

Abstract
Epstein-Barr virus (EBV) is an oncogenic gammaherpesvirus that infects and persists in 95% of adults worldwide and has the potential to cause fatal disease, especially lymphoma, in immunocompromised hosts. Primary immunodeficiencies (PIDs) that predispose to EBV-associated malignancies have provided novel insights into the molecular mechanisms of immune defense against EBV. We have recently characterized a novel PID now named “X-linked immunodeficiency with magnesium defect, EBV infection, and neoplasia” (XMEN) disease characterized by loss-of-function mutations in the gene encoding magnesium transporter 1 (MAGT1), chronic high-level EBV with increased EBV-infected B cells, and heightened susceptibility to EBV-associated lymphomas. The genetic etiology of XMEN disease has revealed an unexpected quantitative role for intracellular free magnesium in immune functions and has led to novel diagnostic and therapeutic strategies. Here, we review the clinical presentation, genetic mutation spectrum, molecular mechanisms of pathogenesis, and diagnostic and therapeutic considerations for this previously unrecognized disease.
Introduction
Epstein-Barr virus (EBV), a B-cell–tropic gammaherpesvirus present in latent form, is widespread and affects the majority of adults and children worldwide.1 While most infections are asymptomatic or cause nonspecific symptoms, about 75% of adolescents and young adults with primary EBV infection develop mononucleosis.2 Moreover, hosts with acquired immunodeficiencies secondary to posttransplantation immunosuppression or HIV are at increased risk of developing EBV-positive B-cell lymphomas and other opportunistic infections. Failure to control EBV along with the potential lethal sequelae associated with persistent active EBV infection, such as EBV-positive B-cell lymphomas, fulminant infectious mononucleosis, chronic active EBV infections (CAEBV), and/or hemophagocytic lymphohistiocytosis, are key pathologic hallmarks of primary immunodeficiencies (PIDs) such as X-linked lymphoproliferative disease type 1 (XLP1), interleukin-2 inducible tyrosine kinase (ITK) deficiency, or CD27 deficiency.3 These PIDs illustrate key proteins in T cells and natural killer (NK) cells that are important for EBV control. We recently identified a new PID associated with chronic high-level EBV and susceptibility to EBV-positive B-cell lymphomas in a cohort of 7 patients, which has now been named “X-linked immunodeficiency with magnesium defect, EBV infection, and neoplasia” (XMEN) disease.4,5 Identification of the gene mutated in XMEN, MAGT1, has revealed an unexpected but essential role of free intracellular magnesium in the control of EBV by T and NK cells.4,5 Here, we review the phenotypic and genotypic features of this disease and how its novel pathogenetic mechanism demonstrates a fundamental role of magnesium transport for immunoregulation and provides new unconventional diagnostic and therapeutic strategies.
Clinical manifestations
We initially identified 3 patients with XMEN disease based on persistent high EBV levels and inverted CD4:CD8 ratio.4 Subsequently, we screened 61 patients for CD4 lymphopenia, chronic high EBV levels comparable to those in CAEBV patients (ranging from 1000 to 100 000), and/or EBV-associated B-cell lymphomas. Although EBV levels detected in XMEN patients vary widely, uncontrolled EBV should be detectable, and extremely high levels of EBV in a male child should raise the index of suspicion of this disease. While we did not find any additional XMEN patients with only inverted CD4:CD8 ratios, we identified 5 additional unrelated XMEN patients with chronic EBV infection and persistent EBV levels in the blood (Table 1).5 All patients also had splenomegaly. The patients were developmentally normal and had no signs of end organ dysfunction except for patient B.1, who presented with CAEBV disease with hepatitis, pancytopenia, and hemophagocytosis.6 We found that, except for the youngest individuals (A.1, A.2, C.1), all postpubertal XMEN patients developed various EBV-associated B-cell lymphoproliferative disorders (Table 1). The most consistent clinical feature among the 7 XMEN patients characterized thus far is a persistent high level of EBV along with increased susceptibility for developing EBV-positive lymphomas.4 All human patients in this study provided written informed consent in accordance with Helsinki principles for enrollment in research protocols that were approved by the institutional review board of the National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH).
Table 1.
Clinical phenotype of XMEN patients
| Patient | |||||||
|---|---|---|---|---|---|---|---|
| A.2 | C.1 | A.1 | D.1 | E.1 | F.1 | B.1* | |
| Age, y | 3 | 4 | 7 | 16 | 16 | 23 | 45 |
| Age at death, y | 23 | 45 | |||||
| MAGT1 mutation | |||||||
| Genomic | g.46668_46677del 10 | g.25009G>A | g.46668_46677del 10 | g.29684C>T | g.43183delC | g.46604G>T | g.29684C>T |
| cDNA | c.859_997del139 | c.172G>A | c.859_997del139 | c.409C>T | c.598delC | c.859_997del139 | c.409C>T |
| Protein | p.Asn287*fs*1 | p.Trp37* | p.Asn287*fs*1 | p.Arg137* | p.Arg200Glyfs*13 | p.Asn287*fs*1 | p.Arg137* |
| Recurrent infections | |||||||
| Epstein-Barr virus | + | + | + | + | + | + | + |
| Herpes simplex virus | + | – | + | – | – | – | – |
| Viral pneumonia | + | – | + | – | – | – | – |
| Otitis media | + | – | + | – | + | + | – |
| Sinusitis | + | + | + | – | – | – | – |
| Streptococcal pharyngitis | – | – | – | + | – | + | – |
| Epiglottitis | – | – | – | + | – | – | – |
| Molluscum contagiosum | – | – | – | – | + | – | – |
| Varicella + recurrent zoster | – | – | – | – | – | + | – |
| Pertussis | – | – | – | – | – | + | – |
| Cancer | |||||||
| Lymphoma | None | None | None | B-cell LPD† | Burkitt’s‡§ | Hodgkin§ | Lymphoma |
| Age at onset, y | 12 | 7, 14 | 17, 22 | 45 | |||
| Vaccination titer | |||||||
| Tetanus toxoid | + | + | – | N/D | + | +/− | N/D |
| Haemophilus influenzae type B | + | N/D | + | N/D | N/D | N/D | N/D |
| Diphtheria | N/D | N/D | + | N/D | + | +/− | N/D |
| Pneumococcal | +/− | + | + | N/D | – | – | N/D |
| Peripheral blood cells, % (range) | |||||||
| T cells | 61.2 (53-75) | N/D | 54.3 (53-75) | 48.6 (53-75) | 53.6 (53-75) | 69 (55-83) | 83.9 (57.3-86.4) |
| CD4 T cells | 27.8 (32-51) | N/D | 13.5 (28-47) | 19.1 (31-47) | 17 (31-47) | 40 (28-57) | 74.4 (28.6-57.2) |
| CD8 T cells | 20.8 (14-30) | N/D | 22.4 (16-30) | 43 (18-35) | 34 (18-35) | 34 (10-39) | 8.6 (12.9-46.9) |
| CD4:CD8 | 0.7 (0.9-3.7) | 0.6 (0.9-3.4) | 0.6 (1.4-1.7) | 0.55 (0.9-3.4) | 0.5 (0.9-3.4) | 1.1 (1.0-3.6) | 8.5 (1.0-3.6) |
| B cells | 26.9 (16-35) | N/D | 37.1 (14-33) | 46 (13-27) | 0 (RITX) | 44 (6-23) | 14.1 (6-23) |
| NK cells | 15 (3-15) | N/D | 5.4 (4-17) | 5 (3-22) | 7 (3-22) | 11 (3-22) | 1.7 (4.6-29.8) |
| Eosinophils | 1.85 (0-4.1) | N/D | 1.2 (0-4.7) | 1.5 (0.8-7) | 1.2 (0.8-7) | 1 (0.8-7) | 0.2 (0.8-7) |
| Neutrophils | 8 (22.4-69) | N/D | 20.4 (28.6-74.5) | 37.9 (28.6-74.5) | 55 (28.6-74.5) | 34 (34-67.9) | 88.2 (34-67.9) |
| Monocytes | 8.1 (4.2-12.2) | N/D | 9.7 (4.2-12.3) | 13 (4.2-12.3) | 9.9 (4.2-12.3) | 7 (4.2-12.3) | 3.4 (4.2-12.3) |
| Immunoglobulin levels (range) | |||||||
| IgG, mg/dL | 286 (424-1051) | 1030 (620-1300) | 1160 (633-1280) | 1690 (639-1349) | 611 (639-1349) | 619 (639-1349) | 734 (642-1730) |
| IgA, mg/dL | 7 (14-23) | 56 (50-200) | 87 (25-154) | 14.8 (45-236) | 35.6 (45-236) | 29.9 (70-312) | 128 (91-499) |
| IgM, mg/dL | 55 (48-1680) | 115 (60-200) | 92 (43-1960) | 29 (56-352) | 87 (56-352) | 38 (56-352) | 14 (34-342) |
| IgE, IU/mL | 2000 (310-2950) | N/D | 1750 (1070-6890) | 2100 (206-1952) | 1500 (206-1952) | 5 (1.53-114) | 5 (0-90) |
+/–, positive for some serotypes and negative for others; LPD, lymphoproliferative disease; N/D, not determined. RITX, rituximab; adapted from Chaigne-Delalande.5
Lymphocyte numbers in peripheral blood were measured a few months prior to death but before chemotherapy and transplantation.
EBV-positive B-cell LPD in the central nervous system.
B-cell lymphomas of two different restrictions: first λ light chain and then κ light chain; no MYC/IGH gene rearrangements were found.
Presumably two independent lymphomas based on the timing of onset.
Two XMEN patients (E.1 and F.1) apparently developed 2 sequential EBV-positive tumors. Two patients (B.1 and F.1) underwent allogeneic hematopoietic stem cell transplantation (HSCT) and died of transplant-related complications shortly after transplantation. Patient F.1 received a 7/8 matched unrelated donor peripheral blood stem cell transplant from a female donor after Campath, fludarabine, and melphalan conditioning. He had 99.8% engraftment by day +30 but died on day +60 after developing staphylococcal bacteremia, hemorrhagic shock, and renal failure. Patient B.1 received a 6/6 matched sibling transplant with cytoxan and fludarabine conditioning but died on day +20 with multiorgan failure, hemophagocytic syndrome, and central pontine demyelination. However, his lymphoma was well controlled and was largely necrotic at the time of death.6
In addition to having elevated EBV levels, two XMEN patients also had excessive childhood infections consistent with an underlying PID. Our index patients (A.1 and A.2) had a history of recurrent otitis media, sinusitis, and diarrhea along with repeated hospitalizations for viral pneumonia. While two XMEN patients had recurrent virus infections, including two episodes of molluscum contagiosum (patient E.1) and severe varicella followed by recurrent zoster (patient F.1), other upper respiratory infections (patients D.1 and F.1), epiglottitis (patient D.1), and pertussis (patient F.1) were observed. None of the patients were noted to have failure to thrive. Autoimmune features were not prominent among XMEN patients, but two patients did have autoimmune cytopenias (patients A.1 and D.1). Remarkably, many of the patients did not come to medical attention until they developed EBV-associated malignancies, sometimes as old as age 45 years.
Although the absolute CD4+ T cell counts were often above the CD4 lymphopenic threshold (<300 cells per microliter or <20% T cells), we observed that all XMEN patients had CD4:CD8 T-cell ratios decreased to ∼1 or less, except for patient B.1 who presented with pancytopenia due to malignancy. This is consistent with our finding that CD31+ cells among the CD4+ CD27+CD45RO– naive T cells were reduced in the patients compared with normal controls, suggesting reduced thymic output of CD4+ T cells.4 The patients also had moderately high B-cell counts, with increased transitional B cells and mild neutropenia, which may be secondary to chronic EBV. Variable deficiencies in immunoglobulin levels (IgG, IgA, and IgM) and vaccination responses were found in some patients (Table 1), which might be attributable to deficiency in T follicular helper cells or an underlying B-cell dysfunction. T-cell proliferation in response to mitogen (phytohemagglutinin, Candida, concanavalin A, or tetanus) stimulation was inconsistently defective. The clinical features of XMEN disease are summarized in Figure 1A.
Figure 1.
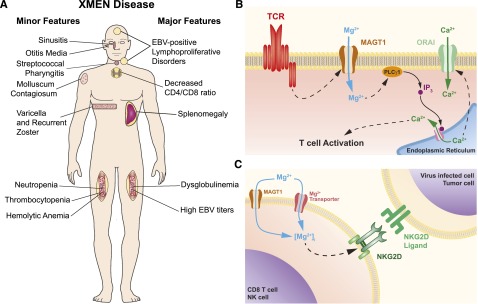
XMEN disease. (A) XMEN disease clinical features. Major features are present in almost all XMEN patients, while minor features are found only in some patients. (B) Model of the role of MAGT1 in TCR activation. In normal T cells, stimulation of the TCR triggers a Mg2+ flux through MAGT1. This transient increase of intracellular Mg2+ is required for the activation of phospholipase C γ1 (PLCγ1) required for the downstream generation of Ca2+ flux through ORAI by promoting the release of Ca2+ from the endoplasmic reticulum via inositol 1,4,5-triphosphate (IP3) and its receptor. (C) Model of role of MAGT1 in Mg2+ homeostasis and NKG2D expression. In XMEN patients, the absence of MAGT1 leads to the chronic reduction of intracellular free Mg2+, which is required to maintain the expression of NKG2D.
Genetic mutation spectrum
XMEN disease is caused by loss-of-function mutations in MAGT1, a 70-kb gene with 10 exons encoding for a 335 amino acid protein. We initially detected a 10–base pair deletion in the index probands (A.1 and A.2) at the exon-intron junction of exon 7. This mutation led to altered splicing, frame shift, early termination, and loss of protein expression by nonsense-mediated decay of the messenger RNA (mRNA).4 Subsequent XMEN patients had different MAGT1 mutations also leading to premature translational termination with loss of mRNA expression. In all cases, there was premature termination of the MAGT1 protein and nonsense-mediated decay of the mRNA leading to deficient gene expression. The mothers of XMEN patients have preferential X chromosome inactivation of the chromosome harboring the mutant allele and appear to be asymptomatic.4 The fact that all patients had mutations leading to the same functional deficiency of MAGT1 but had somewhat different clinical presentations at widely varying ages (3 to 45 years) suggests that the disease is phenotypically heterogeneous. MAGT1 genotype alone does not seem to be predictive for time to onset of lymphoma, which may depend on environmental variables.
Mechanisms of pathogenesis
MAGT1 is an evolutionarily conserved, magnesium-specific transporter expressed ubiquitously in all mammalian cells, with higher expression in certain tissues, including the hematopoietic lineages.7 While it was previously shown to play an essential role in magnesium homeostasis, its function in the immune system was uncharacterized until our discovery of XMEN disease.4 We found unexpected immunoregulatory roles of free (unbound) intracellular Mg2+, which constitutes about 5% of total intracellular Mg2+, most of which is tightly bound to adenosine triphosphate, other polyphosphates, and other intracellular molecules.4,5 While loss of MAGT1 does not alter the bound or total pool of intracellular magnesium, it has 2 consequences on the intracellular free Mg2+: (1) abolition of a rapidly induced, transient T-cell receptor (TCR)–induced Mg2+ flux required for optimal T-cell activation (Figure 1B) and (2) chronic decrease in the basal level of free Mg2+ (Figure 1C). The second defect leads to loss of expression of NKG2D, a receptor on NK and cytotoxic T lymphocytes (CTLs) involved in antiviral and antitumor cytotoxicity.4,5 Remarkably, addition of extracellular Mg2+ can increase intracellular free Mg2+ and restore NKG2D and cytolytic functions that are abnormal in XMEN lymphocytes. This has therapeutic implications as detailed below.
The transient MAGT1-mediated Mg2+ flux induced by TCR stimulation is required to coordinate T-cell signaling during T-cell activation. Loss of this flux leads to delayed phospholipase Cγ1 (PLCγ1) phosphorylation and impaired downstream signaling events resulting in a reduced TCR-gated calcium (Ca2+) flux4 (Figure 1B). Thus, MAGT1 is upstream in the same pathway of Ca2+ regulation that is targeted by conventional immunosuppressants such as cyclosporin A and sirolimus, making it an attractive extracellular therapeutic target. Besides gating the transient Mg2+ flux during T-cell activation, MAGT1 also sets the basal level of intracellular free Mg2+, which is required to maintain the expression of the cytolytic activating receptor NKG2D (Figure 1C). Loss of this activating receptor debilitates specific NKG2D ligand-mediated cytotoxicity functions in NK cells and CTLs, which play essential roles in anti-EBV and antitumor immunity.8 Consequently, while XMEN patients are capable of generating EBV-specific CTLs in vivo, their cells exhibit a profound killing defect for autologous EBV-immortalized lymphoblastoid B-cell lines and other tumor cell line targets.5 XMEN disease is the first PID associated with a specific loss of NKG2D expression. The compromised EBV control in XMEN disease together with the restoration of EBV suppression as NKG2D expression is rescued with Mg2+ supplementation, indicating that there is a secondary, albeit less efficient, Mg2+ transporter that can substitute for MAGT1 in restoring the intracellular free Mg2+ level (Figure 1C). These results also established that NKG2D is important for anti-EBV immunity. EBV, unlike cytomegalovirus, lacks extensive mechanisms of evading NKG2D recognition. Lytic induction in EBV-infected cells can lead to downregulation of HLA class I and upregulation of the NKG2D ligand ULBP1, both of which trigger NK cytotoxicity via NKG2D.9 NKG2D is implicated in tumor immunosurveillance, which may also account for the tumorigenic susceptibility in XMEN disease.
The immune cell cytotoxicity defect in XMEN disease also sheds light on the clinical similarities between XMEN disease and XLP1, another PID manifested by inability to control EBV infection and a high incidence of lymphomas. The unusual susceptibility to EBV in XLP1 has been attributed to the loss of 2B4-mediated cytotoxicity against EBV-infected B cells, which requires SLAM-associated protein–associated receptors for recognition.10-13 Interestingly, NKG2D ligation can synergize with many NK-activating receptors, including 2B4.14 The convergence of the disease pathogenesis of XMEN and XLP1 suggests that the NKG2D-2B4 synergistic cytotoxic pathway may be particularly important for control of EBV and preventing lymphoma development. Thus far, we have observed only EBV-positive tumors in XMEN disease. Whether XMEN patients are also susceptible to developing EBV-negative tumors as in XLP1 remains to be determined.
Diagnostic and therapeutic considerations
The significant mortality associated with XMEN disease due to lymphoma raises the necessity of early diagnosis and close follow-up. Males with unexplained chronic elevated EBV level in the blood and splenomegaly, and/or a past history or family history of either EBV-positive B-cell lymphoproliferative disease or EBV lymphoma, should be considered for XMEN screening. Patients may have EBV serologies indicative of past infection with borderline transaminase elevations (Table 1), but EBV polymerase chain reaction reveals persistently elevated levels of EBV DNA, which is indicative of poor control of EBV replication. Other clinical features that may suggest the diagnosis of XMEN include a reduced CD4:CD8 ratio (<1.3), elevated levels of B cells, dysgammaglobulinemia, or defective T-lymphocyte stimulation responses.
Reduced NKG2D expression can be detected by flow cytometry on whole blood samples in a research laboratory.5 Since NKG2D can also be downregulated in the setting of malignancies,15 a final diagnosis needs to be confirmed by examining MAGT1 mRNA expression or MAGT1 exon sequencing. If no mutations are found, quantitative assessment of MAGT1 mRNA expression is necessary to rule out potential noncoding mutations affecting MAGT1 expression. Although flow cytometry for NKG2D is not currently available in commercial laboratories, MAGT1 sequencing can be performed through the Cincinnati Children’s Molecular Genetics Laboratory or through the National Institutes of Health Clinical Center.
Once the diagnosis is made, treatment depends on the patient’s clinical condition. Rituximab (anti-CD20 antibody) will likely reduce the level of EBV-infected cells in the blood but does not completely deplete CD20+ B cells in the tissues.16 EBV-positive CD20– lymphoproliferative lesions have developed after treatment of EBV-positive CD20+ lesions in patients with CAEBV6; therefore, it is unlikely that rituximab will cure XMEN. Because T cells from patients with XMEN have impaired EBV-specific killing,5 infusions of additional autologous virus-specific T cells would be unlikely to be effective. Although using closely matched third-party EBV-specific T cells is another potential therapeutic option, it might complicate future transplantation due to allo-sensitization. Chemotherapy is useful for treating lymphomas but does not prevent future development of other lymphomas. Two patients with XMEN and lymphoma underwent allogeneic HSCT but did not survive because of transplant-related complications (Table 1). Nonetheless, HSCT might still be curative for patients with a good match. Patients with XMEN disease who have lymphoproliferative disease on biopsy or who are in remission after treatment of lymphoma should be considered for HSCT, especially since these patients have a high incidence of primary and secondary lymphomas. An alternative therapeutic strategy that clinicians can consider is magnesium supplementation. We have observed that oral Mg2+ supplementation restored basal intracellular free Mg2+ along with NKG2D expression and decreased the number of EBV-infected cells in two XMEN patients in vivo.5 The high levels of EBV in XMEN disease, together with the fact that the percentage of EBV-infected cells is suppressed as NKG2D expression is reinstated with Mg2+ supplementation, establish the importance of NKG2D for anti-EBV immunity. Thus far, we have found that continuous oral magnesium threonate supplementation is a safe and well-tolerated treatment.5 Whether this dietary supplement is sufficient to avert the development of lymphoma or promote lymphoma clearance remains to be determined.
Perspectives
The discovery of XMEN disease has provided a novel understanding of magnesium biology for immunoregulation. The pathogenesis of XMEN is consistent with its clinical features, given that it highlights the importance of T-cell activation signaling and immune cell cytotoxicity for controlling EBV pathogenesis similar to other PIDs with poor control of EBV.3 Since the T-cell activation defect is weak and can be overcome with strong or prolonged stimulation, XMEN patients do not present with severe, life-threatening infections like many other T-cell immunodeficiencies. XMEN disease also has some important differences from XLP1 and ITK deficiency. Specifically, XMEN patients do not develop fulminant infectious mononucleosis, rarely present with hemophagocytic lymphohistiocytosis, and have normal levels of NK T cells or invariant NK T cells, which have also been implicated for EBV control.3 The lack of these fatal, dysregulated immune responses against EBV in XMEN disease probably also explains why XLP1 and ITK-deficient patients generally present at a younger age with greater mortality (70% by age 10). While the pathology of XLP1 results from an uncontrolled, overwhelming CD8+ T-cell response triggered by primary EBV infection, XMEN mainly results from the lack of a sufficient immune response to clear EBV. The morbidity and mortality of XMEN disease mainly stems from EBV-associated malignancies, which may not develop until the second decade. This also makes XMEN disease difficult to recognize early in life. Nonetheless, there could be signs of immunodeficiency prior to acquiring EBV, including recurrent infections and decreased CD4 T cells. The long-term disease course of XMEN patients who never acquire EBV remains uncertain at this point, but one may speculate that they may develop lymphoma as a result of the loss of NKG2D-mediated tumor immunosurveillance, as in XLP1 or other types of cancers. The CD4 lymphopenia and T-cell activation defect is mild in XMEN patients, but they may worsen with age and make XMEN patients susceptible to opportunistic infections. Because Mg2+ supplementation may reduce the number of EBV-infected cells and potentially reduce the risk of developing lymphoma, early screening of XMEN disease in males may be beneficial. Besides providing this new therapeutic strategy for XMEN disease, our revelation of the pathologic mechanism of XMEN disease also suggests that MAGT1 is an attractive extracellular therapeutic target for immunomodulation in autoimmune diseases and allogeneic transplantations, given that it plays a specific role in regulating T-cell activation and immune cell cytotoxicity without affecting B-cell activation.
Acknowledgments
The authors would like to thank Drs Stephen Holland, V. Koneti Rao, and Jeffrey Cohen for valuable insights and assistance. We thank our colleagues at Merck for generous collaborative support.
This work was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, NIH (ZO1AI001187-01) and by co-funding through the Office of Disease Prevention, NIH.
Authorship
Contribution: F.-Y.L., B.C.-D., and M.J.L wrote the manuscript; H.S., G.U., and H.M. collected clinical data; and all authors reviewed and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Michael J. Lenardo, Laboratory of Immunology, 10 Center Dr, Building 10, Room 11D14, Bethesda, MD 20892; e-mail: lenardo@nih.gov.
References
- 1.Cohen JI. Epstein-Barr virus infection. N Engl J Med. 2000;343(7):481–492. doi: 10.1056/NEJM200008173430707. [DOI] [PubMed] [Google Scholar]
- 2.Balfour HH, Jr, Odumade OA, Schmeling DO, et al. Behavioral, virologic, and immunologic factors associated with acquisition and severity of primary Epstein-Barr virus infection in university students. J Infect Dis. 2013;207(1):80–88. doi: 10.1093/infdis/jis646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parvaneh N, Filipovich AH, Borkhardt A. Primary immunodeficiencies predisposed to Epstein-Barr virus-driven haematological diseases. Br J Haematol. 2013;162(5):573–586. doi: 10.1111/bjh.12422. [DOI] [PubMed] [Google Scholar]
- 4.Li FY, Chaigne-Delalande B, Kanellopoulou C, et al. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature. 2011;475(7357):471–476. doi: 10.1038/nature10246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaigne-Delalande B, Li FY, O’Connor GM, et al. Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D. Science. 2013;341(6142):186–191. doi: 10.1126/science.1240094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen JI, Jaffe ES, Dale JK, et al. Characterization and treatment of chronic active Epstein-Barr virus disease: a 28-year experience in the United States. Blood. 2011;117(22):5835–5849. doi: 10.1182/blood-2010-11-316745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou H, Clapham DE. Mammalian MagT1 and TUSC3 are required for cellular magnesium uptake and vertebrate embryonic development. Proc Natl Acad Sci USA. 2009;106(37):15750–15755. doi: 10.1073/pnas.0908332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rezaei N, Hedayat M, Aghamohammadi A, Nichols KE. Primary immunodeficiency diseases associated with increased susceptibility to viral infections and malignancies. J Allergy Clin Immunol. 2011;127(6):1329-1341. [DOI] [PubMed]
- 9.Pappworth IY, Wang EC, Rowe M. The switch from latent to productive infection in epstein-barr virus-infected B cells is associated with sensitization to NK cell killing. J Virol. 2007;81(2):474–482. doi: 10.1128/JVI.01777-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakajima H, Cella M, Bouchon A, et al. Patients with X-linked lymphoproliferative disease have a defect in 2B4 receptor-mediated NK cell cytotoxicity. Eur J Immunol. 2000;30(11):3309–3318. doi: 10.1002/1521-4141(200011)30:11<3309::AID-IMMU3309>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 11.Palendira U, Low C, Chan A, et al. Molecular pathogenesis of EBV susceptibility in XLP as revealed by analysis of female carriers with heterozygous expression of SAP. PLoS Biol. 2011;9(11):e1001187. doi: 10.1371/journal.pbio.1001187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parolini S, Bottino C, Falco M, et al. X-linked lymphoproliferative disease. 2B4 molecules displaying inhibitory rather than activating function are responsible for the inability of natural killer cells to kill Epstein-Barr virus-infected cells. J Exp Med. 2000;192(3):337–346. doi: 10.1084/jem.192.3.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharifi R, Sinclair JC, Gilmour KC, et al. SAP mediates specific cytotoxic T-cell functions in X-linked lymphoproliferative disease. Blood. 2004;103(10):3821–3827. doi: 10.1182/blood-2003-09-3359. [DOI] [PubMed] [Google Scholar]
- 14.Bryceson YT, Ljunggren HG, Long EO. Minimal requirement for induction of natural cytotoxicity and intersection of activation signals by inhibitory receptors. Blood. 2009;114(13):2657–2666. doi: 10.1182/blood-2009-01-201632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 2002;419(6908):734–738. doi: 10.1038/nature01112. [DOI] [PubMed] [Google Scholar]
- 16.Genberg H, Hansson A, Wernerson A, Wennberg L, Tydén G. Pharmacodynamics of rituximab in kidney allotransplantation. Am J Transplant. 2006;6(10):2418–2428. doi: 10.1111/j.1600-6143.2006.01497.x. [DOI] [PubMed] [Google Scholar]