

Abstract
Background
Hepatosplenic T cell lymphoma (HSTL) is a rare but very aggressive peripheral T cell lymphoma whose initial silent clinical presentation unfortunately delays the diagnosis and worsens the prognosis of patient survival. Efforts should be aimed at early recognition and treatment.
Methods
We describe a case of a 62-year-old woman who presented at our clinic with a non-palpable purpuric eruption of the face. Investigations revealed thrombocytopenia with hepatosplenomegaly, which showed rapid progression together with accentuation of the purpura. Two months later, a bone marrow biopsy revealed the diagnosis of a HSTL.
Results
The patient received six cycles of CHOP chemotherapy (vincristine, cyclophosphamide, doxorubicin, methylprednisolone) followed by a well-tolerated autologous bone marrow graft. Normalization of the platelet count resulted in regression of the purpuric rash.
Conclusion
To our knowledge, this is the first report of a facial thrombocytopenic purpura as the inaugural symptom of HSTL. It emphasizes the privileged position of the dermatologist for early recognition of potentially lethal HSTL.
Key Words: Purpura, Lymphoma, Sun exposure, Face
Introduction
Hepatosplenic T cell lymphoma (HSTL) is a rare but very aggressive peripheral T cell lymphoma (PTCL) affecting preferentially young men [1]. Hepatosplenic lymphomas are composed of neoplastic mature T cells characterized by CD2, CD3, CD7 and CD16 expression; CD4, CD5 and CD8 as well as B cell surface markers are usually not expressed. HSTLs mostly express the gamma/delta T cell receptor (TCR) (hence the previous designation as gamma/delta T cell lymphoma) although some HSTLs express the alpha/beta TCR. A clonal rearrangement of the TCR can usually be demonstrated. HSTLs infiltrate the sinusoid in the spleen, liver, and bone marrow and affected patients usually present with hepatosplenomegaly without lymphadenopathy. They occasionally complain of fever, night sweat and weight loss. Despite usual response to a CHOP chemotherapy regimen (vincristine, cyclophosphamide, doxorubicin, methylprednisolone), relapses are very common and the median survival of patients with a diagnosis of HSTL is unfortunately less than 2 years [2]. Therefore, efforts should be aimed at early recognition, diagnosis and treatment for HSTL.
Although thrombocytopenia is commonly reported as a main feature of HSTL [3, 4], few cases describe purpuric eruption as a presenting symptom [5, 6]. Here, we report a case of a HSTL revealed by a purpuric eruption initially localized to the face and neck, emphasizing the role of purpura as a possible alerting presenting symptom.
Case Report
A 62-year-old woman presented to our department with an asymptomatic cutaneous eruption of the face extending to the neck that had appeared 2 weeks earlier. She did not report any infectious symptoms in the past weeks. Fever, night sweats or weight loss were denied. There was neither anamnestical atopy nor known allergies. Medication was limited to estradiol (substitution therapy) and occasional ibuprofen intake. Physical examination revealed non-palpable petechial purpura distributed on the face and extending to the neck (fig. 1). Peripheral white blood cell count, repartition and hemoglobin concentration were in the normal range, while platelet count was decreased to 90 G/l. Coagulation tests (aPTT, prothrombin time, fibrinogen) showed no anomaly. Lactate dehydrogenase, aspartate aminotransferase, and alanine aminotransferase were slightly elevated (LDH 316 U/l, ASAT 58 U/l, ALAT 52 U/l); alkaline phosphatase, gamma-glutamyl transferase, total bilirubin and serum creatinine concentrations were all within normal limits. Protein electrophoresis and immune subtraction allowed ruling out a monoclonal gammopathy. Erythrocyte sedimentation rate and serum complement concentrations were within normal limits; antinuclear antibodies, ANCAs (c-ANCA, p-ANCA and x-ANCA) as well as serologies for HIV, HBV, HCV, CMV, EBV and PVB19 were all negative. Histopathological examination of the purpuric macules showed extravasation of erythrocytes and a slight lymphohistiocytic perivascular inflammation in the superficial and mid dermis, without evidence of vasculitis (fig. 2). Amplification of the TCR in the skin biopsy showed no monoclonality. Direct immunofluorescence was negative. Given the particular distribution of the purpura in the upper part of the body, a cervico-thoracoabdominal CT scan was performed, which allowed to rule out a compressive component on the venous flow. It showed a multinodular goiter as well as a slight splenomegaly (12 cm). At that time, the patient was released with a diagnosis of idiopathic thrombocytopenic purpura.
Fig. 1.
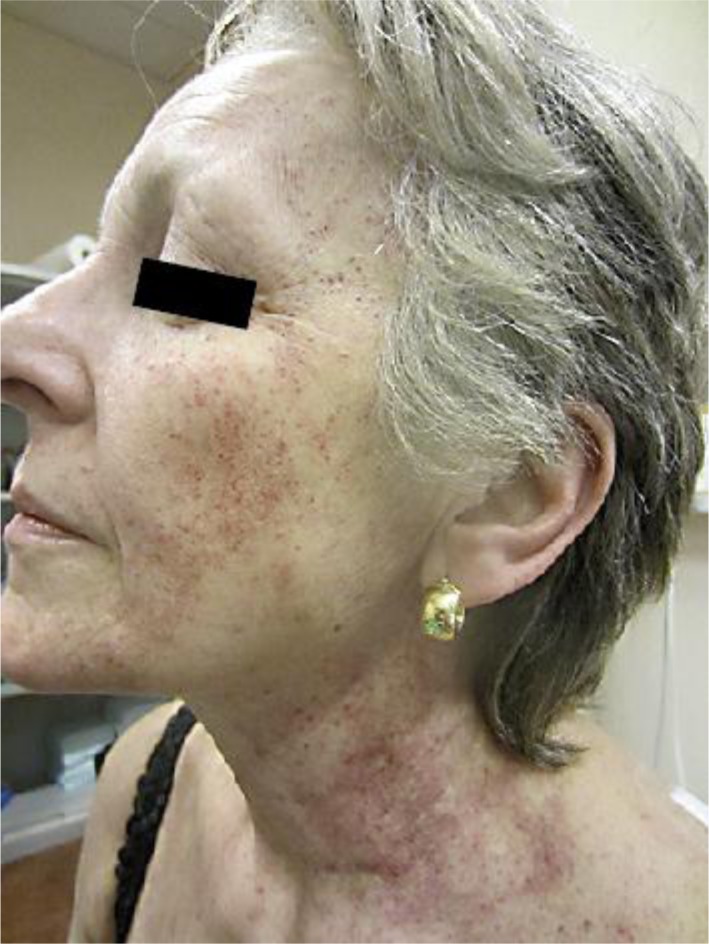
Clinical photographs of the patient at initial presentation. Lateral view showing a purpuric eruption of the face and neck, with sparing of the submental V-shaped area.
Fig. 2.

A 5-mm punch biopsy specimen of the skin. Histopathological analysis showing mild perivascular lymphocytic infiltrates of the superficial dermis and erythrocyte extravasation. a HE, original magnification ×100; b HE, original magnification ×400.
Two months later, the patient complained of night sweats and the purpuric eruption had extended to the whole body (fig. 3). Physical examination revealed a hepatosplenomegaly without palpable adenopathy. Thrombocytopenia had progressed to 30 G/l, LDH was 921 U/l, ALAT 179 U/l, ASAT 236 U/l, alkaline phosphatase 156 U/l, gamma-glutamyl transferase 96 U/l and total bilirubin was 31 μmol/l. Other laboratory examinations were within normal limits. A thoracoabdominal CT scan showed progression of the splenomegaly (from 12 to 16 cm). The examination of the bone marrow biopsy and aspirate showed an atypical lymphoid infiltrate composed of medium-sized cells with oval to irregular nuclei, moderately abundant cytoplasms, featuring a predominantly sinusoidal distribution. Immunophenotyping (immunohistochemistry and flow cytometry) revealed atypical lymphoid cells which were CD2+/CD3+/CD4–/CD5–/CD7–/CD8–/CD30– and focal expression of the T cell-restricted intracellular antigen-1 (TIA1). T cells strongly expressed the gamma/delta TCR and were negative for TCR beta F1. A monoclonal rearrangement of the TCR-gamma gene was demonstrated by PCR. We retained a diagnosis of HSTL and CHOP chemotherapy was administered together with a prophylactic intrathecal mixed chemotherapy (cytarabine, methotrexate, hydrocortisone). After two courses of treatment, the patient reported a diminution of night sweats, although the thoracoabdominal CT scan showed no modification, in particular no change in hepato- and splenomegaly; clinically, the purpuric rash showed a dramatic regression paralleled by a normalization of the platelet count (platelet count 157 G/l). After six courses of chemotherapy, as the infiltration of the bone marrow by neoplastic T cells persisted, the patient underwent a well-tolerated autologous bone marrow graft. Two months later, no recurrence of the purpura was observed. A close clinical, biological and radiological follow-up for early detection of a potential HSTL recurrence is currently ongoing at the oncological unit.
Fig. 3.

Photographs of the clinical evolution 2 months after the initial presentation. The clinical photographs show an intensification of the purpuric eruption of the face (a) as well as an extension of the purpura to the upper chest (b) and legs (c).
Discussion
In this case report, we reported for the first time a rare case of HSTL whose clinical presentation was a petechial non-palpable purpura of the face. Dyscrasia and vasculitis, two common causes of purpuric eruption were ruled out by laboratory findings as well as by histopathological and direct immunofluorescence exams. Blood cell count revealed thrombocytopenia. In our case, association of the purpura to the thrombocytopenia was supported by the progression of the cutaneous eruption, as the thrombocytopenia got worse. Despite the fact that thrombocytopenia has been reported in most HSTL cases [3, 4], the mechanisms implicated remain unclear. Commonly accepted explanations are bone marrow infiltration by lymphomatous T cells as well as marked splenomegaly often presented in HSTL, both were found in our patient. Accordingly, splenectomy was reported to be beneficial for patients with HSTL presenting with severe thrombocytopenia [4]. Cases in which thrombocytopenia was mediated through the production of antiplatelet antibodies were also reported [3, 5]. As gamma/delta T cells are found in increased numbers in several specific autoimmune conditions (e.g., celiac disease, rheumatoid arthritis, systemic lupus erythematosus, type I diabetes, multiples sclerosis), it is hypothesized that gamma/delta lymphomatous T cells could activate the B cell population and the Th1-driven antiplatelet antibody production [7].
Here, we described for the first time a non-palpable purpura developing in the context of HSTL and which was initially limited to the face and neck region. Purpuric eruptions usually develop in the lower extremities, where a higher hydrostatic pressure is found. The atypical presentation of the purpura in the face and neck suggests additional specific contributing factors. A continuous or intermittent increase of the hydrostatic pressure in the face and neck vasculature could represent a possible explanation. Based on the cervico-thoracoabdominal CT, radiologists ruled out any compression of the venous outflow from the face and neck area, in particular by the described multinodular goiter. Furthermore, the patient confidently and repeatedly denied either Valsalva-like spikes (repetitive coughing, vomiting or obstipation) or regular exercise [8]. Periorbital purpura has been associated with AL type amyloidosis [9], although rather presenting as large purpuric macules (ecchymosis). However, in our case histopathological analysis revealed no sign of amyloidosis. Primary cutaneous gamma/delta lymphoma may also occur in the skin [10], and purpuric lesions may result from intradermal infiltration by lymphoma T cells. These T cells may directly trigger vascular destruction via the release of granzyme B [11]. Nevertheless, histopathology did not show evidence of neoplastic T cell infiltration in our patient, which was confirmed by molecular studies which failed to demonstrate T cell clonality in the skin. Finally, a purpuric eruption in the context of a viral activation seemed rather unlikely as serologies for HIV, HBV, HCV, CMV, EBV and PVB19 were all negative.
Petechial purpura limited to sun-exposed areas has been previously described after solar exposure [12, 13, 14]. Histological analyses revealed erythrocyte extravasation, without signs of vasculitis but with mild lymphocytic perivascular infiltrates of the superficial and mid dermis, referred to as a ‘solar capillaritis’ in the literature [14]. Our case is suggestive of solar purpura as the purpura was initially limited to sun-exposed areas, with typical sparing of the submental V-shaped area (fig. 1). Furthermore, histological analysis confirmed the presence of a discrete perivascular lymphocytic infiltrate (fig. 2) reminiscent of solar capillaritis. The patient admitted regular solar exposure during the summer months, especially during the weeks preceding the cutaneous eruption. Interestingly, previous reports identified an association between solar purpura and additional predisposing factors, such as erythropoietic protoporphyria [15], high-dose aspirin intake [12] or thrombocytopenia [13]. Similarly, we believe that the particular presentation of the purpura to the face and neck in our patient is the result of the synergistic effect of HSTL-induced thrombocytopenia and superficial capillaritis induced by solar exposure.
In conclusion, our case illustrates how thrombocytopenic purpura should be considered as a possible presenting feature of rare but devastating HSTL, allowing early recognition and rapid therapeutic intervention. An atypical facial presentation should prompt the clinician to look for additional contributing factors like local obstruction of the venous outflow, amyloidosis, neoplastic T cell infiltration of the skin or sun exposure.
Disclosure Statement
No funding was obtained for this study. The authors have no conflicts of interest to disclose.
References
- 1.Belhadj K, Reyes F, Farcet JP, Tilly H, Bastard C, Angonin R, Deconinck E, Charlotte F, Leblond V, Labouyrie E, Lederlin P, Emile JF, Delmas-Marsalet B, Arnulf B, Zafrani ES, Gaulard P. Hepatosplenic gammadelta T-cell lymphoma is a rare clinicopathologic entity with poor outcome: Report on a series of 21 patients. Blood. 2003;102:4261–4269. doi: 10.1182/blood-2003-05-1675. [DOI] [PubMed] [Google Scholar]
- 2.Wong KF, Chan JK, Matutes E, McCarthy K, Ng CS, Chan CH, Ma SK. Hepatosplenic gamma delta T-cell lymphoma. A distinctive aggressive lymphoma type. Am J Surg Pathol. 1995;19:718–726. doi: 10.1097/00000478-199506000-00013. [DOI] [PubMed] [Google Scholar]
- 3.Motta G, Vianello F, Menin C, De Nicolo A, Agata S, Altavilla G, Pietrogrande F, Girolami A. Hepatosplenic gammadelta T-cell lymphoma presenting with immune-mediated thrombocytopenia and hemolytic anemia (Evans’ syndrome) Am J Hematol. 2002;69:272–276. doi: 10.1002/ajh.5555. [DOI] [PubMed] [Google Scholar]
- 4.Gumbs AA, Zain J, Neylon E, MacGregor-Cortelli B, Patterson M, O'Connor OA. Importance of early splenectomy in patients with hepatosplenic T-cell lymphoma and severe thrombocytopenia. Ann Surg Oncol. 2009;16:2014–2017. doi: 10.1245/s10434-009-0470-0. [DOI] [PubMed] [Google Scholar]
- 5.Garderet L, Aoudjhane M, Bonte H, Lagrange M, Isnard F, Fouillard L, Najman A. Immune thrombocytopenic purpura: first symptom of gamma/delta t-cell lymphoma. Am J Med. 2001;111:242–243. doi: 10.1016/s0002-9343(01)00775-6. [DOI] [PubMed] [Google Scholar]
- 6.Perfetto F, Tarquini R, Mancuso F, Di Lollo S, Tozzini S, Bellesi G, Laffi G. Hepato-splenic lymphoma: a rare entity mimicking acute hepatitis: a case report. World J Gastroenterol. 2003;9:1381–1384. doi: 10.3748/wjg.v9.i6.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yin Z, Craft J. Gamma delta T-cells in autoimmunity. Springer Semin Immunopathol. 2000;22:311–320. doi: 10.1007/s002810000048. [DOI] [PubMed] [Google Scholar]
- 8.Hwa C, Brauer JA, Mundi JP, Wu JM, Patel RR, Greenspan A, Stein JA. Exercise-induced progressive pigmentary purpura of the forehead. J Am Acad Dermatol. 2011;65:e149–e150. doi: 10.1016/j.jaad.2011.07.027. [DOI] [PubMed] [Google Scholar]
- 9.Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med. 1997;337:898–909. doi: 10.1056/NEJM199709253371306. [DOI] [PubMed] [Google Scholar]
- 10.Toro JR, Beaty M, Sorbara L, Turner ML, White J, Kingma DW, Raffeld M, Jaffe ES. Gamma delta T-cell lymphoma of the skin: a clinical, microscopic, and molecular study. Arch Dermatol. 2000;136:1024–1032. doi: 10.1001/archderm.136.8.1024. [DOI] [PubMed] [Google Scholar]
- 11.Shimauchi T, Hirokawa Y, Tokura Y. Purpuric adult T-cell leukaemia/lymphoma: expansion of unusual CD4/CD8 double-negative malignant T-cells expressing CCR4 but bearing the cytotoxic molecule granzyme B. Br J Dermatol. 2005;152:350–352. doi: 10.1111/j.1365-2133.2004.06281.x. [DOI] [PubMed] [Google Scholar]
- 12.Latenser BA, Hempstead RW. Exercise-associated solar purpura in an atypical location. Cutis. 1985;35:365–366. [PubMed] [Google Scholar]
- 13.Leung AK. Purpura associated with exposure to sunlight. J R Soc Med. 1986;79:423–424. doi: 10.1177/014107688607900712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waters AJ, Sandhu DR, Green CM, Ferguson J. Solar capillaritis as a cause of solar purpura. Clin Exp Dermatol. 2009;34:e821–e824. doi: 10.1111/j.1365-2230.2009.03567.x. [DOI] [PubMed] [Google Scholar]
- 15.Torinuki W, Miura T. Erythropoietic protoporphyria showing solar purpura. Dermatologica. 1983;167:220–222. doi: 10.1159/000249785. [DOI] [PubMed] [Google Scholar]