

Abstract
Background
The intestinal microbiota has been implicated in the pathophysiology of Irritable Bowel Syndrome (IBS). Due to the variable resolutions of techniques used to characterize the intestinal microbiota, and the heterogeneity of IBS, the defined alterations of the IBS intestinal microbiota are inconsistent. We analyzed the composition of the intestinal microbiota in a defined subgroup of IBS patients (diarrhea-predominant IBS, D-IBS) using a technique that provides the deepest characterization available for complex microbial communities.
Methods
Fecal DNA was isolated from 23 D-IBS patients and 23 healthy controls (HC). Variable regions V1-V3 and V6 of the 16S rRNA gene were amplified from all samples. PCR products were sequenced using 454 high throughput sequencing. The composition, diversity and richness of microbial communities were determined and compared between D-IBS and HC using the Quantitative Insights Into Microbial Ecology pipeline.
Key Results
The contribution of bacterial groups to the composition of the intestinal microbiota differed between D-IBS and HC. D-IBS patients had significantly higher levels of Enterobacteriaceae (p = 0.03), and lower levels of Fecalibacterium genera (p = 0.04) compared to HC. β-diversity values demonstrated significantly lower levels of UniFrac distances in HC compared to D-IBS patients. The richness of 16S rRNA sequences was significantly decreased in D-IBS patients (p < 0.04).
Conclusions and Inferences
Our 16S rRNA sequence data demonstrates a community-level dysbiosis in D-IBS. The altered composition of the intestinal microbiota in D-IBS is associated with significant increases in detrimental and decreases in beneficial bacterial groups, and a reduction in microbial richness.
Keywords: Irritable Bowel Syndrome, Intestinal Microbiota, 16S rRNA gene, Pyro-Sequencing, Faecalibacteium prausnitzii
Background & Aims
Functional gastrointestinal disorders (FGID) are the most prevalent gastrointestinal disorders (GI) in the US[1], and are associated with major patient disability[2], impaired quality of life[3], and a significant economic burden[4]. Irritable Bowel Syndrome (IBS) is the most common disorder among FGID[5], affecting 10-20% of adults and adolescents in the US[6]. Traditionally, IBS has been viewed as a disorder of altered intestinal motility and visceral hypersensitivity, in which the clinical presentation and the severity of the symptoms can be influenced by various psychosocial factors[7]. IBS can present as diarrhea, constipation, or mixed bowel type predominant. Despite intensive research over the past two decades the etiology of IBS remains poorly understood, thus leading to limited effective treatments for patients with these disorders[8].
Animal and clinical human studies have provided evidence that the intestinal microbiota plays an important role in maintaining normal GI function, and that alterations in the composition of the intestinal microbial community can contribute to the development of functional GI symptoms[9-13]. For example, it has been reported that altering the composition of the intestinal microbiota with an antibiotic in mice can increases visceral sensation and that this antibiotic-induced hypersensitivity can be reversed using a probiotic[14]. Furthermore, studies show that manipulation of the intestinal microbiota in humans with probiotics or antibiotics may be effective in alleviating IBS symptoms[15-18].
Several studies have shown differences in the composition of the microbiota between patients with IBS and healthy controls. However, the results of these studies are not consistent, and a consensus has not been reached regarding the specific differences in the microbiota between clinically-defined IBS subgroups of patients and healthy controls[13, 19]. This can be partly ascribed to methodological biases, and limited depth of characterization of complex microbial communities in the intestine. To better understand the role of the intestinal microbiota in IBS we used high throughput 454 pyro-sequencing of the 16S rRNA gene to characterize the composition and diversity of the complex intestinal microbial community in an unbiased manner[20-22]. Since IBS is a heterogeneous disorder, and it is likely that differences in intestinal functions between different IBS subgroups may further affect the composition of the intestinal microbiota, the current study specifically focused on diarrhea-predominant IBS (D-IBS).
Methods and Methods
Study Population
We studied 23 patients that met the Rome III criteria for D-IBS and 23 healthy controls. Subjects were recruited from the Chapel Hill general population and from the University of North Carolina (UNC) healthcare outpatient clinics by advertising.
Inclusion criteria consisted of subjects that were 18 years or older, and of any gender, race, or ethnicity. Healthy controls had no recurring GI symptoms. IBS subjects with a history of GI tract surgery (other than appendectomy or cholecystectomy), a history of inflammatory bowel diseases (IBD), celiac disease, lactose malabsorption, or any other diagnosis that could explain chronic or recurring bowel symptoms were excluded from the study. In addition, participants were excluded if they had a history of treatment with antibiotics, anti-inflammatory agents including aspirin, non-steroid anti-inflammatory drugs (NSAIDs or steroids), or if they had intentionally consumed probiotics two months prior to the study. An eight-week wash-out period was required for subjects who reported intentional consumption of probiotics prior to enrollment. All subjects were evaluated by a physician to exclude an alternative diagnosis to IBS. The study was approved by the UNC Internal Review Board (IRB) and all subjects provided written consent prior to participation in the study.
Sample Collection and Preparation
Fresh stool samples were collected from all 46 subjects on site when possible during a single study visit at UNC. Subjects unable to provide stool samples at the visit were instructed to collect a specimen at home and return it to study staff at the same morning. Each fecal sample was immediately transferred to the laboratory where it was homogenized, divided into aliquots and stored at -80°C for future DNA isolation and molecular microbiological analysis.
Isolation of DNA
Bacterial DNA was isolated from a total of 46 fecal samples (which included 37 samples from our previously reported studies [healthy controls = 21, D-IBS = 16]) using a phenol/chloroform extraction method combined with physical disruption of bacterial cells and a DNA clean-up kit (Qiagen DNeasy® Blood and Tissue extraction kit [Qiagen, Valencia, CA]) as previously described[23]. Briefly, 100 mg of frozen feces was suspended in 750 μl of sterile bacterial lysis buffer (200 mM NaCl, 100 mM Tris [pH 8.0], 20 mM EDTA, 20 mg mL-1 lysozyme) and incubated at 37°C for 30 min. Next, 40 μl of proteinase K (20 mg mL-1) and 85 μl of 10% SDS was added to the mixture and incubated at 65°C for 30 min. 300 mg of 0.1 mm zirconium beads (BioSpec Products, Bartlesville, OK) was then added and the mixture and homogenized in a bead beater (BioSpec Products, Bartlesville, OK) for 2 min. The homogenized mixture was cooled on ice and then centrifuged at 14,000 rpm for 5 min. The supernatant was transferred to a new 1.5 ml microfuge tube and fecal DNA was further extracted by phenol/chloroform/iso-amyl alcohol (25:24:1) and then chloroform/iso-amyl alcohol (24:1). Following extraction the supernatant was precipitated by absolute ethanol at -20°C for 1 hour. The precipitated DNA was suspended in DNase free H2O and then cleaned using the DNeasy® Blood and Tissue extraction kit (Qiagen, Valencia, CA) from step 3 as per the manufacturer's instructions.
454 pyro-sequencing of 16S rRNA genes
Bacterial community composition in isolated DNA samples was characterized by amplification of the V1-3 (forward, 8f:5′-AGAGTTTGATCMTGGCTCAG-3′; reverse 518r: 5′-ATTACCGCGGCTGCTGG-3′) and V6 (forward, 8f:5′-AGGATTAGATACCCTGGTA-3′; reverse 518r: 5′-CRRCACGAGCTGACGAC-3′) variable regions of the 16S rRNA gene by polymerase chain reaction (PCR). Forward primers were tagged with 10 bp unique barcode labels at the 5′ end along with the adaptor sequence (5′-CCATCTCATCCCTGCGTGTCTCCGACTCAG-3′) to allow multiple samples to be included in a single 454 GS FLX Titanium sequencing plate as previously described[24, 25]. 16S rRNA PCR products were quantified, pooled, and purified for the sequencing reaction. 454 GS FLX Titanium sequencing was performed on a 454 Life Sciences Genome Sequencer FLX machine (Roche, Florence, SC) at the microbiome core at UNC-Chapel Hill (http://www.med.unc.edu/microbiome).
Analysis of 16S rRNA sequences using the QIIME pipeline
16S rRNA sequence data generated by the 454 GS FLX Titanium sequencer was processed by the quantitative insights into microbial ecology (QIIME) pipeline[26]. Briefly, sequences that were less than 200 bp or greater than 1,000 bp in length, contained incorrect primer sequences, or contained more than 1 ambiguous base were discarded. The remaining sequences were assigned to D-IBS and HC groups based on their unique nucleotide barcodes, including error-correction[24]. Chimeric sequences were removed using ChimeraSlayer[27]. Sequences were clustered into Operational Taxonomic Units (OTUs) based on their sequence similarity at 97% sequence similarity (similar to species level) using UCLUST[28]. A representative sequence for each OTU was chosen for downstream analysis based on the most abundant sequence from each OTU. PyNAST was used to align sequences with a minimum length of 150 bp and a minimum percent identity of 75.0[29]. OTUs were assigned to a taxonomy using the Ribosomal Database Project (RDP) Naive Bayes classifier[30]. The RDP classifies a 16S rRNA sequence based on Bergey's manual of determinative bacteriology[31] (a main resource for determining bacterial species). A bacterial group is designated as unclassified if the 16S rRNA gene sequence from an organism does not show significant homology to any known classified bacterium in the RDP. α-diversity (diversity within samples) was assessed and rarefaction curves were generated for each experimental group. β-diversity (diversity between groups of samples) was used to generate principal coordinate plots for each sample using un-weighted and weighted UniFrac distances[32-34]. α- and β-diversity measurements were combined with each group (D-IBS and HC) to produce visualizations (α-diversity - rarefaction plots; β-diversity - Principal Co-ordinate Analysis [PCoA] plots, and OTU network plots) that allow composition of the intestinal microbiota between groups to be readily interpreted. α-rarefaction was used to measure the distribution of bacterial taxa richness in sample groups. PCoA plots were used to visualize the similarities or dissimilarities of variables that best represent the pair-wise distances between sample groups. An OTU network-based analysis was used to visualize OTU sharing between sample groups where a bipartite network was generated in which D-IBS and HC samples were designated as one node type, and bacterial OTUs were designated as a second node type. A given sample was connected to a given bacterial OTU node through a line (edge) if that OTU was detected in the subject.
Quantitative PCR
qPCR was performed using the SYBR ® Green PCR master mix (Applied Biosystems, Carlsbad, CA) with primers that amplify the genes encoding 16S rRNA from Faecaibacterium prausnitzii (forward, 5′-GATGGCCTCGCGTCCGATTAG-3′; reverse, 5′-CCGAAGACCTTCTTCCTCC-3′) and all bacteria (forward, 5′-GTGSTGCAYGGYTGTCGTCA-3′; reverse, 5′- ACGTCRTCCMCACCTTCCTC-3′). qPCR assays were conducted in 96-well plates on an Eppendorf Realplex2 mastercycler thermocycler (Eppendorf, Hauppauge, NY). Each PCR was carried out in a final volume of 25 μl and contained the following: 1 × SYBR green master mix, 0.5 μM of each primer and 10 ng of purified fecal DNA. PCR conditions were as follows: 10 min at 95°C, followed by 40 cycles of 95°C for 15 s, 20 s at 50°C, and 72°C for 1 min. Each plate included duplicate reactions per DNA sample, the appropriate set of standards and a ‘no template’ negative control for each primer set. qPCR standards were generated by PCR amplification of target sequences from genomic DNA of an appropriate positive control strain. Analysis of melting curves confirmed that the fluorescence signal originated from specific PCR products and not from primer-dimers or other artifacts.
Statistical Analysis
Before statistical comparisons were made all data sets were assessed for normality using the D'Agostino and Pearson omnibus normality test. When a data set was identified as not having a normal distribution it was transformed by square root and retested for normality. Data sets normally distributed were compared using a Student's t-test. All statistical comparisons were carried out using GraphPad software (v4.0a; Prism, San Diego, CA). We used taxon and phylogenetic-based analyses to compare the 16S rRNA gene sequences between D-IBS patients and healthy controls. Taxon-based: The means and standard deviations of abundances of bacterial groups (Phylum, Class, Order, Family, and genus) were calculated and compared between D-IBS patients and healthy controls. A p value of less than 0.05 was considered significant. Phylogenetic-based: Phylogenetic trees for D-IBS patients and healthy controls were generated using the QIIME pipeline[26]. Each tree was subjected to weighted and un-weighted UniFrac analysis[32-34] through the QIIME pipeline. UniFrac distances represent the fraction of branch length that is shared by any two samples' communities in a phylogenetic tree built from 16S rRNA sequence data from all samples. A ‘weighted’ UniFrac analysis indicates microbiota community differences between sample groups due to differences in relative taxon abundance, whereas an ‘un-weighted’ UniFrac analysis indicates microbiota community differences between sample groups based on the presence or absence of taxa[32-34]. UniFrac distances (weighted and un-weighted) were compiled into matrices and average UniFac distances were calculated for each group and compared using a student's t test. For qPCR the percentage of F. prausnitzii was determined in all fecal samples ([copies 16S rRNA gene for F. prausnitzii/copies of 16S rRNA gene for all bacteria] ×100). qPCR data was transformed to normality by square root and means were compared using a Student's t-test.
Results
I. Study Population
A total of 46 subjects (D-IBS n=23, HC n=23) were investigated. All subjects provided fresh fecal samples. The study population consisted of 76% females with a mean age of 34 years. Demographics and body mass index (BMI) were similar in the two study groups (Table 1).
Table 1. Characteristics of D-IBS patients and healthy controls.
| D-IBS patients | Healthy Controls | |
|---|---|---|
| Number of subjects | 23 | 23 |
| Age (yr): mean (range) | 35 (23-70) | 34 (21-58) |
| % Female: | 74 | 78 |
| BMI (kg/m2): mean (range) | 29 (18.7-43.8) | 25 (18.1-40.5) |
II. Characterization of the D-IBS and HC fecal microbiota
A total of 378,693 and 303,213 16S rRNA sequences with acceptable quality were obtained from the V1-3 and V6 16S rRNA regions, respectively, with an average of 8,232 and 6,591 reads per sample (range: V1-3 2,939-19,305; V6 1,556-13,027). The number of sequence reads did not significantly differ between D-IBS and HC groups. In order to determine the number and abundances of different bacterial groups in each sample we used 3% dissimilarity between 16S rRNA gene sequences as an indicator of a ‘genus level’ OTU. Using this procedure, we found a total of 8,340 and 2,754 OTUs from the V1-3 and V6 regions, respectively, in samples obtained from the 46 subjects.
III. Contribution of bacterial taxa to the composition of the fecal microbiota
The abundance of different bacterial taxa identified by 16S rRNA V1-3 and V6 region OTU sequence alignments were found to differ between D-IBS patients and HC (Figures 1 A&B for V1-3 region, and supplementary figure S1 for V6 region). The levels of several specific bacterial groups, ranging from Phylum to genus, were found to significantly differ between D-IBS patients and HC (Table 2). V1-3 and V6 16S rRNA sequence analysis found that unclassified Enterobacteriaceae (genus level) were significantly increased in D-IBS patients when compared to HC. This significant increase was also observed in higher taxonomic levels that encompass unclassified Enterobacteriaceae (Class – Gammaproteobacteria, Order – Enterobacteriales, Family – Enterobacteriaceae), indicating a structural shift in the microbiota of D-IBS patients. This significant change was also found at the Phylum level (Proteobacteria) with V6 16S rRNA sequences.
Figure 1.
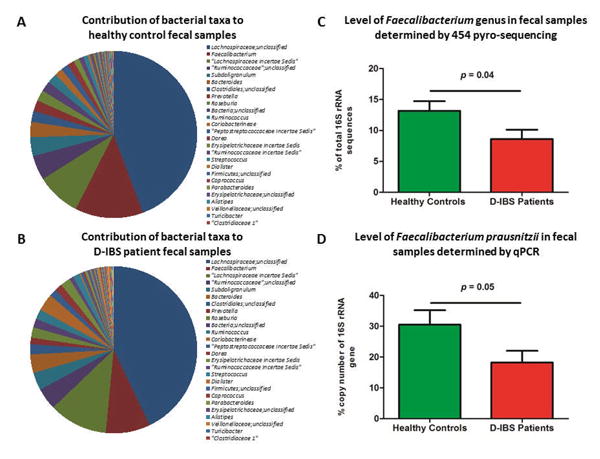
Percent contribution of bacterial genera, identified by 454 pyro-sequencing of the V1-3 16S rRNA gene region, to the fecal microbiota in (A) healthy controls, and (B) D-IBS patients. (C) Average abundance of the Faecalibacterium genus in fecal samples from D-IBS patients and healthy controls, based on V1-3 454 pyro-sequencing data. (D) Concentrations of Faecalibacterium prausnitzii in the fecal microbiota of D-IBS patients and healthy controls using qPCR. Statistical comparisons were carried out on the square root means of each group.
Table 2. Bacterial taxa that differ between D-IBS patients and healthy controls.
| 16S rRNA Region | Taxon | D-IBS patients | Healthy Controls | ¥p | *Fold Change | ||||
|---|---|---|---|---|---|---|---|---|---|
| Phylum | Class | Order | Family | Genus | Abundance of bacterial taxa (%) | ||||
| V3 | Gammaproteobacteria | 0.11 | 0.04 | 0.05 | ↑ 2.9 | ||||
| Enterobacteriales | 0.09 | 0.02 | 0.03 | ↑ 3.8 | |||||
| Enterobacteriaceae | 0.09 | 0.02 | 0.03 | ↑ 3.8 | |||||
| Unclassified Enterobacteriaceae | 0.07 | 0.02 | 0.03 | ↑ 3.4 | |||||
| Clostridiales | Incertae Sedis XII | 0 | 4.40×10-3 | ||||||
| Peptococcaceae | 1.41×10-3 | 0 | |||||||
| Ruminococcaceae | Faecalibacterium | 8.61 | 13.17 | 0.04 | ↓1.5 | ||||
| Fusobacteriales | 0.01 | 0 | |||||||
| Fusobacteriaceae | 0.01 | 0 | |||||||
| Fusobacterium | 0.01 | 0 | |||||||
| Pseudomonadales | 2.02×10-5 | 0 | |||||||
| Pseudomonadaceae | 2.02×10-3 | 0 | |||||||
| Lactobacillaceae | Unclassified Lactobacillaceae | 3.30×10-3 | 0 | ||||||
| Pediococcus | 0.016 | 0 | |||||||
| V6 | Proteobacteria | 0.11 | 0.07 | 0.03 | ↑1.6 | ||||
| Gammaproteobacteria | 0.02 | 4.93×10-4 | 0.03 | ↑43.1 | |||||
| Enterobacteriales | 0.02 | 4.93×10-4 | 0.03 | ↑43.1 | |||||
| Enterobacteriaceae | 0.02 | 4.93×10-4 | 0.03 | ↑43.1 | |||||
| Unclassified Enterobacteriaceae | 0.02 | 4.93×10-4 | 0.03 | ↑43.1 | |||||
| Lactobacillales | Enterococcaceae | 4.01×10-3 | 0 | ||||||
| Enterococcus | 4.01×10-3 | 0 | |||||||
| Ruminococcaceae | Ethanoligenens | 0.51 | 0.07 | 0.01 | ↑7.6 | ||||
| Veillonellaceae | Veillonella | 5.54×10-3 | 0 | ||||||
Unclassified refers to 16S rRNA sequences that could not be assigned to known bacterial groups. ‘Unclassified Enterobacteriaceae’ refers to 16S rRNA sequences that are assigned to the Enterobacteriaceae Family but these sequences do not show sufficient homology with any known genus within this order.
P values were determined by comparing the square root of means between groups.
Fold change with respect to healthy controls.
Bold entries indicate a statistically significant change in a bacterial group between D-IBS patients and healthy controls.
As shown in Figure 1C we found a significant 1.5 fold decrease in the Faecalibacterium genus in D-IBS patients when compared to HC when analyzing V1-3 16S rRNA sequences. A decrease (1.2 fold) in the Faecalibacterium genus in D-IBS patients was also observed with V6 16S rRNA sequences, however this this not reach statistical significance (p = 0.41). Thus, to validate the observation in V1-3 16S rRNA sequences we investigated the concentration of the only known member of the Faecalibacterium genus (Faecalibacterium prausnitzii) using qPCR. Our analysis found a significant 1.7 fold decrease (p = 0.05) in the concentration of F. prausnitzii in samples from D-IBS patients when compared to HC (Figure 1D).
IV. Differentiation of the Healthy and D-IBS intestinal microbiotas
To compare the global composition of the microbiota between D-IBS patients and HC we used two different yet complementary methods. In the first approach weighted and un-weighted UniFrac[32-34] distances between samples were calculated and compared. The Principal Coordinate Analysis (PCoA) of weighted and un-weighted D-IBS patients and HC UniFrac distances revealed an overlap between D-IBS and HC sample clusters. However, comparison of average UniFrac distances from V6 16S rRNA sequences revealed a significant decrease in HC samples when compared to the D-IBS group in both weighted (D-IBS = 0.19, HC = 0.16, p = 0.01) and un-weighted (D-IBS = 0.58, HC = 0.55, p = 0.04) analyses (Figure 2 A&B), indicating the microbiotas of HC are more similar to each other when compared to D-IBS patients. A decrease in UniFrac distances were also observed with V1-3 16S rRNA sequences, however these differences did not reach statistical significance (weighted: D-IBS = 0.22, HC = 0.21, P = 0.25) and un-weighted (D-IBS = 0.74, HC = 0.73, p = 0.25).
Figure 2.
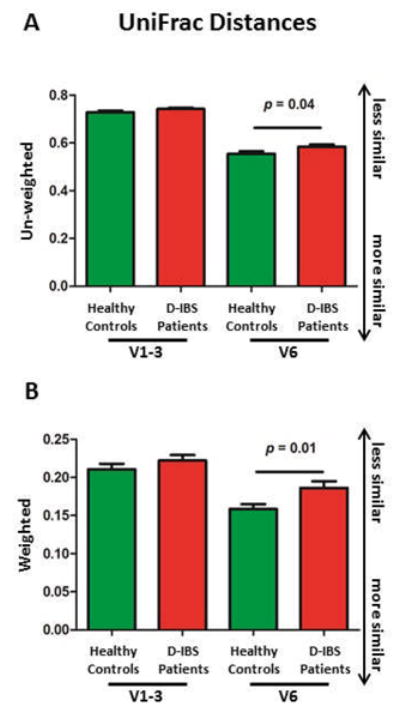
Variation in bacterial community composition between D-IBS patients and healthy controls. A significant decrease in average UniFrac distances was found in healthy controls when compared to the D-IBS group using weighted (A) and un-weighted (B) values for the V6 16S rRNA. All analyses represent the relationship between the microbiota in D-IBS and healthy control in fecal samples based on their β-diversity. Statistical comparisons were carried out on the square root means of each group.
In our second approach a bipartite network of D-IBS and HC samples was generated. In figure 3 D-IBS and HC samples are represented by one node type and bacterial OTUs are designated as a second node type. If a sample from a particular group contains a specific OTU they are connected via an ‘edge’ to create a pattern/network of shared OTUs. This OTU network, based on V1-3 16S rRNA gene sequences, demonstrates that D-IBS patients share more OTUs with other D-IBS patients than with HC; similarly HC subjects share more OTUs with other HC subjects than with D-IBS patients (Figure 3). Analogous results were found for V6 16S rRNA sequences (supplementary figure S2).
Figure 3.

Operational Taxonomic Unit (OTU) network analysis of bacterial communities from D-IBS patient and healthy control fecal samples for the V1-3 16S rRNA region. Nodes represent individual D-IBS samples (red circles), individual healthy control samples (green circles), and OTUs (white circles). Edges (lines) connecting D-IBS nodes (red edges) or healthy control nodes (green edges) to OTUs indicate whether a given OTU was found in that sample. The pattern of green and red edges indicates that although both groups share common OTUs, D-IBS samples share more OTUs in common and segregate from healthy control shared OTUs.
V. Bacterial richness in the intestinal microbiota
Bacterial richness was assessed by rarefraction of OTUs found in D-IBS and HC samples. From 3,670 sampled sequences microbial richness was found to be significantly lower in D-IBS patients when compared to HC (p < 0.04), based on the V1-3 16S rRNA variable region (Figure 4). Microbial richness was slightly lower in D-IBS patients when compared to HC based on the V6 16S rRNA variable region; however this reduction did not reach statistical significance (Supplemental Figure 3). Although a relatively large number of 16S rRNA gene sequence reads were generated from each sample, these rarefaction curves indicate that species discovery is still increasing at this sequence depth. Thus, it is important to note that differences between groups of organisms with abundance lower than 0.1% may not be detected.
Figure 4.
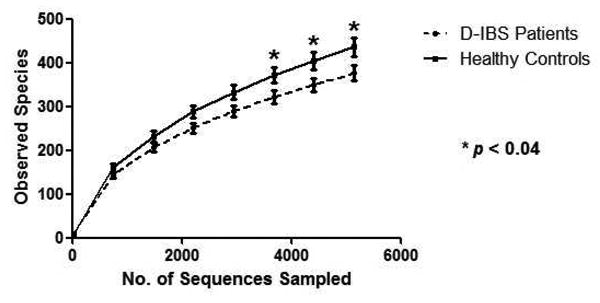
Rarefaction curves of D-IBS (broken line) patient and healthy control (solid line) fecal samples. Curves are based on α-diversity within sample groups for the V1-3 16S rRNA region.
*Indicates significant differences between groups (p < 0.04). Statistical comparisons were carried out on the square root means of each group.
Discussion
In the current study we characterize and compare the fecal microbiota in a well-characterized subgroup of patients with D-IBS and healthy controls using high throughput 454 pyro-sequencing of the 16S rRNA gene. The 16S rRNA gene contains nine variable regions, all of which have been used to characterize complex microbial communities[35]. The depth of our analyses is strengthened by the investigation of four variable regions of the 16S rRNA gene (V1-3 and V6). We chose to investigate the V1-3 regions of the 16S rRNA gene as it has been reported that combining variable regions to make longer sequences (specifically V1-3) provides a more accurate resolution of the intestinal microbiota[36]. We also chose to use the shorter V6 region of the 16S rRNA gene as it has been used previously to investigate the intestinal microbiota[37], and also to provide comparative and complementary data for the longer V1-3 region. Using this approach we investigated over 680,000 16S rRNA gene sequences with an average of over 8,000 and 6,500 sequences per sample for the V1-3 and V6 regions, respectively. The results of our study indicate an intestinal microbial dysbiosis in D-IBS patients, with alterations in specific bacterial groups (ranging from genus to Phylum level), and microbial community diversity. Taxonomic analysis revealed a significant increase in unclassified Enterobacteriaceae in D-IBS patients. Unclassifed Enterobacteriaceae refers to bacterial genera encompassed by the Enterobacteriaceae Family, yet little is known about this group or their function. The Enterobacteriaceae Family contains thirty seven genera which include several pathogenic bacteria such as; Escherichia, Shigella, Salmonella, and Yersinia. The significant increase of unclassified Enterobacteriaceae in our D-IBS samples raised the possibility of recent infection or a post-infections etiology (PI-IBS). However, none of our patients reported the onset of IBS symptoms after intestinal infection (in response to specific question at screening), and all our study subjects were screened for intestinal infections by routine stool culture for intestinal pathogens. In regard to this, it should be noted that the Enterobacteriaceae Family does not solely encompass pathogenic bacteria.
At the genus level, our study also found a significant reduction in the concentrations of the Fecalibacterium genus in D-IBS patients when compared to healthy controls. Currently, it is thought that the Faeclibacterium genus consists of one bacterial species; Faecalibacterium prausnitzii, a major constituent of the Clostridium leptum group[38-40]. Interestingly, reduced numbers of this organism has been shown in ileal Crohn's disease[41, 42], suggesting protective and anti-inflammatory properties of this organism. Our finding of reduced numbers of F. prausnitzii in patients with D-IBS further supports the possibility of an inflammatory component in this condition. Given the finding of increased unclassified Enterobacteriaceae and a reduction in the Faeclibacterium genus, it can be speculated that the intestinal microbial dysbiosis in D-IBS is associated with an imbalance in the ratio of detrimental and beneficial bacterial groups, a phenomenon also reported in inflammatory bowel disease[20]. Our study used a deep and comprehensive sequencing approach to characterize the intestinal microbiota, and as such we have identified additional bacterial groups not yet reported as being altered in D-IBS compared to healthy controls. At the genus level we found a significant increase in the concentration of Ethanoligenens in D-IBS patients. Additionally, Enterococcus, Fusobacterium, Pediococcus, unclassified Lactobacillaceae and Veilonella species were found in D-IBS patients but were below detection limits in healthy controls. The biological relevance for these organisms and their association with D-IBS remains to be determined.
Complex microbial communities are also typically compared based on their biodiversity[43]. Changes in α-diversity (diversity within a sample) and β-diversity (diversity between collections of samples) are important independent measures for complex microbial communities. Diversity measures provide additional important information on the overall structure of intestinal microbial communities. Our study found a significant reduction in α-diversity measurements (rarefaction analysis) in the fecal microbiota of D-IBS patients suggesting that the fecal microbiota of D-IBS patients has lower microbial richness. In addition, we found a significant reduction in β-diversity measurements (UniFrac distance analysis) in the fecal microbiota of healthy controls indicating that the fecal microbiotas in healthy controls are more similar to each other than to the fecal microbiotas of D-IBS patients. Intestinal microbial richness is a result of co-evolution between host and microbe [44], and has been shown to be stable over time in healthy individuals [45] and reduced in disease conditions such as ileal Crohn's disease[20]. Thus, a departure from the normal microbial richness of the intestinal microbiota may reflect a disease, possibly inflammatory, state in D-IBS. Given the significant differences in bacterial taxa and α- and β-diversity measurements between the microbiotas in D-IBS patients and healthy controls, it can be speculated that a microbial intestinal dysbiosis exists in D-IBS patients that is a result of the increase or decrease in specific bacterial groups and changes in the global structure of the intestinal microbiota[34].
Previous studies have investigated the intestinal microbiota in patients with IBS and reported various alterations in specific bacterial groups[46-50], changes in fingerprint patterns[23, 49, 51-54], and global compositional differences in the microbiota based on clone libraries[55, 56]. However, many of these studies have used molecular techniques that provide a limited resolution of complex intestinal microbial communities. Two recently published studies have used advanced molecular techniques based on the 16S rRNA gene that provide deep characterization of the intestinal microbiota in IBS patients and healthy controls. In the first study Saulnier et al.[57] used high throughput sequencing of the 16S rRNA gene to characterize the microbiota in a mixed group of 22 children with IBS and 22 healthy controls. Differences in specific bacterial taxa were reported between the groups suggesting an intestinal microbial dysbiosis in pediatric patients with IBS. γ-proteobacteria were found to be increased in the microbiota of IBS patients, and a novel Ruminococcus-like microbe was also enriched pediatric patients with this disorder. The study population from this report significantly differed to our population as we investigated adult patients with a defined sub-group of D-IBS. In the second study Rajilić-Stojanović et al.[58] used a 16S rRNA gene microarray (the Human Intestinal Tract Chip – HITChip) to compare the intestinal microbiotas between a mixed group of adult IBS patients (n=62) and healthy controls (n=46). Interestingly, consistent with our finding, this group also reported a significant decrease in the Faeclibacterium genus in their IBS patients. Similarly to the Saulnier et al.[57] study, our current study differs from this report as it focuses on a specific subgroup of IBS patients. Our decision to focus on a single subgroup of patients with D-IBS was based on the assumption that differences in bowel habits (e.g., diarrhea and constipation) could potentially mask differences in the levels of bacterial taxa and diversity between specific subtype of IBS and healthy controls.
Our analyses the V1-3 and V6 regions of the 16S rRNA gene yielded different and complementary results. For example, our sequence data found a significant increase in unclassified Enterobacteriaceae with both V1-3 and V6 regions of the 16S rRNA gene. In contrast, the V1-3 region found that the Fecalibacterium genus was significantly reduced in D-IBS patients; however a decrease in the Fecalibacterium genus was also found for sequences from the V6 region that did not reach statistical significance. Additionally, a significant decrease in average weighted and un-weighted UniFrac distances was found when analyzing the V6 16S rRNA region in healthy controls. A decrease in average weighted and un-weighted UniFrac distances was also found when analyzing the V1-3 16S rRNA region in healthy controls; however this did not reach statistical significance. Thus, based on our sequence data investigation of four variable 16S rRNA regions has provided a detailed and comprehensive view of the fecal microbiota in patients with D-IBS and its differences from the microbiota in healthy individuals.
Our study investigates fecal material to represent the intestinal microbiota in D-IBS patients and healthy controls. However, it is important to note that the fecal and mucosal-associated microbiota differ in composition and diversity[23, 59, 60]. Using the terminal-restriction fragment length polymorphism (T-RFLP) fingerprinting technique we have recently demonstrated compositional differences in both, fecal- and mucosal-associated microbiota between patients with D-IBS and healthy controls. Thus, emphasizing the importance of investigating both of these intestinal niches in this disorder[23].
In conclusion, we report an intestinal dysbiosis in D-IBS patients that is a result of taxonomic, structural, and diversity differences in the intestinal microbiota in these patients when compared to healthy controls. Furthermore, the findings of our taxonomic analysis indicating higher levels of Enterobacteriaceae Family and lower levels of Faecalibacterium prausnitzii suggests that the intestinal ‘dysbiosis’ in D-IBS may be associated with an imbalance in the ratio of detrimental and beneficial intestinal bacteria. Although our study does not answer the question of causality or confirm an etiological role for the altered microbiota in this disorder, our study findings emphasizes the importance and provides the rationale for further investigation of the role of the intestinal microbiota in the pathogenesis of IBS. Future studies will need to address the association between intestinal functions (such as motility) and the gut microbiota, and how these factors influence IBS symptoms. This investigation will advance our understanding of the disorder and lead to new therapeutic options for patients with functional bowel disorders.
Supplementary Material
Supplemental figure S1. Percent contribution of bacterial genera, identified by 454 pyro-sequencing of the V6 16S rRNA gene, to the fecal microbiota in (A) healthy controls, and (B) D-IBS patients.
{kind=link}
Supplemental figure S2. Operational Taxonomic Unit (OTU) network analysis of bacterial communities from D-IBS patient and healthy control fecal samples for the V6 16S rRNA region.
{kind=link}
Supplemental figure S3. Rarefaction curves of D-IBS patient (broken line) and healthy control (solid line) fecal samples. Curves are based on α-diversity within sample groups for the V6 16S rRNA region.
{kind=link}
Acknowledgments
The authors would like to thank Sheena Neil her contribution to this study. This study was funded by DK084294 and DK075621 grants from the National Institutes of Health awarded to YR. This study was also supported by Award Number UL1RR025747 from the National Center for Research Resources. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Footnotes
Author Contribution: Ian M. Carroll – study design, molecular techniques, data analysis, interpretation of data, scribe of ms.
Tamar Ringel-Kulka – study design, interpretation of data, scribe of ms.
Jennica P. Siddle – study design, sample acquisition.
Yehuda Ringel – study design, subjects' eligibility evaluation, interpretation of data, funding acquisition, study supervision, scribe of ms.
Disclosures: Ian M. Carroll – None to declare.
Tamar Ringel-Kulka – None to declare.
Jennica P. Siddle – None to declare.
Yehuda Ringel – None to declare.
References
- 1.Saito YA, Schoenfeld P, Locke GR., 3rd The epidemiology of irritable bowel syndrome in North America: a systematic review. Am J Gastroenterol. 2002;97(8):1910–5. doi: 10.1111/j.1572-0241.2002.05913.x. [DOI] [PubMed] [Google Scholar]
- 2.Hungin AP, Chang L, Locke GR, Dennis EH, Barghout V. Irritable bowel syndrome in the United States: prevalence, symptom patterns and impact. Aliment Pharmacol Ther. 2005;21(11):1365–75. doi: 10.1111/j.1365-2036.2005.02463.x. [DOI] [PubMed] [Google Scholar]
- 3.El-Serag HB, Olden K, Bjorkman D. Health-related quality of life among persons with irritable bowel syndrome: a systematic review. Aliment Pharmacol Ther. 2002;16(6):1171–85. doi: 10.1046/j.1365-2036.2002.01290.x. [DOI] [PubMed] [Google Scholar]
- 4.Maxion-Bergemann S, Thielecke F, Abel F, Bergemann R. Costs of irritable bowel syndrome in the UK and US. Pharmacoeconomics. 2006;24(1):21–37. doi: 10.2165/00019053-200624010-00002. [DOI] [PubMed] [Google Scholar]
- 5.Ringel Y, Drossman DA. Irritable Bowel Syndrome. In: Runge MS, Greganti MA, editors. Netter's textbook of Internal Medicine. 2nd. Vol. 59. Sauders Elsevier; 2008. pp. 419–25. [Google Scholar]
- 6.Longstreth GF, Thompson WG, Chey WD, Houghton LA, Mearin F, Spiller RC. Functional bowel disorders. Gastroenterology. 2006;130(5):1480–91. doi: 10.1053/j.gastro.2005.11.061. [DOI] [PubMed] [Google Scholar]
- 7.Ringel Y, Drossman DA, Leserman JL, Suyenobu BY, Wilber K, Lin W, et al. Effect of abuse history on pain reports and brain responses to aversive visceral stimulation: an FMRI study. Gastroenterology. 2008;134(2):396–404. doi: 10.1053/j.gastro.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 8.Brandt LJ, Chey WD, Foxx-Orenstein AE, Schiller LR, Schoenfeld PS, Spiegel BM, et al. An evidence-based position statement on the management of irritable bowel syndrome. Am J Gastroenterol. 2009;104(Suppl 1):S1–35. doi: 10.1038/ajg.2008.122. [DOI] [PubMed] [Google Scholar]
- 9.Caenepeel P, Janssens J, Vantrappen G, Eyssen H, Coremans G. Interdigestive myoelectric complex in germ-free rats. Dig Dis Sci. 1989;34(8):1180–4. doi: 10.1007/BF01537265. [DOI] [PubMed] [Google Scholar]
- 10.Husebye E, Hellstrom PM, Sundler F, Chen J, Midtvedt T. Influence of microbial species on small intestinal myoelectric activity and transit in germ-free rats. Am J Physiol Gastrointest Liver Physiol. 2001;280(3):G368–80. doi: 10.1152/ajpgi.2001.280.3.G368. [DOI] [PubMed] [Google Scholar]
- 11.Lesniewska V, Rowland I, Laerke HN, Grant G, Naughton PJ. Relationship between dietary-induced changes in intestinal commensal microflora and duodenojejunal myoelectric activity monitored by radiotelemetry in the rat in vivo. Exp Physiol. 2006;91(1):229–37. doi: 10.1113/expphysiol.2005.031708. [DOI] [PubMed] [Google Scholar]
- 12.Arebi N, Gurmany S, Bullas D, Hobson A, Stagg A, Kamm M. Review article: the psychoneuroimmunology of irritable bowel syndrome--an exploration of interactions between psychological, neurological and immunological observations. Aliment Pharmacol Ther. 2008;28(7):830–40. doi: 10.1111/j.1365-2036.2008.03801.x. [DOI] [PubMed] [Google Scholar]
- 13.Ringel Y, Carroll IM. Alterations in the intestinal microbiota and functional bowel symptoms. Gastrointest Endosc Clin N Am. 2009;19(1):141–50. vii. doi: 10.1016/j.giec.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 14.Verdu EF, Bercik P, Verma-Gandhu M, Huang XX, Blennerhassett P, Jackson W, et al. Specific probiotic therapy attenuates antibiotic induced visceral hypersensitivity in mice. Gut. 2006;55(2):182–90. doi: 10.1136/gut.2005.066100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brenner DM, Chey WD. Bifidobacterium infantis 35624: a novel probiotic for the treatment of irritable bowel syndrome. Rev Gastroenterol Disord. 2009;9(1):7–15. [PubMed] [Google Scholar]
- 16.Pimentel M, Lembo A, Chey WD, Zakko S, Ringel Y, Yu J, et al. Rifaximin therapy for patients with irritable bowel syndrome without constipation. N Engl J Med. 2011;364(1):22–32. doi: 10.1056/NEJMoa1004409. [DOI] [PubMed] [Google Scholar]
- 17.Pimentel M, Chatterjee S, Chow EJ, Park S, Kong Y. Neomycin improves constipation-predominant irritable bowel syndrome in a fashion that is dependent on the presence of methane gas: subanalysis of a double-blind randomized controlled study. Dig Dis Sci. 2006;51(8):1297–301. doi: 10.1007/s10620-006-9104-6. [DOI] [PubMed] [Google Scholar]
- 18.Sharara AI, Aoun E, Abdul-Baki H, Mounzer R, Sidani S, Elhajj I. A randomized double-blind placebo-controlled trial of rifaximin in patients with abdominal bloating and flatulence. Am J Gastroenterol. 2006;101(2):326–33. doi: 10.1111/j.1572-0241.2006.00458.x. [DOI] [PubMed] [Google Scholar]
- 19.Lee KJ, Tack J. Altered intestinal microbiota in irritable bowel syndrome. Neurogastroenterol Motil. 2010;22(5):493–8. doi: 10.1111/j.1365-2982.2010.01482.x. [DOI] [PubMed] [Google Scholar]
- 20.Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z, et al. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139(6):1844–54 e1. doi: 10.1053/j.gastro.2010.08.049. [DOI] [PubMed] [Google Scholar]
- 21.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480–4. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andersson AF, Lindberg M, Jakobsson H, Backhed F, Nyren P, Engstrand L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE. 2008;3(7):e2836. doi: 10.1371/journal.pone.0002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carroll IM, Ringel-Kulka T, Keku TO, Chang YH, Packey CD, Sartor RB, et al. Molecular analysis of the luminal- and mucosal-associated intestinal microbiota in diarrhea-predominant irritable bowel syndrome. Am J Physiol Gastrointest Liver Physiol. 2011;301(5):G799–807. doi: 10.1152/ajpgi.00154.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat Methods. 2008;5(3):235–7. doi: 10.1038/nmeth.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fierer N, Hamady M, Lauber CL, Knight R. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci U S A. 2008;105(46):17994–9. doi: 10.1073/pnas.0807920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21(3):494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–1. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 29.Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010;26(2):266–7. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–7. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holt JG. Bergey's Manual of Determinative Bacteriology. 9th. Baltimore: 1994. Holt JG. [Google Scholar]
- 32.Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71(12):8228–35. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: an effective distance metric for microbial community comparison. Isme J. 2011;5(2):169–72. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol. 2007;73(5):1576–85. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robinson CJ, Bohannan BJ, Young VB. From structure to function: the ecology of host-associated microbial communities. Microbiol Mol Biol Rev. 2010;74(3):453–76. doi: 10.1128/MMBR.00014-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schloss PD. The effects of alignment quality, distance calculation method, sequence filtering, and region on the analysis of 16S rRNA gene-based studies. PLoS Comput Biol. 2010;6(7):e1000844. doi: 10.1371/journal.pcbi.1000844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–31. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 38.Barcenilla A, Pryde SE, Martin JC, Duncan SH, Stewart CS, Henderson C, et al. Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl Environ Microbiol. 2000;66(4):1654–61. doi: 10.1128/aem.66.4.1654-1661.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suau A, Bonnet R, Sutren M, Godon JJ, Gibson GR, Collins MD, et al. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol. 1999;65(11):4799–807. doi: 10.1128/aem.65.11.4799-4807.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hold GL, Schwiertz A, Aminov RI, Blaut M, Flint HJ. Oligonucleotide probes that detect quantitatively significant groups of butyrate-producing bacteria in human feces. Appl Environ Microbiol. 2003;69(7):4320–4. doi: 10.1128/AEM.69.7.4320-4324.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sokol H, Seksik P, Furet JP, Firmesse O, Nion-Larmurier I, Beaugerie L, et al. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. 2009;15(8):1183–9. doi: 10.1002/ibd.20903. [DOI] [PubMed] [Google Scholar]
- 42.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. 2008;105(43):16731–6. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326(5960):1694–7. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307(5717):1915–20. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 45.Zoetendal EG, Akkermans AD, De Vos WM. Temperature gradient gel electrophoresis analysis of 16S rRNA from human fecal samples reveals stable and host-specific communities of active bacteria. Appl Environ Microbiol. 1998;64(10):3854–9. doi: 10.1128/aem.64.10.3854-3859.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Balsari A, Ceccarelli A, Dubini F, Fesce E, Poli G. The fecal microbial population in the irritable bowel syndrome. Microbiologica. 1982;5(3):185–94. [PubMed] [Google Scholar]
- 47.Si JM, Yu YC, Fan YJ, Chen SJ. Intestinal microecology and quality of life in irritable bowel syndrome patients. World J Gastroenterol. 2004;10(12):1802–5. doi: 10.3748/wjg.v10.i12.1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carroll IM, Chang YH, Park J, Sartor RB, Ringel Y. Luminal and Mucosal-Associated Intestinal Microbiota in Patients with Diarrhea-Predominant Irritable Bowel Syndrome. Gut Pathog. 2010;2(1):19. doi: 10.1186/1757-4749-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Malinen E, Rinttila T, Kajander K, Matto J, Kassinen A, Krogius L, et al. Analysis of the fecal microbiota of irritable bowel syndrome patients and healthy controls with real-time PCR. Am J Gastroenterol. 2005;100(2):373–82. doi: 10.1111/j.1572-0241.2005.40312.x. [DOI] [PubMed] [Google Scholar]
- 50.Lyra A, Rinttila T, Nikkila J, Krogius-Kurikka L, Kajander K, Malinen E, et al. Diarrhoea-predominant irritable bowel syndrome distinguishable by 16S rRNA gene phylotype quantification. World J Gastroenterol. 2009;15(47):5936–45. doi: 10.3748/wjg.15.5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Codling C, O'Mahony L, Shanahan F, Quigley EM, Marchesi JR. A Molecular Analysis of Fecal and Mucosal Bacterial Communities in Irritable Bowel Syndrome. Dig Dis Sci. 2009;55:392–7. doi: 10.1007/s10620-009-0934-x. [DOI] [PubMed] [Google Scholar]
- 52.Maukonen J, Satokari R, Matto J, Soderlund H, Mattila-Sandholm T, Saarela M. Prevalence and temporal stability of selected clostridial groups in irritable bowel syndrome in relation to predominant faecal bacteria. J Med Microbiol. 2006;55(Pt 5):625–33. doi: 10.1099/jmm.0.46134-0. [DOI] [PubMed] [Google Scholar]
- 53.Noor SO, Ridgway K, Scovell L, Kemsley EK, Lund EK, Jamieson C, et al. Ulcerative colitis and irritable bowel patients exhibit distinct abnormalities of the gut microbiota. BMC Gastroenterol. 2010;10:134. doi: 10.1186/1471-230X-10-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ponnusamy K, Choi JN, Kim J, Lee SY, Lee C. Microbial community and metabolomic comparison of irritable bowel syndrome faeces. J Med Microbiol. 2011;60(pt 6):817–27. doi: 10.1099/jmm.0.028126-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krogius-Kurikka L, Lyra A, Malinen E, Aarnikunnas J, Tuimala J, Paulin L, et al. Microbial community analysis reveals high level phylogenetic alterations in the overall gastrointestinal microbiota of diarrhoea-predominant irritable bowel syndrome sufferers. BMC Gastroenterol. 2009;9:95. doi: 10.1186/1471-230X-9-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kerckhoffs AP, Samsom M, van der Rest ME, de Vogel J, Knol J, Ben-Amor K, et al. Lower Bifidobacteria counts in both duodenal mucosa-associated and fecal microbiota in irritable bowel syndrome patients. World J Gastroenterol. 2009;15(23):2887–92. doi: 10.3748/wjg.15.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saulnier DM, Riehle K, Mistretta TA, Diaz MA, Mandal D, Raza S, et al. Gastrointestinal microbiome signatures of pediatric patients with irritable bowel syndrome. Gastroenterology. 2011;141(5):1782–91. doi: 10.1053/j.gastro.2011.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rajilic-Stojanovic M, Biagi E, Heilig HG, Kajander K, Kekkonen RA, Tims S, et al. Global and deep molecular analysis of microbiota signatures in fecal samples from patients with irritable bowel syndrome. Gastroenterology. 2011;141(5):1792–801. doi: 10.1053/j.gastro.2011.07.043. [DOI] [PubMed] [Google Scholar]
- 59.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science. 2005;308(5728):1635–8. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zoetendal EG, von Wright A, Vilpponen-Salmela T, Ben-Amor K, Akkermans AD, de Vos WM. Mucosa-associated bacteria in the human gastrointestinal tract are uniformly distributed along the colon and differ from the community recovered from feces. Appl Environ Microbiol. 2002;68(7):3401–7. doi: 10.1128/AEM.68.7.3401-3407.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figure S1. Percent contribution of bacterial genera, identified by 454 pyro-sequencing of the V6 16S rRNA gene, to the fecal microbiota in (A) healthy controls, and (B) D-IBS patients.
Supplemental figure S2. Operational Taxonomic Unit (OTU) network analysis of bacterial communities from D-IBS patient and healthy control fecal samples for the V6 16S rRNA region.
Supplemental figure S3. Rarefaction curves of D-IBS patient (broken line) and healthy control (solid line) fecal samples. Curves are based on α-diversity within sample groups for the V6 16S rRNA region.