

Abstract
The taxanes are a class of chemotherapeutic agents that are widely used in the treatment of various solid tumors. Although taxanes are highly effective in cancer treatment, their use is associated with serious complications attributable to large interindividual variability in pharmacokinetics and a narrow therapeutic window. Unpredictable toxicity occurrence necessitates close patient monitoring while on therapy and adverse effects frequently require decreasing, delaying or even discontinuing taxane treatment. Currently, taxane dosing is based primarily on body surface area, ignoring other factors that are known to dictate variability in pharmacokinetics or outcome. This article discusses three potential strategies for individualizing taxane treatment based on patient information that can be collected before or during care. The clinical implementation of pharmacogenetics, enzyme probes or therapeutic drug monitoring could enable clinicians to personalize taxane treatment to enhance efficacy and/or limit toxicity.
Keywords: docetaxel, enzyme probe, paclitaxel, personalized medicine, pharmacogenetics, therapeutic drug monitoring
Taxanes are commonly used in the treatment of various solid tumors, such as breast cancer, non-small-cell lung cancer and prostate cancer. They work by binding to the microtubule β-tubulin subunit and interfering with microtubule depolymerization, which inhibits cell division [1,2]. The first taxane, paclitaxel, was approved by the US FDA in 1992 [201], followed by docetaxel 4 years later [202]. The search for improved formulations and new taxanes has resulted in FDA approval of albumin-bound paclitaxel [3] and cabazitaxel [4], and continues to this day [5,203].
Despite the initial success and continued optimization of taxanes, their use is associated with serious limitations. Taxanes have a narrow therapeutic window and a broad adverse event profile, of which hematopoietic and neurologic toxicities are most notable. Although the overall profiles are generally similar, the incidence of specific toxicities is quite different between the two agents and the dose-limiting toxicities of paclitaxel and docetaxel, neurotoxicity [6] and (febrile) neutropenia [7], respectively, are not the same.
Currently, taxane dosing is based primarily on body surface area (BSA) and in some cases diminished liver function is considered [8,9]. Using this approach to initial dose selection, large interindividual differences in pharmacokinetics (PK) are observed [10,11] and a subpopulation of the treated population experiences severe, treatment-limiting adverse events.
The taxanes are primarily eliminated via hepatic metabolism and biliary elimination (Figure 1) [12-14]. The proteins involved in hepatic uptake and intracellular metabolism influence taxane PK and may have an indirect role in determining taxane outcome. Taxanes enter the hepatocytes through rapid passive diffusion, due to their lipophilic character, and by hepatocellular uptake by OATP1B3 [15,16]. Both taxanes are substrates for the highly promiscuous CYP3A4/3A5 metabolic system [17,18], but the formation of paclitaxel metabolites is also highly dependent on CYP2C8 activity [19]. The efflux proteins P-gp and MRP2 are responsible for excreting taxanes and their metabolites into the bile [20-22].
Figure 1. Intracellular metabolism of docetaxel and paclitaxel.
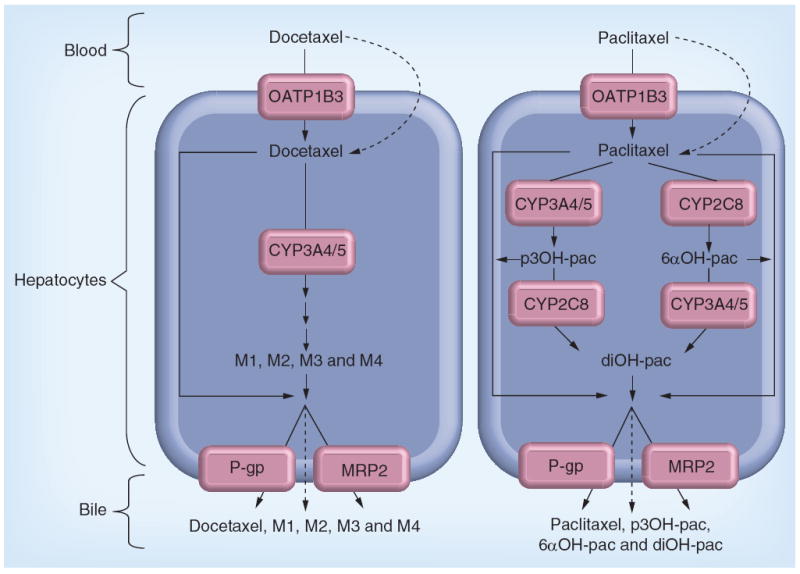
Dashed lines indicate passive diffusion. The short series of arrows before the formation of the docetaxel metabolites (M1, M2, M3 and M4) designates the possible involvement of multiple enzyme reactions not presented in this figure.
6αOH-pac: 6α-hydroxypaclitaxel; diOH-pac: 6α-p-3′-dihydroxypaclitaxel; p3OH-pac: p-3′-hydroxypaclitaxel.
Currently, there are no methods for identifying patients who are at high risk of toxicity before treatment, thus all patients receiving taxanes need to be closely monitored by their physician. In this article we will describe the use of three potential strategies (pharmacogenetics, enzyme probes and therapeutic drug monitoring [TDM]), as tools to individualize treatment with paclitaxel or docetaxel.
The germline genome may be a principal source of variability in drug PK or patient susceptibility to adverse events. Discovery and validation of pharmacogenetic markers that modify drug PK or patient sensitivity could enable appropriate drug or dose selection before treatment initiation. A second approach to optimize initial dose selection is the use of enzyme probes. Enzyme probes may be useful for estimating the patient’s metabolic activity prior to treatment initiation, enabling selection of the optimal taxane starting dose for an individual patient. The final approach, TDM, would allow clinicians to optimize taxane doses during therapy. TDM is the systematic use of estimated drug exposure during previous cycles to select ideal doses in subsequent cycles. In this article we will summarize the rationale, describe preliminary attempts at implementation, discuss specific challenges to clinical translation, and make recommendations for reasonable next steps in the development of each of these three potential tools for individualizing taxane treatment.
Pharmacogenetics
Occurrence of adverse events and response to taxane treatment vary greatly among individuals [10,11]. These interindividual differences are in part a result of differences in drug exposure [23-25], but also reflect differences in patient sensitivity. Variation in the patient’s germline genome is a major factor that influences drug exposure and patient sensitivity. Genotyping specific SNPs can identify patients at elevated risk of adverse events or lack of efficacy, enabling clinicians to make more informed decisions for patients being treated with those chemotherapeutic agents [26]. Pharmacogenetics has been translated into clinical practice to enable individualization of therapy with 6-mercaptopurine and irinotecan based on TPMT [27] and UGT [28] polymorphisms, respectively. There may be individual SNPs that are valuable pretreatment biomarkers to individualize taxane therapy; however, no polymorphisms are currently being used to guide treatment decisions.
Candidate SNPs in genes relevant to taxane PK
Early pharmacogenetic studies typically used a candidate gene approach in which the investigator selects SNPs in genes that are likely to be important based on known biology of the drug or phenotype of interest. Because paclitaxel and docetaxel have comparable pharmacology and adverse event profiles it is likely that there is overlap between the polymorphisms that influence treatment outcomes. A few studies have been published that investigated the direct influence of SNPs on taxane PK, reporting inconsistent results [29-36]. For example, Bergmann et al. [34] reported lower paclitaxel clearance in individuals carrying the CYP2C8*3 variant and Fransson et al. [35] found a relationship between ABCB1 2677G>T/A polymorphisms and decreased clearance of the 6α-hydroxypaclitaxel metabolite. Henningsson et al. [33] and Marsh et al. [36] also investigated the influence of these polymorphisms on paclitaxel PK, but could not reproduce these findings.
At this time it is unclear whether there are any SNPs that could be useful for guiding taxane treatment decisions because the pharmacogenetic literature is highly inconsistent. This issue is caused by the frequent reporting of studies in small, heterogeneous cohorts of patients, and the accepted practice of evaluating multiple SNPs and end points without appropriate statistical correction. Thus, there is a high likelihood that published results reflect both false-positive and false-negative findings. This section will not be a comprehensive review of all SNPs or studies, we instead direct readers interested in that topic to previous reviews on taxane pharmacogenetics [37,38]. In this section we will highlight SNPs that are most likely to be associated with taxane treatment efficacy or toxicity based on multiple reports with consistent findings. In order to maximize reader interpretability, all results will be described as the effect of the variant compared with the wild-type genotype. An overview of successfully replicated associations between polymorphisms and taxane outcomes is displayed in Table 1.
Table 1.
Replicated pharmacogenetic associations for taxane treatment outcomes from candidate gene studies.
| Gene variant | SNP information | Study (year) | Total patients (n), M:F | Ethnicity | Cancer type | Taxane treatment and concomitant therapy | Outcome | Measure of association | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| CYP3A5*3 | 6986A>G (rs776746, splicing defect) | Tsai et al. (2009) | 59, 0:59 | Taiwanese | BC | Docetaxel 75 mg/m2 + epirubicin + cyclophosphamide | ↓ pleural effusion | p = 0.077 (trend) | [39] |
| ↓ febrile neutropenia | p = 0.030 | ||||||||
| Gandara et al. (2009) | 381, 252:129 | Japanese and American | NSCLC | Paclitaxel 225 mg/m2 + carboplatin | ↑ OS | HR: 1.64; p = 0.07 (trend) | [43] | ||
| ↑ PFS | HR: 1.56; p = 0.09 (trend) | ||||||||
| Gréen et al. (2011) | 33, 0:33 | NR | OC | Paclitaxel 135 or 175 mg/m2 + carboplatin | ↓ leukopenia | p = 0.01 | [40] | ||
| ↑ leukocyte nadir | p = 0.07 (trend) | ||||||||
| Leskelä et al. (2011) | 118, 42:76 | Caucasian | Solid tumors | Paclitaxel 80–90 or 150–175 mg/m2 + various agents | ↓ neurotoxicity | HR per allele: 0.51; p = 0.012 | [42] | ||
|
| |||||||||
| CYP2C8*3 | 1196A>G (rs11572080, R139K) | Leskelä et al. (2011) | 118, 42:76 | Caucasian | Solid tumors | Paclitaxel 80–90 or 150–175 mg/m2 + various agents | ↑ neuropathy | HR: 1.72; p = 0.032 | [42] |
| 416G>A (rs10509681, K399R) | Hertz et al. (2012) | 111, 0:111 | Primarily Caucasian | BC | Paclitaxel 80–90 or 175 mg/m2 + various agents | ↑ clinical complete response | OR = 3.92; p = 0.0066 | [44] | |
| ↑ neuropathy | OR = 3.13; p = 0.075 (trend) | ||||||||
|
| |||||||||
| ABCB1*2† | 1236C>T (rs1128503, G412G); 2677G>T/A (rs2032582, A893S/T); 3435C>T (rs1045642, I1145I) | Tran et al. (2006) | 58, 29:29 | NR | Solid tumors | Docetaxel 75–100 mg/m2 | ↑ neutropenia | p = 0.046 | [30] |
| Sissung et al. (2008) | 73, 73:0 | NR | PC | Docetaxel 30 mg/m2 single agent or + thalidomide | ↓ OS | p = 0.0017 | [49] | ||
| ↑ neuropathy | p = 0.035 | ||||||||
| ↑ neutropenia | p = 0.053 (trend) | ||||||||
| Pan et al. (2009) | 54, 38:16 | Chinese | NSCLC | Docetaxel 75 mg/m2 + cisplatin | ↓ clinical response | p = 0.015 | [54] | ||
| Kim et al. (2009) | 118, 0:118 | Korean | OC | Docetaxel 75 or paclitaxel 175 mg/m2 + cisplatin or carboplatin | ↑ hematological and GI toxicity | p = 0.01 | [50] | ||
| Kim et al. (2012) | 218, 0:218 | Korean | BC | Docetaxel 100 mg/m2 | ↑ neutropenia | p = 0.015 | [41] | ||
| Sissung et al. (2006) | 26, NR | NR | Solid tumors | Paclitaxel 100 mg/m2 | ↑ neuropathy | p = 0.09 (trend) | [53] | ||
| Greater neutrophil decrease | p = 0.02 | ||||||||
| Gréen et al. (2006) | 53, 0:53 | Caucasian | OC | Paclitaxel 175 mg/m2 or 135 mg/m2 + carboplatin | ↑ clinical complete response | p < 0.05 | [57] | ||
| Johnatty et al. (2008) | 309, 0:309 | NR | OC | Paclitaxel 175 mg/m2 or 135 mg/m2 + carboplatin | ↑ PFS | p = 0.001 | [58] | ||
| Grau et al. (2009) | 47, 4:43 | Caucasian | H&N | Paclitaxel 80 mg/m2 | ↑ response rate | p < 0.001 | [56] | ||
| ↑ OS | p = 0.039 | ||||||||
| Chang et al. (2010) | 43, 26:17 | Korean | Gastric cancer | Paclitaxel 175 mg/m2 + leucovorin + 5-FU | ↓ PFS | p = 0.001 | [51] | ||
| ↑ diarrhea | p = 0.034 | ||||||||
| ↑ mucositis | p = 0.004 | ||||||||
| Bergmann et al. (2011) | 92, 0:92 | Caucasian | OC | Paclitaxel 175 mg/m2 + carboplatin | Greater neutrophil decrease | p = 0.02 | [52] | ||
|
| |||||||||
| CYP1B1*3 | 4326C>G (rs1056836, L432V) | Figg et al. (2007) | 20, 20:0 | Primarily Caucasian | PC | Docetaxel 30 mg/m2 + estramustine + thalidomide | ↓ survival | p = 0.013 | [69] |
| Sissung et al. (2008) | 52, 52:0 | NR | PC | Docetaxel 30 mg/m2 ± estamustine and thalidomide or prednisone | ↓ OS | p = 0.0004 | [68] | ||
| Rizzo et al. (2010) | 95, 1:94 | Caucasian | BC | Docetaxel 75 or 100 mg/m2 or paclitaxel 80 mg/m2 | ↓ hypersensitivity reactions | OR: 0.1361, p = 0.0008 | [130] | ||
| Pastina et al. (2010) | 60, 60:0 | NR | PC | Docetaxel 75 mg/m2 or 30 mg/m2 | ↓ response rate | p = 0.014 | [70] | ||
| ↓ PFS | p = 0.032 | ||||||||
| ↓ OS | p < 0.001 | ||||||||
| Marsh et al. (2007) | 84, 0:84 | Primarily Caucasian | BC | Paclitaxel 575–775 mg/m2 + doxorubicin + cyclophosphamide | ↓ PFS | p = 0.037 | [36] | ||
Assessed any or all of the ABCB1*2 SNPs that are in linkage disequilibrium.
↑: Increase; ↓: Decrease; 5-FU: 5-fluorouracil; BC: Breast cancer; F: Female; GI: Gastrointestinal; H&N: Head and neck cancer; HR: Hazard ratio; M: Male; NR: Not reported; NSCLC: Non-small-cell lung cancer; OC: Ovarian cancer; OR: Odds ratio; OS: Overall survival; PC: Prostate cancer; PFS: Progression-free survival.
CYP3A4 & CYP3A5
Both taxanes are substrates for the highly promiscuous CYP3A4/3A5 metabolic system [17,18]. There is a strong linkage between the functional CYP3A5*1 allele and CYP3A4*1B allele, and presence of these alleles together has been related to increased docetaxel clearance [30,31]. A few studies have reported associations between the low-activity CYP3A5*3 (6986A>G, rs776746) SNP and a lower risk of hematological toxicity in patients treated with docetaxel [39] or paclitaxel [40], however, the opposite finding has also been reported [41].
Paclitaxel is only partially metabolized by CYP3A4/3A5, so it is somewhat less likely that this polymorphism will have adequate predictive power to be clinically useful in guiding paclitaxel treatment decisions. Regardless, it is interesting that the low-activity CYP3A5*3 variant has been demonstrated to decrease neurotoxicity [42] and increase survival [43] in paclitaxel-treated patients.
CYP2C8
Systemic elimination of paclitaxel occurs primarily by CYP2C8-mediated hepatic metabolism [27]. Secondary analyses of three small studies [32,40,44] suggested an increase in neurotoxicity risk of patients carrying the low-activity CYP2C8*3 variant, which consists of two polymorphisms that occur in almost complete linkage disequilibrium (416G>A, R139K, rs11572080; and 1196A>G, K399R, rs10509681). This has recently been replicated in analyses of independent patient cohorts [42,45]. Hertz et al. also reported a corresponding increase in neoadju-vant clinical complete response from 23 to 55% in breast cancer patients who carried the *3 variant [44]. These reports of increased toxicity and efficacy are consistent with the finding that patients carrying the *3 variant have 11% lower paclitaxel clearance and, consequently, greater drug exposure [34].
ABCB1
The ABCB1 gene encodes P-gp (also known as MDR1), expressed in the liver and intestine. Three polymorphisms (3435C>T, I1145I rs1045642; 2677G>T/A, A893S/T, rs2032582; and 1236C>T, G412G, rs1128503) in moderate linkage disequilibrium in the ABCB1 gene have been extensively studied for an association with taxane treatment outcomes, however, it is unclear which, if any, SNP is causing the observed effect on outcome [46]. These three SNPs have been looked at as a haplotype, sometimes referred to as ABCB1*2, as well as being studied individually. It was originally hypothesized that the nonsynonymous triallelic (2677G>T/A) SNP is causative but more recent data suggest that the two silent polymorphisms (3435C>T and 1236C>T) may alter protein function [47,48].
The ABCB1 variant alleles have been associated with a higher risk of neutropenia in multiple studies with single-agent docetaxel [30,41,49]. In addition, an association between an ABCB1 variant allele and a higher risk of hematological and gastrointestinal toxicities has also been reported [50]. These findings correspond with the study of Bosch et al., in which patients homozygous for the variant alleles had higher area under the curve (AUC) due to decreased docetaxel elimination [29]. Consistent with their shared pharmacology, multiple studies have reported an increase in hematological or gastrointestinal toxicities for paclitaxel-treated patients carrying ABCB1 variants [50-53].
The three frequently studied polymorphisms in ABCB1 (3435C>T, 2677G>T/A and 1236C>T) have also been associated with taxane response or survival. The previously mentioned study of Sissung et al. found that docetaxel-treated patients carrying variant alleles had decreased overall survival (p = 0.0017) [49] and Pan et al. observed a poorer response in these patients [54]. The association with efficacy of paclitaxel treatment is somewhat less clear; ABCB1 variant alleles have been associated with shorter progression-free survival, overall survival and lower disease control rates in Koreans [51,55], but greater response rate [56,57] and progression-free survival [58] in other populations.
Other SNPs in genes relevant to taxane PK
Associations with taxane treatment outcomes have been reported for other SNPs in a variety of transporters or phase II metabolic enzymes, such as the hepatocellular uptake and efflux proteins OATP1B3 and MRP2 [59]. The GST system has also been extensively investigated, but in most cases these patients were treated with taxane–platinum combination therapy and the findings are most likely relevant to the pharmacogenetics of the platinum compound [30,50,60-62].
Candidate SNPs in genes relevant to taxane pharmacodynamics
CYP1B1
CYP1B1 is an enzyme that is not expressed in normal tissue and does not contribute to taxane PK, however, it has been detected in various tumors [63]. The *3 variant of the CYP1B1 enzyme (4326C>G, L432V, rs1056836) has shown increased catalytic activity toward its substrates, including endogenous hormones such as 17β-estradiol [64-66]. This altered enzyme activity may influence taxane efficacy by binding directly to taxanes or producing an estrogen metabolite that antagonizes the taxane mechanism of action [49,67], providing a plausible explanation for the multiple studies that have demonstrated inferior survival or response to taxanes for patients carrying the CYP1B1*3 genotype [36,68-70].
β-tubulin
Taxanes exert their cytotoxic effect by binding to β-tubulin in the cellular microtubules. Recently, Leandro-García et al. identified two linked polymorphisms in the proximal promoter of TUBB2A (-101T>C and -112A>G) that increase gene transcription. Patients carrying these variants may be at decreased risk of developing paclitaxel-induced neurotoxicity [71]. Similarly, a polymorphism in TUBB1 may modulate a patient’s risk of taxane-induced thrombocytopenia [72], however, replication of these associations has not yet been attempted to our knowledge.
It is worth pointing out that β-tubulin somatic mutations have also been investigated for an influence on paclitaxel response [73], however, the importance of these variations has been controversial. The β-tubulin genes are known to be highly conserved across species [74] and genotyping of this gene has been problematic owing to interference of β-tubulin pseudogenes [75].
Other SNPs in genes involved in taxane pharmacodynamics
Testing the hypothesis that taxane sensitivity may be related to the activity of proteins involved in the repair of DNA damage, one study reported a significant association with a tagSNP in the FANCD2 gene that increased gene expression and neuropathy risk [76]. This association awaits replication in an independent patient cohort.
Genome-wide & noncandidate gene association studies
An alternative method for discovering polymorphisms that influence treatment outcome is the genome-wide association study (GWAS). This approach enables the simultaneous interrogation of a huge amount of the known genetic variation in humans. Baldwin et al. published the first GWAS of a taxane clinical end point, reporting an association between an FGD4 polymorphism and the onset of peripheral sensory neuropathy in a large paclitaxel-treated discovery cohort and two independent replication cohorts [77]. This polymorphism in FGD4 was found to increase the risk of neuropathy by 57% in the discovery cohort (hazard ratio [HR]: 1.57; 95% CI: 1.30–1.91; p = 2.6 × 10-6) and an even larger increase in risk was detected in independent cohorts of European (HR: 1.72; 95% CI: 1.06–2.80; p = 0.013) and African–American (HR: 1.93; 95% CI: 1.13–3.28; p = 6.7 × 10-3) patients. FGD4 encodes for the protein Frabin, a widely expressed guanine nucleotide exchange factor for Cdc42, a small rhoGTPases that regulates cellular morphogenesis, including myelination. FGD4 has previously been linked with the congenital Charcot–Marie–Tooth disease, a condition that resembles taxane-induced sensory peripheral neuropathy, providing a plausible biological explanation for their finding. Other intriguing SNPs related to the onset or severity of neuropathy in this study were found in EPHA5 (rs7349683) and FZD3 (rs10771973).
No GWAS of docetaxel outcomes has been published, however, one study used the Affymetrix DMET 1.0 platform (Affymetrix Inc., CA, USA) to simultaneously interrogate nearly 2000 variants in 225 genes that may be relevant to drug PK [78]. Interesting associations of SNPs with docetaxel response (PPAR-δ, SLUT1C2 and CHST3) and toxicity (SPG7, CHST3, CYP2D6, NAT2, ABCC6, ATP7A, CYP4B1 and SLC10A2) were reported, however, replication of these findings has yet to be presented so it is unclear whether these variants have a true effect on docetaxel treatment outcome.
Challenges of pharmacogenetics
The primary challenge to the field of pharmacogenetics at this time is the difficulty of validating associations. Even the most highly studied and biologically reasonable effects, such as the relationship between CYP2D6 genotype and tamoxifen outcomes, continue to be debated in the literature [79]. Validation of pharmacogenetic associations requires carefully planned studies in large, independent patient cohorts. After validation, prospective genotype-guided studies are required to demonstrate a clinical benefit of modifying treatment in patients who carry a specific SNP. This potential benefit will need to be weighed against the additional cost of genotyping patients to identify potentially rare variants of interest. Finally, variability in treatment outcome may be the result of the interplay of many genetic factors, including modifications in the epigenome or the proteome. Thus, the explanatory value of any one SNP may be limited, and assessment of a set of SNPs may be required to to guide taxane treatment decisions.
Future work for pharmacogenetics
Several associations have been reported between SNPs and taxane treatment outcomes, although none have been consistently demonstrated in multiple independent cohorts to warrant clinical implementation. Still, there are a few promising gene variants, such as the polymorphisms in ABCB1, CYP2C8, CYP1B1, and FGD4, which based on the available data should be prioritized for replication in large, independent, prospectively collected patient cohorts. After replication, these markers should be tested in prospective, genotype-guided studies to ascertain whether the clinical implementation of pharmacogenetics would improve patient outcomes at a reasonable cost to the system. Finally, future work should seek to understand the influence of dynamic changes upstream or downstream of the genome on the static changes found within the genome.
Enzyme probes
Activity of metabolic enzymes is a major determinant of drug exposure. There are many factors that could influence enzymatic activity for a given patient, such as drug interactions, comorbidities, or genetic mutations. Instead of attempting to account for each factor individually, it may be best to directly measure the metabolic activity for the patient using a probe marker for the enzyme of interest. The ideal probe would measure enzyme activity in a patient sample ex vivo, however, for taxane metabolism only in vivo probes have been investigated. An ideal in vivo enzymatic probe is a safe, conveniently administered and quickly interpretable marker agent that shares the metabolic pathway of the drug of interest, enabling estimation of the expected rate of metabolism of the drug [80]. Use of a pretreatment probe could help clinicians select a more appropriate dose in order to target a desired level of exposure, potentially preventing toxicity or ineffectiveness.
The FDA maintains a list of substrates that can be used in drug development to measure in vivo activity of the major CYP450 enzymes involved in drug metabolism, including CYP2D6, CYP3A4, and CYP2C9 [204]. Currently, the use of enzyme probes in clinical practice is limited; one example is the use of an enzyme probe to predict TPMT activity ex vivo in patients who will receive thiopurine drugs. However, there could be many more applications, particularly for CYP3A4, which is involved in the metabolism of the majority of drugs used clinically. In this article we will refer to CYP3A because CYP3A4 and CYP3A5 have highly overlapping substrate specificity [81], which makes it difficult to determine their individual contribution to drug metabolism. CYP3A is involved in the metabolism of both docetaxel and paclitaxel [82], and paclitaxel is also metabolized by CYP2C8, therefore, this section will focus on the clinical translation of probes for CYP3A and CYP2C8 activity.
Docetaxel probes
Erythromycin breath test
The erythromycin breath test (ERMBT) is a validated in vivo assay for CYP3A activity that has been studied as a potential probe for docetaxel PK. For this test 14C-labelled erythromycin is administered intravenously and the amount of 14C-labelled CO2 exhaled is measured at specific time points, yielding an estimate of CYP3A activity [83]. Several studies have reported a significant correlation between the ERMBT and docetaxel clearance [31,84], with ERMBT explaining as much as 67% of the docetaxel PK variability [85]. The utility of the ERMBT has also been demonstrated in groups of patients in whom docetaxel dose selection is particularly challenging or would be particularly beneficial, such as the elderly [86] or patients with impaired liver function [87]. However, not all studies of the ERMBT were able to detect a significant correlation with docetaxel PK [88].
Alternative probes for CYP3A activity
Several CYP3A probes other than the ERMBT have been investigated for a relationship with docetaxel PK and toxicity (Table 2). In the study from Michael et al., which did not detect an association with the ERMBT, the clearance of anti-pyrine was correlated with docetaxel clearance (r2 = 79.49%; p = 0.007) [88]. Dexamethasone plasma clearance has also been studied, though the correlation was unexpectedly limited to females only [89,90]. The utility of midazolam plasma clearance as a CYP3A probe is uncertain based on several conflicting reports [91-93]. Finally, the urinary excretion of metabolites from exogenous cortisol has shown promise; a correlation between urinary 6-β-hydroxycortisol (6-β-OHF) and docetaxel clearance has been reported (r = 0.867; p < 0.001) [94]. Yamamoto et al. published the first attempt to prospectively use an enzyme probe to guide taxane dosing. In this study the docetaxel dose was selected based on 6-β-OHF, resulting in a significant decrease in docetaxel PK variability when compared with standard BSA-based dosing [95]. Prospective validation of this approach makes a compelling case for further research into the use of 6-β-OHF to guide docetaxel dosing to improve therapeutic outcomes.
Table 2.
Summary of probe studies estimating taxane pharmacokinetic parameters.
| Probe | Study (year) | Total patients (n), M:F | Cancer type | Taxane dose and infusion time | Probe parameter | Taxane pharmacokinetic parameter | Correlation | p-value | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| ERMBT | Baker et al. (2009)† | 92, 51:41 | Solid tumors | 20–75 mg/m2, various infusion times | ERMBT C20 min | CL (l/h) | r2 = 0.072 | p = 0.036 | [31] |
| Slaviero et al. (2004)† | 54, 31:23 | Solid tumors | 40 mg/m2, 2 h | ERMBT C20 min | CL (l/h) | r2 = 0.019 | p = 0.0005 | [84] | |
| Hirth et al. (2000)† | 21, 12:9 | Bone or soft tissue sarcoma | 100 mg/m2, 1 h | Ln ERMBT C20 min | BSA-adjusted CL (l/h/m2) | r2 = 0.67 | p = 0.0001 | [85] | |
| Hurria et al. (2006)† | 19‡, 8:12 | Breast, prostate or lung cancer | 35 mg/m2, 30 min | ERMBT C20 min | AUCinf (μg/ml × h) | NR | p = 0.02 | [86] | |
| AUCu inf (μg/ml × h) | NR | p = 0.01 | |||||||
| CLu (l/h) | NR | p = 0.04 | |||||||
| Hooker et al. (2008)† | 77, 41:36 | Solid tumors | 40–75 mg/m2, 1 h | ERMBT C20 min | Ln CLu | r2 = 0.035 | p = 0.177 | [87] | |
| 22§, NR | Solid tumors | 40–75 mg/m2, 1 h | ERMBT C20 min | Ln CLu | r2 = 0.603 | p < 0.0001 | |||
| Hertz et al. (2012)¶ | 14, 3:11 | Solid tumors | 75–90 mg/m2, 1 h | Ln ERMBT AUC0–1h | BSA-adjusted Ln AUC0–6 h | NR | NS | [96] | |
|
| |||||||||
| Dexamethasone | Puisset et al. (2004)† | 21, 10:11 | Solid tumors | 75–100 mg/m2, 1 h | CLdexamethasone (l/h) | CLdocetaxel (l/h) | r2 = 0.34 | NR | [89] |
| Puisset et al. (2007)† | 17, 10:7 | Solid tumors | 75–100 mg/m2, 1 h | CLdexamethasone (l/h) | CLdocetaxel (l/h) | r2 = 0.78 | p = 0.01 | [90] | |
| 38, 20:18 | Solid tumors | 75–100 mg/m2, 1 h | CL (l/h) | CL (l/h) | NR | p = 0.001 | |||
|
| |||||||||
| Hydrocortisone | Yamamoto et al. (2000)† | 29, 10:19 | Advanced NSCLC | 60 mg/m2, 1 h | Total urinary 6-β-OHF over 24 h | CL (l/h) | r2 = 0.752 | p < 0.001 | [94] |
|
| |||||||||
| Antipyrine | Michael et al. (2012)† | 19‡, 12:8 | Advanced breast or NSCLC | 75 or 36 mg/m2, 1 h | CL (ml/min) | CL (l/h) | r2 = 0.795 | p = 0.007 | [88] |
|
| |||||||||
| Midazolam | Goh et al. (2002)† | 31‡, 16:16 | Solid tumors | 75 or 100 mg/m2, 1 h | CL (ml/min) | CL (l/h) | r2 = 0.36 | p = 0.005 | [91] |
| Hilli et al. (2011)† | 20, 0:20 | High-risk breast cancer | 80 mg/m2, 1 h | Midazolam-1-OH AUCinf | CL (l/h) | r2 = 0.012 | NS | [92] | |
| Midazolam AUC ratio | CL (l/h) | r2 = 0.005 | NS | ||||||
| Zamboni et al. (2011)† | 29, 0:29 | Ovarian or peritoneal cancer | 75 mg/m2, 1 h | CL (ml/min) | CL (l/h) | r2 = 0.05 | NS | [93] | |
|
| |||||||||
| Quinidine | Gréen et al. (2009)¶ | 38, 0:38 | Ovarian or peritoneal cancer | 175 or 135 mg/m2, 3 h | MR (quinidine/3-hydroxyquinidine | AUC0–24 h (μg/ml × h)# | r2 = 0.450 | p < 0.001 | [32] |
|
| |||||||||
| Rosiglitazone | Hertz et al. (2012)¶ | 14, 3:11 | Solid tumors | 75–90 mg/m2, 1 h | BSA-adjusted Ln 3 h concentration | BSA-adjusted Ln AUC0–6 h | r2 = 0.38 | p = 0.018 | [96] |
Docetaxel studies.
One patient of unknown gender was excluded from the pharmacokinetic analyses.
Subanalysis in patients with liver impairment.
Paclitaxel studies.
AUC0–24 h refers to the AUC of the metabolite 6α-hydroxypaclitaxel.
6-β-OHF: 6-β-hydroxycortisol; AUC: Area under the concentration–time curve of total drug; AUCinf: Area under the concentration–time curve from zero to infinity; AUCu inf: Area under the concentration–time curve of unbound drug from zero to infinity; BSA: Body surface area; C20 min: Flux of 14CO2 at 20 min after administration; CL: Plasma clearance; CLu: Plasma clearance of unbound drug; ERMBT: Erythromycin breath test; F: Female; Ln: Natural log; M: Male; Midazolam AUC ratio: Area under the concentration–time curve midazolam-1-OH: area under the concentration–time curve midazolam; MR: Metabolic ratio; NR: Not reported; NS: Not significant; NSCLC: Non-small-cell lung cancer.
Paclitaxel probes
The use of a probe for paclitaxel dosing, as compared with docetaxel, is complicated by the multiple routes of metabolism. The first attempt to predict paclitaxel clearance with a probe was performed by Gréen and colleagues, who attempted to use quinidine as a CYP3A probe, but no correlation was observed with paclitaxel clearance (Table 2) [32]. However, low enzyme activity was correlated with high 6α-hydroxypaclitaxel AUC, the metabolite formed by CYP2C8. This suggests that when CYP3A enzyme activity is diminished, CYP2C8 may compensate by increasing its metabolism of paclitaxel.
Our group attempted to explain the variability in paclitaxel exposure by utilizing separate probes for each enzyme. We reported for the first time that rosiglitazone may have value as an in vivo probe of CYP2C8 activity; a single concentration of rosiglitazone at 3 h explained approximately 38% of the variability in paclitaxel AUC0–6 h (p = 0.018), however, the inclusion of ERMBT as a CYP3A probe did not meaningfully contribute to the explanation of the variability in paclitaxel exposure [96].
Relationship between probes & clinical outcome
There is reasonable evidence of a relationship between CYP3A probes and docetaxel PK, however, the association between the probe and clinical outcome has not been as comprehensively documented [86,95,97]. In one study the antipyrine disappearance rate correlated with neutrophil nadir and risk of grade 3+ neutropenia [97]. In the previously mentioned prospective study of cortisol metabolite-based dosing, nominally fewer patients in the individualized dosing arm experienced grade 3–4 neutropenia when compared with the BSA-based dosing arm (86 vs 93%), though the small magnitude of effect suggests that this may not be a clinically useful approach for preventing neutropenia [95].
Challenges of enzyme probes
In most cases the PK of a drug is determined not by a single enzyme, but by the interplay of a variety of factors such as enzymes, transporters and formulation vehicles. For instance, both taxanes are substrates for a variety of uptake (OATP1B3) [15,16] and efflux (P-gp) transporters [20,21]. The influence of these transporters on the results of probe assays has only been recently recognized and is not well characterized [31,98]. In addition, the vehicle Cremophor® EL (BASF Corp., Ludwigshafen, Germany) might interfere with the ability of probes to estimate paclitaxel metabolism since Cremophor EL influences the unbound paclitaxel concentration [99], but further research is required to assess this possible effect.
Another challenge for the development of enzyme probes is substrate dependence, the idea that a change in the enzyme–substrate interaction or the system within which the interaction takes place will not have a consistent effect on the probe and the drug of interest. For example, imatinib, a CYP3A4 inhibitor, decreases ERMBT-estimated CYP3A activity but does not change docetaxel clearance [100]. Substrate dependency has also been reported in relation to certain genotypes, such as CYP2C8*3, which was described as a low-activity variant in the previous section, but seems to have increased activity toward other substrates, including rosiglitazone [101] and pioglitazone [102]. The influence of transporters, supplementary metabolic pathways, and substrate dependence may explain the lack of correlation seen when patients are administered multiple probes of the same enzyme [103].
If an enzyme probe that accurately predicts in vivo taxane exposure is validated, logistical challenges to clinical implementation will still exist. Sample collection and analysis, particularly for the ERMBT which requires specialized collection equipment, will introduce new costs to the healthcare system and potential inconvenience to patients. Each potential probe has specific challenges but the overall benefit of a validated probe that could accurately predict taxane exposure is likely to outweigh these limitations and have widespread clinical utility.
Future work for enzyme probes
The existing studies suggest that an in vivo probe may explain a significant portion of the interindividual variability in taxane PK. There is more work to be done to translate enzyme probes to clinical application, starting with determination of the probe that best reflects the PK of each taxane. Once the optimal probe is identified, prospective studies are needed to verify the a priori improvement in taxane dose selection from probe usage. Then, prospective studies comparing therapeutic outcomes using probe versus BSA-based dosing are needed to validate the clinical utility of enzyme probes, and the costs of this approach must be weighed against these benefits, particularly in subgroups of patients in whom empirical dose selection is challenging.
TDM
TDM is an approach to individualizing therapy that employs systematic drug-level monitoring to adjust future dosages. TDM enables targeting of an exposure level in order to improve the likelihood of response, minimize the probability of toxicity, or both. In general, TDM is considered useful for drugs that have shown extensive PK variability, a narrow therapeutic window and a well-defined relationship between systemic exposure (PK) and toxicity or response (pharmacodynamics [PD]) [104,105]. Most of the current anticancer drugs, including taxanes, meet the first two criteria [104], but it is challenging to define a drug’s PK–PD relationship. Over the last 10 years, tremendous progress has been made in describing the PK–PD for taxanes. In this section we will briefly review these PK–PD relationships and describe the potential and challenges of using TDM as a tool for individualizing docetaxel or paclitaxel therapy.
Docetaxel
PK–PD relationship
The toxicity profile of docetaxel is predominately hematological, with other less common toxicities including fluid retention and neuropathy. Several studies have reported a relationship between docetaxel PK parameters, particularly clearance or AUC, and these toxicities [23,25,106-111]. Our understanding of the relationship between docetaxel PK and treatment efficacy is more limited with only a few studies demonstrating an association between PK and progression or survival (Table 3) [23,25,106].
Table 3.
Summary of studies on docetaxel pharmacokinetic–pharmacodynamic relationships.
| Study (year) | Total patients (n), M:F | Cancer type | Docetaxel dose, infusion duration and schedule | PK parameter | Clinical outcome | Statistical association | Ref. |
|---|---|---|---|---|---|---|---|
| Bruno et al. (1998) | 640, 270:370 | Solid tumors | 75 or 100 mg/m2, 1 h, 3-weekly | 50% lower CL | 4.3-fold increase in odds of experiencing neutropenia | p < 0.0001 | [106] |
| Plasma concentration >0.20 μM for over 4 h | 40% greater risk of fluid retention | p = 0.0029 | |||||
| AUC | Risk of progression decreased as AUC increase in NSCLC patients | p = 0.0232 | |||||
|
| |||||||
| Charles et al. (2006) | 68, 39:29 | Solid tumors | 40 mg/m2, 1 h, weekly | CL < 30 l/h | Odds ratio of hematological toxicity = 9.1 | p = 0.008 | [107] |
| AUC > 2.3 μM | Odds ratio of hematological toxicity = 6.9 | p = 0.02 | |||||
|
| |||||||
| Bruno et al. (2003) | 180, 118:62 | NSCLC | 100 mg/m2, 1 h, 3-weekly | AUC | Predictor of severe toxicity during first cycle | p < 0.0001 | [25] |
|
| |||||||
| Ozawa et al. (2008) | 200, 86:114 | Solid tumors | 60 mg/m2, 1 h, 3-weekly | AUC | Predictor of febrile neutropenia | p = 0.001 | [108] |
|
| |||||||
| Baker et al. (2005) | 55, 26:29 | Solid tumors | 75 or 50 mg/m2, 1 h, 3-weekly | AUCu and AUC | Correlation with percentage decrease in ANC | p = 0.002 and 0.029 | [111] |
| AUCu | Correlation with occurrence of grade 4 neutropenia | p = 0.013 | |||||
|
| |||||||
| Minami et al. (2006) | 69, 12:57 | Solid tumors | 20–60 mg/m2, 1 h, 3-weekly | AUCu | Correlation with occurrence of grade 4 neutropenia | p = 0.05 | [109] |
| Cmaxu | Correlation with occurrence of grade 4 neutropenia | p = 0.01 | |||||
|
| |||||||
| Sandstrom et al. (2005) | 44, 0:44 | Breast cancer | Docetaxel 70 mg/m2, 1 h, 3-weekly + epirubicin | AUC | Correlation with white blood cell survival fraction | NR | [110] |
ANC: Absolute neutrophil count; AUC: Area under the concentration–time curve of total drug; AUCu: Area under the concentration–time curve of unbound drug; CL: Plasma clearance; Cmaxu: Peak plasma concentration of unbound drug; F: Female; M: Male; NR: Not reported; NSCLC: Non-small-cell lung cancer; PK: Pharmacokinetics.
Clinical attempts at TDM
Only one published study has explored the potential for individualizing docetaxel dosing through TDM [112]. Engels et al. utilized a limited PK sampling strategy and a validated population PK model with a predefined target AUC of 4.9 mg/l × h. Fifteen patients were treated with at least one course of PK-guided docetaxel and were compared with a group of 15 patients receiving conventional (BSA-based) docetaxel therapy. TDM-guided dosing resulted in a decrease in interindividual variability (standard deviation of natural log-transformed AUC) of 39%; somewhat lower than the study’s predefined objective of a 50% decrease. Even though the differences in percentage decrease in white blood cell or absolute neutrophil count were also not statistically significant, PK-guided dosing successfully decreased the interindividual variability in these measures by approximately 50%. Despite the lack of statistical significance, this small study suggests that AUC-targeted dosing may attenuate the interindividual variability in drug exposure and hematological toxicity, and demonstrates the clinical feasibility of docetaxel TDM with limited PK sampling.
Paclitaxel
PK–PD relationships
Numerous studies have found a relationship between paclitaxel exposure and toxicity [113-122] or efficacy (Table 4) [116,118,123]. Based on these studies the most important parameter of paclitaxel PK seems to be the time that the systemic concentration remains above a threshold of 0.05 or 0.1 μM. This parameter has been associated with a variety of adverse events, such as neutropenia and neuropathy, and measures of treatment effectiveness.
Table 4.
Summary of studies on paclitaxel pharmacokinetic–pharmacodynamic relationships.
| Study (year) | Total patients (n), M:F | Cancer type | Paclitaxel dose, infusion duration and schedule | PK parameter | Outcome | Statistical association | Ref. |
|---|---|---|---|---|---|---|---|
| Gianni et al. (1995) | 30, 0:30 | OC and BC | 135 or 175 mg/m2 (OC) or 225 mg/m2 (BC), 3 h, 3-weekly | T > 0.05 μM > 24 h | ↑ neutropenia | p < 0.05 | [115] |
|
| |||||||
| Ohtsu et al. (1995) | 27, 18:9 | Solid tumors | 105–270 mg/m2, 3 or 24 h, 3-weekly | T > 0.05 μM = 14.3 h† | Corresponds to a 50% ↓ in granulocyte count | H = 1.88 | [117] |
| p < 0.05 | |||||||
|
| |||||||
| Mielke et al. (2005) | 24, 12:12 | Solid tumors | 100 mg/m2, 1 or 3 h, weekly | T > 0.05 μM ≥ 10.6 h | ↑ neuropathy | p = 0.023 | [119] |
| AUC | Correlation with neuropathy | p = 0.002 | |||||
| AUCu | Correlation with neuropathy | p = 0.003 | |||||
|
| |||||||
| Joerger et al. (2007) | 105, 0:105 | OC | 175 mg/m2, 3 h, 3-weekly + carboplatin | T > 0.05 μM (continuous) | Correlation with tumor complete response | p = 0.02 | [116] |
| T > 0.05 μM (continuous) | Correlation with tumor partial response | p = 0.05 | |||||
| T > 0.05 μM > 61.4 h | ↑ severe neutropenia | p = 0.01 | |||||
| T > 0.05 μM > 61.4 h | ↑ time to progression | p = 0.05 | |||||
|
| |||||||
| Miller et al. (2004) | 29, 0:29 | Solid tumors | 360 mg, 3 h, 3-weekly | T > 0.05 μM (continuous) | Inverse correlation with nadir ANC | p = 0.04 | [120] |
|
| |||||||
| Huizing et al. (1993) | 18, 0:18 | OC | 175 or 135 mg/m2, 3 or 24 h, 3-weekly | T > 0.1 μM = 11.16 h† | Corresponds to a 50% ↓ in ANC | H = 2.73 | [113] |
| T > 0.1 μM = 15.16 h† | Corresponds to a 50% ↓ in WBC count | H = 2.16 | |||||
|
| |||||||
| Huizing et al. (1997) | 55, 41:14 | NSCLC | 100–250 mg/m2, 3 h + carboplatin | T > 0.1 μM > 15 h | ↑ survival | p = 0.05 | [123] |
|
| |||||||
| Jiko et al. (2007) | 16, 16:0 | Urogenital cancers | 175 or 150 mg/m2, 3 h, 3-weekly + carboplatin (n = 10) or gemcitabine (n = 6) | T > 0.1 μM (continuous; in patients receiving gemcitabine) | Correlated with percentage ↓ in neutrophils and platelets | p = 0.018 | [114] |
|
| |||||||
| Mould et al. (2006) | 160, 0:160 | Endometrial cancer | Paclitaxel 150 mg/m2, 24 h, 3-weekly + doxorubicin | Linear binned AUC | Correlation with granulocytopenia | p < 0.001 | [118] |
| AUC | Inverse correlation with overall survival | p = 0.055 | |||||
|
| |||||||
| Nakajima et al. (2005) | 23, 0:23 | OC | 180 mg/m2, 3 h, 3-weekly + carboplatin | AUC | Correlation with leukocytopenia | p < 0.05 | [121] |
|
| |||||||
| Kobayashi et al. (2007) | 33, 22:11 | Gastric cancer | 60–90 mg/m2, 1.5 h, weekly + 5-FU or cisplatin | AUC (in patients receiving 90 mg/m2) | Correlation with hematological toxicity | p = 0.01 | [122] |
| Correlation with dose-limiting toxicity | p = 0.03 | ||||||
Value calculated in a sigmoid E-max model representing the time above threshold corresponding to a 50% decrease in the pharmacodynamic parameter.
↑: Increase; ↓: Decrease 5-FU: 5-fluorouracil; ANC: Absolute neutrophil count; AUC: Area under the concentration–time curve; AUCu: Area under the concentration–time curve of unbound drug; BC: Breast cancer; F: Female; H: Hill constant; M: Male; NSCLC: Non-small-cell lung cancer; OC: Ovarian cancer; PK: Pharmacokinetics; T > 0.05 μM: Amount of time paclitaxel plasma concentration exceeds 0.05 μM; T > 0.1 μM: Amount of time paclitaxel plasma concentration exceeds 0.1 μM; WBC: White blood cell.
Clinical attempts at TDM
Woo et al. reported the initial attempt at utilizing paclitaxel TDM, targeting a range for systemic exposure in seven children with recurrent acute leukemia receiving 24-h infusions [124]. In this single-arm study the infusion rate was adjusted based on clearance estimated 8 h into infusion. Five of the seven children reached AUCs between 75 and 125% of the target, whereas none were projected to reach target AUC without adjustment, demonstrating that adjusting the paclitaxel dose based on a target AUC range is clinically feasible for a 24-h infusion. In another study de Jonge et al. targeted a paclitaxel threshold of >0.1 μM for ≥15 h in 25 non-small-cell lung cancer patients [125]. In the first cycle a standard regimen of 175 mg/m2 was given to all patients and subsequent doses were adjusted based on their time above threshold. In the first cycle more than one-third of the patients (nine out of 25; 36%) had suboptimal exposure, but using TDM this decreased to 23% (five out of 22) and 11% (one out of nine) by cycles 2 and 6, respectively.
Challenges of TDM
The previously described studies demonstrate that taxane TDM is clinically feasible. However, there are serious challenges to its widespread implementation. TDM requires the development of an assay for the drug of interest that quickly returns accurate results. As with the other techniques described, TDM will introduce new healthcare costs by requiring collection and analysis of patient samples, and highly trained staff to interpret the PK results and recommend appropriate dose adjustments. It could also be burdensome on the patient, who will need to remain at the treatment center or return to the facility for sample collection. However, these challenges have been overcome in other therapeutic areas such as infectious disease or neurology, both of which employ TDM in certain treatment situations.
Future work for TDM
Pilot studies of the use of TDM suggest that this approach is clinically feasible and can successfully limit taxane interindividual variability or enable clinicians to target specific parameters of drug exposure. Though the connection between taxane exposure and treatment outcome has been an area of rich investigation, additional research is needed to more explicitly define these PK–PD relationships. Application of this developing knowledge will facilitate the design and execution of prospective TDM studies. One approach to this, reported recently by Joerger et al., is to perform PK–PD simulations to establish TDM-based dosing algorithms and estimate the clinical benefit of implementation [126]. Their proposed algorithm was simulated in 1000 patients and resulted in a reduction of grade 4 neutropenia in cycle 1 from 15 to 7%, and a further reduction to 4% in cycle 2. These simulations could assist in the design of large, prospective studies to compare TDM and empirical dosing with the primary aim of demonstrating an improvement in relevant clinical outcomes. The cost–effectiveness of TDM will also need to be considered before clinical uptake of TDM for taxane therapy is realized.
Conclusion
This article examined the potential use of pharmacogenetics, enzyme probes and TDM as tools for individualizing taxane therapy. Of the three approaches, TDM may, when ready for implementation, have the greatest value for clinical practice. Until then, pharmacogenetics and enzyme probes could assist in treatment decision-making in the near future. All three tools require continued validation and prospective studies with predefined, clinically relevant end points and robust statistical power, in addition to recognition of the added costs and potential inconvenience of clinical implementation.
It is important to note that these tools are not mutually exclusive; ultimately taxane treatment may incorporate elements of each approach into an adaptive treatment paradigm centered on the individual patient (Figure 2). Pharmacogenetics, in parallel with our rapidly advancing ability to select drugs based on tumor characteristics [127,128], could be used to select patients most likely to benefit or least likely to experience severe toxicity for treatment with a taxane. An enzyme probe could then be combined with other patient characteristics known to influence taxane exposure, such as age, gender and bilirubin levels, in order to design an initial dosing regimen for that patient [11,129]. Finally, TDM could guide iterative dose adjustment to ensure the patient’s taxane exposure is optimized. After 20 years of experience with taxanes in clinical practice it is time to look beyond BSA-based dosing and utilize the available tools to establish a revolutionary, individualized taxane treatment paradigm, ushering in the long-awaited era of personalized cancer therapy.
Figure 2. Integrated approach to taxane treatment that utilizes pharmacogenetics, enzyme probes and therapeutic drug monitoring to enhance efficacy and limit toxicity of therapy for cancer patients.
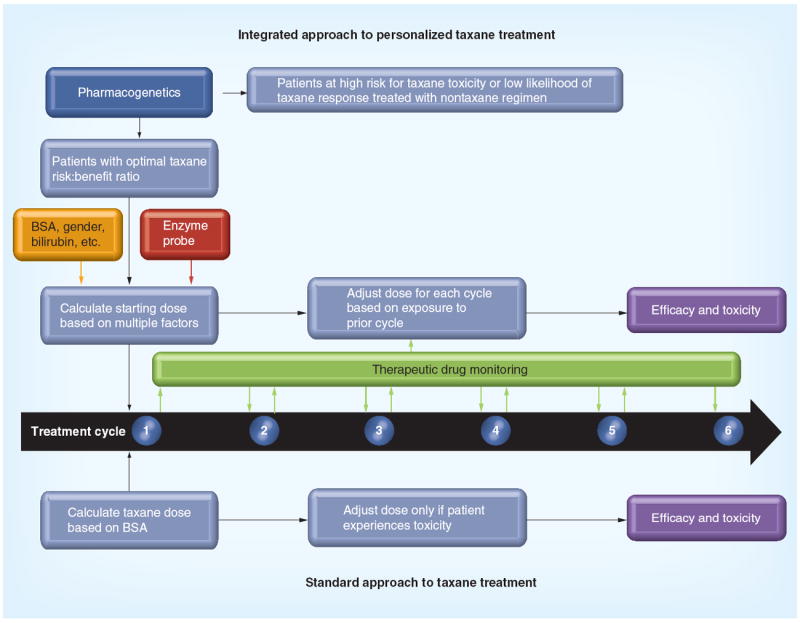
These tools can help screen out patients who are not good candidates for taxane treatment, assist in estimating a starting dose for taxane-treated patients and enable ongoing optimization of taxane dosing throughout treatment.
BSA: Body surface area in mg/m2.
Future perspective
The one-size-fits-all approach to taxane dosing, which almost exclusively uses the patient’s BSA to determine what dose they will receive throughout their course of treatment, will eventually be replaced by an individualized, adaptive dosing scheme. The SNPs that appreciably influence a patient’s likelihood of response or toxicity will be used in conjunction with somatic genetic information to screen out patients who are likely to experience suboptimal treatment outcomes. Initial doses of taxanes will be estimated using complex algorithms that combine standard patient factors with in vivo assays that predict drug exposure. Actual exposure will be monitored during treatment to enable adjustment of subsequent doses to achieve target concentrations that maximize patient outcomes. Initial validation and translation of these tools will likely be limited to specific treatment settings, but ultimately these incremental steps will coalesce into a revolutionary treatment paradigm that deploys these complementary tools to maximize efficacy and minimize toxicity of taxane treatment.
Executive summary.
Background
-
■
Taxane dosing is primarily based on body surface area, but this does not reduce the large interindividual variability in pharmacokinetics and unpredictability of treatment response and toxicity.
-
■
Tools that have been used to individualize treatment with other drugs, such as pharmacogenetics, enzyme probes and therapeutic drug monitoring, are being investigated for their potential role in taxane treatment.
-
■
This article highlights the progress and potential for each tool and the challenges and next steps for clinical translation.
Pharmacogenetics
-
■
Variants in genes relevant to pharmacology or biology may influence a patient’s response to therapy or risk of side effects.
-
■
Numerous studies have been published on associations between pharmacogenetic variants and taxane treatment outcomes, with highly inconsistent results.
-
■
Some associations have been consistently replicated in multiple independent patient populations (CYP3A5*3, CYP2C8*3, ABCB1*2 and CYP1B1*3), suggesting that there may be a true association between the variant and treatment outcome.
-
■
These variants should be prioritized for validation in large patient cohorts, followed by prospective evaluation of genotype-guided taxane treatment.
Enzyme probes
-
■
Drug exposure is largely determined by the activity of metabolic enzymes.
-
■
Enzyme probes may be able to estimate the in vivo activity of the enzyme of interest, enabling selection of an appropriate dose to achieve a target exposure.
-
■
Multiple CYP3A probes (erythromycin breath test, antipyrine and cortisol) have demonstrated an in vivo correlation with docetaxel pharmacokinetics.
-
■
Rosiglitazone is the only CYP2C8 probe that has been reported to have an in vivo correlation with paclitaxel pharmacokinetics.
-
■
Translation of a probe into clinical practice will require prospective confirmation that probe-based dosing confers a clinically relevant benefit.
Therapeutic drug monitoring
-
■
The taxanes have high interindividual pharmacokinetic variability and a narrow therapeutic window, making them ideal for therapeutic drug monitoring.
-
■
Pharmacokinetic–pharmacodynamic studies have demonstrated that certain parameters of drug exposure are associated with likelihood of toxicity or efficacy from taxane therapy.
-
■
Therapeutic drug monitoring has been piloted for both docetaxel and paclitaxel, demonstrating clinical feasibility and success in achieving targets of drug exposure.
-
■
Prospective evaluation of the benefit of therapeutic drug monitoring is necessary before widespread adoption into clinical use.
Conclusion
-
■
The three tools outlined in this article have great potential for improving taxane treatment; however, none are ready for clinical implementation at this time.
-
■
Each will need to improve patient outcomes in large, well-designed prospective studies before integration into standard clinical practice.
-
■
The benefit to patients will need to be balanced with the additional expense and potential for patient inconvenience when determining the clinical role for each tool.
-
■
In the future these three tools will be used complementarily to individualize each patient’s taxane treatment to maximize efficacy and limit toxicity.
Acknowledgments
This work was supported in part by NIH grants UL1 RR025747 and P01CA142538 and the UNC University Cancer Research Fund. DL Hertz is an AFPE fellow and SD Krens received a student grant from the Dutch Cancer Society and the Royal Dutch Pharmacists Association.
Footnotes
Financial & competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
-
■
of interest
-
■■
of considerable interest
- 1.Huizing MT, Misser VH, Pieters RC, et al. Taxanes: a new class of antitumor agents. Cancer Invest. 1995;13(4):381–404. doi: 10.3109/07357909509031919. [DOI] [PubMed] [Google Scholar]
- 2.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4(4):253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 3.Socinski MA, Bondarenko I, Karaseva NA, et al. Weekly nab-paclitaxel in combination with carboplatin versus solvent-based paclitaxel plus carboplatin as first-line therapy in patients with advanced non-small-cell lung cancer: final results of a Phase III trial. J Clin Oncol. 2012;30(17):2055–2062. doi: 10.1200/JCO.2011.39.5848. [DOI] [PubMed] [Google Scholar]
- 4.Galsky MD, Dritselis A, Kirkpatrick P, Oh WK. Cabazitaxel. Nat Rev Drug Discov. 2010;9(9):677–678. doi: 10.1038/nrd3254. [DOI] [PubMed] [Google Scholar]
- 5.Saif MW, Sarantopoulos J, Patnaik A, Tolcher AW, Takimoto C, Beeram M. Tesetaxel, a new oral taxane, in combination with capecitabine: a Phase I, dose-escalation study in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2011;68(6):1565–1573. doi: 10.1007/s00280-011-1639-3. [DOI] [PubMed] [Google Scholar]
- 6.Lee JJ, Swain SM. Peripheral neuropathy induced by microtubule-stabilizing agents. J Clin Oncol. 2006;24(10):1633–1642. doi: 10.1200/JCO.2005.04.0543. [DOI] [PubMed] [Google Scholar]
- 7.Pazdur R, Newman RA, Newman BM, et al. Phase I trial of Taxotere: five-day schedule. J Natl Cancer Inst. 1992;84(23):1781–1788. doi: 10.1093/jnci/84.23.1781. [DOI] [PubMed] [Google Scholar]
- 8.Taxol® (paclitaxel) Injection Package Insert. Bristol-Myers Squibb; Princeton, NJ, USA: 2011. [Google Scholar]
- 9.Taxotere® (docetaxel) Injection Concentrate Package Insert. Sanofi-Aventis; Bridgewater, NJ, USA: 2010. [Google Scholar]
- 10.Steed H, Sawyer MB. Pharmacology, pharmacokinetics and pharmacogenomics of paclitaxel. Pharmacogenomics. 2007;8(7):803–815. doi: 10.2217/14622416.8.7.803. [DOI] [PubMed] [Google Scholar]
- 11.Baker SD, Sparreboom A, Verweij J. Clinical pharmacokinetics of docetaxel : recent developments. Clin Pharmacokinet. 2006;45(3):235–252. doi: 10.2165/00003088-200645030-00002. [DOI] [PubMed] [Google Scholar]
- 12.Monsarrat B, Mariel E, Cros S, et al. Taxol metabolism. Isolation and identification of three major metabolites of taxol in rat bile. Drug Metab Dispos. 1990;18(6):895–901. [PubMed] [Google Scholar]
- 13.Wiernik PH, Schwartz EL, Einzig A, Strauman JJ, Lipton RB, Dutcher JP. Phase I trial of taxol given as a 24-hour infusion every 21 days: responses observed in metastatic melanoma. J Clin Oncol. 1987;5(8):1232–1239. doi: 10.1200/JCO.1987.5.8.1232. [DOI] [PubMed] [Google Scholar]
- 14.Sparreboom A, Van Tellingen O, Scherrenburg EJ, et al. Isolation, purification and biological activity of major docetaxel metabolites from human feces. Drug Metab Dispos. 1996;24(6):655–658. [PubMed] [Google Scholar]
- 15.van de Steeg E, van Esch A, Wagenaar E, et al. High impact of Oatp1a/1b transporters on in vivo disposition of the hydrophobic anticancer drug paclitaxel. Clin Cancer Res. 2011;17(2):294–301. doi: 10.1158/1078-0432.CCR-10-1980. [DOI] [PubMed] [Google Scholar]
- 16.Smith NF, Acharya MR, Desai N, Figg WD, Sparreboom A. Identification of OATP1B3 as a high-affinity hepatocellular transporter of paclitaxel. Cancer Biol Ther. 2005;4(8):815–818. doi: 10.4161/cbt.4.8.1867. [DOI] [PubMed] [Google Scholar]
- 17.Oshiro C, Marsh S, McLeod H, Carrillo M, Klein T, Altman R. Taxane pathway. Pharmacogenet Genomics. 2009;19(12):979–983. doi: 10.1097/FPC.0b013e3283335277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shou M, Martinet M, Korzekwa KR, Krausz KW, Gonzalez FJ, Gelboin HV. Role of human cytochrome P450 3A4 and 3A5 in the metabolism of taxotere and its derivatives: enzyme specificity, interindividual distribution and metabolic contribution in human liver. Pharmacogenetics. 1998;8(5):391–401. doi: 10.1097/00008571-199810000-00004. [DOI] [PubMed] [Google Scholar]
- 19.Dai D, Zeldin DC, Blaisdell JA, et al. Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics. 2001;11(7):597–607. doi: 10.1097/00008571-200110000-00006. [DOI] [PubMed] [Google Scholar]
- 20.van Waterschoot RA, Lagas JS, Wagenaar E, Rosing H, Beijnen JH, Schinkel AH. Individual and combined roles of CYP3A, P-glycoprotein (MDR1/ABCB1) and MRP2 (ABCC2) in the pharmacokinetics of docetaxel. Int J Cancer. 2010;127(12):2959–2964. doi: 10.1002/ijc.25279. [DOI] [PubMed] [Google Scholar]
- 21.Kwak JO, Lee SH, Lee GS, et al. Selective inhibition of MDR1 (ABCB1) by HM30181 increases oral bioavailability and therapeutic efficacy of paclitaxel. Eur J Pharmacol. 2010;627(1–3):92–98. doi: 10.1016/j.ejphar.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 22.Lagas JS, Vlaming ML, van Tellingen O, et al. Multidrug resistance protein 2 is an important determinant of paclitaxel pharmacokinetics. Clin Cancer Res. 2006;12(20 Pt 1):6125–6132. doi: 10.1158/1078-0432.CCR-06-1352. [DOI] [PubMed] [Google Scholar]
- 23.Bruno R, Vivier N, Veyrat-Follet C, Montay G, Rhodes GR. Population pharmacokinetics and pharmacokinetic–pharmacodynamic relationships for docetaxel. Invest New Drugs. 2001;19(2):163–169. doi: 10.1023/a:1010687017717. [DOI] [PubMed] [Google Scholar]
- 24.Persohn E, Canta A, Schoepfer S, et al. Morphological and morphometric analysis of paclitaxel and docetaxel-induced peripheral neuropathy in rats. Eur J Cancer. 2005;41(10):1460–1466. doi: 10.1016/j.ejca.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 25.Bruno R, Olivares R, Berille J, et al. Alpha-1-acid glycoprotein as an independent predictor for treatment effects and a prognostic factor of survival in patients with non-small cell lung cancer treated with docetaxel. Clin Cancer Res. 2003;9(3):1077–1082. [PubMed] [Google Scholar]
- 26.Weinshilboum R, Wang L. Pharmacogenomics: bench to bedside. Nat Rev Drug Discov. 2004;3(9):739–748. doi: 10.1038/nrd1497. [DOI] [PubMed] [Google Scholar]
- 27.McLeod HL, Coulthard S, Thomas AE, et al. Analysis of thiopurine methyltransferase variant alleles in childhood acute lymphoblastic leukaemia. Br J Haematol. 1999;105(3):696–700. doi: 10.1046/j.1365-2141.1999.01416.x. [DOI] [PubMed] [Google Scholar]
- 28.Innocenti F, Undevia SD, Iyer L, et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol. 2004;22(8):1382–1388. doi: 10.1200/JCO.2004.07.173. [DOI] [PubMed] [Google Scholar]
- 29.Bosch TM, Huitema AD, Doodeman VD, et al. Pharmacogenetic screening of CYP3A and ABCB1 in relation to population pharmacokinetics of docetaxel. Clin Cancer Res. 2006;12(19):5786–5793. doi: 10.1158/1078-0432.CCR-05-2649. [DOI] [PubMed] [Google Scholar]
- 30.Tran A, Jullien V, Alexandre J, et al. Pharmacokinetics and toxicity of docetaxel: role of CYP3A, MDR1, and GST polymorphisms. Clin Pharmacol Ther. 2006;79(6):570–580. doi: 10.1016/j.clpt.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 31.Baker SD, Verweij J, Cusatis GA, et al. Pharmacogenetic pathway analysis of docetaxel elimination. Clin Pharmacol Ther. 2009;85(2):155–163. doi: 10.1038/clpt.2008.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gréen H, Soderkvist P, Rosenberg P, et al. Pharmacogenetic studies of paclitaxel in the treatment of ovarian cancer. Basic Clin Pharmacol Toxicol. 2009;104(2):130–137. doi: 10.1111/j.1742-7843.2008.00351.x. [DOI] [PubMed] [Google Scholar]
- 33.Henningsson A, Marsh S, Loos WJ, et al. Association of CYP2C8, CYP3A4, CYP3A5, and ABCB1 polymorphisms with the pharmacokinetics of paclitaxel. Clin Cancer Res. 2005;11(22):8097–8104. doi: 10.1158/1078-0432.CCR-05-1152. [DOI] [PubMed] [Google Scholar]
- 34.Bergmann TK, Brasch-Andersen C, Gréen H, et al. Impact of CYP2C8*3 on paclitaxel clearance: a population pharmacokinetic and pharmacogenomic study in 93 patients with ovarian cancer. Pharmacogenomics J. 2011;11(2):113–120. doi: 10.1038/tpj.2010.19. [DOI] [PubMed] [Google Scholar]
- 35.Fransson MN, Gréen H, Litton JE, Friberg LE. Influence of Cremophor EL and genetic polymorphisms on the pharmacokinetics of paclitaxel and its metabolites using a mechanism-based model. Drug Metab Dispos. 2011;39(2):247–255. doi: 10.1124/dmd.110.035394. [DOI] [PubMed] [Google Scholar]
- 36.Marsh S, Somlo G, Li X, et al. Pharmacogenetic analysis of paclitaxel transport and metabolism genes in breast cancer. Pharmacogenomics J. 2007;7(5):362–365. doi: 10.1038/sj.tpj.6500434. [DOI] [PubMed] [Google Scholar]
- 37.Rodriguez-Antona C. Pharmacogenomics of paclitaxel. Pharmacogenomics. 2010;11(5):621–623. doi: 10.2217/pgs.10.32. [DOI] [PubMed] [Google Scholar]
- 38.Marsh S. Pharmacogenomics of taxane/platinum therapy in ovarian cancer. Int J Gynecol Cancer. 2009;19(Suppl. 2):S30–S34. doi: 10.1111/IGC.0b013e3181c10513. [DOI] [PubMed] [Google Scholar]
- 39.Tsai SM, Lin CY, Wu SH, et al. Side effects after docetaxel treatment in Taiwanese breast cancer patients with CYP3A4, CYP3A5, and ABCB1 gene polymorphisms. Clin Chim Acta. 2009;404(2):160–165. doi: 10.1016/j.cca.2009.03.038. [DOI] [PubMed] [Google Scholar]
- 40.Gréen H, Khan MS, Jakobsen-Falk I, Avall-Lundqvist E, Peterson C. Impact of CYP3A5*3 and CYP2C8-HapC on paclitaxel/carboplatin-induced myelosuppression in patients with ovarian cancer. J Pharm Sci. 2011;100(100):4205–4209. doi: 10.1002/jps.22680. [DOI] [PubMed] [Google Scholar]
- 41.Kim KP, Ahn JH, Kim SB, et al. Prospective evaluation of the drug-metabolizing enzyme polymorphisms and toxicity profile of docetaxel in Korean patients with operable lymph node-positive breast cancer receiving adjuvant chemotherapy. Cancer Chemother Pharmacol. 2012;69(5):1221–1227. doi: 10.1007/s00280-011-1816-4. [DOI] [PubMed] [Google Scholar]
- 42.Leskelä S, Jara C, Leandro-García LJ, et al. Polymorphisms in cytochromes P450 2C8 and 3A5 are associated with paclitaxel neurotoxicity. Pharmacogenomics J. 2011;11(2):121–129. doi: 10.1038/tpj.2010.13. [DOI] [PubMed] [Google Scholar]
- 43.Gandara DR, Kawaguchi T, Crowley J, et al. Japanese–US common-arm analysis of paclitaxel plus carboplatin in advanced non-small-cell lung cancer: a model for assessing population-related pharmacogenomics. J Clin Oncol. 2009;27(21):3540–3546. doi: 10.1200/JCO.2008.20.8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hertz DL, Motsinger-Reif AA, Drobish A, et al. CYP2C8*3 predicts benefit/risk profile in breast cancer patients receiving neoadjuvant paclitaxel. Breast Cancer Res Treat. 2012;134(1):401–410. doi: 10.1007/s10549-012-2054-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hertz DL, Roy S, Motsinger-Reif AA, et al. CYP2C8*3 increases risk of neuropathy in breast cancer patients treated with paclitaxel. Ann Oncol. 2013 doi: 10.1093/annonc/mdt018. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kroetz DL, Pauli-Magnus C, Hodges LM, et al. Sequence diversity and haplotype structure in the human ABCB1 (MDR1, multidrug resistance transporter) gene. Pharmacogenetics. 2003;13(8):481–494. doi: 10.1097/00008571-200308000-00006. [DOI] [PubMed] [Google Scholar]
- 47.Kimchi-Sarfaty C, Oh JM, Kim IW, et al. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315(5811):525–528. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- 48.Fung KL, Gottesman MM. A synonymous polymorphism in a common MDR1 (ABCB1) haplotype shapes protein function. Biochim Biophys Acta. 2009;1794(5):860–871. doi: 10.1016/j.bbapap.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sissung TM, Baum CE, Deeken J, et al. ABCB1 genetic variation influences the toxicity and clinical outcome of patients with androgen-independent prostate cancer treated with docetaxel. Clin Cancer Res. 2008;14(14):4543–4549. doi: 10.1158/1078-0432.CCR-07-4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim HS, Kim MK, Chung HH, et al. Genetic polymorphisms affecting clinical outcomes in epithelial ovarian cancer patients treated with taxanes and platinum compounds: a Korean population-based study. Gynecol Oncol. 2009;113(2):264–269. doi: 10.1016/j.ygyno.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 51.Chang H, Rha SY, Jeung HC, et al. Association of the ABCB1 3435C>T polymorphism and treatment outcomes in advanced gastric cancer patients treated with paclitaxel-based chemotherapy. Oncol Rep. 2010;23(1):271–278. [PubMed] [Google Scholar]
- 52.Bergmann TK, Brasch-Andersen C, Gréen H, et al. Impact of ABCB1 variants on neutrophil depression: a pharmacogenomic study of paclitaxel in 92 women with ovarian cancer. Basic Clin Pharmacol Toxicol. 2011;100(2):199–204. doi: 10.1111/j.1742-7843.2011.00802.x. [DOI] [PubMed] [Google Scholar]
- 53.Sissung TM, Mross K, Steinberg SM, et al. Association of ABCB1 genotypes with paclitaxel-mediated peripheral neuropathy and neutropenia. Eur J Cancer. 2006;42(17):2893–2896. doi: 10.1016/j.ejca.2006.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pan JH, Han JX, Wu JM, Huang HN, Yu QZ, Sheng LJ. MDR1 single nucleotide polymorphism G2677T/A and haplotype are correlated with response to docetaxel–cisplatin chemotherapy in patients with non-small-cell lung cancer. Respiration. 2009;78(1):49–55. doi: 10.1159/000158454. [DOI] [PubMed] [Google Scholar]
- 55.Chang H, Rha SY, Jeung HC, et al. Association of the ABCB1 gene polymorphisms 2677G>T/A and 3435C>T with clinical outcomes of paclitaxel monotherapy in metastatic breast cancer patients. Ann Oncol. 2009;20(2):272–277. doi: 10.1093/annonc/mdn624. [DOI] [PubMed] [Google Scholar]
- 56.Grau JJ, Caballero M, Campayo M, et al. Gene single nucleotide polymorphism accumulation improves survival in advanced head and neck cancer patients treated with weekly paclitaxel. Laryngoscope. 2009;119(8):1484–1490. doi: 10.1002/lary.20254. [DOI] [PubMed] [Google Scholar]
- 57.Gréen H, Soderkvist P, Rosenberg P, Horvath G, Peterson C. mdr-1 single nucleotide polymorphisms in ovarian cancer tissue: G2677T/A correlates with response to paclitaxel chemotherapy. Clin Cancer Res. 2006;12(3 Pt 1):854–859. doi: 10.1158/1078-0432.CCR-05-0950. [DOI] [PubMed] [Google Scholar]
- 58.Johnatty SE, Beesley J, Paul J, et al. ABCB1 (MDR 1) polymorphisms and progression-free survival among women with ovarian cancer following paclitaxel/carboplatin chemotherapy. Clin Cancer Res. 2008;14(17):5594–5601. doi: 10.1158/1078-0432.CCR-08-0606. [DOI] [PubMed] [Google Scholar]
- 59.Kiyotani K, Mushiroda T, Kubo M, Zembutsu H, Sugiyama Y, Nakamura Y. Association of genetic polymorphisms in SLCO1B3 and ABCC2 with docetaxel-induced leukopenia. Cancer Sci. 2008;99(5):967–972. doi: 10.1111/j.1349-7006.2008.00765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Medeiros R, Pereira D, Afonso N, et al. Platinum/paclitaxel-based chemotherapy in advanced ovarian carcinoma: glutathione S-transferase genetic polymorphisms as predictive biomarkers of disease outcome. Int J Clin Oncol. 2003;8(3):156–161. doi: 10.1007/s10147-003-0318-8. [DOI] [PubMed] [Google Scholar]
- 61.Sun N, Sun X, Chen B, et al. MRP2 and GSTP1 polymorphisms and chemotherapy response in advanced non-small cell lung cancer. Cancer Chemother Pharmacol. 2010;65(3):437–446. doi: 10.1007/s00280-009-1046-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mir O, Alexandre J, Tran A, et al. Relationship between GSTP1 Ile(105)Val polymorphism and docetaxel-induced peripheral neuropathy: clinical evidence of a role of oxidative stress in taxane toxicity. Ann Oncol. 2009;20(4):736–740. doi: 10.1093/annonc/mdn698. [DOI] [PubMed] [Google Scholar]
- 63.Murray GI, Taylor MC, McFadyen MC, et al. Tumor-specific expression of cytochrome P450 CYP1B1. Cancer Res. 1997;57(14):3026–3031. [PubMed] [Google Scholar]
- 64.Shimada T, Watanabe J, Kawajiri K, et al. Catalytic properties of polymorphic human cytochrome P450 1B1 variants. Carcinogenesis. 1999;20(8):1607–1613. doi: 10.1093/carcin/20.8.1607. [DOI] [PubMed] [Google Scholar]
- 65.Li DN, Seidel A, Pritchard MP, Wolf CR, Friedberg T. Polymorphisms in P450 CYP1B1 affect the conversion of estradiol to the potentially carcinogenic metabolite 4-hydroxyestradiol. Pharmacogenetics. 2000;10(4):343–353. doi: 10.1097/00008571-200006000-00008. [DOI] [PubMed] [Google Scholar]
- 66.Hanna IH, Dawling S, Roodi N, Guengerich FP, Parl FF. Cytochrome P450 1B1 (CYP1B1) pharmacogenetics: association of polymorphisms with functional differences in estrogen hydroxylation activity. Cancer Res. 2000;60(13):3440–3444. [PubMed] [Google Scholar]
- 67.Bournique B, Lemarie A. Docetaxel (Taxotere) is not metabolized by recombinant human CYP1B1 in vitro, but acts as an effector of this isozyme. Drug Metab Dispos. 2002;30(11):1149–1152. doi: 10.1124/dmd.30.11.1149. [DOI] [PubMed] [Google Scholar]
- 68.Sissung TM, Danesi R, Price DK, et al. Association of the CYP1B1*3 allele with survival in patients with prostate cancer receiving docetaxel. Mol Cancer Ther. 2008;7(1):19–26. doi: 10.1158/1535-7163.MCT-07-0557. [DOI] [PubMed] [Google Scholar]
- 69.Figg WD, Li H, Sissung T, et al. Pre-clinical and clinical evaluation of estramustine, docetaxel and thalidomide combination in androgen-independent prostate cancer. BJU Int. 2007;99(5):1047–1055. doi: 10.1111/j.1464-410X.2007.06763.x. [DOI] [PubMed] [Google Scholar]
- 70.Pastina I, Giovannetti E, Chioni A, et al. Cytochrome 450 1B1 (CYP1B1) polymorphisms associated with response to docetaxel in castration-resistant prostate cancer (CRPC) patients. BMC Cancer. 2010;10:511. doi: 10.1186/1471-2407-10-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leandro-García LJ, Leskelä S, Jara C, et al. Regulatory polymorphisms in beta-tubulin IIa are associated with paclitaxel-induced peripheral neuropathy. Clin Cancer Res. 2012;18(16):4441–4448. doi: 10.1158/1078-0432.CCR-12-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Leandro-García LJ, Leskelä S, Inglada-Pérez L, et al. Hematological beta-tubulin VI isoform exhibits genetic variability that influences paclitaxel toxicity. Cancer Res. 2012;72(18):4744–4752. doi: 10.1158/0008-5472.CAN-11-2861. [DOI] [PubMed] [Google Scholar]
- 73.Monzo M, Rosell R, Sanchez JJ, et al. Paclitaxel resistance in non-small-cell lung cancer associated with beta-tubulin gene mutations. J Clin Oncol. 1999;17(6):1786–1793. doi: 10.1200/JCO.1999.17.6.1786. [DOI] [PubMed] [Google Scholar]
- 74.Lamendola DE, Duan Z, Penson RT, Oliva E, Seiden MV. Beta tubulin mutations are rare in human ovarian carcinoma. Anticancer Res. 2003;23(1B):681–686. [PubMed] [Google Scholar]
- 75.Berrieman HK, Lind MJ, Cawkwell L. Do beta-tubulin mutations have a role in resistance to chemotherapy? Lancet Oncol. 2004;5(3):158–164. doi: 10.1016/S1470-2045(04)01411-1. [DOI] [PubMed] [Google Scholar]
- 76.Sucheston LE, Zhao H, Yao S, et al. Genetic predictors of taxane-induced neurotoxicity in a SWOG Phase III intergroup adjuvant breast cancer treatment trial (S0221) Breast Cancer Res Treat. 2011;130(3):993–1002. doi: 10.1007/s10549-011-1671-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77■■.Baldwin RM, Owzar K, Zembutsu H, et al. A genome-wide association study identifies novel loci for paclitaxel-induced sensory peripheral neuropathy in CALGB 40101. Clin Cancer Res. 2012;18(18):5099–5109. doi: 10.1158/1078-0432.CCR-12-1590. Pharmacogenetic study that discovered and validated the influence of FGD4 on paclitaxel-induced neuropathy risk using independent, prospectively accrued patient cohorts. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Deeken JF, Cormier T, Price DK, et al. A pharmacogenetic study of docetaxel and thalidomide in patients with castration-resistant prostate cancer using the DMET genotyping platform. Pharmacogenomics J. 2010;10(3):191–199. doi: 10.1038/tpj.2009.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hertz DL, McLeod HL, Irvin WJ., Jr Tamoxifen and CYP2D6: a contradiction of data. Oncologist. 2012;17(5):620–630. doi: 10.1634/theoncologist.2011-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Frye RF. Probing the world of cytochrome P450 enzymes. Mol Interv. 2004;4(3):157–162. doi: 10.1124/mi.4.3.5. [DOI] [PubMed] [Google Scholar]
- 81.Liu YT, Hao HP, Liu CX, Wang GJ, Xie HG. Drugs as CYP3A probes, inducers, and inhibitors. Drug Metab Rev. 2007;39(4):699–721. doi: 10.1080/03602530701690374. [DOI] [PubMed] [Google Scholar]
- 82.Fujita K. Cytochrome P450 and anticancer drugs. Curr Drug Metab. 2006;7(1):23–37. doi: 10.2174/138920006774832587. [DOI] [PubMed] [Google Scholar]
- 83.Watkins PB, Hamilton TA, Annesley TM, Ellis CN, Kolars JC, Voorhees JJ. The erythromycin breath test as a predictor of cyclosporine blood levels. Clin Pharmacol Ther. 1990;48(2):120–129. doi: 10.1038/clpt.1990.126. [DOI] [PubMed] [Google Scholar]
- 84.Slaviero KA, Clarke SJ, McLachlan AJ, Blair EY, Rivory LP. Population pharmacokinetics of weekly docetaxel in patients with advanced cancer. Br J Clin Pharmacol. 2004;57(1):44–53. doi: 10.1046/j.1365-2125.2003.01956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hirth J, Watkins PB, Strawderman M, Schott A, Bruno R, Baker LH. The effect of an individual’s cytochrome CYP3A4 activity on docetaxel clearance. Clin Cancer Res. 2000;6(4):1255–1258. [PubMed] [Google Scholar]
- 86.Hurria A, Fleming MT, Baker SD, et al. Pharmacokinetics and toxicity of weekly docetaxel in older patients. Clin Cancer Res. 2006;12(20 Pt 1):6100–6105. doi: 10.1158/1078-0432.CCR-06-0200. [DOI] [PubMed] [Google Scholar]
- 87.Hooker AC, Ten Tije AJ, Carducci MA, et al. Population pharmacokinetic model for docetaxel in patients with varying degrees of liver function: incorporating cytochrome P4503A activity measurements. Clin Pharmacol Ther. 2008;84(1):111–118. doi: 10.1038/sj.clpt.6100476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88■.Michael M, Cullinane C, Hatzimihalis A, et al. Docetaxel pharmacokinetics and its correlation with two in vivo probes for cytochrome P450 enzymes: the C(14)-erythromycin breath test and the antipyrine clearance test. Cancer Chemother Pharmacol. 2012;69(1):125–135. doi: 10.1007/s00280-011-1676-y. Compared multiple CYP3A4 probes and determined that antipyrine clearance was the most highly correlated with docetaxel clearance. [DOI] [PubMed] [Google Scholar]
- 89.Puisset F, Chatelut E, Dalenc F, et al. Dexamethasone as a probe for docetaxel clearance. Cancer Chemother Pharmacol. 2004;54(3):265–272. doi: 10.1007/s00280-004-0823-0. [DOI] [PubMed] [Google Scholar]
- 90.Puisset F, Chatelut E, Sparreboom A, et al. Dexamethasone as a probe for CYP3A4 metabolism: evidence of gender effect. Cancer Chemother Pharmacol. 2007;60(2):305–308. doi: 10.1007/s00280-006-0385-4. [DOI] [PubMed] [Google Scholar]
- 91.Goh BC, Lee SC, Wang LZ, et al. Explaining interindividual variability of docetaxel pharmacokinetics and pharmacodynamics in Asians through phenotyping and genotyping strategies. J Clin Oncol. 2002;20(17):3683–3690. doi: 10.1200/JCO.2002.01.025. [DOI] [PubMed] [Google Scholar]
- 92.Hilli J, Sailas L, Jyrkkio S, Pyrhonen S, Laine K. NCT01110291: induction of CYP3A activity and lowered exposure to docetaxel in patients with primary breast cancer. Cancer Chemother Pharmacol. 2011;67(6):1353–1362. doi: 10.1007/s00280-010-1426-6. [DOI] [PubMed] [Google Scholar]
- 93.Zamboni WC, Combest AJ, DeLoia JA, et al. Pharmacologic and phenotypic study of docetaxel in patients with ovarian or primary peritoneal cancer. Cancer Chemother Pharmacol. 2011;68(5):1255–1262. doi: 10.1007/s00280-011-1609-9. [DOI] [PubMed] [Google Scholar]
- 94.Yamamoto N, Tamura T, Kamiya Y, Sekine I, Kunitoh H, Saijo N. Correlation between docetaxel clearance and estimated cytochrome P450 activity by urinary metabolite of exogenous cortisol. J Clin Oncol. 2000;18(11):2301–2308. doi: 10.1200/JCO.2000.18.11.2301. [DOI] [PubMed] [Google Scholar]
- 95■■.Yamamoto N, Tamura T, Murakami H, et al. Randomized pharmacokinetic and pharmacodynamic study of docetaxel: dosing based on body-surface area compared with individualized dosing based on cytochrome P450 activity estimated using a urinary metabolite of exogenous cortisol. J Clin Oncol. 2005;23(6):1061–1069. doi: 10.1200/JCO.2005.11.036. Only reported attempt at prospectively using enzyme probes to select a taxane dose. Demonstrated that probe-based dosing results in significantly lower docetaxel pharmacokinetic variability than standard body surface area-based dosing. [DOI] [PubMed] [Google Scholar]
- 96.Hertz DL, Walko CM, Bridges AS, et al. Pilot study of rosiglitazone as an in vivo probe of paclitaxel exposure. Br J Clin Pharmacol. 2012;74(1):197–200. doi: 10.1111/j.1365-2125.2012.04165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nishio M, Matsuda M, Ohyanagi F, et al. Antipyrine test predicts pharmacodynamics in docetaxel and cisplatin combination chemotherapy. Lung Cancer. 2005;49(2):245–251. doi: 10.1016/j.lungcan.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 98.Kurnik D, Wood AJ, Wilkinson GR. The erythromycin breath test reflects P-glycoprotein function independently of cytochrome P450 3A activity. Clin Pharmacol Ther. 2006;80(3):228–234. doi: 10.1016/j.clpt.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 99.van Tellingen O, Huizing MT, Panday VR, Schellens JH, Nooijen WJ, Beijnen JH. Cremophor EL causes (pseudo-) non-linear pharmacokinetics of paclitaxel in patients. Br J Cancer. 1999;81(2):330–335. doi: 10.1038/sj.bjc.6690696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Connolly RM, Rudek MA, Garrett-Mayer E, et al. Docetaxel metabolism is not altered by imatinib: findings from an early phase study in metastatic breast cancer. Breast Cancer Res Treat. 2011;127(1):153–162. doi: 10.1007/s10549-011-1413-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kirchheiner J, Thomas S, Bauer S, et al. Pharmacokinetics and pharmacodynamics of rosiglitazone in relation to CYP2C8 genotype. Clin Pharmacol Ther. 2006;80(6):657–667. doi: 10.1016/j.clpt.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 102.Tornio A, Niemi M, Neuvonen PJ, Backman JT. Trimethoprim and the CYP2C8*3 allele have opposite effects on the pharmacokinetics of pioglitazone. Drug Metab Dispos. 2008;36(1):73–80. doi: 10.1124/dmd.107.018010. [DOI] [PubMed] [Google Scholar]
- 103.Kinirons MT, O’Shea D, Kim RB, et al. Failure of erythromycin breath test to correlate with midazolam clearance as a probe of cytochrome P4503A. Clin Pharmacol Ther. 1999;66(3):224–231. doi: 10.1016/S0009-9236(99)70029-9. [DOI] [PubMed] [Google Scholar]
- 104.Galpin AJ, Evans WE. Therapeutic drug monitoring in cancer management. Clin Chem. 1993;39(11 Pt 2):2419–2430. [PubMed] [Google Scholar]
- 105.Rousseau A, Marquet P. Application of pharmacokinetic modelling to the routine therapeutic drug monitoring of anticancer drugs. Fundam Clin Pharmacol. 2002;16(4):253–262. doi: 10.1046/j.1472-8206.2002.00086.x. [DOI] [PubMed] [Google Scholar]
- 106.Bruno R, Hille D, Riva A, et al. Population pharmacokinetics/pharmacodynamics of docetaxel in Phase II studies in patients with cancer. J Clin Oncol. 1998;16(1):187–196. doi: 10.1200/JCO.1998.16.1.187. [DOI] [PubMed] [Google Scholar]
- 107.Charles KA, Rivory LP, Stockler MR, et al. Predicting the toxicity of weekly docetaxel in advanced cancer. Clin Pharmacokinet. 2006;45(6):611–622. doi: 10.2165/00003088-200645060-00004. [DOI] [PubMed] [Google Scholar]
- 108.Ozawa K, Minami H, Sato H. Logistic regression analysis for febrile neutropenia (FN) induced by docetaxel in Japanese cancer patients. Cancer Chemother Pharmacol. 2008;62(3):551–557. doi: 10.1007/s00280-007-0648-8. [DOI] [PubMed] [Google Scholar]
- 109.Minami H, Kawada K, Sasaki Y, et al. Pharmacokinetics and pharmacodynamics of protein-unbound docetaxel in cancer patients. Cancer Sci. 2006;97(3):235–241. doi: 10.1111/j.1349-7006.2006.00166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sandstrom M, Lindman H, Nygren P, Lidbrink E, Bergh J, Karlsson MO. Model describing the relationship between pharmacokinetics and hematologic toxicity of the epirubicin–docetaxel regimen in breast cancer patients. J Clin Oncol. 2005;23(3):413–421. doi: 10.1200/JCO.2005.09.161. [DOI] [PubMed] [Google Scholar]
- 111.Baker SD, Li J, ten Tije AJ, et al. Relationship of systemic exposure to unbound docetaxel and neutropenia. Clin Pharmacol Ther. 2005;77(1):43–53. doi: 10.1016/j.clpt.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 112.Engels FK, Loos WJ, van der Bol JM, et al. Therapeutic drug monitoring for the individualization of docetaxel dosing: a randomized pharmacokinetic study. Clin Cancer Res. 2011;17(2):353–362. doi: 10.1158/1078-0432.CCR-10-1636. [DOI] [PubMed] [Google Scholar]
- 113.Huizing MT, Keung AC, Rosing H, et al. Pharmacokinetics of paclitaxel and metabolites in a randomized comparative study in platinum-pretreated ovarian cancer patients. J Clin Oncol. 1993;11(11):2127–2135. doi: 10.1200/JCO.1993.11.11.2127. [DOI] [PubMed] [Google Scholar]
- 114.Jiko M, Yano I, Sato E, et al. Pharmacokinetics and pharmacodynamics of paclitaxel with carboplatin or gemcitabine, and effects of CYP3A5 and MDR1 polymorphisms in patients with urogenital cancers. Int J Clin Oncol. 2007;12(4):284–290. doi: 10.1007/s10147-007-0681-y. [DOI] [PubMed] [Google Scholar]
- 115.Gianni L, Kearns CM, Giani A, et al. Nonlinear pharmacokinetics and metabolism of paclitaxel and its pharmacokinetic/pharmacodynamic relationships in humans. J Clin Oncol. 1995;13(1):180–190. doi: 10.1200/JCO.1995.13.1.180. [DOI] [PubMed] [Google Scholar]
- 116.Joerger M, Huitema AD, Richel DJ, et al. Population pharmacokinetics and pharmacodynamics of paclitaxel and carboplatin in ovarian cancer patients: a study by the European organization for research and treatment of cancer-pharmacology and molecular mechanisms group and new drug development group. Clin Cancer Res. 2007;13(21):6410–6418. doi: 10.1158/1078-0432.CCR-07-0064. [DOI] [PubMed] [Google Scholar]
- 117.Ohtsu T, Sasaki Y, Tamura T, et al. Clinical pharmacokinetics and pharmacodynamics of paclitaxel: a 3-hour infusion versus a 24-hour infusion. Clin Cancer Res. 1995;1(6):599–606. [PubMed] [Google Scholar]
- 118.Mould DR, Fleming GF, Darcy KM, Spriggs D. Population analysis of a 24-h paclitaxel infusion in advanced endometrial cancer: a gynaecological oncology group study. Br J Clin Pharmacol. 2006;62(1):56–70. doi: 10.1111/j.1365-2125.2006.02718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mielke S, Sparreboom A, Steinberg SM, et al. Association of paclitaxel pharmacokinetics with the development of peripheral neuropathy in patients with advanced cancer. Clin Cancer Res. 2005;11(13):4843–4850. doi: 10.1158/1078-0432.CCR-05-0298. [DOI] [PubMed] [Google Scholar]
- 120.Miller AA, Rosner GL, Egorin MJ, Hollis D, Lichtman SM, Ratain MJ. Prospective evaluation of body surface area as a determinant of paclitaxel pharmacokinetics and pharmacodynamics in women with solid tumors: Cancer and Leukemia Group B Study 9763. Clin Cancer Res. 2004;10(24):8325–8331. doi: 10.1158/1078-0432.CCR-04-1078. [DOI] [PubMed] [Google Scholar]
- 121.Nakajima M, Fujiki Y, Kyo S, et al. Pharmacokinetics of paclitaxel in ovarian cancer patients and genetic polymorphisms of CYP2C8, CYP3A4, and MDR1. J Clin Pharmacol. 2005;45(6):674–682. doi: 10.1177/0091270005276204. [DOI] [PubMed] [Google Scholar]
- 122.Kobayashi M, Oba K, Sakamoto J, et al. Pharmacokinetic study of weekly administration dose of paclitaxel in patients with advanced or recurrent gastric cancer in Japan. Gastric Cancer. 2007;10(1):52–57. doi: 10.1007/s10120-006-0411-6. [DOI] [PubMed] [Google Scholar]
- 123.Huizing MT, Giaccone G, van Warmerdam LJ, et al. Pharmacokinetics of paclitaxel and carboplatin in a dose-escalating and dose-sequencing study in patients with non-small-cell lung cancer. The European Cancer Centre. J Clin Oncol. 1997;15(1):317–329. doi: 10.1200/JCO.1997.15.1.317. [DOI] [PubMed] [Google Scholar]
- 124.Woo MH, Relling MV, Sonnichsen DS, et al. Phase I targeted systemic exposure study of paclitaxel in children with refractory acute leukemias. Clin Cancer Res. 1999;5(3):543–549. [PubMed] [Google Scholar]
- 125■■.de Jonge ME, van den Bongard HJ, Huitema AD, et al. Bayesian pharmacokinetically guided dosing of paclitaxel in patients with non-small cell lung cancer. Clin Cancer Res. 2004;10(7):2237–2244. doi: 10.1158/1078-0432.ccr-03-0060. Single-arm study that successfully used paclitaxel therapeutic drug monitoring. The percentage of patients who had suboptimal paclitaxel exposure decreased from 36% in cycle 1 to 11% by cycle 6. [DOI] [PubMed] [Google Scholar]
- 126■.Joerger M, Kraff S, Huitema AD, et al. Evaluation of a pharmacology-driven dosing algorithm of 3-weekly paclitaxel using therapeutic drug monitoring: a pharmacokinetic–pharmacodynamic simulation study. Clin Pharmacokinet. 2012;51(9):607–617. doi: 10.1007/BF03261934. Illustration of the use of pharmacokinetic–pharmacodynamic simulation to develop an algorithm for future prospective therapeutic drug-monitoring studies. [DOI] [PubMed] [Google Scholar]
- 127.Hugh J, Hanson J, Cheang MC, et al. Breast cancer subtypes and response to docetaxel in node-positive breast cancer: use of an immunohistochemical definition in the BCIRG 001 trial. J Clin Oncol. 2009;27(8):1168–1176. doi: 10.1200/JCO.2008.18.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Roychowdhury S, Iyer MK, Robinson DR, et al. Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med. 2011;3(111):111ra121. doi: 10.1126/scitranslmed.3003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mielke S. Individualized pharmacotherapy with paclitaxel. Curr Opin Oncol. 2007;19(6):586–589. doi: 10.1097/CCO.0b013e3282a08f8c. [DOI] [PubMed] [Google Scholar]
- 130.Rizzo R, Spaggiari F, Indelli M, et al. Association of CYP1B1 with hypersensitivity induced by taxane therapy in breast cancer patients. Breast Cancer Res Treat. 2010;124(2):593–598. doi: 10.1007/s10549-010-1034-5. [DOI] [PubMed] [Google Scholar]
Websites
- 201. [26 June 2012];Drugs @ FDA: FDA Approved Drug Products, Taxol. 2012 www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.DrugDetails.
- 202. [26 June 2012];Drugs @ FDA: FDA Approved Drug Products, Taxotere. 2012 www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.DrugDetails.
- 203. [26 June 2012];ClinicalTrials.gov: Taxanes search results. http://clinicaltrials.gov/ct2/results?term=taxanes&pg=2%29.
- 204.US FDA. [26 June 2012];Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. 2012 www.fda.gov/drugs/developmentapprovalprocess/developmentresources/druginteractionslabeling/ucm093664.htm.