

Abstract
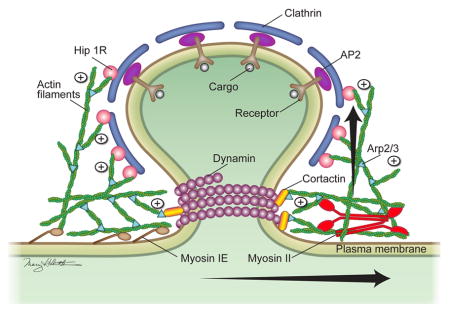
Clathrin-mediated endocytosis (CME) is essential to cell functions including nutrient uptake, receptor-mediated signaling, and membrane recycling. The requirement for actin in CME remains unclear and may vary due to regional or cellular differences in membrane tension. Previous studies have investigated the role of actin polymerization in counteracting this tension. Here we examined the role of the actin motor protein, myosin II (MII), during constitutive receptor-mediated endocytosis. In fibroblasts from embryonic MIIB knockout (MIIB KO) mice, we observed clear defects in CME: internalization of transferrin was significantly decreased, and the surface lifetime of YFP-clathrin light chain was increased. In addition, acute blebbistatin treatment to inactivate MII, shRNA-mediated knockdown of MIIB or MIIA, and treatment with nM latruculin A also inhibited transferrin uptake. Electron microscopy of MIIB KO cells or cells treated with blebbistatin revealed an increased percentage of shallow coated pits. Moreover, highly-invaginated coated pits were distorted and asymmetric. Our results indicate that MII activity is critical for coated pit progression during CME. Loss of MII function results in significant decreases in the probability of clathrin-dependent internalization. We conclude that CME is actomyosin-dependent, and that through its role in scaffolding, actin supports MII-driven force generation that regulates membrane bending and scission.
Keywords: endocytosis, clathrin, actin, myosin II
Introduction
Clathrin-mediated endocytosis (CME) is a core process by which cells internalize membrane, cell surface receptors, and paracrine and endocrine signals (1, 2). A recent synthetic biology study has demonstrated the minimum set of molecular components required for CME progression in a cell free system (3). While the core process appears to be highly conserved across many cell types, the specific components contributing to the core process and its regulation for different specialized functions can vary. For instance, the presence of the membrane bending and structural proteins N-BAR/F-BAR and Epsin may under some circumstances compensate for the absence of dynamin, a major component involved in vesicle scission (4–6). Similarly actin may be required for CME when membranes are under tension (7).
The exact mechanism by which actin contributes to CME has been a subject of long-standing debate due to conflicting results (8, 9) (10). In polarized mammalian cells, actin is considered essential for CME on the apical surface, which has a thick cortical actin layer, but not on the basal surface, which has a relatively thin cortical actin layer (11, 12), presumably due to differences in cortical actin-induced membrane tension (7). One study has shown that the barbed end of polymerizing actin is oriented toward and interacts with the Hip1r-cortactin complex at clathrin-coated pits (CCPs) (13). The interaction of the barbed end with this complex is thought to block further actin polymerization (14). Other studies found that CCP invagination correlates with the arrival of BAR proteins and a burst of actin polymerization (8, 15, 16). Based on these findings, the current model for actin-dependent CME has the following 4 steps: 1) the clathrin coat and associated adaptor proteins recruit actin; 2) membrane bending proteins (BAR proteins, epsin and dynamin) generate the forces required for coated pit curvature and initial formation of the neck; 3) intercalated actin polymerization (forming an actin “plume”) originating between the cortical actin network and the developing neck of a CCP pushes the forming vesicle inward (17). The pulling activity of myosin VI may also assist the inward movement (18), and the membrane binding activity of myosin 1E may help anchor the actin to the plasma membrane (19). And 4) the constricting activity of dynamin at the neck combined with the tension generated by the pushing activity of polymerizing actin causes scission (20, 21). This modeling assumes that actin polymerization alone provides the extra force generation necessary for endocytosis in situ. It does not ascribe a role to actin-MII contractility; however, recent evidence suggests that MII participates in vesicle fission from the Golgi network by acting on actin at the site of a budding vesicle (22). Because plasma membrane tension is a function of both the degree of attachment between the lipid bilayer and cortical actin and the contractile state of the cortex (23), we hypothesized that actomyosin-based local force generation may facilitate membrane invagination (step 2) and fission mechanisms at CCPs (step 4). To test this hypothesis, we examined the role of MII in constitutive receptor-mediated (clathrin-dependent) endocytosis. Here we present the first genetic and cellular evidence for MII having a critical role in CME.
Results and Discussion
Myosin II is essential for clathrin-dependent receptor mediated endocytosis
To test the hypothesis that MII regulates CME, we evaluated the internalization/uptake of biotin-tagged or fluorescently-conjugated transferrin in primary mouse embryonic fibroblasts (MEFs) isolated from Wt or MIIB KO mice. MIIB KO fibroblasts express only MIIA at normal levels (Figure 1A). In the first set of experiments, initial binding of transferrin (biotin-conjugated, 50 μg/ml) was performed on serum-starved cells at 4°C, followed by warming and internalization at 37 °C for different time intervals (Figure 1 B). In order to remove non-internalized transferrin from the membrane surface, we used an iron chelation-acid wash protocol (24). We then did a short saturating wash with unlabeled transferrin to outcompete any remaining non-specific binding (with the iron chelator still present) and fixed the cells. After labeling with Cy3-strepavidin, the mean fluorescence intensity per cell was measured to determine the amount of surface-bound (at 4 °C) or internalized (37 °C) transferrin. The amount of surface bound transferrin was significantly higher on Wt cells than on MIIB KO cells (Figure 1C). The amount of internalization (normalized to the amount initially bound) was more than two-fold higher for Wt cells than for MIIB KO cells at 15 min (Figure 1B). The difference in internalization was also confirmed by comparing the brightness of label in confocal optical sections taken at midsection planes through the cells (Supplemental Figure 1A).
Figure 1.

Transferrin uptake is defective when MII activity is inhibited or decreased. (A) An immunoblot comparing the levels of MIIA, MIIB and MIIC in mouse lung, Wt and MIIB KO fibroblasts. MIIC is undetectable in fibroblasts. MIIA levels appear to be the same in Wt and MIIB KO fibroblasts (see lower panel). (B) Analysis of biotin labeled transferrin uptake. Cells were serum starved for 1 hr and then labeled at 4°C for 10 min with 50 μg/ml biotin transferrin. The cultures were rinsed and then warmed to 37 °C for the indicated times. Cultures were washed with the acidic iron chelation solutions prior to fixation. They were then processed for Cy3-strepavidin labeling. The difference at 15 min was significant. T-test, P<0.0001, Wt, N= 138, MIIB KO, N=125. (C) Comparison of the fluorescence intensity of transferrin binding at 4 °C. Serum starvation results in a significant difference (*, t-test, P<0.001, N=18–20 cells per condition from two separate cultures) in the levels of biotin transferrin binding between the Wt and MIIB KO cells. (D) Quantitative analysis of fluorescent transferrin (Alexa 488) uptake at 15 min as in Supplemental Figure 1C comparing Wt cells to MIIB KO cells (+/− blebbistatin treatment) and knockdown of MIIA and MIIB (see Figure 1E for details of knockdown). Blebbistatin was preincubated for 15 min a 37 °C and then readded during transferrin labeling (4 °C) and was maintained during warmup (15 min). Transferrin uptake is significantly decreased after KO or acute inhibition of MII activity (ANOVA, P<0.01, N> 100 cells per condition from 3–5 cultures). Error bars=S.E.M. (E) MII knockdown: Stable MIIA and MIIB knockdown cell lines were generated by infecting MEFs with a lentivirus expressing a shRNA targeted to either mouse MIIA or MIIB heavy chain. MIIA and MIIB protein levels in stable myosin II knockdown MEFs were reduced by 95% and 97%, respectively, compared to control (WT) MEFs. Knockdown of MIIA had no affect on MIIB content while depletion of MIIB had a minimal affect on MIIA content (15%). MII protein levels were routinely monitored to ensure MIIA and MIIB knockdown was consistent between experiments.
Recent work indicates that surface levels of transferrin receptors may increase (especially in mitotic cells) with serum starvation or with warming after labeling at 4 °C (25); therefore, as a control, we repeated the experiments to measure binding and internalization of biotin-tagged transferrin. In the absence of serum starvation we found no difference between Wt and MIIB KO cells in the amount of biotin-tagged transferrin binding at 4 °C (Figure 1C). As an additional control, we used Alexa-488 conjugated transferrin to monitor uptake. The advantage of this label is that it can also be used to monitor internalization in live cells. Unlike biotin tagged transferrin, Alex-488 conjugated transferrin showed a progressive increase in fluorescence brightness with internalization at 37 °C (compared to the fluorescence intensity at 4 °C) (Supplemental Figure 1B, C). Similar to biotin tagged transferrin, low concentrations (25 μg/ml) of Alexa-488 conjugated transferrin showed a higher level of binding of transferrin to Wt cells compared to MIIB KO following serum starvation (Supplemental Figure 1B). However when higher concentrations (100 μg/ml) of Alexa-488 conjugated transferrin were used, we did not observe a difference (Supplemental Figure 1C). Wt cells showed greater internalization of Alexa-488 conjugated transferrin at both low and high labeling concentrations compared to MIIB KO cells (Supplemental Figures 1B–D). The increase in brightness of the Alexa-488 conjugated transferrin label upon internalization was confirmed by imaging live cells at 4 °C followed by warming and imaging the same cells after 5 min. (Supplemental Figure 1E). To test whether the effect on transferrin uptake was directly a result of the loss of MII activity (as opposed to downstream developmental defects), we also acutely applied the small molecule inhibitor blebbistatin to inhibit MII during binding and internalization of transferrin. Blebbistatin treatment caused significant decreases in transferrin uptake in both Wt and MIIB KO cells (Figure 1D, Supplemental Figure 1D). The inhibition observed in the MIIB KO cells suggests that both MIIB and MIIA are involved in CME. Next, in order to test the contribution of each MII isoform in the endocytic pathway, we performed knockdown (KD) of MIIA or MIIB in the Wt fibroblasts (Figure 1E) and then quantified transferrin uptake per cell (Figure 1D). Knockdown of the MIIB isoform resulted in decreased transferrin uptake at 15 min comparable to that of MIIB KO cells (Figure 1D, Supplemental Figure 1D). Knockdown of the MIIA isoform also inhibited internalization of transferrin, although the effect was slightly less severe than that seen in MIIB KO or MIIB KD cells (Figure 1D, Supplemental Figure 1D). This result is consistent with MII having an important role in CME in that, similar to the knockout of MIIB, acute inhibition of MII compromises transferrin uptake.
To determine if the decreased rate of transferrin uptake resulted from defects in receptor recycling or altered kinetics of uptake, we analyzed the steady state uptake of Alexa-488 conjugated transferrin (100 μg/ml) in cells that were continuously binding and internalizing transferrin at 37 °C (Figure 2A). The cells were fixed at specific time intervals without acid washing. Thus the measured fluorescence represents the total surface-bound and internalized transferrin at the different time points. Although the MIIB KO cells showed somewhat slower uptake at the initial time points, both the Wt and MIIB-KO cells appeared to reach a steady state by 25 min; notably, the maximum steady state fluorescence intensity of the MIIB KO cells was approximately 1/3 that of the Wt cells. Because the amount of surface-bound transferrin appears to be the same on MIIB KO cells as on Wt cells (at 4 °C; Supplemental Figure 1C), the decreased fluorescence intensity suggests that the MIIB KO cells internalize less transferrin.
Figure 2.

Steady state transferrin uptake is defective in MIIB KO fibroblasts and can also be influenced by latrunculin A treatment; transferrin receptor levels are normal. (A) Comparison of Alexa-488 transferrin uptake in Wt and MIIB KO cells incubated with the label at 37 °C. Cells were rinsed and then fixed at the indicated times. MIIB KO cells had lower fluorescence at every time point. Both Wt and MIIB KO cells appear to reach a steady state between internalization and processing (exocytosis of fluorescent transferrin) at about 25 min. N values (cells) at the different time points (2.5, 5, 10, 25 min) from 3 experiments, Wt= 204, 135, 152, 130, MIIB KO=140, 166, 115, 145. (B) Treatment with a low concentration of latrunculin A (250 nM) reduces the uptake of transferrin in both Wt and MIIB KO cells. Cells were pre-incubated with latrunculin A for 30 min at 37 °C (without serum starvation) and then incubated with Alexa-488 conjugated transferrin (± latrunculin A) for the indicated times at 37 °C. The cultures were washed with the acidic iron chelation solutions prior to fixation. Wt cells showed a significant decrease in transferrin uptake at both 5 and 15 min (t-test, P<0.001, N≥70 cells per time point). MIIB KO cells showed a significant decrease in transferrin uptake at 15 min (t-test, P=0.02, N≥70 cells per time point).(C) MIIB KO cells show decreased capacity to bind and internalize dye labeled transferrin after initial loading with unlabeled transferrin. Cells were incubated at 37 °C for 15 min with unlabeled transferrin (100 μg/ml), rinsed and then labeled at 37 °C with Alexa-488 transferrin for 10 min. Cells were rinsed 4× and then fixed. Wt cells are more than 2× brighter indicating a much higher capacity to bind and internalize labeled transferring after binding and processing unlabeled transferrin. Difference was significant (t-test, P<0.001, Wt, N=121, MIIB KO, N=118). (D) Comparison of transferrin receptor immunofluorescence labeling (without transferrin labeling or serum starvation). Cells were labeled with an antibody to the transferrin receptor and then imaged by TIRF microscopy. There was no difference in the brightness between Wt and MIIB KO cells. (t-test, P=0.8, N=10 cells each). (E) An immunoblot of the transferrin receptor. Equal amounts of protein were loaded in each lane. The bands were of equivalent intensity. (F) Dual channel TIRF microscopy of transferrin receptor immunofluorescence (green) and biotin transferrin (red) from cells fixed after 5 min at 37 °C. RGB line profiles of lines drawn diagonally (left to right) through the images are shown below the TRIF images. In the Wt, spots show good correspondence between intensities in both channels. In the MIIB KO cells some receptor spots show relatively low levels of transferrin label (arrows). Bar=8 μm.
MII is known to regulate tension in the actin cortex of cells (26). The mechanical properties of cells are sensitive to low levels of latrunculin, an actin polymerization inhibitor (27, 28). Latrunculin treatment reduces cell stiffness consistent with a reduction in cortical tension. To determine if reduced cortical tension may contribute to the decrease in transferrin uptake observed following MII inhibition, we treated cells with low concentrations (250 nM) of latrunculin for 30 min and then assayed the steady state transferrin uptake at 37 °C (no serum starvation) using Alexa-488 conjugated transferrin. Cells were rinsed with the acid wash/iron chelation solutions prior to fixation to remove surface-bound label. At 250 nM, latrunculin treatment did not produce a detectable change in morphology when cells were observed for 30 min by time-lapse microscopy (Supplemental Figure 1A). 250 nM latrunculin A caused some minor changes in both actin and MIIB organization detected by rhodamine-phalloidin staining and MIIB immunofluorescence (Supplemental Figure 2B, C). Latrunculin treatment caused a significant decrease in transferrin uptake by Wt cells at 5 and 15 min (Figure 2B). The treatment had less of an effect on the MIIB KO cells, but it was roughly proportional to the reduced amount of MII present in the KO cells (~50% of that in Wt cells). A significant decrease in transferrin uptake was observed at 15 min.
The effect of latrunculin A treatment on Wt cell transferrin uptake is relatively small compared to the effect of MIIB inhibition. This is likely because the low concentration of latrunculin A caused some changes in the organization of the more labile cortical actin filaments, but they remained largely intact (Supplemental Figure 2B). Thus, myosin II could still act on actin scaffolds and thereby produce enough force to assist CME. Consistent with this possibility is the observation from Eliot Elson’s lab that treatment with latrunculin B has a dose-dependent effect on cell stiffness. Minimum stiffness (maximum effect) was only achieved with treatment at a concentration ≥ 600 nM (27). Thus it is likely that actomyosin interactions continue to contribute to cortical tension and have other more local effects when latrunculin A is applied at a concentration of 250 nM. Another possibility is that myosin II preferentially interacts with formin regulated cortical actin filaments. If this is the case, then it is likely that actomyosin interactions that aid in CME will persist even after treatment with higher concentrations of latrunculin A because it has been shown that this population of filaments is resistant to actin-depolymerization drugs such as latrunculin or cytochalasin (29). Further experiments will be required to distinguish between these possibilities.
To test whether the decreases in transferrin uptake by MIIB KO cells were possibly a result of abnormal processing of Alexa-dye conjugated transferrin, we incubated cells with non-fluorescent transferrin for 15 min followed by the fluorescent transferrin for 10 min (Figure 2C). The difference in fluorescence intensity was approximately the same as that observed with the steady state binding of Alex-488 conjugated transferrin at 25 min (Figure 2A) indicating that processing of dye-labeled transferrin was normal.
To test whether recycling of the transferrin-receptor complex was defective, Wt and MIIB KO cells were incubated (15 min) sequentially with two different colors of fluorescent-dye conjugated transferrin (Alexa 546 followed by Alexa 488 with intervening acid washes to remove surface-bound transferrin). After fixation, MIIB KO cells were found to have decreased intensities of both colors of fluorescent transferrin at 30 min (Wt, R= 3±0.4, G=5.2±0.4, N=30, MIIB KO R=1±0.1, G=2.4±0.3, N=24). The ratio of intensities (MIIB KO/Wt) was less for Alexa-546 conjugated transferrin than for Alexa-488 conjugated transferrin (0.32 vs 0.46) indicating that processing of the sequentially applied transferrin did not appear to be delayed.
To determine if the level of transferrin receptors differed between Wt and MIIB KO cells we compared the fluorescence levels at or near the basal surface using TIRF microscopy of immunofluorescence, as well as immunoblotting. Both tests showed that Wt and MIIB KO cells have approximately the same number of receptors (Figure 2D, E). If cells were not serum starved, biotin-tagged transferrin labeling for 5 min (at 37 °C) still resulted in a large difference in the amount of transferrin internalized in Wt versus MIIB KO cells (a 51±3% lower fluorescence intensity in MIIB KO cells; Wt= 652±25, MIIB KO=335±27, significantly different, t-test, P<0.001, Wt, N=23, MIIB KO, N=18). This indicates that MIIB KO cells have a defect in internalization that can be observed whether or not the amount of surface transferrin binding at 4 °C differs between Wt and MIIB KO cells.
To further investigate potential transferrin receptor defects, we tested whether there was any difference in the distribution of surface transferrin receptors between Wt and MIIB KO cells. Following biotin-tagged transferrin uptake for 5 min at 37 °C, cells were fixed and immunolabeled for the transferrin receptor. The basal surface was imaged using dual channel TIRF microscopy (Figure 2F). In Wt cells the receptor staining and the biotin-tagged transferrin labeling were highly correlated. However in the MIIB KO cells, areas with strong receptor staining frequently did not show commensurate transferrin label intensity. This suggests that the binding of transferrin to areas containing high levels of receptor in the MIIB KO cells may be compromised and may lead to the differences in transferrin binding observed with lower concentrations of the ligand (see Supplemental Figure 1B).
Using dual-channel TIRF microscopy, we also compared the receptor staining of cells fixed at 4 °C to that of cells labeled and fixed after 5 min at 37 °C (no serum starvation; without iron chelation or acid washing). Both Wt and MIIB KO cells showed no change in the levels of receptor fluorescence staining (slight decreases that were not significant; t-test, Wt P=0.1, MIIB KO, P=0.7) suggesting that surface receptor levels are unaffected under these conditions. These results strongly suggest that the decrease in MII activity severely compromises constitutive receptor-mediated CME independent of any effects on surface receptor levels and that the primary defect may lie in the early internalization steps.
Myosin II regulates coated pit dynamics
To determine the effect of MII loss on CME progression, we imaged surface dynamics of YFP-clathrin light chain (CLC) in Wt and MIIB KO cells using TIRF microscopy. We found that YFP-CLC had reduced dynamics in the MIIB KO cells compared to the Wt cells (Figure 3). Using kymograph analysis we found that the average lifetime of fluorescent CLC spots on the cell surface was more than double for MIIB KO cells than for Wt cells and that lateral movements were attenuated (maximum displacement Wt=1.3±0.14 μm, KO=0.7±0.07 μm) (Figure 3 A, B, supplemental movies 3S1–3). In addition, a greater percentage (Wt<1% vs MIIB KO=2–10%) of CLC spots on the MIIB KO cells persisted during the entire recording time (5 min) suggesting that they may have stalled. Expression of the Wt MIIB heavy chain in the KO cells partially rescued the phenotype as indicated by the increase in the percentage of dynamic YFP-CLC spots (including the lateral movements) (Figure 3B). Interestingly, incubation of MIIB KO cells with transferrin (cargo loading) caused a further decrease in dynamics in MIIB KO cells, whereas Wt cells were insensitive to cargo loading (Figure 3C). The latter is consistent with previous studies that showed cargo loading is not rate limiting for CCP maturation (30). In the absence of MIIB, perhaps cargo loading is rate limiting for CCP maturation because rapid progression of curvature requires MII ATPase activity and increased tension. To test whether MII ATPase activity is essential for MII’s role in regulating the coated pit dynamics, we co-expressed either Wt (GFP-MIIB) or a point mutant heavy chain of MIIB (GFP MIIB R709C) that has compromised ATPase activity (31) and RFP-CLC in the MIIB KO cells. TIRF microscopy showed that the point mutant was unable to rescue the coated pit dynamics (surface lifetimes) in the MIIB KO cells compared to the Wt GFP MIIB heavy chain, which was efficient at decreasing lifetimes (Figure 3D,E). These results suggest that MII activity is important for controlling the rate of clathrin-coated pit formation and, in the absence of MII, the probability of internalization is decreased.
Figure 3.

TIRF microscopy of clathrin light chain (YFP-CLC or RFP-CLC) reveals an increased lifetime that is accentuated by transferrin labeling (cargo loading) on the basal surface of fibroblasts in MIIB KO cells. (A) Panels on left show kymographs of YFP-CLC in Wt and MIIB KO cells and also in MIIB KO cells following partial rescue by transient expression of the MIIB heavy chain. The MIIB KO shows greater persistence of spots (longer streaks). Images were taken at 5 s intervals. Panels on right show recordings from regions of interest containing a few spots. In the MIIB KO some spots (circled in red) persist for prolonged periods. Time is in seconds. Note that time 0 represents the beginning of the example sequence not the initiation of recording (the spots appeared during the recording). (B) Quantitative analysis of YFP-CLC lifetime. The average lifetime per cell (±S.E.M) is shown along with the average for the group (thick horizontal black line). At minimum, 50 spots were analyzed in each cell. MIIB KO cells show a significant increase in lifetime (t-test; P=0.02, the number of cells is represented by the points on the graph; from 3–4 cultures). This is an underrepresentation of the difference because 2–10% of the YFP-CLC spots in the MIIB KO cells persisted throughout the entire recording and were not included in the analysis. Rescue by transient expression of the MIIB heavy chain in the MIIB KO cells was done blindly (using an adenovirus vector to infect cells) because the vector did not express a fluorescent marker protein. Typical infection rates were 75–80% and most cells showed a shorter lifetime compared to MIIB KO cells. A few cells had longer lifetimes and were negative for MIIB heavy chain expression as determined by post hoc fixation and immunolabeling. (C) Comparison of lifetimes in transferrin-labeled and unlabeled cells. YFP-CLC lifetimes in the MIIB KO cells increase after transferrin labeling. Cells were labeled with Alexa-546 transferrin (100 μg/ml) and then recorded. The difference was not significant in the Wt cells (t-test, P=0.09, N=8 cells each from 3 cultures) but was significant in the MIIB KO cells (t-test, P=0.02, N=8 each from 3 cultures). (D) On left are examples of kymographs for Wt and MIIB KO cells expressing RFP-CLC. On the right are RFP-CLC spots from cells analyzed for lifetime by TIRF microscopy. Co-expression of either GFP-MIIB or GFP-R709C MIIB with RFP-CLC decreased the signal to noise. (E) Quantitative analysis of lifetimes from kymographs. Expression of the Wt MIIB decreased lifetimes MIIB KO cells (rescue), but expression of the mutated MIIB had no detectable affect on lifetimes. Difference were significant, t-test left to right * P≤0.001, Wt, N=12 cells from 4 cultures, MIIB KO N=11 cells from 4 cultures, ** P≤0.001, GFP-MIIB expression in MIIB KO cells N=8 cells from 3 cultures, ***P≤0.001, GFP-R790C expression in MIIB KO cells, N=8 cells from 3 cultures.
Coated pits show associations with actin and MIIB
If MIIB contributes to the regulation of coated pit internalization by locally exerting force or reorganizing actin, then it should at least partially colocalize with coated pits. To test this idea, we labeled fixed cells with antibodies to MIIB and clathrin heavy chain (CHC). With wide-field fluorescence microscopy, colocalization was difficult to detect because of a high background signal associated with the staining for MIIB (Figure 4A). However intensely stained stress fibers appeared to exclude CHC spots. With TIRF microscopy, close association could be observed between some YFP-CLC spots and distinct MIIB spots. The YFP-CLC spots sometimes aligned adjacent to MIIB staining associated with basal stress fibers. Similarly, we also observed partial close association between CHC and MIIB spots (up to 60%) in images of unroofed fibroblasts obtained using wide-field fluorescence microscopy. In most cases, the CHC and MIIB spots were not superimposed but adjacent to each other. This spatial relationship was confirmed by confocal imaging of YPF-CLC and MIIB staining (Figure 4B). In addition, we found MIIA staining to have a similar close association (Figure 4B). Rotary shadow EM of unroofed Wt fibroblasts showed coated pits inter-digitated between actin stress fibers and closely associated with an intervening meshwork of actin filaments (Figure 4C). We observed a similar association between coated pits and actin filaments in MIIB KO cells, although the coated pits tended to be larger and less symmetrical (Figure 4D). Immunogold EM labeling of MIIB in unroofed Wt fibroblasts revealed MIIB closely associated with coated pits. Frequently the label was found in association with actin filaments contacting coated pits on one or two sides and within 150 nm of the coated pit surface (Figure 5A). Although unroofing causes variable amounts of actin filament removal, densely branched actin filaments are often found to be associated with coated pit structures following unroofing (13, 32, 33). Our results show that MII is also associated with these patches of branched actin filaments.
Figure 4.

Morphological relationship between coated pits, actin and MIIB. (A) Wide-field (WF) fluorescence microscopy (left panel) of YFP-CLC and immunofluorescence staining shows an apparent interdigitation of coated pits (green) with MIIB positive (red) stress fibers labeled with an antibody to the MIIB heavy chain. Bar=3 μm. TIRF microscopy (middle panel) shows that some of the coated pits (YFP-CLC, green) associate with bright spots of MIIB immunofluorescence (red) on the basal surface in intact cells. Bar=2 μm. Wide field microscopy of unroofed fibroblasts processed for immunofluorescence. In areas largely devoid of membrane associated stress fibers, a close relationship is observed between many coated pits (green) labeled with an antibody to the clathrin heavy chain and bright MIIB spots labeled with an antibody to the MIIB heavy chain (red). The high magnifications show the percentage that coated pits associated with the MIIB spots in different configurations (top to bottom); a. clathrin heavy chain spots unassociated with MIIB, b. associated on one side, c. associated on two sides, d. associated on more than two sides. Bar=4μm. (B) Confocal (single optical slice) images taken close to the substrate using a 60X 1.4 N.A. lens with low chromatic aberration. The association between YFP-CLC (green) and MIIB (left panel, red)) or MIIA (right panel, red). 38±3% of the YFP-CLC spots overlapped or contacted MIIB spots, while 33±4% showed the same association with MIIA spots. (C) Deep etch, rotary shadow images of unroofed Wt fibroblasts showing the relationship between actin filaments and coated pits. In low magnification images (top panel) coated pits appear to interdigitate between stress fiber associated actin. Bar=175 nm. At higher magnification (bottom panel) individual or branched networks of actin filaments (arrowheads) closely associate with curved coated pits. Bar=100 nm. (D) A similar comparison for MIIB KO cells. Coated pits appeared more asymmetric (double arrow) and show associations with branched networks of actin filaments (arrowheads). Top, Bar=175 nm, bottom, Bar=150 nm.
Figure 5.

MIIB is closely associated with coated pits in the Wt and clathrin-coated pit (CCP) structure is abnormal in MIIB KO cells. (A) Immunogold labeling for MIIB in Wt cells. Particles are circled in red for better visibility. Gold particles were identified in anaglyphs. From two separate preparations: on left, 18 nm gold, on right, 12 nm gold. Bar=90 nm. (B) Comparison of highly-invaginated pits in thin section (left) and in rotary shadowed replicas (right arrows indicate coated pit). Far right panel is an anaglyph. (C) The same comparison in MIIB KO cells. Note that the coated pits are distorted. Bar=150 nm. (D) CCP profiles in thin sections from Wt cells. S= shallow, C=curved, I=Invaginated, CCV=clathrin coated vesicle. (E) CCP profiles in MIIB KO cells. Arrowheads indicate distorted pits. Bar=100 nm. (F) Distribution of CCP profiles in Wt cells as a percentage of total (N=142 from 20 cells) observed. (G) Distribution of CCP profiles (N=132 from 15 cells) in MIIB KO cells as compared to Wt. The frequency of shallow and curved coated pits is increased, whereas the frequency of CCVs is greatly decreased. (H) Blebbistatin treatment results in a greater frequency of shallow and curved coated pits compared to untreated fibroblasts. The frequency of CCV’s is decreased. Untreated Ct, N=138 from 18 cells, bleb treated, N=144 from 16 cells. (I) Shallow coated pits measured in rotary shadowed replicas from unroofed fibroblasts are larger in diameter in MIIB KO cells. (T-test, P<0.001, N=20 each). The same result was obtained from measurements made from thin sections, except that the diameters for shallow coated pits in both Wt and MIIB KO cells were approximately 30% less than in replicas. (J) Histograms showing the distribution of coated pit symmetry (width in x divided by width in y) measured in rotary shadowed preparations. Coated pits are more asymmetrical in MIIB KO cells.
MII is required for coated pit curvature progression and normal symmetry during CME
We compared the ultrastructure and actin-association of coated pits in thin sections and in replicas of unroofed fibroblasts prepared by freeze-etch and rotary shadowing (Figure 5B, C). Highly-invaginated coated pits were distorted in the MIIB KO cells. To quantify the difference in morphology, we compared the size of shallow coated pits (Figure 5I) and the degree of asymmetry (Figure 5J). Coated pits in the MIIB KO cells were larger in diameter and more asymmetric. The actin filaments associated with coated pits, including the population of short branched filaments that might be involved in vesicle internalization (33), appeared to have a relatively normal distribution. In the MIIB KO cells, coated pits reside on the surface for longer times. To determine if a specific stage of coated pit maturation was delayed, we cut thin sections perpendicular to the substrate and scored the percentage of coated pits at each stage of maturation (Figure 5D, E). The MIIB KO cells had fewer coated vesicles and a greater percentage of shallow and curved coated pits compared to the Wt cells suggesting that MIIB contributes to the early stages of coated pit curvature (Figure 5 F, G). Highly invaginated coated pits were rare in MIIB KO cells but when found were distorted (Figure 5E). Wt cells treated with blebbistatin had a similar increase in shallow and curved coated pits (Figure 5H). MII can generate tension on actin filaments adjacent to coated pits that might aid in the progression of coated pit curvature necessary for the fission process. Recent evidence suggests that MII-based force generation is required for recruitment of membrane sculpting N-BAR proteins to the plasma membrane (34). Our results indicate that, in the absence of MII activity, curvature can still occur, but it progresses more slowly and generates an asymmetric invaginated coated pit that may compromise the final steps of the fission process.
MII is a primary regulator of tension in the cortical actin network. The effect of the tension can be local or global depending upon the cell state and its environment (23, 35). Cortical actin turns over at different rates (29). Latrunculin A treatment may have the greatest effect on more labile actin, which predominates and may be the primary form involved in actomyosin-generated cortical tension. Thus perturbation of either MII or actin may decrease cortical tension. A simple interpretation of our results is that local tension generated in the plane of the plasma membrane by MII regulates coated pit symmetry and progression. Our results show that MII is integrated into the membrane-associated actin network located immediately adjacent to coated pits. In the absence of MII or its ATPase activity, shallow coated pits reside on the membrane surface for longer times. If they do become highly invaginated, they are likely to be asymmetric. MII-based force generation is required for recruitment of membrane sculpting N-BAR proteins to the plasma membrane (34). Therefore recruitment of these proteins may be compromised in the absence of MII activity. Alternatively, MII activity may be required for these proteins to fully coordinate their actions to promote normal membrane deformation and scission. The activity of dynamin and its interaction with the actin cytoskeleton may be similarly compromised. Because of its integration into the cortical actin network, MII is uniquely positioned to regulate tension at levels that are optimal for endocytosis. Recently it has been shown that actin polymerization is important for endocytosis on the apical membrane of polarized cells that are under tension, but not the basal surface (7). Because actin polymerization also contributes to the thicker cortex and specializations of the apical surface, it remains unclear how membrane tension is regulated or how actin polymerization facilitates endocytosis without also increasing membrane tension. Our findings indicate that the role of MII in regulating CME is likely as important, or more important than the role of actin polymerization. MII-dependent tension appears to regulate CME in nonpolarized cells and, so, may have an essential role in most mammalian cell types. Moreover, it may have an especially important role in cells that require rapid endocytosis to compensate for membrane addition following intense exocytosis. The phenomenon of synaptic vesicle recycling following evoked release of neurotransmitter from hippocampal neuron synapses is one such example (36).
While MII clearly has a role in regulating CME, the exact mechanism by which it facilitates coated pit progression and internalization remain to be clarified. One possibility is that actomyosin works in a manner analogous to a purse-string around the coated pit. If cortical tension is below normal (e.g., due to membrane expansion) then contraction of the network around the coated pit increases the tension (pulls the strings). If the tension is above normal (e.g., due to increased actin polymerization), then the network relaxes (loosens the strings). Thus, MII may act to keep tension within an optimal range as required for membrane curving. The MII contraction then adds tension to aid coated pit progression and recruitment of membrane curvature proteins to form the neck. MII may then stiffen the cortex around the mouth (in the plane of the plasma membrane) to maintain tension as actin polymerization “pushes” the invaginating membrane internally. This may allow the relatively weak forces generated by dynamin or BAR proteins to further deform the membrane until the tension increases to a level required for scission at the dynamin/membrane interface (21). Further work will be required to reveal the details of this mechanism as well as the upstream regulatory elements for specialized cell functions that are CME-dependent such as compensatory endocytosis and constitutive receptor-mediated endocytosis.
Materials and Methods
Cell Culture
The genotype of embryos was determined by a combination of PCR against the PGK-Neo cassette and immunoblotting (37, 38). Fibroblast cultures were prepared from fibroblasts isolated from Wt or MIIB KO embryos (39). Cells were maintained in DMEM +10% FCS on cell culture treated dishes or flasks. For experiments, cells were plated on polylysine- and fibronectin-coated glass bottom dishes (Fluorodishes, WPI). All procedures involving use of animals have been approved by the Washington University Animal Studies Committee.
Fluorescent transferrin uptake
Unless indicated, fibroblasts were serum starved for 1 hr and then labeled with fluorescent transferrin using either Alexa-546 or -488 conjugates at 4 °C for either 10 min (100 μg/ml) or 30 min (25 μg/ml). They were rinsed 4× with cold medium (1% serum) and 3× with room temperature medium (1% serum). Warm medium (10% serum) was then added after pre-equilibrating it with CO2. Biotin tagged transferrin was used at 50 μg/ml and unless noted was incubated for 10 min at 4 °C. The cultures were incubated in a 37 °C CO2 incubator for the indicated times prior to fixation or live imaging (using a stage top incubator). To remove surface-bound transferrin, the cells were rinsed prior to fixation with an acidic solution containing an iron chelator (deferoxamine mesylate) followed by a solution containing both the iron chelator and unlabeled transferrin (100 μg/ml) (24). To detect the biotin tagged transferrin, fixed cells were permeabilized with 0.2% Triton, incubated with a blocking solution and then labeled with Cy3-strepavidin for 1 hr. For blebbistatin treatment or latrunculin A treatment cultures were preincubated with the drugs as described (see figure legends) prior to adding fluorescent conjugated transferrin. The drug was maintained in the medium during the incubation with the conjugated transferrin.
Electron Microscopy
For thin section EM fibroblast cultures were fixed with 2.5% glutaraldehyde and then postfixed with 0.5% OsO4+ 0.5% K+ ferricyanide for 15 min. After reaction with 0.1% tannic acid (15 min) cultures were stained en bloc with 50 mM uranyl acetate in acetate buffer for 15 min. Dehydration and embedding in Araldite was done using conventional procedures. For rotary shadow EM, cultures were subjected to sonication to remove the apical membrane and then immediately fixed (40). They were then rapid frozen and freeze-etched prior to rotary shadowing with platinum. Images were taken on a JOEL 1400 transmission electron microscope equipped with 4K AMT digital camera. Images were processed in Adobe Photoshop to adjust brightness and contrast but not gamma.
Knockdown of MIIA and MIIB in Mouse Embryonic Fibroblasts
Knockdown of MIIA and MIIB in MEFs was achieved using a shRNA-MIIA and MIIB heavy chain lentiviral vector obtained from Open Biosystems (Clone#-071504 and 123074 respectively; Thermo Scientific, Waltham, MA). Lentiviruses were produced in 293T/17 cells as outlined previously using 2nd generation transfer plasmids (41, 42). MEFs (4×105 cells) were infected with shRNA-MIIA or MIIB virus for 72 hrs. Cells were grown to confluence, split and incubated in the presence of 5 μg/ml puromycin to establish stable cell lines. Knockdown of MIIA and MIIB protein levels was verified by Western blot analysis and immunofluorescence confocal microscopy.
Transfection and Immunolabeling
The cultures were transfected with plasmids expressing YFP-CLC (Addgene plasmid #20921) or RFP-CLC (Addgene plasmid 14435), GFP-MIIB heavy chain or the GFP-R709C mutated MIIB heavy chain (31) by Lipofectamine 2000. Imaging was done within 18 hrs of transfection. For rescue experiments cells were infected with an adenovirus construct (VQAd5-CMV) with an insert that encoded the MIIB heavy chain. Fixed cultures were labeled with polyclonal antibodies to the MIIB or MIIA heavy chains (CPI), transferrin receptor (BD), and monoclonal antibodies to clathrin heavy chain (BD) and transferrin receptor (Invitrogen) as described previously (37).
Light microscopy
For monitoring transferrin uptake, cultures were imaged on either a Zeiss LSM 510 NLO microscope system equipped with an inverted stand and a stage-top incubator or an Olympus FV1200 confocal similarly equipped. Both fluorescence and DIC images were taken for each field. For live imaging on the Zeiss LSM system or for capture of total fluorescence from fixed cells, single-plane confocal images were acquired using a 40X water-immersion lens (N.A. 1.2) with the pinhole size adjusted to produce an optical section that was roughly the thickness of well spread cells (2.2 μm). To allow repeated imaging of the same cells over time, the stage coordinates were recorded. For live imaging on the FV1200 LSM system, a 60X 1.3 N.A silicon oil objective was used. For fixed cultures on the Zeiss LSM, a 63× oil-immersion lens (N.A. 1.4) was used. For multi-plane confocal imaging, the pinhole was set to give an optical section thickness of 0.8 μm and the Z-step size was set to 0.6 μm. For confocal imaging of fixed cultures on the FV1200 LSM system, two lenses were used. For optical sectioning we used a 60X, 1.4 N.A lens (PLAPON60XOSC) with reduced chromatic aberration. The point spread function for green and red fluorescence was checked using 0.3 μm beads and showed symmetrical distribution of fluorescence in both channels. The pinhole was set to give a 0.4 μm optical section and the Z-step was also set to 0.4 μm. For lower magnification images, a 40× 1.25 N.A silicone oil lens was used with a wide pinhole setting (2.8 μm).
An Olympus IX71 inverted light microscope equipped with a 60× oil-immersion lens (N.A. 1.4) and a Sensicam CCD camera (Cooke Corp) was used for epifluorescence imaging of fixed cells. A second Olympus IX71 inverted microscope equipped with two laser lines (488 and 543 nm), a 60× oil-immersion TIRF objective (N.A. 1.46), a stage-top incubator, and a Hamamatsu EMCCD camera was used for TIRF microscopy. Fluorescence images of live cells were acquired using serum-containing medium (free of phenol red) following serum starvation. Addition of labeled transferrin to monitor endocytosis was used where indicated. Kymographs were generated in ImageJ using Multikymograph and analyzed using published criteria where the lifetime was defined as an intensity with a threshold equal to 20% of the maximum (43). At least 3 kymographs were generated for each cell. To verify the accuracy of the kymograph analysis, some spots were analyzed manually using the time-lapse sequences. Fixed cells were imaged in phosphate buffered saline. Image brightness and contrast were adjusted using Adobe Photoshop. Gamma settings were not changed.
Measurements of fluorescence intensity were performed using ImageJ. To determine the average fluorescence intensity per pixel for each cell, cells were outlined by hand using the DIC image as a guide. Overlapping cells were avoided. The background was measured in an adjacent (noncellular) area of the substrate and subtracted from the average intensity value measured for each cell. For intensity measurements using TIRF microscopy, a modified procedure was used; a ROI with set dimensions was measured in the brightest area of each TIRF image of a cell.
Supplementary Material
Movie 3S-1. YFP clathrin light chain dynamics in a Wt fibroblast imaged with TIRF microscopy. 5 s intervals.
Movie 3S-2. YFP clathrin light chain dynamics in a MIIB KO fibroblast imaged with TIRF microscopy. 5 s intervals.
Movie 3S-3 YFP clathrin light chain dynamics in a MIIB KO fibroblast imaged with TIRF microscopy. The cell was infected with an adenovirus construct containing the MIIB heavy chain to rescue the KO phenotype. 5 s intervals.
Supplemental Figure 1. (A) Comparison of the uptake of biotin transferrin in single confocal optical slices taken at the middle equatorial plane after 15 min at 37 °C. Cells were not serum starved and were incubated with the biotin transferrin at 37 °C prior to rinsing, iron chelation/acid washing and fixation. MIIB KO cells show fewer spots and spots show lower fluorescence intensities. Bar=8 μm. Quantitative analysis and validation of the Alexa-dye labeled transferrin uptake assay. Uptake is decreased in MIIB KO cells compared to Wt controls. Cells were serum starved for 1 hr and then labeled at 4 °C for 30 min with 25 μg/ml (B) or 10 min with 100 μg/ml (C) Alexa-488 conjugated transferrin. They were then rinsed extensively and fixed at 4 °C (first time point) or rinsed and warmed by adding prequilibrated medium (10% FBS) and incubated for the indicated times at 37 °C. Prior to fixation the warmed cells were acid washed with solutions containing an iron chelator to remove surface bound transferrin followed by a saturating concentration of unlabeled transferrin with the iron chelator still present (24). The amount of surface labeling differed between Wt and MIIB KO cells when the concentration of Alexa-488 transferrin used was 25 μg/ml, but was the same when the concentration of label was raised to 100 μg/ml as indicated by the fluorescence intensities in cells fixed at 4 °C (t-test, 25 μg/ml, P<0.0001, Wt, N=140, MIIB KO, N=138P=0.08, 100 μg/ml, Wt, N=63 cells, MIIKO, N=49 cells). The increase in fluorescence intensity after warming indicates that internalized fluorescently labeled transferrin increases in brightness. This increase may be a result of decreased fluorescence quenching by iron; the metal ion is released from transferrin starting within 3 min of internalization (44, 45). The differences were significant for both concentrations at 5 min and 15 min (t-test, 25 μg/ml, 5 min 37 °C P<0.0001, Wt, N=102, MIIB KO N=62; 15 min 37 °C, P<0.0001, Wt, N= 95, MIIB KO, N=72, 100 μg/ml, 5 min, P<0.0001, Wt, N=90 cells, MIIB KO, N= 39 cells, 15 min, t-test, P<0.0001, Wt, N= 107 cells, MIIB KO, N=47 cells). Three cultures were used for each category. (D) Examples of fluorescent transferrin uptake in fixed cells at 15 min in Wt and MIIB KO fibroblasts (+/− blebbistatin) and following knockdown of MIIA and MIIB. Bar=20 μm.(E) Increase in the brightness of Alexa-488 conjugated transferrin label following warm-up of live cells. Cells were labeled for 10 min at 4 °C with 100 μg/ml Alexa-488 conjugated transferrin, washed extensively with cold medium and then placed on the microscope for imaging with high sensitive detectors (Olympus FV1200). The left panel shows part of a field from an image taken at 4 °C. Warm medium pre-equilbrated with CO2 was then added and the stage heater turned on. After 5 min another image of the same field was taken using the same imaging parameters (right panel). The Alexa-488 conjugated transferrin label increased in brightness by an average of 21% per cell (n=8).
Supplemental Figure 2. Validation of latrunculin A treatment for tests of transferrin uptake. Latrunculin has varying effects on cultures depending upon cell type and adhesive conditions. Therefore low concentrations of latrunculin A (50–500 nM) were tested on Wt cultures to determine the maximum concentration that could be used for a 30 min treatment without having large effects on morphology that could complicate the fluorescence microscopy analysis of transferrin uptake. Cultures were monitored by time-lapse DIC microscopy (1 min intervals) during treatment with different concentrations of latrunculin (A). Only the first and last images in the sequence are shown. Controls showed some minor changes in morphology resulting from cell dynamics (locomotion) and shape changes. Treatment with either 50 or 250 nM latrunculin did not cause detectable changes in cell behavior or morphology. However treatment with 500 nM latrunculin A stopped locomotion and caused morphological changes in 88% of the cells observed (5% rounded up, 83% retracted their periphery-arrowheads). Field width=160 μm. (B) Changes in actin organization with latrunculin A treatment. Cultures were treated for 30 min, fixed and stained with rhodamin-phalloidin. Confocal optical sections were taken with a high N.A. lens (60x, 1.4) at a small pin hole setting adjacent to the substrate. The control cells (Ct) had actin stress fibers and a dense actin network (left panel). Following treatment with 250 nM latrunculin (Lat A) internal stress fibers appeared to be shorter and the dense network of actin was partially disrupted and replaced with bright spots in some areas. However most of actin cortex remained largely intact. Treatment with 500 nM latrunculin disrupted most stress fibers leaving some internal short densely stained fibers. The actin network was disrupted and bright spots of actin were widely distributed. In addition, actin free areas appeared (arrows) and were sometimes rimmed with actin bundles. Bar=8 μm. (C) Changes in MIIB organization with latrunculin A treatment. Cells were immunostained with an isoform specific antibody to MIIB and imaged as in B. Control cells had both peripheral and internal stress fibers associated with MIIB. Treatment with 250 nM latrunculin decreased the MIIB staining of internal stress fibers and increased the frequency of bright spots. However cortical actin associated staining remained in most areas. Peripheral stress fiber staining usually appeared normal. Treatment with 500 nM latrunculin greatly decreased overall staining for MIIB (stress fibers and networks), although numerous bright spots were retained. Bar=14 μm. Inset: comparison of MIIB staining along stress fibers. The staining decreases and becomes less compact with increasing concentrations of latrunculin.
Acknowledgments
This work was supported by grants to P. C. Bridgman from NIH (R21 MH081260, R21EB9776) and in part by the Bakewell Neuroimaging Core, supported by the Bakewell Family Foundation and the National Institutes of Health Neuroscience Blueprint Interdisciplinary Center Core Grant P30 (NS057105) to Washington University. Support was also provided by NHLB (DIR) to R.S. Adelstein and grants to R. B. Wysolmerski (HL-090937, P20RR016440) from NIH. We thank Robyn Roth and Dr. John E. Heuser for their help with the electron microscopy and Marcy Hartstein for the table of contents graphic.
Footnotes
The authors have no conflict of interest to declare.
References
- 1.Stahl P, Schwartz AL. Receptor-mediated endocytosis. J Clin Invest. 1986 Mar;77:657–62. doi: 10.1172/JCI112359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2011 Aug;12:517–33. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- 3.Dannhauser PN, Ungewickell EJ. Reconstitution of clathrin-coated bud and vesicle formation with minimal components. Nat Cell Biol. 2012 Jun;14:634–9. doi: 10.1038/ncb2478. [DOI] [PubMed] [Google Scholar]
- 4.Ferguson SM, Raimondi A, Paradise S, Shen H, Mesaki K, Ferguson A, et al. Coordinated actions of actin and BAR proteins upstream of dynamin at endocytic clathrin-coated pits. Dev Cell. 2009 Dec;17:811–22. doi: 10.1016/j.devcel.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boucrot E, Pick A, Camdere G, Liska N, Evergren E, McMahon HT, et al. Membrane Fission Is Promoted by Insertion of Amphipathic Helices and Is Restricted by Crescent BAR Domains. Cell. 2012 Mar 30;149:124–36. doi: 10.1016/j.cell.2012.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meinecke M, Boucrot E, Camdere G, Hon WC, Mittal R, McMahon HT. Cooperative recruitment of dynamin and BIN/amphiphysin/Rvs (BAR) domain-containing proteins leads to GTP-dependent membrane scission. J Biol Chem. 2013 Mar 1;288:6651–61. doi: 10.1074/jbc.M112.444869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boulant S, Kural C, Zeeh JC, Ubelmann F, Kirchhausen T. Actin dynamics counteract membrane tension during clathrin-mediated endocytosis. Nat Cell Biol. 2011 Sep;13:1124–31. doi: 10.1038/ncb2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yarar D, Waterman-Storer CM, Schmid SL. A dynamic actin cytoskeleton functions at multiple stages of clathrin-mediated endocytosis. Mol Biol Cell. 2005 Feb;16:964–75. doi: 10.1091/mbc.E04-09-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lamaze C, Fujimoto LM, Yin HL, Schmid SL. The actin cytoskeleton is required for receptor-mediated endocytosis in mammalian cells. J Biol Chem. 1997 Aug 15;272:20332–5. doi: 10.1074/jbc.272.33.20332. [DOI] [PubMed] [Google Scholar]
- 10.Fujimoto LM, Roth R, Heuser JE, Schmid SL. Actin assembly plays a variable, but not obligatory role in receptor-mediated endocytosis in mammalian cells. Traffic. 2000 Feb;1:161–71. doi: 10.1034/j.1600-0854.2000.010208.x. [DOI] [PubMed] [Google Scholar]
- 11.Da Costa SR, Sou E, Xie J, Yarber FA, Okamoto CT, Pidgeon M, et al. Impairing actin filament or syndapin functions promotes accumulation of clathrin-coated vesicles at the apical plasma membrane of acinar epithelial cells. Mol Biol Cell. 2003 Nov;14:4397–413. doi: 10.1091/mbc.E03-05-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gottlieb TA, Ivanov IE, Adesnik M, Sabatini DD. Actin microfilaments play a critical role in endocytosis at the apical but not the basolateral surface of polarized epithelial cells. J Cell Biol. 1993 Feb;120:695–710. doi: 10.1083/jcb.120.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engqvist-Goldstein AE, Warren RA, Kessels MM, Keen JH, Heuser J, Drubin DG. The actin-binding protein Hip1R associates with clathrin during early stages of endocytosis and promotes clathrin assembly in vitro. J Cell Biol. 2001 Sep 17;154:1209–23. doi: 10.1083/jcb.200106089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le Clainche C, Pauly BS, Zhang CX, Engqvist-Goldstein AE, Cunningham K, Drubin DG. A Hip1R-cortactin complex negatively regulates actin assembly associated with endocytosis. EMBO J. 2007 Mar 7;26:1199–210. doi: 10.1038/sj.emboj.7601576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Merrifield CJ, Feldman ME, Wan L, Almers W. Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat Cell Biol. 2002 Sep;4:691–8. doi: 10.1038/ncb837. [DOI] [PubMed] [Google Scholar]
- 16.Taylor MJ, Perrais D, Merrifield CJ. A high precision survey of the molecular dynamics of mammalian clathrin-mediated endocytosis. PLoS Biol. 2011 Mar;9:e1000604. doi: 10.1371/journal.pbio.1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ungewickell EJ, Hinrichsen L. Endocytosis: clathrin-mediated membrane budding. Curr Opin Cell Biol. 2007 Aug;19:417–25. doi: 10.1016/j.ceb.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 18.Spudich G, Chibalina MV, Au JS, Arden SD, Buss F, Kendrick-Jones J. Myosin VI targeting to clathrin-coated structures and dimerization is mediated by binding to Disabled-2 and PtdIns(4,5)P2. Nat Cell Biol. 2007 Feb;9:176–83. doi: 10.1038/ncb1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng J, Grassart A, Drubin DG. Myosin 1E coordinates actin assembly and cargo trafficking during clathrin-mediated endocytosis. Mol Biol Cell. 2012 Aug;23:2891–904. doi: 10.1091/mbc.E11-04-0383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roux A, Uyhazi K, Frost A, De Camilli P. GTP-dependent twisting of dynamin implicates constriction and tension in membrane fission. Nature. 2006 May 25;441:528–31. doi: 10.1038/nature04718. [DOI] [PubMed] [Google Scholar]
- 21.Morlot S, Galli V, Klein M, Chiaruttini N, Manzi J, Humbert F, et al. Membrane shape at the edge of the dynamin helix sets location and duration of the fission reaction. Cell. 2012 Oct 26;151:619–29. doi: 10.1016/j.cell.2012.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miserey-Lenkei S, Chalancon G, Bardin S, Formstecher E, Goud B, Echard A. Rab and actomyosin-dependent fission of transport vesicles at the Golgi complex. Nat Cell Biol. 2010 Jul;12:645–54. doi: 10.1038/ncb2067. [DOI] [PubMed] [Google Scholar]
- 23.Diz-Munoz A, Fletcher DA, Weiner OD. Use the force: membrane tension as an organizer of cell shape and motility. Trends Cell Biol. 2013 Feb;23:47–53. doi: 10.1016/j.tcb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh RN, Gelman DL, Maxfield FR. Quantification of low density lipoprotein and transferrin endocytic sorting HEp2 cells using confocal microscopy. J Cell Sci. 1994 Aug;107:2177–89. doi: 10.1242/jcs.107.8.2177. [DOI] [PubMed] [Google Scholar]
- 25.Tacheva-Grigorova SK, Santos AJ, Boucrot E, Kirchhausen T. Clathrin-mediated endocytosis persists during unperturbed mitosis. Cell Rep. 2013 Aug 29;4:659–68. doi: 10.1016/j.celrep.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salbreux G, Charras G, Paluch E. Actin cortex mechanics and cellular morphogenesis. Trends Cell Biol. 2012 Oct;22:536–45. doi: 10.1016/j.tcb.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 27.Wakatsuki T, Schwab B, Thompson NC, Elson EL. Effects of cytochalasin D and latrunculin B on mechanical properties of cells. J Cell Sci. 2001 Mar;114:1025–36. doi: 10.1242/jcs.114.5.1025. [DOI] [PubMed] [Google Scholar]
- 28.Liu AP, Loerke D, Schmid SL, Danuser G. Global and local regulation of clathrin-coated pit dynamics detected on patterned substrates. Biophys J. 2009 Aug 19;97:1038–47. doi: 10.1016/j.bpj.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fritzsche M, Lewalle A, Duke T, Kruse K, Charras G. Analysis of turnover dynamics of the submembranous actin cortex. Mol Biol Cell. 2013 Mar;24:757–67. doi: 10.1091/mbc.E12-06-0485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu AP, Aguet F, Danuser G, Schmid SL. Local clustering of transferrin receptors promotes clathrin-coated pit initiation. J Cell Biol. 2010 Dec 27;191:1381–93. doi: 10.1083/jcb.201008117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma X, Kawamoto S, Hara Y, Adelstein RS. A point mutation in the motor domain of nonmuscle myosin II-B impairs migration of distinct groups of neurons. Mol Biol Cell. 2004 Jun;15:2568–79. doi: 10.1091/mbc.E03-11-0836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Engqvist-Goldstein AE, Zhang CX, Carreno S, Barroso C, Heuser JE, Drubin DG. RNAi-mediated Hip1R silencing results in stable association between the endocytic machinery and the actin assembly machinery. Mol Biol Cell. 2004 Apr;15:1666–79. doi: 10.1091/mbc.E03-09-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Collins A, Warrington A, Taylor KA, Svitkina T. Structural organization of the actin cytoskeleton at sites of clathrin-mediated endocytosis. Curr Biol. 2011 Jul 26;21:1167–75. doi: 10.1016/j.cub.2011.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galic M, Jeong S, Tsai FC, Joubert LM, Wu YI, Hahn KM, et al. External push and internal pull forces recruit curvature-sensing N-BAR domain proteins to the plasma membrane. Nat Cell Biol. 2012 Aug;14:874–81. doi: 10.1038/ncb2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gauthier NC, Masters TA, Sheetz MP. Mechanical feedback between membrane tension and dynamics. Trends Cell Biol. 2012 Oct;22:527–35. doi: 10.1016/j.tcb.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 36.Chandrasekar I, Huettner JE, Turney SG, Bridgman PC. Myosin II Regulates Activity Dependent Compensatory Endocytosis at Central Synapses. J Neurosci. 2013 Oct 9;33:16131–45. doi: 10.1523/JNEUROSCI.2229-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bridgman PC, Dave S, Asnes CF, Tullio AN, Adelstein RS. Myosin IIB is required for growth cone motility. J Neurosci. 2001 Aug 15;21:6159–69. doi: 10.1523/JNEUROSCI.21-16-06159.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tullio AN, Accili D, Ferrans VJ, Yu ZX, Takeda K, Grinberg A, et al. Nonmuscle myosin II-B is required for normal development of the mouse heart. Proc Natl Acad Sci U S A. 1997 Nov 11;94:12407–12. doi: 10.1073/pnas.94.23.12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conner DA. Mouse Embryonic Fibroblast (MEF) Feeder Cell Preparation. Current Protocols in Molecular Biology. 2000;23(Supplement 51):23.2–7. doi: 10.1002/0471142727.mb2302s51. [DOI] [PubMed] [Google Scholar]
- 40.Heuser J. The production of ‘cell cortices’ for light and electron microscopy. Traffic. 2000 Jul;1:545–52. doi: 10.1034/j.1600-0854.2000.010704.x. [DOI] [PubMed] [Google Scholar]
- 41.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998 Nov;72:8463–71. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, McManus M. Lentivirus production. J Vis Exp. 2009:e1499. doi: 10.3791/1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Loerke D, Mettlen M, Yarar D, Jaqaman K, Jaqaman H, Danuser G, et al. Cargo and dynamin regulate clathrin-coated pit maturation. PLoS Biol. 2009 Mar 17;7:e57. doi: 10.1371/journal.pbio.1000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Breuer W, Cabantchik ZI. A fluorescence-based one-step assay for serum non-transferrin-bound iron. Anal Biochem. 2001 Dec 15;299:194–202. doi: 10.1006/abio.2001.5378. [DOI] [PubMed] [Google Scholar]
- 45.Bakkeren DL, de Jeu-Jaspars CM, Kroos MJ, van Eijk HG. Release of iron from endosomes is an early step in the transferrin cycle. Int J Biochem. 1987;19:179–86. doi: 10.1016/0020-711x(87)90329-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie 3S-1. YFP clathrin light chain dynamics in a Wt fibroblast imaged with TIRF microscopy. 5 s intervals.
Movie 3S-2. YFP clathrin light chain dynamics in a MIIB KO fibroblast imaged with TIRF microscopy. 5 s intervals.
Movie 3S-3 YFP clathrin light chain dynamics in a MIIB KO fibroblast imaged with TIRF microscopy. The cell was infected with an adenovirus construct containing the MIIB heavy chain to rescue the KO phenotype. 5 s intervals.
Supplemental Figure 1. (A) Comparison of the uptake of biotin transferrin in single confocal optical slices taken at the middle equatorial plane after 15 min at 37 °C. Cells were not serum starved and were incubated with the biotin transferrin at 37 °C prior to rinsing, iron chelation/acid washing and fixation. MIIB KO cells show fewer spots and spots show lower fluorescence intensities. Bar=8 μm. Quantitative analysis and validation of the Alexa-dye labeled transferrin uptake assay. Uptake is decreased in MIIB KO cells compared to Wt controls. Cells were serum starved for 1 hr and then labeled at 4 °C for 30 min with 25 μg/ml (B) or 10 min with 100 μg/ml (C) Alexa-488 conjugated transferrin. They were then rinsed extensively and fixed at 4 °C (first time point) or rinsed and warmed by adding prequilibrated medium (10% FBS) and incubated for the indicated times at 37 °C. Prior to fixation the warmed cells were acid washed with solutions containing an iron chelator to remove surface bound transferrin followed by a saturating concentration of unlabeled transferrin with the iron chelator still present (24). The amount of surface labeling differed between Wt and MIIB KO cells when the concentration of Alexa-488 transferrin used was 25 μg/ml, but was the same when the concentration of label was raised to 100 μg/ml as indicated by the fluorescence intensities in cells fixed at 4 °C (t-test, 25 μg/ml, P<0.0001, Wt, N=140, MIIB KO, N=138P=0.08, 100 μg/ml, Wt, N=63 cells, MIIKO, N=49 cells). The increase in fluorescence intensity after warming indicates that internalized fluorescently labeled transferrin increases in brightness. This increase may be a result of decreased fluorescence quenching by iron; the metal ion is released from transferrin starting within 3 min of internalization (44, 45). The differences were significant for both concentrations at 5 min and 15 min (t-test, 25 μg/ml, 5 min 37 °C P<0.0001, Wt, N=102, MIIB KO N=62; 15 min 37 °C, P<0.0001, Wt, N= 95, MIIB KO, N=72, 100 μg/ml, 5 min, P<0.0001, Wt, N=90 cells, MIIB KO, N= 39 cells, 15 min, t-test, P<0.0001, Wt, N= 107 cells, MIIB KO, N=47 cells). Three cultures were used for each category. (D) Examples of fluorescent transferrin uptake in fixed cells at 15 min in Wt and MIIB KO fibroblasts (+/− blebbistatin) and following knockdown of MIIA and MIIB. Bar=20 μm.(E) Increase in the brightness of Alexa-488 conjugated transferrin label following warm-up of live cells. Cells were labeled for 10 min at 4 °C with 100 μg/ml Alexa-488 conjugated transferrin, washed extensively with cold medium and then placed on the microscope for imaging with high sensitive detectors (Olympus FV1200). The left panel shows part of a field from an image taken at 4 °C. Warm medium pre-equilbrated with CO2 was then added and the stage heater turned on. After 5 min another image of the same field was taken using the same imaging parameters (right panel). The Alexa-488 conjugated transferrin label increased in brightness by an average of 21% per cell (n=8).
Supplemental Figure 2. Validation of latrunculin A treatment for tests of transferrin uptake. Latrunculin has varying effects on cultures depending upon cell type and adhesive conditions. Therefore low concentrations of latrunculin A (50–500 nM) were tested on Wt cultures to determine the maximum concentration that could be used for a 30 min treatment without having large effects on morphology that could complicate the fluorescence microscopy analysis of transferrin uptake. Cultures were monitored by time-lapse DIC microscopy (1 min intervals) during treatment with different concentrations of latrunculin (A). Only the first and last images in the sequence are shown. Controls showed some minor changes in morphology resulting from cell dynamics (locomotion) and shape changes. Treatment with either 50 or 250 nM latrunculin did not cause detectable changes in cell behavior or morphology. However treatment with 500 nM latrunculin A stopped locomotion and caused morphological changes in 88% of the cells observed (5% rounded up, 83% retracted their periphery-arrowheads). Field width=160 μm. (B) Changes in actin organization with latrunculin A treatment. Cultures were treated for 30 min, fixed and stained with rhodamin-phalloidin. Confocal optical sections were taken with a high N.A. lens (60x, 1.4) at a small pin hole setting adjacent to the substrate. The control cells (Ct) had actin stress fibers and a dense actin network (left panel). Following treatment with 250 nM latrunculin (Lat A) internal stress fibers appeared to be shorter and the dense network of actin was partially disrupted and replaced with bright spots in some areas. However most of actin cortex remained largely intact. Treatment with 500 nM latrunculin disrupted most stress fibers leaving some internal short densely stained fibers. The actin network was disrupted and bright spots of actin were widely distributed. In addition, actin free areas appeared (arrows) and were sometimes rimmed with actin bundles. Bar=8 μm. (C) Changes in MIIB organization with latrunculin A treatment. Cells were immunostained with an isoform specific antibody to MIIB and imaged as in B. Control cells had both peripheral and internal stress fibers associated with MIIB. Treatment with 250 nM latrunculin decreased the MIIB staining of internal stress fibers and increased the frequency of bright spots. However cortical actin associated staining remained in most areas. Peripheral stress fiber staining usually appeared normal. Treatment with 500 nM latrunculin greatly decreased overall staining for MIIB (stress fibers and networks), although numerous bright spots were retained. Bar=14 μm. Inset: comparison of MIIB staining along stress fibers. The staining decreases and becomes less compact with increasing concentrations of latrunculin.