

Abstract
Macrophages are highly plastic hematopoietic cells with diversified functions related to their anatomic location and differentiation states. A number of recent studies have examined the role of macrophages in solid organ transplantation. These studies show that macrophages can induce allograft injury but, conversely, can also promote tissue repair in ischemia-reperfusion injury and acute rejection. Therapeutic strategies that target macrophages to improve outcomes in solid organ transplant recipients are being examined in preclinical and clinical models. In this review, we discuss the role of macrophages in different types of injury and rejection, with a focus on macrophage-mediated tissue injury, specifically vascular injury, repair and remodeling. We also discuss emerging macrophage-centered therapeutic opportunities in solid organ transplantation.
Keywords: Macrophage, Transplantation, Ischemia reperfusion injury, Acute rejection, Graft vascular disease
Introduction
Macrophages are ancient cells in metazoan phylogeny that play critical roles in detecting and eliminating harmful pathogens. They were first identified and described more than a century ago by Élie Metchnikoff [1] and are an essential component of the innate immune system, forming the first line of defense against infectious agents [2,3]. In response to pathogens, macrophages accumulate in tissues both through the recruitment and differentiation of circulating monocytes as well as through in situ proliferation [4,5]. There, they bind to Toll-like receptor (TLR) ligands such as lipopolysaccharide (LPS) or interferon-γ (IFN)-γ and are induced into an activation state that is characterized by a shift from aerobic metabolism to anaerobic glycolysis, increased production of proinflammatory mediators, augmented expression of inducible nitric oxide synthase (iNOS) and synthesis of reactive oxygen (ROS) and nitrogen species (RNS). This phenotype leads to efficient digestion of engulfed pathogens and is known as the classically-activated macrophage (CAM) [3,6]. In addition to their participation in host defense, macrophages have also been shown to play key roles in a range of physiological processes, including development, homeostasis, tissue repair, as well as pathologic processes including fibrosis, obesity and malignancy. These macrophages are induced by exposure to interleukin (IL)-4 and IL-13 and have a phenotype that is different from the CAMs. They are classified as alternatively-activated macrophages (AAMs) [2,3,6-9]. Finally, the regulatory macrophage (Mreg) has been recognized for its anti-inflammatory properties [10] and may play a protective role in transplant recipients.
CAMs and AAMs are routinely classified as ‘M1’ and ‘M2’ respectively [9]. However, they display tremendous heterogeneity, changing their phenotypes dramatically in response to cues from the microenvironment. To address these different phenotypes, M2-polarized macrophages have been sub-classified into M2a, M2b and M2c to discriminate their differentiation status in recent years [11]. In reality, even this more recent classification scheme does not clearly represent the very wide array of macrophage types manifesting highly diverse functions and phenotypes. While a more detailed classification of macrophages based on gene expression profiles or differentiation status will facilitate target identification for therapeutic interventions in various pathological conditions [2,9], the fact that macrophages are so highly mutable is a persistent consideration in scientific explorations of these cells. A recent landmark study evaluating the transcriptome of human macrophages induced by a variety of stimuli revealed an extraordinary spectrum of macrophage activation states that far extend the current M1 versus M2-polarization model [12]. For these reasons, this review generally avoids the M classification scheme and focuses instead on macrophage phenotype and function.
It has been recognized since the 1970s that macrophages are involved in the rejection of solid organ transplants [13,14]. Macrophages have been shown to play a role in cell- and antibody-mediated rejection as well as in the development of graft vascular disease (GVD), a manifestation of chronic rejection [15,16]. Macrophages may promote the development of acute rejection by producing ROS, eicosanoids and cytokines such as IL-1, tumor necrosis factor (TNF)-α and IL-18 [17,18]. On the other hand, macrophages may also dampen the alloimmune response by acquiring a regulatory phenotype that has been recently described [18]. Lastly, monocytes/macrophages may also help repair injured allograft microvasculature by producing proangiogenic factors [19].
Currently, immunosuppressive therapy regimens in organ transplantation primarily target T cells. As transplant outcomes continue to be suboptimal [20,21], identification and characterization of macrophages with distinct phenotypes may provide novel therapeutic targets to improve transplant survival. In this review, we will highlight studies that provide new insights into the role that macrophage play in different types of allograft injury and rejection and conclude with potential therapeutic strategies to promote allograft health.
Macrophages in ischemia reperfusion injury (IRI)
Ischemia – reperfusion describes the condition of an organ during the procurement and transplantation process. When an organ is harvested, the blood flow to the organ is cut off and cooled with physiologically-buffered solution - a state known as cold ischemia. Reperfusion occurs after the organ is transplanted and blood perfusion and oxygenation is restored. Reperfusion exacerbates the initial ischemia-induced tissue injury by triggering adaptive and innate immune responses [22] including macrophages (as described in greater detail below). The pathophysiological features of IRI include: 1) impaired endothelial barrier function with increased vascular permeability and leakage, 2) promotion of donor cell death programs, including apoptosis, autophagy-induced cell death and necrosis, 3) transcriptional reprogramming of donor cells, by upregulation of hypoxia inducible factor (HIF) and nuclear factor κB (NF-κB)- induced gene expression, 4) activation of the complement system and 5) activation of TLRs on macrophages and donor parenchymal cells [22-25].
IRI of the transplanted organ has long been recognized as a non-allogeneic factor that influences graft function and survival [26] and macrophages are key components of this pathology [22]. Macrophages have been shown to accumulate during the early phase of IRI in kidney and liver transplants [27,28]. Recently, it was shown that inhibition of sphingosine kinase-2 (SK-2) led to decreased macrophage accumulation in liver transplants, an effect that correlated with attenuated graft IRI [29]. In cardiac transplantation, decreased infiltration of macrophages during IRI also correlated with improved microvascular health and graft survival [30]. Also, alveolar macrophages are thought to be essential in the initiation of IRI in lung transplantation. They have been shown to induce tissue injury through the production of ROS and proinflammatory cytokines including IL-8, IL-12, IL-18, TNF-α and platelet activating factor (PAF) [31]. Furthermore, it has been demonstrated that increased macrophage recruitment into rat lung allografts induced by IRI is associated with impaired endothelial cell (EC) barrier function, and EC injury can be diminished when macrophage infiltration is decreased [32]. These studies provide compelling evidence that macrophages play a deleterious role in IRI and contribute to microvascular EC injury following transplantation. Moreover, macrophages accumulated during IRI may also help boost and maintain the adaptive T cell response by producing proinflammatory mediators and by acting as antigen presenting cells [33].
TLR signaling has been shown to be essential for macrophage activation [34]. In a study of spinal cord injury, it was shown that TLR4 deficiency protected the spinal cord from IRI in mice. This study went on to show that hypoxia and deprivation of glucose induced TLR4 expression on macrophages and that TLR4-deficient macrophages produced much lower levels of TNF-α and IL-6 [35]. These findings suggest a mechanism for TLR4-dependent macrophage-induced IRI. In a cardiac IRI study, TLR4-dependent, high-mobility group box-1 (HMGB-1)-activated macrophages produce IL-23, which in turn induced IL-17 production and caused heart allograft injury [36]. This result provides a mechanistic link between macrophages and IL-17-mediated IRI. In brain IRI, peroxiredoxin family proteins were also shown to induce IL-23 production in macrophages through activation of TLR2 and TLR4 signaling [37]. These recent studies from both the transplant and non-transplant animal models further demonstrate that activation of macrophage TLRs is required for macrophage-induced IRI and suggest that TLR-mediated macrophage activation likely contributes to IRI in newly-transplanted organs.
While macrophages have been shown to be harmful and cause allograft injury, they have also been shown to play a reparative role in both transplant and non-transplant IRI. In kidney, a macrophage-specific deletion of wnt7b significantly hindered tissue repair and regeneration following IRI [38]; this study suggests that wnt7b may also play a protective role in organ transplantation. In another study of acute renal injury and repair, it was shown that colony stimulating factor-1 (CSF-1), a hematopoietic growth factor, promotes tissue repair by enhancing tubular cell proliferation and diminishing its apoptosis and that this effect partially depends on the function of macrophages [39]. In lung allografts, polarization of macrophages by prednisone preconditioning diminished IRI and prolonged graft survival [40], suggesting that macrophages with an anti-inflammatory phenotype may also be beneficial during the late stage of IRI. Another recent study showed that treatment with human umbilical cord-derived stromal cells reduces renal IRI and that the beneficial effect depends not only on the presence of macrophages, but also on polarization in the later repair phase [41]. This result further supports the notion that in contrast to the deleterious effect of infiltrating macrophages seen in early IRI (1-3 days post transplant), Macrophages may act in a reparative role in late IRI (3-5 days) [42]. The leukotriene B4 receptor type-1 (BLT1) was recently shown to facilitate macrophage recruitment to the IR injured liver and BLT1 deficiency leads to decreased EGF production and impaired tissue repair [43], suggesting a role of macrophage-produced growth factor in tissue regeneration. These studies collectively demonstrate that phenotypically-distinct macrophages exist in different IRI phases and differential targeting strategies, such as depletion or phenotypic polarization, are needed to harness the macrophage as a therapeutic target to prevent or attenuate IRI in solid organ transplants.
Macrophages in acute allograft rejection
Acute rejection (AR) is a result of the alloimmune attack against the graft and is characterized by an inflammatory pathology that is generally reversible with early immunosuppressive intervention [33]. EC injury and vascular damage is a well-known phenomenon in AR [44,45] and macrophages are increasingly appreciated as an important player in both cellular and antibody-mediated AR [16]. Here we first highlight recent advances in macrophage biology in the setting of solid organ transplantation and then discuss in detail how the ECs of the allograft microvasculature can be damaged as well as repaired by different types of macrophages during AR.
In a clinical study, CD68+ macrophages but not T cell infiltration was shown to be associated with renal allograft dysfunction during AR [46]. Consistent with this finding, a pre-clinical study of kidney transplantation showed that macrophage depletion with liposomal-clodronate significantly attenuated graft damage during AR [47]. More recently, inhibition of Rho kinase was demonstrated to promote allograft function and this beneficial effect was associated with decreased macrophage infiltration in renal transplants [48]. Additionally, intravascular macrophage accumulation has been observed in cardiac allografts undergoing antibody-mediated rejection [49]. Furthermore, the accumulation of intravascular macrophages in early human cardiac transplantation also predicts the presence of donor specific antibodies (DSA), C4d deposition and symptoms of antibody-mediated rejection [50]. Collectively, these studies suggest that macrophages play an important role in both cellular and antibody-mediated rejection.
As stated above, macrophages accumulate within a tissue by recruitment of monocytes from the circulation and through proliferation of resident cells. The mechanisms associated with macrophage accumulation in solid organ transplants have been extensively studied. CD99 expressed on ECs is required for monocyte migration through EC junctions [51] and EC expression of P-selectin is also needed for macrophage accumulation in cardiac allografts during antibody-mediated rejection [52], suggesting that EC expression of adhesion molecules is required for monocyte/macrophage extravasation and subsequent tissue accumulation. Chemokines such as monocyte chemoattractant protein-1 (MCP-1), macrophage colony-stimulating factor (M-CSF, also known as CSF-1) and macrophage migration inhibitory factor (MIF) were shown to be positively associated with the number of infiltrated CD68+ or ED1+ macrophages in renal allografts [53-56]; additionally, chemokine receptors such as CX3C chemokine receptor 1 (CX3CR1), CC chemokine receptor 5 (CCR5) and CXC chemokine receptor 3 (CXCR3) have also been shown to mediate macrophage recruitment in renal grafts [57,58]. Interestingly, while the RANTES/CCR5 pathway also contributes to macrophage accumulation in cardiac transplants, CCR5 blockade only modestly prolonged allograft survival likely because the recruitment of regulatory T cells also requires this signaling pathway [59,60]. These studies collectively demonstrate that multiple chemokines are involved in the recruitment of macrophages into allografts during AR and blocking chemokine-induced signaling pathways may be a promising therapeutic strategy. Other studies have shown that local macrophages proliferate in AR [55,56,61], suggesting another mechanism for increasing the number of macrophages in allografts undergoing AR.
Once in the allograft, macrophages have been shown to promote inflammation, induce tissue damage and secrete inflammatory mediators. CAMs have been shown to produce ROS and RNS, which are probably the primary mediators of tissue damage in AR [47,62-67]. Cytokines such as IL-1β, IL-12, IL-18, TNF-α and IFN-γ have also been shown to be secreted by macrophages. These cytokines not only activate ECs and promote cytotoxic T cell generation, but also induce the production of chemokines such as CSF-1 and MCP-1 [68].
To explain why macrophage-mediated microvascular EC injury is relevant to transplantation, it is useful to consider several lines of evidence demonstrating the relevance of microvascular health in allograft function and why macrophage injury to the microvasculature may negatively affect the transplant. In a mouse orthotopic tracheal transplantation model, our group has shown that EC death and loss of the graft microvasculature during AR precedes (and is likely a key driver for) the development of airway fibrosis (i.e. chronic rejection) [69]. We subsequently demonstrated that the activation of the complement system and CD4+ T cells (but not CD8+ T cells) independently induces airway microvascular loss following transplantation [70]. We also showed that donor cell-expressed HIF-1α is associated with airway microvascular health and augmented expression of HIF-1α using adenovirus-mediated gene transfer prolongs EC survival, promotes vascular repair, and results in delayed and attenuated airway fibrosis [19]. These data are consistent with clinical studies which show that microvascular loss precedes and, consequently, may play a causal role in chronic rejection [71-74]. This concept emphasizes the importance of understanding how microvascular ECs are injured and developing new therapeutic targets to protect them during AR. Macrophages have been shown to induce EC apoptosis through activation of the Wnt pathway in patterning the eye vasculature during development [75]. Macrophages can also induce EC death through iNOS-derived nitric oxide [76]. We recently demonstrated that the lipid mediator leukotriene B4 (LTB4) produced by macrophages in pulmonary hypertension lungs induced EC apoptosis; LTB4 was found to induce significant EC apoptotic death in a dose-dependent manner within 24 hours of culture [77]. By extension, it is possible that macrophage- produced LTB4 may also induce allograft EC apoptosis during AR. Thus, macrophages may directly or indirectly induce EC death through production of cytotoxic molecules or proinflammatory mediators during AR (Figure 1).
Figure 1.
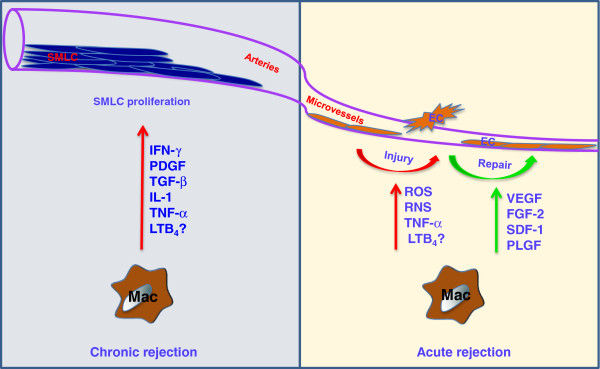
Macrophages and graft vasculature. During acute rejection, macrophages induce microvascular EC injury through the production of ROS, RNS, TNF-α and possibly LTB4. On the other hand, macrophages can also promote microvascular repair through the production of angiogenic factors, such as VEGF, FGF-2, SDF-1 and PLGF. During chronic rejection, macrophages promote SMLC proliferation by producing IFN-γ, PDGF, TNF-α, IL-1, TGF-β and possibly LTB4. Abbreviations: IFN, interferon; ROS, reactive oxygen species; RNS, reactive nitrogen species; TNF, tumor necrosis factor; PDGF, platelet-derived growth factor; IL, interleukin; TGF, transforming growth factor; LTB4, leukotriene B4; SMLCs, smooth muscle-like cells, VEGF, vascular endothelial growth factor; FGF, fibroblast growth factor; SDF, stromal cell-derived factor; PLGF, placental growth factor.
Despite the injurious effects on ECs, macrophages have also been shown to promote vessel growth in tumors [78-80] and angiogenesis in the hind limb ischemia model [81]. These macrophages are identified by expression of the Tie2 receptor. In the airway transplantation model, our group has also shown that Tie2-expressing monocytes/macrophages contribute to donor microvascular repair during AR [19]. Tie2-expressing monocytes/macrophage may promote graft microvascular repair by producing proangiogenic growth factors such as vascular endothelial cell growth factor (VEGF), placental growth factor (PLGF), stromal cell-derived factor (SDF)-1 and fibroblast growth factor (FGF)-2 [19,82]. In addition, increased expression of HIF through Von Hippel–Lindau (VHL) knockdown in recipient derived Tie2 lineage cells promotes donor vascular regeneration and limits graft invasion by aspergillus [83]. These studies suggest that during AR, a subpopulation of macrophages may help repair the injured graft microvasculature and therefore, deeper characterization of macrophages in AR is needed for efficient therapeutic targeting. Figure 1 demonstrates the myriad of effects that graft-infiltrating macrophages may have on donor microvessels and arteries.
In addition to graft-protective functions likely conferred by angiogenic macrophages, macrophages with regulatory function capable of quelling maladaptive inflammation likely serve a protective role in transplantation. Research in the last decade has identified numerous mechanisms that can induce Mregs both in vitro and in preclinical animal models [11], including macrophage stimulation by M-CSF, IL-10, vitamin D3, glucocorticoids and prostaglandin E2 [84-88] as well as macrophage repetitive stimulation by TLRs [89,90]. The human Mreg has also recently been generated by culturing CD14+ peripheral blood monocytes for 7 days in the presence of M-CSF and 10% human serum plus a 24-hour IFN-γ pulse [91]. These Mregs have been shown to be able to potently suppress T cell proliferation through IFN-γ-induced indoleaminepyrrole 2,3-dioxygenase (IDO) production and contact-dependent depletion of activated T cells [92]. In addition, a recent in depth phenotypic and functional characterization of the mouse Mregs revealed that these cells belong to a subset of suppressor macrophages expressing markers that distinguish them from the M1- and M2-polarized states [93]. In vitro, these Mregs completely suppress polyclonal T cell proliferation in an iNOS-dependent and allospecific fashion and administration of in vitro-derived Mregs significantly reduces acute rejection and prolongs the survival of the mouse cardiac allografts [93]. This study suggested that macrophages may also protect the vascular EC by differentiating into a regulatory subtype and consequently suppressing alloreactive T cells. This study also demonstrated that Mregs may be produced in vitro and could potentially be used as a source of cellular therapy for tolerance induction with a reduced dosage of immunosuppressive drugs in solid organ transplantation.
Macrophages in GVD
Chronic rejection is the leading cause of graft rejection, which is manifested by transplant tissue fibrosis and/or GVD [15,19,33]. GVD is the single most important limitation to long-term survival of transplanted solid organs [15]. It is traditionally seen in the arterioles and the arteries and may affect the entire length of the arterial vasculature in transplants. It is characterized by a concentric vascular intimal lesion comprised of smooth muscle-like cells (SMLC) and abnormally laid extracellular matrix and may simply be considered a result of abnormal stereotypic healing following alloimmune induced vascular injury [15,94].
Numerous studies show that macrophages are associated with the development of GVD; these cells have been observed in the lesions of GVD [95-99]. Macrophage depletion, but not inhibition of their ability to phagocytose, suppressed the development of cardiac graft vascular disease [100], suggesting that macrophages likely promote GVD through the production of proinflammatory, cytotoxic and trophic mediators but not their function as antigen-presenting cells. A study in kidney transplants showed that treatment with a macrophage inhibitor prevented progressive glomerulosclerosis, interstitial fibrosis, and arterial obliteration [101]. A more recent clinical study revealed that in heart transplants with very late rejection (> 7 years following transplantation), the presence of intravascular macrophages and donor specific antibodies are robust predictors of the development of more severe GVD [102]. Therapies effective in reducing GVD have also been shown to be associated with a significantly deceased macrophage infiltration [103,104]. These recent studies further confirmed that macrophages play a role in the pathogenesis of GVD.
Numerous mechanisms have been identified by which macrophages may promote the development of GVD. They may act as the predominant effector cells in CD4+ T cell-mediated delayed type hypersensitivity and have been shown to induce tissue and vascular damage through the production of eicosanoids, deleterious proteases, ROS and nitric oxide [15]. Macrophages may also promote GVD through the production of proinflammatory cytokines including IL-1, TNF-α, IFN-γ, platelet-derived growth factor (PDGF) and transforming growth factor (TGF)-β [15] (Figure 1). Double knockouts of both TNF-α receptor-1 and -2 in the graft significantly attenuated GVD in heart transplants [105], suggesting that TNF-α mediated signaling also contributes to the development of GVD. IFN-γ is also an important cytokine in the development of chronic rejection. In a heart transplant model, IFN-γ was shown to be both necessary and sufficient to drive the development of GVD [106]. Following IFN-γ stimulation, it has also been shown that macrophages produce IL-12 and IL-18, which further activates CD4+ T cell production of IFN-γ, thus forming a positive feedback loop [107]. Additionally, SMLCs also produce IFN-γ following IL-12 and IL-18 stimulation [108], demonstrating that macrophages and SMLCs may work together to promote the development of GVD.
SMLCs that display a synthetic phenotype are the primary cells that populate the lesions of GVD [15]. SMLCs with both donor and recipient origins have been described [109,110]. Numerous chemokine receptors including CXCR3, CXCR4, CCR1, CCR2, CCR3 and CCR5 are expressed on SMLCs [111-114]. Thus, macrophages producing cognate chemokines may promote recruitment and retention of recipient derived SMLCs, which may then facilitate neointimal formation and the development of GVD [15,94,115]. In an endothelial injury model of fulminant pulmonary arterial hypertension, our group showed that macrophages are the prominent producers of LTB4 (described above) and, in addition to causing EC apoptosis, also promote vascular smooth muscle cell proliferation and vascular remodeling. Blockade of LTB4 production by inhibition of the enzyme LTA4 hydrolase efficiently reopens obstructed pulmonary arterioles and reverses severe pulmonary arterial hypertension [77]. Together, these studies suggested that blockade of the chemokine signaling involved in macrophage recruitment and its production of the proinflammatory mediator may prevent/reverse GVD.
Of note, despite increasing appreciation that microvascular loss in solid organ transplants may play a causal role in the development of graft fibrosis and chronic rejection [19,30,69,71-74], GVD does not describe the pathology of the capillary loss and subsequent abnormal angiogenesis (e.g. microvascular loss in the airway transplant undergoing rejection as we described [19]). Interestingly, we recently found that macrophage infiltration is nearly absent around the remodeled capillaries of the chronically rejected airway transplants (unpublished observation). It is therefore possible that macrophages may only have significant effects on the microvasculature during IRI and AR.
Concluding remarks
Macrophages, historically thought of as ‘accessory cells’ with a poorly-described secondary function, are now emerging as an important cell type in solid organ transplantation. Compelling preclinical and clinical studies have shown that macrophages not only promote graft injury and GVD, but also participate in tissue repair, including microvascular repair, in different types of transplant related injury. Strategies for macrophage-centered therapeutics may include macrophage depletion or polarization to a reparative phenotype. Depletion may be achieved by direct killing through antagonism of CSF-1R or CSF-1 [85] or by blockade of recruitment by targeting CCR and CXCR mediated chemotactic pathways [57-60,116-118]. In transplantation rejection, when injurious and reparative classes coexist within the allograft, polarization of macrophages to a reparative phenotype may be a better strategy. Indeed, commonly used immunosuppressive drugs, such as glucocorticoids and mammalian target of rapamycin (mTOR) inhibitors, in addition to antagonizing T cells, are known to polarize macrophages to a suppressive phenotype [119,120]. More recently, ex vivo expanded regulatory macrophages were characterized and used in clinical trials, which may represent a promising therapeutic modality to prolong graft survival [92].
In summary, macrophages play numerous roles in solid organ transplant injury and rejection. A better understanding of how macrophages both damage and repair the allograft circulatory system in different types of transplant injury and rejection is required to further promote this cell, in all its myriad manifestations, as a promising therapeutic target. With advances in the fields of genomic analysis and systems biology, an improving delineation of macrophage subtypes is already occurring and opening new doors of investigation.
Abbreviations
AAM: Alternatively-activated macrophage; AR: Acute rejection; BLT1: Leukotriene B4 receptor type-1; CAM: Classically-activated macrophage; CCR: CC chemokine receptor; CSF-1: Colony stimulating factor-1; CXCR: CXC chemokine receptor; CX3CR: CX3C chemokine receptor; DSA: Donor specific antibody; EC: Endothelial cell; FGF: Fibroblast growth factor; GVD: Graft vascular disease; HIF: Hypoxia inducible factor; HMGB-1: High-mobility group box-1; IDO: Indoleaminepyrrole 2,3-dioxygenase; IFN: Interferon; IL: Interleukin; iNOS: Inducible nitric oxide synthase; IRI: Ischemia reperfusion injury; LPS: Lipopolysaccharide; LTB4: Leukotriene B4; MCP-1: Monocyte chemoattractant protein-1; M-CSF: Macrophage-colony stimulating factor; MIF: Migration inhibitory factor; Mreg: Regulatory macrophage; mTOR: Mammalian target of rapamycin; NF-κB: Nuclear factor κB; PAF: Platelet activating factor; PDGF: Platelet-derived growth factor; PLGF: Placental growth factor; RNS: Reactive nitrogen species; ROS: Reactive oxygen species; SDF: Stromal cell-derived factor; SK-2: Sphingosine kinase-2; SMLCs: Smooth muscle-like cells; TGF: Transforming growth factor; TLR: Toll-like receptor; TNF: Tumor necrosis factor; VHL: Von Hippel–Lindau; VEGF: Vascular endothelial growth factor.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XJ, WT, YS, JQ and MN prepared and wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Xinguo Jiang, Email: xinguoj@stanford.edu.
Wen Tian, Email: amytian@stanford.edu.
Yon K Sung, Email: yonsung@stanford.edu.
Jin Qian, Email: jinq@stanford.edu.
Mark R Nicolls, Email: mnicolls@stanford.edu.
Acknowledgements
This study was supported by NIH grant HL095686 and by a Veterans Affairs Merit Award BX000509 to M.R. Nicolls.
References
- Metchnikoff É. Immunity in Infective Diseases. London: Cambridge University Press; 1905. [Google Scholar]
- Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol. 2013;31:317–343. doi: 10.1146/annurev-immunol-032712-095906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11(11):750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, MacDonald AS, Allen JE. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011;332(6035):1284–1288. doi: 10.1126/science.1204351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176(1):287–292. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32(5):593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broichhausen C, Riquelme P, Geissler EK, Hutchinson JA. Regulatory macrophages as therapeutic targets and therapeutic agents in solid organ transplantation. Curr Opin Organ Transplant. 2012;17(4):332–342. doi: 10.1097/MOT.0b013e328355a979. [DOI] [PubMed] [Google Scholar]
- Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, Ganesan H, Nino-Castro A, Mallmann MR, Labzin L, Theis H, Kraut M, Beyer M, Latz E, Freeman TC, Ulas T, Schultze JL. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40(2):274–288. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom TB, Tilney NL, Carpenter CB, Busch GJ. Identity and cytotoxic capacity of cells infiltrating renal allografts. N Engl J Med. 1975;292(24):1257–1263. doi: 10.1056/NEJM197506122922402. [DOI] [PubMed] [Google Scholar]
- von Willebrand E, Hayry P. Composition and in vitro cytotoxicity of cellular infiltrates in rejecting human kidney allografts. Cell Immunol. 1978;41(2):358–372. doi: 10.1016/0008-8749(78)90233-2. [DOI] [PubMed] [Google Scholar]
- Mitchell RN. Graft vascular disease: immune response meets the vessel wall. Annu Rev Pathol. 2009;4:19–47. doi: 10.1146/annurev.pathol.3.121806.151449. [DOI] [PubMed] [Google Scholar]
- Magil AB. Monocytes/macrophages in renal allograft rejection. Transplant Rev (Orlando) 2009;23(4):199–208. doi: 10.1016/j.trre.2009.06.005. [DOI] [PubMed] [Google Scholar]
- Wyburn KR, Jose MD, Wu H, Atkins RC, Chadban SJ. The role of macrophages in allograft rejection. Transplantation. 2005;80(12):1641–1647. doi: 10.1097/01.tp.0000173903.26886.20. [DOI] [PubMed] [Google Scholar]
- Mannon RB. Macrophages: contributors to allograft dysfunction, repair, or innocent bystanders? Curr Opin Organ Transplant. 2012;17(1):20–25. doi: 10.1097/MOT.0b013e32834ee5b6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Khan MA, Tian W, Beilke J, Natarajan R, Kosek J, Yoder MC, Semenza GL, Nicolls MR. Adenovirus-mediated HIF-1alpha gene transfer promotes repair of mouse airway allograft microvasculature and attenuates chronic rejection. J Clin Invest. 2011;121(6):2336–2349. doi: 10.1172/JCI46192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber DJ, Wilkes DS. The role of autoimmunity in obliterative bronchiolitis after lung transplantation. Am J Physiol Lung Cell Mol Physiol. 2013;304(5):L307–L311. doi: 10.1152/ajplung.00378.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay D, Jameson J. Kidney transplantation and the ageing immune system. Nat Rev Nephrol. 2012;8(12):700–708. doi: 10.1038/nrneph.2012.242. [DOI] [PubMed] [Google Scholar]
- Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med. 2011;17(11):1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364(7):656–665. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361(16):1570–1583. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10(12):826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins PN, Chandraker A, Tullius SG. Modifying graft immunogenicity and immune response prior to transplantation: potential clinical applications of donor and graft treatment. Trans Int Official J Eur Soc Org Trans. 2006;19(5):351–359. doi: 10.1111/j.1432-2277.2006.00301.x. [DOI] [PubMed] [Google Scholar]
- Jo SK, Sung SA, Cho WY, Go KJ, Kim HK. Macrophages contribute to the initiation of ischaemic acute renal failure in rats. Nephrol Dial Transplant. 2006;21(5):1231–1239. doi: 10.1093/ndt/gfk047. [DOI] [PubMed] [Google Scholar]
- Ke B, Shen XD, Zhang Y, Ji H, Gao F, Yue S, Kamo N, Zhai Y, Yamamoto M, Busuttil RW, Kupiec-Weglinski JW. Keap1-Nrf2 Complex in Ischemia-Induced Hepatocellular Damage of Mouse Liver Transplants. J Hepatol. 2013;59(6):1200–7. doi: 10.1016/j.jhep.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Rehman H, Shi Y, Krishnasamy Y, Lemasters JJ, Smith CD, Zhong Z. Inhibition of sphingosine kinase-2 suppresses inflammation and attenuates graft injury after liver transplantation in rats. PLoS One. 2012;7(7):e41834. doi: 10.1371/journal.pone.0041834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuuminen R, Syrjala S, Krebs R, Arnaudova R, Rouvinen E, Nykanen AI, Lemstrom KB. Combined donor simvastatin and methylprednisolone treatment prevents ischemia-reperfusion injury in rat cardiac allografts through vasculoprotection and immunomodulation. Transplantation. 2013;95(9):1084–1091. doi: 10.1097/TP.0b013e3182881b61. [DOI] [PubMed] [Google Scholar]
- Zhao M, Fernandez LG, Doctor A, Sharma AK, Zarbock A, Tribble CG, Kron IL, Laubach VE. Alveolar macrophage activation is a key initiation signal for acute lung ischemia-reperfusion injury. Am J Physiol Lung Cell Mol Physiol. 2006;291(5):L1018–1026. doi: 10.1152/ajplung.00086.2006. [DOI] [PubMed] [Google Scholar]
- Kohno M, Watanabe M, Goto T, Kamiyama I, Ohtsuka T, Tasaka S, Sawafuji M. Attenuation of Lung Ischemia-Reperfusion Injury by Rho-Associated Kinase Inhibition in a Rat Model of Lung Transplantation. Ann Thorac Cardiovasc Surg. 2013. In Press. [DOI] [PubMed]
- Moreau A, Varey E, Anegon I, Cuturi MC. Effector mechanisms of rejection. Cold Spring Harb Perspect Med. 2013;3:a015461. doi: 10.1101/cshperspect.a015461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy CE, O'Neill LA. The role of toll-like receptors in macrophages. Front Biosci. 2008;13:62–70. doi: 10.2741/2660. [DOI] [PubMed] [Google Scholar]
- Bell MT, Puskas F, Agoston VA, Cleveland JC Jr, Freeman KA, Gamboni F, Herson PS, Meng X, Smith PD, Weyant MJ, Fullerton DA, Reece TB. Toll-like receptor 4-dependent microglial activation mediates spinal cord ischemia-reperfusion injury. Circulation. 2013;128(26 Suppl 1):S152–S156. doi: 10.1161/CIRCULATIONAHA.112.000024. [DOI] [PubMed] [Google Scholar]
- Zhu H, Li J, Wang S, Liu K, Wang L, Huang L. Hmgb1-TLR4-IL-23-IL-17A axis promote ischemia-reperfusion injury in a cardiac transplantation model. Transplantation. 2013;95(12):1448–1454. doi: 10.1097/TP.0b013e318293b7e1. [DOI] [PubMed] [Google Scholar]
- Shichita T, Hasegawa E, Kimura A, Morita R, Sakaguchi R, Takada I, Sekiya T, Ooboshi H, Kitazono T, Yanagawa T, Ishii T, Takahashi H, Mori S, Nishibori M, Kuroda K, Akira S, Miyake K, Yoshimura A. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat Med. 2012;18(6):911–917. doi: 10.1038/nm.2749. [DOI] [PubMed] [Google Scholar]
- Lin SL, Li B, Rao S, Yeo EJ, Hudson TE, Nowlin BT, Pei H, Chen L, Zheng JJ, Carroll TJ, Pollard JW, McMahon AP, Lang RA, Duffield JS. Macrophage Wnt7b is critical for kidney repair and regeneration. Proc Natl Acad Sci USA. 2010;107(9):4194–4199. doi: 10.1073/pnas.0912228107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke J, Iwata Y, Rabacal WA, Basu R, Yeung YG, Humphreys BD, Wada T, Schwarting A, Stanley ER, Kelley VR. CSF-1 signals directly to renal tubular epithelial cells to mediate repair in mice. J Clin Invest. 2009;119(8):2330–2342. doi: 10.1172/JCI39087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus P, Holfeld J, Urbschat A, Mutlak H, Ockelmann PA, Tacke S, Zacharowski K, Reissig C, Stay D, Scheller B. Prednisolone as preservation additive prevents from ischemia reperfusion injury in a rat model of orthotopic lung transplantation. PLoS One. 2013;8(8):e73298. doi: 10.1371/journal.pone.0073298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Zhang Q, Wang M, Wu H, Mao F, Zhang B, Ji R, Gao S, Sun Z, Zhu W, Qian H, Chen Y, Xu W. Macrophages are involved in the protective role of human umbilical cord-derived stromal cells in renal ischemia-reperfusion injury. Stem Cell Res. 2013;10(3):405–416. doi: 10.1016/j.scr.2013.01.005. [DOI] [PubMed] [Google Scholar]
- Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi BS, Ruhrberg C, Cantley LG. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol. 2011;22(2):317–326. doi: 10.1681/ASN.2009060615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkubo H, Ito Y, Minamino T, Mishima T, Hirata M, Hosono K, Shibuya M, Yokomizo T, Shimizu T, Watanabe M, Majima M. Leukotriene B4 type-1 receptor signaling promotes liver repair after hepatic ischemia/reperfusion injury through the enhancement of macrophage recruitment. FASEB J. 2013;27(8):3132–3143. doi: 10.1096/fj.13-227421. [DOI] [PubMed] [Google Scholar]
- Ponticelli C. The mechanisms of acute transplant rejection revisited. J Nephrol. 2012;25(2):150–158. doi: 10.5301/jn.5000048. [DOI] [PubMed] [Google Scholar]
- Drachenberg CB, Papadimitriou JC. Endothelial injury in renal antibody-mediated allograft rejection: a schematic view based on pathogenesis. Transplantation. 2013;95(9):1073–1083. doi: 10.1097/TP.0b013e31827e6b45. [DOI] [PubMed] [Google Scholar]
- Girlanda R, Kleiner DE, Duan Z, Ford EA, Wright EC, Mannon RB, Kirk AD. Monocyte infiltration and kidney allograft dysfunction during acute rejection. Am J Trans Official J Am Soc Trans Am Soc Trans Surg. 2008;8(3):600–607. doi: 10.1111/j.1600-6143.2007.02109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose MD, Ikezumi Y, van Rooijen N, Atkins RC, Chadban SJ. Macrophages act as effectors of tissue damage in acute renal allograft rejection. Transplantation. 2003;76(7):1015–1022. doi: 10.1097/01.TP.0000083507.67995.13. [DOI] [PubMed] [Google Scholar]
- Poosti F, Yazdani S, Dolman ME, Kok RJ, Chen C, Ding G, Lacombe M, Prakash J, van den Born J, Hillebrands JL, van Goor H, de Borst MH. Targeted inhibition of renal Rho kinase reduces macrophage infiltration and lymphangiogenesis in acute renal allograft rejection. Eur J Pharmacol. 2012;694(1–3):111–119. doi: 10.1016/j.ejphar.2012.08.010. [DOI] [PubMed] [Google Scholar]
- Fishbein GA, Fishbein MC. Morphologic and immunohistochemical findings in antibody-mediated rejection of the cardiac allograft. Human Immunol. 2012;73(12):1213–1217. doi: 10.1016/j.humimm.2012.07.011. [DOI] [PubMed] [Google Scholar]
- Fedrigo M, Feltrin G, Poli F, Frigo AC, Benazzi E, Gambino A, Tona F, Caforio AL, Castellani C, Toscano G, Gerosa G, Thiene G, Angelini A. Intravascular macrophages in cardiac allograft biopsies for diagnosis of early and late antibody-mediated rejection. J Heart Lung Transplant Official Pub Int Soc Heart Transplantat. 2013;32(4):404–409. doi: 10.1016/j.healun.2012.12.017. [DOI] [PubMed] [Google Scholar]
- Schenkel AR, Mamdouh Z, Chen X, Liebman RM, Muller WA. CD99 plays a major role in the migration of monocytes through endothelial junctions. Nat Immunol. 2002;3(2):143–150. doi: 10.1038/ni749. [DOI] [PubMed] [Google Scholar]
- Valenzuela NM, Hong L, Shen XD, Gao F, Young SH, Rozengurt E, Kupiec-Weglinski JW, Fishbein MC, Reed EF. Blockade of p-selectin is sufficient to reduce MHC I antibody-elicited monocyte recruitment in vitro and in vivo. Am J Transplant Official J Am Soc Transplant Am Soc Transplant Surg. 2013;13(2):299–311. doi: 10.1111/ajt.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandaliano G, Gesualdo L, Ranieri E, Monno R, Stallone G, Schena FP. Monocyte chemotactic peptide-1 expression and monocyte infiltration in acute renal transplant rejection. Transplantation. 1997;63(3):414–420. doi: 10.1097/00007890-199702150-00015. [DOI] [PubMed] [Google Scholar]
- Lan HY, Yang N, Brown FG, Isbel NM, Nikolic-Paterson DJ, Mu W, Metz CN, Bacher M, Atkins RC, Bucala R. Macrophage migration inhibitory factor expression in human renal allograft rejection. Transplantation. 1998;66(11):1465–1471. doi: 10.1097/00007890-199812150-00009. [DOI] [PubMed] [Google Scholar]
- Le Meur Y, Jose MD, Mu W, Atkins RC, Chadban SJ. Macrophage colony-stimulating factor expression and macrophage accumulation in renal allograft rejection. Transplantation. 2002;73(8):1318–1324. doi: 10.1097/00007890-200204270-00022. [DOI] [PubMed] [Google Scholar]
- Jose MD, Le Meur Y, Atkins RC, Chadban SJ. Blockade of macrophage colony-stimulating factor reduces macrophage proliferation and accumulation in renal allograft rejection. Am J Transplant. 2003;3(3):294–300. doi: 10.1034/j.1600-6143.2003.00068.x. [DOI] [PubMed] [Google Scholar]
- Hoffmann U, Bergler T, Segerer S, Rummele P, Kruger B, Banas MC, Reinhold S, Banas B, Kramer BK. Impact of chemokine receptor CX3CR1 in human renal allograft rejection. Transplant Immunol. 2010;23(4):204–208. doi: 10.1016/j.trim.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Kakuta Y, Okumi M, Miyagawa S, Tsutahara K, Abe T, Yazawa K, Matsunami K, Otsuka H, Takahara S, Nonomura N. Blocking of CCR5 and CXCR3 suppresses the infiltration of macrophages in acute renal allograft rejection. Transplantation. 2012;93(1):24–31. doi: 10.1097/TP.0b013e31823aa585. [DOI] [PubMed] [Google Scholar]
- Schroder C, Pierson RN 3rd, Nguyen BN, Kawka DW, Peterson LB, Wu G, Zhang T, Springer MS, Siciliano SJ, Iliff S, Ayala JM, Lu M, Mudgett JS, Lyons K, Mills SG, Miller GG, Singer II, Azimzadeh AM, DeMartino JA. CCR5 blockade modulates inflammation and alloimmunity in primates. J Immunol. 2007;179(4):2289–2299. doi: 10.4049/jimmunol.179.4.2289. [DOI] [PubMed] [Google Scholar]
- Azzawi M, Hasleton PS, Geraghty PJ, Yonan N, Krysiak P, El-Gammal A, Deiraniya AK, Hutchinson IV. RANTES chemokine expression is related to acute cardiac cellular rejection and infiltration by CD45RO T-lymphocytes and macrophages. J Heart Lung Transplant Official Pub Int Soc Heart Transplant. 1998;17(9):881–887. [PubMed] [Google Scholar]
- Kajiwara I, Kawamura K, Takebayashi S. An analysis of monocyte/macrophage subsets and granulocyte-macrophage colony-stimulating factor expression in renal allograft biopsies. Nephron. 1996;73(4):536–543. doi: 10.1159/000189137. [DOI] [PubMed] [Google Scholar]
- Worrall NK, Lazenby WD, Misko TP, Lin TS, Rodi CP, Manning PT, Tilton RG, Williamson JR, Ferguson TB Jr. Modulation of in vivo alloreactivity by inhibition of inducible nitric oxide synthase. J Exp Med. 1995;181(1):63–70. doi: 10.1084/jem.181.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis I, Hellinger A, Dehmlow C, Rauen U, Erhard J, Eigler FW, De Groot H. Evidence for increased nitric oxide production after liver transplantation in humans. Transplantation. 1995;59(9):1293–1297. doi: 10.1097/00007890-199505000-00012. [DOI] [PubMed] [Google Scholar]
- Roza AM, Cooper M, Pieper G, Hilton G, Dembny K, Lai CS, Lindholm P, Komorowski R, Felix C, Johnson C, Adams M. NOX 100, a nitric oxide scavenger, enhances cardiac allograft survival and promotes long-term graft acceptance. Transplantation. 2000;69(2):227–231. doi: 10.1097/00007890-200001270-00006. [DOI] [PubMed] [Google Scholar]
- Holan V, Krulova M, Zajicova A, Pindjakova J. Nitric oxide as a regulatory and effector molecule in the immune system. Mol Immunol. 2002;38(12–13):989–995. doi: 10.1016/s0161-5890(02)00027-5. [DOI] [PubMed] [Google Scholar]
- Takeiri M, Tachibana M, Kaneda A, Ito A, Ishikawa Y, Nishiyama S, Goto R, Yamashita K, Shibasaki S, Hirokata G, Ozaki M, Todo S, Umezawa K. Inhibition of macrophage activation and suppression of graft rejection by DTCM-glutarimide, a novel piperidine derived from the antibiotic 9-methylstreptimidone. Inflamm Res. 2011;60(9):879–888. doi: 10.1007/s00011-011-0348-z. [DOI] [PubMed] [Google Scholar]
- Kipari T, Cailhier JF, Ferenbach D, Watson S, Houlberg K, Walbaum D, Clay S, Savill J, Hughes J. Nitric oxide is an important mediator of renal tubular epithelial cell death in vitro and in murine experimental hydronephrosis. Am J Pathol. 2006;169(2):388–399. doi: 10.2353/ajpath.2006.050964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadban SJ, Wu H, Hughes J. Macrophages and kidney transplantation. Semin Nephrol. 2010;30(3):278–289. doi: 10.1016/j.semnephrol.2010.03.009. [DOI] [PubMed] [Google Scholar]
- Babu AN, Murakawa T, Thurman JM, Miller EJ, Henson PM, Zamora MR, Voelkel NF, Nicolls MR. Microvascular destruction identifies murine allografts that cannot be rescued from airway fibrosis. J Clin Invest. 2007;117(12):3774–3785. doi: 10.1172/JCI32311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MA, Jiang X, Dhillon G, Beilke J, Holers VM, Atkinson C, Tomlinson S, Nicolls MR. CD4+ T cells and complement independently mediate graft ischemia in the rejection of mouse orthotopic tracheal transplants. Circ Res. 2011;109(11):1290–1301. doi: 10.1161/CIRCRESAHA.111.250167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop GA, Waugh JA, Landers DV, Krensky AM, Hall BM. Microvascular destruction in renal transplant rejection. Transplantation. 1989;48(3):408–414. doi: 10.1097/00007890-198909000-00011. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, McCaughan GW, Painter DM, Bishop GA. Evidence that portal tract microvascular destruction precedes bile duct loss in human liver allograft rejection. Transplantation. 1993;56(1):69–75. doi: 10.1097/00007890-199307000-00012. [DOI] [PubMed] [Google Scholar]
- Luckraz H, Goddard M, McNeil K, Atkinson C, Charman SC, Stewart S, Wallwork J. Microvascular changes in small airways predispose to obliterative bronchiolitis after lung transplantation. J Heart Lung Transplant Official Pub Int Soc Heart Transplant. 2004;23(5):527–531. doi: 10.1016/j.healun.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Luckraz H, Goddard M, McNeil K, Atkinson C, Sharples LD, Wallwork J. Is obliterative bronchiolitis in lung transplantation associated with microvascular damage to small airways? Ann Thorac Surg. 2006;82(4):1212–1218. doi: 10.1016/j.athoracsur.2006.03.070. [DOI] [PubMed] [Google Scholar]
- Lobov IB, Rao S, Carroll TJ, Vallance JE, Ito M, Ondr JK, Kurup S, Glass DA, Patel MS, Shu W, Morrisey EE, McMahon AP, Karsenty G, Lang RA. WNT7b mediates macrophage-induced programmed cell death in patterning of the vasculature. Nature. 2005;437(7057):417–421. doi: 10.1038/nature03928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adair A, Mitchell DR, Kipari T, Qi F, Bellamy CO, Robertson F, Hughes J, Marson LP. Peritubular capillary rarefaction and lymphangiogenesis in chronic allograft failure. Transplantation. 2007;83(12):1542–1550. doi: 10.1097/01.tp.0000266689.93615.cd. [DOI] [PubMed] [Google Scholar]
- Tian W, Jiang X, Tamosiuniene R, Sung YK, Qian J, Dhillon G, Gera L, Farkas L, Rabinovitch M, Zamanian RT, Inayathullah M, Fridlib M, Rajadas J, Peters-Golden M, Voelkel NF, Nicolls MR. Blocking macrophage leukotriene b4 prevents endothelial injury and reverses pulmonary hypertension. Sci Transl Med. 2013;5(200):200ra117. doi: 10.1126/scitranslmed.3006674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 2013;23(3):277–286. doi: 10.1016/j.ccr.2013.02.013. [DOI] [PubMed] [Google Scholar]
- Welford AF, Biziato D, Coffelt SB, Nucera S, Fisher M, Pucci F, Di Serio C, Naldini L, De Palma M, Tozer GM, Lewis CE. TIE2-expressing macrophages limit the therapeutic efficacy of the vascular-disrupting agent combretastatin A4 phosphate in mice. J Clin Invest. 2011;121(5):1969–1973. doi: 10.1172/JCI44562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nucera S, Biziato D, De Palma M. The interplay between macrophages and angiogenesis in development, tissue injury and regeneration. Int J Dev Biol. 2011;55(4–5):495–503. doi: 10.1387/ijdb.103227sn. [DOI] [PubMed] [Google Scholar]
- Patel AS, Smith A, Nucera S, Biziato D, Saha P, Attia RQ, Humphries J, Mattock K, Grover SP, Lyons OT, Guidotti LG, Siow R, Ivetic A, Egginton S, Waltham M, Naldini L, De Palma M, Modarai B. TIE2-expressing monocytes/macrophages regulate revascularization of the ischemic limb. EMBO Mol Med. 2013;5(6):858–869. doi: 10.1002/emmm.201302752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X. Harnessing the immune system for the treatment of breast cancer. J Zhejiang Univ Sci B. 2014;15(1):1–15. doi: 10.1631/jzus.B1300264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Hsu JL, Tian W, Yuan K, Olcholski M, de Jesus PV, Semenza GL, Nicolls MR. Tie2-dependent VHL knockdown promotes airway microvascular regeneration and attenuates invasive growth of Aspergillus fumigatus. J Mol Med (Berl) 2013;91(9):1081–93. doi: 10.1007/s00109-013-1063-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeke F, Takiishi T, Korf H, Gysemans C, Mathieu C. Vitamin D: modulator of the immune system. Curr Opin Pharmacol. 2010;10(4):482–496. doi: 10.1016/j.coph.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Hume DA, MacDonald KP. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood. 2012;119(8):1810–1820. doi: 10.1182/blood-2011-09-379214. [DOI] [PubMed] [Google Scholar]
- Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002;169(5):2253–2263. doi: 10.4049/jimmunol.169.5.2253. [DOI] [PubMed] [Google Scholar]
- Rickard AJ, Young MJ. Corticosteroid receptors, macrophages and cardiovascular disease. J Mole Endocrinol. 2009;42(6):449–459. doi: 10.1677/JME-08-0144. [DOI] [PubMed] [Google Scholar]
- Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol. 2012;188(1):21–28. doi: 10.4049/jimmunol.1101029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor AL, Baban B, Chandler PR, Manlapat A, Kahler DJ, Munn DH. Cutting edge: CpG oligonucleotides induce splenic CD19+ dendritic cells to acquire potent indoleamine 2,3-dioxygenase-dependent T cell regulatory functions via IFN Type 1 signaling. J Immunol. 2005;175(9):5601–5605. doi: 10.4049/jimmunol.175.9.5601. [DOI] [PubMed] [Google Scholar]
- Dillon S, Agrawal S, Banerjee K, Letterio J, Denning TL, Oswald-Richter K, Kasprowicz DJ, Kellar K, Pare J, van Dyke T, Ziegler S, Unutmaz D, Pulendran B. Yeast zymosan, a stimulus for TLR2 and dectin-1, induces regulatory antigen-presenting cells and immunological tolerance. J Clin Invest. 2006;116(4):916–928. doi: 10.1172/JCI27203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson JA, Riquelme P, Geissler EK, Fandrich F. Human regulatory macrophages. Methods Mole Biol. 2011;677:181–192. doi: 10.1007/978-1-60761-869-0_13. [DOI] [PubMed] [Google Scholar]
- Hutchinson JA, Riquelme P, Sawitzki B, Tomiuk S, Miqueu P, Zuhayra M, Oberg HH, Pascher A, Lutzen U, Janssen U, Broichhausen C, Renders L, Thaiss F, Scheuermann E, Henze E, Volk HD, Chatenoud L, Lechler RI, Wood KJ, Kabelitz D, Schlitt HJ, Geissler EK, Fändrich F. Cutting Edge: Immunological consequences and trafficking of human regulatory macrophages administered to renal transplant recipients. J Immunol. 2011;187(5):2072–2078. doi: 10.4049/jimmunol.1100762. [DOI] [PubMed] [Google Scholar]
- Riquelme P, Tomiuk S, Kammler A, Fandrich F, Schlitt HJ, Geissler EK, Hutchinson JA. IFN-gamma-induced iNOS expression in mouse regulatory macrophages prolongs allograft survival in fully immunocompetent recipients. Mole Therapy J Am Soc Gene Therapy. 2013;21(2):409–422. doi: 10.1038/mt.2012.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell RN. Learning from rejection: What transplantation teaches us about (other) vascular pathologies. J Autoimmun. 2013;45:80–89. doi: 10.1016/j.jaut.2013.05.006. [DOI] [PubMed] [Google Scholar]
- Libby P, Tanaka H. The pathogenesis of coronary arteriosclerosis ("chronic rejection") in transplanted hearts. Clin Transplant. 1994;8(3 Pt 2):313–318. [PubMed] [Google Scholar]
- Russell ME, Wallace AF, Hancock WW, Sayegh MH, Adams DH, Sibinga NE, Wyner LR, Karnovsky MJ. Upregulation of cytokines associated with macrophage activation in the Lewis-to-F344 rat transplantation model of chronic cardiac rejection. Transplantation. 1995;59(4):572–578. doi: 10.1097/00007890-199559040-00023. [DOI] [PubMed] [Google Scholar]
- Hutchinson IV. Cardiac allograft vasculopathy--the cellular attack. Z Kardiol. 2000;89(Suppl 9):X/16–X/20. doi: 10.1007/s003920070020. [DOI] [PubMed] [Google Scholar]
- Libby P, Pober JS. Chronic rejection. Immunity. 2001;14(4):387–397. doi: 10.1016/S1074-7613(01)00119-4. [DOI] [PubMed] [Google Scholar]
- Huibers M, De Jonge N, Van Kuik J, Koning ES, Van Wichen D, Dullens H, Schipper M, De Weger R. Intimal fibrosis in human cardiac allograft vasculopathy. Transplant Immunol. 2011;25(2–3):124–132. doi: 10.1016/j.trim.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Kitchens WH, Chase CM, Uehara S, Cornell LD, Colvin RB, Russell PS, Madsen JC. Macrophage depletion suppresses cardiac allograft vasculopathy in mice. Am J Transplant Official J Am Soc Transplant Am Soc Transplant Surg. 2007;7(12):2675–2682. doi: 10.1111/j.1600-6143.2007.01997.x. [DOI] [PubMed] [Google Scholar]
- Azuma H, Nadeau KC, Ishibashi M, Tilney NL. Prevention of functional, structural, and molecular changes of chronic rejection of rat renal allografts by a specific macrophage inhibitor. Transplantation. 1995;60(12):1577–1582. doi: 10.1097/00007890-199560120-00034. [DOI] [PubMed] [Google Scholar]
- Loupy A, Cazes A, Guillemain R, Amrein C, Hedjoudje A, Tible M, Pezzella V, Fabiani JN, Suberbielle C, Nochy D, Hill GS, Empana JP, Jouven X, Bruneval P, Duong Van Huyen JP. Very late heart transplant rejection is associated with microvascular injury, complement deposition and progression to cardiac allograft vasculopathy. Am J Transplant Official J Am Soc Transplant Am Soc Transplant Surg. 2011;11(7):1478–1487. doi: 10.1111/j.1600-6143.2011.03563.x. [DOI] [PubMed] [Google Scholar]
- Lourenco-Filho DD, Maranhao RC, Mendez-Contreras CA, Tavares ER, Freitas FR, Stolf NA. An artificial nanoemulsion carrying paclitaxel decreases the transplant heart vascular disease: a study in a rabbit graft model. J Thorac Cardiovasc Surg. 2011;141(6):1522–1528. doi: 10.1016/j.jtcvs.2010.08.032. [DOI] [PubMed] [Google Scholar]
- Waanders F, Rienstra H, Boer MW, Zandvoort A, Rozing J, Navis G, van Goor H, Hillebrands JL. Spironolactone ameliorates transplant vasculopathy in renal chronic transplant dysfunction in rats. Am J Physiol Renal Physiol. 2009;296(5):F1072–1079. doi: 10.1152/ajprenal.90643.2008. [DOI] [PubMed] [Google Scholar]
- Suzuki J, Cole SE, Batirel S, Kosuge H, Shimizu K, Isobe M, Libby P, Mitchell RN. Tumor necrosis factor receptor -1 and -2 double deficiency reduces graft arterial disease in murine cardiac allografts. Am J Transplant Official J Am Soc Transplant Am Soc Transplant Surg. 2003;3(8):968–976. doi: 10.1034/j.1600-6143.2003.00164.x. [DOI] [PubMed] [Google Scholar]
- Nagano H, Mitchell RN, Taylor MK, Hasegawa S, Tilney NL, Libby P. Interferon-gamma deficiency prevents coronary arteriosclerosis but not myocardial rejection in transplanted mouse hearts. J Clin Invest. 1997;100(3):550–557. doi: 10.1172/JCI119564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellides G, Pober JS. Interferon-gamma axis in graft arteriosclerosis. Circ Res. 2007;100(5):622–632. doi: 10.1161/01.RES.0000258861.72279.29. [DOI] [PubMed] [Google Scholar]
- Gerdes N, Sukhova GK, Libby P, Reynolds RS, Young JL, Schonbeck U. Expression of interleukin (IL)-18 and functional IL-18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for atherogenesis. J Exp Med. 2002;195(2):245–257. doi: 10.1084/jem.20011022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillebrands JL, Klatter FA, van den Hurk BM, Popa ER, Nieuwenhuis P, Rozing J. Origin of neointimal endothelium and alpha-actin-positive smooth muscle cells in transplant arteriosclerosis. J Clin Invest. 2001;107(11):1411–1422. doi: 10.1172/JCI10233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser R, Lu MM, Narula N, Epstein JA. Smooth muscle cells, but not myocytes, of host origin in transplanted human hearts. Circulation. 2002;106(1):17–19. doi: 10.1161/01.CIR.0000021923.58307.8F. [DOI] [PubMed] [Google Scholar]
- Goldberg SH, van der Meer P, Hesselgesser J, Jaffer S, Kolson DL, Albright AV, Gonzalez-Scarano F, Lavi E. CXCR3 expression in human central nervous system diseases. Neuropathol App Neurobiol. 2001;27(2):127–138. doi: 10.1046/j.1365-2990.2001.00312.x. [DOI] [PubMed] [Google Scholar]
- Sakihama H, Masunaga T, Yamashita K, Hashimoto T, Inobe M, Todo S, Uede T. Stromal cell-derived factor-1 and CXCR4 interaction is critical for development of transplant arteriosclerosis. Circulation. 2004;110(18):2924–2930. doi: 10.1161/01.CIR.0000146890.93172.6C. [DOI] [PubMed] [Google Scholar]
- Veillard NR, Steffens S, Pelli G, Lu B, Kwak BR, Gerard C, Charo IF, Mach F. Differential influence of chemokine receptors CCR2 and CXCR3 in development of atherosclerosis in vivo. Circulation. 2005;112(6):870–878. doi: 10.1161/CIRCULATIONAHA.104.520718. [DOI] [PubMed] [Google Scholar]
- Kodali RB, Kim WJ, Galaria II, Miller C, Schecter AD, Lira SA, Taubman MB. CCL11 (Eotaxin) induces CCR3-dependent smooth muscle cell migration. Arterioscler Thromb Vasc Biol. 2004;24(7):1211–1216. doi: 10.1161/01.ATV.0000131654.90788.f5. [DOI] [PubMed] [Google Scholar]
- Mitchell RN, Libby P. Vascular remodeling in transplant vasculopathy. Circ Res. 2007;100(7):967–978. doi: 10.1161/01.RES.0000261982.76892.09. [DOI] [PubMed] [Google Scholar]
- Gladue RP, Brown MF, Zwillich SH. CCR1 antagonists: what have we learned from clinical trials. Curr Top Med Chem. 2010;10(13):1268–1277. doi: 10.2174/156802610791561237. [DOI] [PubMed] [Google Scholar]
- Zhao Q. Dual targeting of CCR2 and CCR5: therapeutic potential for immunologic and cardiovascular diseases. J Leukoc Biol. 2010;88(1):41–55. doi: 10.1189/jlb.1009671. [DOI] [PubMed] [Google Scholar]
- Khatri P, Roedder S, Kimura N, De Vusser K, Morgan AA, Gong Y, Fischbein MP, Robbins RC, Naesens M, Butte AJ, Sarwal MM. A common rejection module (CRM) for acute rejection across multiple organs identifies novel therapeutics for organ transplantation. J Exp Med. 2013;210(11):2205–2221. doi: 10.1084/jem.20122709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnquist HR, Raimondi G, Zahorchak AF, Fischer RT, Wang Z, Thomson AW. Rapamycin-conditioned dendritic cells are poor stimulators of allogeneic CD4+ T cells, but enrich for antigen-specific Foxp3+ T regulatory cells and promote organ transplant tolerance. J Immunol. 2007;178(11):7018–7031. doi: 10.4049/jimmunol.178.11.7018. [DOI] [PubMed] [Google Scholar]
- Ehrchen J, Steinmuller L, Barczyk K, Tenbrock K, Nacken W, Eisenacher M, Nordhues U, Sorg C, Sunderkotter C, Roth J. Glucocorticoids induce differentiation of a specifically activated, anti-inflammatory subtype of human monocytes. Blood. 2007;109(3):1265–1274. doi: 10.1182/blood-2006-02-001115. [DOI] [PubMed] [Google Scholar]