs). Cancer Microenviron. 5, 133–149 10.1007/s12307-011-0091-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims J. E., Nicklin M. J., Bazan J. F., Barton J. L., Busfield S. J., Ford J. E., et al. (2001). A new nomenclature for IL-1-family genes. Trends Immunol. 22, 536–537 10.1016/S1471-4906(01)02040-3 [DOI] [PubMed] [Google Scholar]
- Sims J. E., Smith D. E. (2010). The IL-1 family: regulators of immunity. Nat. Rev. Immunol. 10, 89–102 10.1038/nri2691 [DOI] [PubMed] [Google Scholar]
- Song X., Krelin Y., Dvorkin T., Bjorkdahl O., Segal S., Dinarello C. A., et al. (2005). CD11b+/Gr-1+ immature myeloid cells mediate suppression of T Cells in mice bearing tumors of IL-1{beta}-secreting cells. J. Immunol. 175, 8200–8208 [DOI] [PubMed] [Google Scholar]
- Song X., Voronov E., Dvorkin T., Fima E., Cagnano E., Benharroch D., et al. (2003). Differential effects of IL-1 alpha and IL-1 beta on tumorigenicity patterns and invasiveness. J. Immunol. 171, 6448–6456 [DOI] [PubMed] [Google Scholar]
- Stavri G. T., Zachary I. C., Baskerville P. A., Martin J. F., Erusalimsky J. D. (1995). Basic fibroblast growth factor upregulates the expression of vascular endothelial growth factor in vascular smooth muscle cells. Synergistic interaction with hypoxia. Circulation 92, 11–14 10.1161/01.CIR.92.1.11 [DOI] [PubMed] [Google Scholar]
- Stylianou E., Saklatvala J. (1998). Interleukin-1. Int. J. Biochem. Cell Biol. 30, 1075–1079 10.1016/S1357-2725(98)00081-8 [DOI] [PubMed] [Google Scholar]
- Suswam E. A., Nabors L. B., Huang Y., Yang X., King P. H. (2005). IL-1beta induces stabilization of IL-8 mRNA in malignant breast cancer cells via the 3' untranslated region: involvement of divergent RNA-binding factors HuR, KSRP and TIAR. Int. J. Cancer 113, 911–919 10.1002/ijc.20675 [DOI] [PubMed] [Google Scholar]
- Thacker S. G., Berthier C. C., Mattinzoli D., Rastaldi M. P., Kretzler M., Kaplan M. J. (2010). The detrimental effects of IFN-alpha on vasculogenesis in lupus are mediated by repression of IL-1 pathways: potential role in atherogenesis and renal vascular rarefaction. J. Immunol. 185, 4457–4469 10.4049/jimmunol.1001782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas K. A. (1996). Vascular endothelial growth factor, a potent and selective angiogenic agent. J. Biol. Chem. 271, 603–606 10.1074/jbc.271.2.603 [DOI] [PubMed] [Google Scholar]
- Thornton P., McColl B. W., Greenhalgh A., Denes A., Allan S. M., Rothwell N. J. (2010). Platelet interleukin-1alpha drives cerebrovascular inflammation. Blood 115, 3632–3639 10.1182/blood-2009-11-252643 [DOI] [PubMed] [Google Scholar]
- Tu S., Bhagat G., Cui G., Takaishi S., Kurt-Jones E. A., Rickman B., et al. (2008). Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 14, 408–419 10.1016/j.ccr.2008.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnquist H. R., Sumpter T. L., Tsung A., Zahorchak A. F., Nakao A., Nau G. J., et al. (2008). IL-1beta-driven ST2L expression promotes maturation resistance in rapamycin-conditioned dendritic cells. J. Immunol. 181, 62–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbich C., Heeschen C., Aicher A., Dernbach E., Zeiher A. M., Dimmeler S. (2003). Relevance of monocytic features for neovascularization capacity of circulating endothelial progenitor cells. Circulation 108, 2511–2516 10.1161/01.CIR.0000096483.29777.50 [DOI] [PubMed] [Google Scholar]
- Verheul H. M., Pinedo H. M. (2007). Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition. Nat. Rev. Cancer 7, 475–485 10.1038/nrc2152 [DOI] [PubMed] [Google Scholar]
- Voronov E., Carmi Y., Apte R. N. (2007). Role of IL-1-mediated inflammation in tumor angiogenesis. Adv. Exp. Med. Biol. 601, 265–270 10.1007/978-0-387-72005-0_28 [DOI] [PubMed] [Google Scholar]
- Voronov E., Dotan S., Gayvoronsky L., White R. M., Cohen I., Krelin Y., et al. (2010a). IL-1-induced inflammation promotes development of leishmaniasis in susceptible BALB/c mice. Int. Immunol. 22, 245–257 10.1093/intimm/dxq006 [DOI] [PubMed] [Google Scholar]
- Voronov E., Dotan S., Krelin Y., Song X., Elkabets M., Carmi Y., et al. (2013). Unique Versus Redundant Functions of IL-1alpha and IL-1beta in the Tumor Microenvironment. Front. Immunol. 4:177 10.3389/fimmu.2013.00177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronov E., Reich E., Dotan S., Dransh P., Cohen I., Huszar M., et al. (2010b). Effects of IL-1 molecules on growth patterns of 3-MCA-induced cell lines: an interplay between immunogenicity and invasive potential. J. Immunotoxicol. 7, 27–38 10.3109/15476910903405528 [DOI] [PubMed] [Google Scholar]
- Voronov E., Shouval D. S., Krelin Y., Cagnano E., Benharroch D., Iwakura Y., et al. (2003). IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 100, 2645–2650 10.1073/pnas.0437939100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreich D. M., Elaraj D. M., Puhlmann M., Hewitt S. M., Carroll N. M., Feldman E. D., et al. (2003). Effect of interleukin 1 receptor antagonist gene transduction on human melanoma xenografts in nude mice. Cancer Res. 63, 5957–5961 [PubMed] [Google Scholar]
- Wels J., Kaplan R. N., Rafii S., Lyden D. (2008). Migratory neighbors and distant invaders: tumor-associated niche cells. Genes Dev. 22, 559–574 10.1101/gad.1636908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witz I. P. (2008). Tumor-microenvironment interactions: dangerous liaisons. Adv. Cancer Res. 100, 203–229 10.1016/S0065-230X(08)00007-9 [DOI] [PubMed] [Google Scholar]
- Zheng Y., Humphry M., Maguire J. J., Bennett M. R., Clarke M. C. (2013). Intracellular interleukin-1 receptor 2 binding prevents cleavage and activity of interleukin-1alpha, controlling necrosis-induced sterile inflammation. Immunity 38, 285–295 10.1016/j.immuni.2013.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W., Guo S., Gonzalez-Perez R. R. (2011). Leptin pro-angiogenic signature in breast cancer is linked to IL-1 signalling. Br. J. Cancer 104, 128–137 10.1038/sj.bjc.6606013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegelhoeffer T., Fernandez B., Kostin S., Heil M., Voswinckel R., Helisch A., et al. (2004). Bone marrow-derived cells do not incorporate into the adult growing vasculature. Circ. Res. 94, 230–238 10.1161/01.RES.0000110419.50982.1C [DOI] [PubMed] [Google Scholar]