

Abstract
Oncolytic virotherapy is a promising strategy to reduce tumor burden through selective virus replication in rapidly proliferating cells. However, the lysis of slowly replicating cancer stem cells (CSC), which maintain neoplastic clonality, is relatively modest and the potential contribution of programmed cell death (PCD) pathways to oncolytic activity is still poorly understood. We show that the oncolytic virus ΔPK lyses CSC-enriched breast cancer and melanoma 3D spheroid cultures at low titers (0.1pfu/cell) and without resistance development and it inhibits the 3D growth potential (spheroids and agarose colonies) of melanoma and breast cancer cells. ΔPK induces calpain activation in both melanoma and breast cancer 3D cultures as determined by the loss of the p28 regulatory subunit, and 3D growth is restored by treatment with the calpain inhibitor PD150606. In melanoma, ΔPK infection also induces LC3-II accumulation and p62/SQSTM1 clearance, both markers of autophagy, and 3D growth is restored by treatment with the autophagy inhibitor chloroquine (CQ). However, expression of the autophagy-required protein Atg5 is not altered and CQ does not restore p62/SQSTM1 expression, suggesting that the CQ effect may be autophagy-independent. PD150606 restores expression of p62/SQSTM1 in ΔPK infected melanoma cultures, suggesting that calpain activation induces anti-tumor activity through p62/SQSTM1 clearance.
Keywords: Virotherapy, oncolytic HSV, CSC, spheroid cultures, breast cancer
INTRODUCTION
Oncolytic virotherapy is an innovative, targeted strategy designed to reduce tumor burden through selective virus replication in the tumor cells and the generation of infectious progeny that spreads throughout the tumor mass. Platforms have been developed from virtually all the virus families and they have their respective advantages and disadvantages. However, clinical efficacy is modest 1. This was attributed to low levels of virus replication resulting from the deletions used to achieve tumor selectivity, the induction of antiviral immunity and the failure to effectively eradicate cancer stem cells (CSC), which are believed to initiate tumor formation1-4. From a clinical perspective CSC eradication is crucial, but direct studies of CSC lysis are relatively scant. Evolving complexities include the recent recognition that tumors can harbor multiple phenotypically and/or genetically distinct CSCs and the CSC phenotype can vary substantially between patients 5,6, suggesting that oncolytic efficacy could differ for distinct tumor cell types and/or oncolytic viruses .
The oncolytic herpes simplex viruses (oHSV) are primarily based on HSV-1 and are deleted in ICP34.5 and the large subunit of ribonucleotide reductase (R1). Tumor selectivity is due to the increased ribonucleotide reductase activity in tumor as compared to normal cells and the reduced activity of the ICP34.5 target double stranded RNA dependent protein kinase (PKR).7 oHSV may also be deleted in ICP47, which is responsible for immune evasion, and/or be armed with a pro-inflammatory cytokine gene.8 An oHSV armed with the cytokine granulocyte macrophage colony stimulating factor (GM-CSF), known as talimogene laherparepvec, showed promise in a recent clinical study of metastatic melanoma9. Another oHSV deleted in R1, ICP34.5 and ICP47 (G47Δ) lysed human glioblastoma (CD133+) CSC-85 like cells (GSC)2 and targeted mouse GSC in vivo when armed with murine IL-12 10. oHSV prevented tumor formation by neuroblastoma cells, which presumably reflects CSC targeting11, and lysed CD133+ myogenically primitive rhabdomyosarcoma cells with CSC-like properties12. Engineered oHSVs that lyse GSC include those designed to have increased tumor penetration, for example through the addition of an endoangio fusion gene (endostatin and angiostatin are angiogenesis inhibitors) or a chondroitinase adenosine triphosphate-binding cassette that removes the chondroitin sulfate from the tumor extracellular matrix proteoglycans. The latter oHSV spread throughout the glioma spheroids more efficiently than the unmodified virus, and had enhanced replication and antitumor activity in vivo 13-15 . A multi-mutated oHSV, MG18L (Us3 and R1-negative), replicated well in GSC and synergized with PI3K/Akt pathway inhibitors in killing the CSC through enhanced apoptosis16 and an oHSV containing the expression cassette for human GM-CSF lysed Doxorubicin-enriched, ALDH (br) mouse breast CSC 17. Because virus replication is optimal in highly proliferating cells, it is becoming increasingly evident that the ability to induce programmed cell death (PCD) increases the therapeutic potential of the oncolytic viruses for CSC, which are largely quiescent cells 6,18-20. However, the contribution of the tumor cell type to CSC lysis and the ability of the virus mutants to override such potential limitations, if any, are still poorly understood.
ΔPK is a HSV-2 mutant that is only deleted in the R1 protein kinase domain (known as ICP10PK), which is not conserved in HSV-1. ICP10PK activates Ras signaling pathways and is required for virus growth in slowly replicating normal cells, which unlike tumor cells, have low levels of Ras activity 21,22. The loss of ICP10PK imparts tumor selectivity and favors virus induction of multiple death pathways 18, thereby presumably increasing the clinical efficacy of ΔPK as an oncolytic agent 19,20. ΔPK is therapeutically promising because in addition to being safe in intracranially and intraperitoneally injected animals 18,23,24, it was well tolerated in phase I/II human clinical trials 25. We have previously shown that ΔPK has strong in vivo oncolytic activity for melanoma, with xenograft-bearing animals remaining tumor-free for at least 1 year after treatment 18. While this implies that CSC are not resistant to ΔPK, their lysis has not been documented and the role of virus-induced PCD pathways and tumor cell type, if any, are still poorly understood. These questions are clinically relevant because some pathways that may function in cell death, notably autophagy, are critically involved in maintaining normal physiological processes that counteract oncolytic activity in the CSC 26
RESULTS
ΔPK lyses CSC-enriched breast cancer and melanoma cultures
Two series of experiments were done in order to examine the ability of ΔPK to lyse cells with CSC-like properties. In the first series of experiments, we used breast cancer (HS578T) and melanoma (A2058 and A375) cells grown as 3-Dimensional (3D) multi-cellular tumor spheroids. These cultures include a gradient of proliferating cells similar to that found in tumor avascular micro-regions, reflect the tumor microenvironment and are often used as in vitro surrogates of tumorigenesis 5,27-30. While the heterogeneity of CSC phenotypic markers is increasingly recognized 5,6,31, our cultures are significantly enriched for cells with the widely distributed markers CD44+CD24-/low (breast cancer) and CD271 (melanoma) 5,31-33 (99.8% and 86.3% positive cells, respectively) [Supplementary Material (Fig. 1S)]. The 3D cultures were infected with ΔPK (moi =1) or mock-infected with PBS and examined for cell death by regular microscopy and staining with the cell death dye propidium iodide (PI) followed by flow cytometry (FCM). The ΔPK, but not mock-infected spheroids were largely reduced to debris (Fig. 1a) and most of the cells stained with PI, confirming cell death (Fig. 1b).
Figure 1. ΔPK has oncolytic activity in breast cancer and melanoma spheroid cultures.

(a) HS578T spheroid cultures were mock- or ΔPK-infected (moi=1) and examined at 48 hrs p.i. (20×). Similar results were obtained for A2058 and A375 cells. (b) Mock- or ΔPK-infected A2058 spheroid cultures were dissociated with accutase stained with PI and analyzed by FCM. Similar results were obtained for A375 and HS578T cells. (c) HS578T and A2058 cells mock infected or infected with ΔPK (moi=1; 48 hrs) were assayed for growth in spheroid culture and the results are expressed as No. of spheroids/1×104 cells ± SD. Similar results were obtained for A375 cells and for colony forming potential in soft agar.
In the second series of experiments we examined the ability of ΔPK to prevent growth under 3D conditions that include spheroid growth and colony formation in soft agar. 2D cultures of HS578T, A2058 and A375 cells were infected with ΔPK (moi =1) or mock-infected with PBS and examined for spheroid and colony formation at 7 and 14 days p.i., respectively. The results, expressed as spheroids or colonies/104 cells±SD, indicate that ΔPK-infected cells do not grow under these conditions (Fig. 1c and Supplementary Material Fig. 2S). Collectively, the data confirm that ΔPK lyses melanoma and breast cancer cultures that are CSC-like growth potential.
Low ΔPK titers lyse 3D spheroid cultures without resistance development
Modest clinical efficacy of oncolytic virotherapy was attributed to poor tumor penetration due to low levels of virus replication and the presence of cell subpopulations with innate or acquired resistance 3,34. To examine the effect of virus titers and resistance on the ability of ΔPK to lyse 3D tumor-like cultures, spheroids were infected with ΔPK at different moi (10-0.1 pfu/cell) and examined for cell death by PI staining and the failure to establish fresh spheroid cultures when sub-cultured in virus-free medium. As shown in Fig. 2 for A2058 cultures, virtually all the cells (95-99%) in the spheroids infected with 10 pfu/cell of ΔPK stained with PI at 48hrs p.i. and the cells collected at 4-5 days p.i. failed to establish new cultures. Penetration was poorer in the cultures given 0.1 pfu/cell of ΔPK, with PI staining at 48hrs p.i., seen only at the spheroid periphery However, at 10 days p.i the cultures were fully disrupted and ≥ 95% of the cells stained with PI. Cultures established from the few remaining clusters of live (PI negative) cells (Fig. 2A arrow) were equally susceptible to ΔPK-mediated lysis and all the cells were lost through 4 iterations of infection with 0.1 pfu/cell. Similar results were obtained for A375 and HS578T cells. The data indicate that low titers of ΔPK can penetrate and lyse spheroids without resistance development.
Figure 2. Low titers of ΔPK penetrate and lyse 3-D spheroids without resistance development.
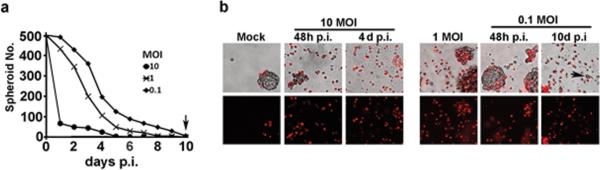
(a) A2058 spheroid cultures were infected with ΔPK at moi= 10, 1 or 0.1, virus was removed by centrifugation and the spheroids were re-plated in fresh, virus-free medium and counted as described in Materials and methods over a period of 10 days p.i. Spheroid counts decreased with time p.i. and were no longer seen by day 10 p.i. (arrow) (b) Spheroids mock-infected or infected with ΔPK at moi=10 and 0.1 were stained with PI at the listed times p.i. and imaged under phase contrast (top panels) and red fluorescence microscopy (bottom panels). Representative images are shown and similar results were obtained for A375 and HS578T cells. Arrow indicates a remaining viable cell.
ΔPK-infected 3D cultures evidence extensive cell lysis in the presence of low virus titers
Deletions that impart tumor selectivity are known to reduce virus growth 1-3. This was also shown for ΔPK, the titers of which in 2D cultures are 600-1000 fold lower than those of the wild type virus (1-1.5 vs 1000-1200 pfu/cell, respectively) 18,35. To examine whether this affects the ability of ΔPK to lyse CSC-enriched 3D cultures, HS578T and A2058 spheroids were infected with ΔPK (moi=1; 48hrs) or mock-infected with PBS and stained in double immunofluorescence with antibodies to the respective CSC phenotypic markers (CD44/CD24 and CD271 for HS578T and A2058, respectively) and PI (to measure cell death). Co-staining with antibody to the major virus capsid protein VP5 and plaque forming assays were used to determine virus growth. Co-staining cells were counted in five randomly selected fields (≥250 cells each) and the % calculated relative to the total number of imaged cells and virus titers were expressed as pfu/cell. The majority of the cells (total and expressing the respective CSC-associated phenotypes) in the ΔPK-infected cultures stained with PI (≥ 80-92%; Fig. 3a) indicating that ΔPK induces extensive cell death in these 3D, CSC-enriched cultures. However, the maximal virus titers were only 1.6 pfu/cell (Fig. 3d) and only one-third (≥ 25-35%) of the cells (total or positive for CSC markers) co-stained with VP5 antibody (Fig. 3b, c). Collectively, the data suggest that virus-induced PCD pathways are likely to contribute to its oncolytic activity.
Figure 3. Lysis of spheroid cultures independent of virus replication.
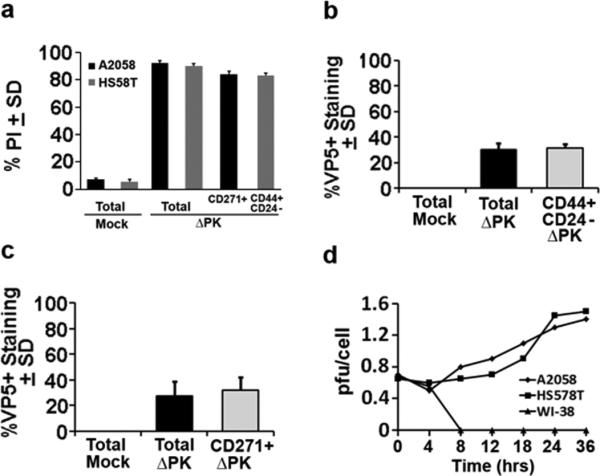
(a) HS578T and A2058 spheroid cultures were infected with ΔPK (moi=1; 48hrs) or mock-478 infected with PBS and stained with PI or double stained with PI and antibodies to the CSC phenotypic markers CD271 (A2058) or CD44/CD24 (HS578T) and examined by FCM using gates drawn based on forward and side scatter and isotype control staining patterns. Results are expressed as % PI+ cells calculated relative to the total number of cells in the spheroid cultures (total) or the CD44+/CD24low/- or CD271+ gated cells ± SD. (b,c). Dissociated spheroid cultures mock or ΔPK infected as in (a) were co-stained with antibodies to VP5 and the respective CSC markers CD44/CD24 (b) or CD271 (c) and the % staining cells counted as described in Materials and Methods. Results are expressed as % VP5+ cells calculated relative to the total number of cells in the spheroid cultures (total) or the % marker+ cells ± SEM. (d) A2058, HS578T and WI-38 cells were infected with ΔPK (moi = 1), examined for virus growth in serum-free medium and virus titers were determined by plaque assay. Results are expressed as mean pfu/cell (burst size). ΔPK is growth restricted in A2058 and HS578T cells and does not grow in WI-38 cells12.
Calpain activation contributes to the ΔPK-mediated inhibition of 3D growth
Activated PCD pathways increase the efficacy of oncolytic virotherapy 6,18-20. To better understand the contribution of death pathways to the oncolytic activity of ΔPK for CSC-enriched 3D cultures, A2058, A375, and HS578T cells were mock-infected with PBS or infected with ΔPK (moi = 1; 48 hrs) in the absence or presence of the calpain-specific inhibitor PD150606 (100μM) or the pancaspase inhibitor ZVAD-fmk (20μM) and assayed for growth in soft agar or spheroid culture. We focused on these two PCD pathways because they are involved in the ΔPK-mediated killing of 2D melanoma cultures 18. Results are expressed as % colonies or spheroids ± SD calculated relative to the mock-infected cells (100%) and are shown in Fig. 4 for A2058 colony formation. ΔPK infected cells failed to grow in 3D culture, as also shown in Fig. 1c. The calpain-specific inhibitor PD150606 restored 3D growth, but growth was not restored by treatment with the pancaspase inhibitor ZVAD-fmk and the inhibitors did not affect the 3D growth of the mock-infected cultures. Similar results were obtained for spheroid growth and in all the cell types. The data indicate that calpain, but not caspase activation contributes to the ability of ΔPK to inhibit 3D growth in both melanoma and breast cancer. Interestingly, however, the restoration of 3D growth was approximately 2-fold higher in melanoma than breast cancer cultures (52.4±6.2% for A2058; 67.8±4.7% for A375; and 30.5±3.8% for HS578T) (Fig. 4a-c), suggesting that the contribution of the death pathways to the oncolytic activity of ΔPK is malignant cell type specific.
Figure 4. Cell type-specific death pathways contribute to ΔPK oncolytic activity.

(a-c). A2058, A375 and HS578T 2-D cultures were mock-infected or infected with ΔPK (moi = 1; 48hrs) in the absence or presence of PD150606 (100μM), CQ (10μM) or zVAD-fmk (20μM) and plated under soft agar as described in Materials and methods. Colonies were counted and the results are expressed as % colonies ± SD calculated relative to the mock-infected cultures (100%). Similar results were obtained for growth in spheroid cultures. (d). A2058 cultures infected with ΔPK in the absence or presence of the inhibitors were assayed for virus growth by plaque assay at 4-36 hrs p.i. and the results are expressed as pfu/cell ± SD. Similar results were obtained for A375 and HS578T cells.
CQ restores the 3D growth of ΔPK-infected melanoma, but not breast cancer cells
Having previously shown that ΔPK induces the expression of the autophagy protein Beclin-1 in melanoma xenografts 18, we wanted to know whether autophagy is also associated with the ΔPK-induced cell lysis seen in our 3D cultures. A2058, A375 and HS578T cells were mock-infected with PBS or infected with ΔPK (moi=1; 48 hrs) in the absence or presence of CQ (10μM), which is an established autophagy inhibitor 36,37 , and assayed for growth in soft agar or spheroid conditions. CQ did not affect the 3D growth potential of the uninfected cultures, but it rescued the 3D growth of the ΔPK-infected melanoma cells. Rescue was cell type-specific, being 2-fold higher in A375 than A2058 melanoma cells (70.8±7.8% and 41.1±4.1%, respectively) and it was not seen in the breast cancer (HS578T) cells (Fig. 4a-c). Cell type specificity is not due to differential modulation of virus replication, because replication was nearly identical in all cultures. This is shown in Fig. 4d for A2058 cells and similar results were obtained in A375 and HS578T cells (0.9-1.1 pfu/cell at 24hrs p.i). The data indicate that CQ, used at the concentration at which it is known to inhibit autophagy36,37 restores the 3D growth potential of the ΔPK-infected melanoma cells, thereby associating the ΔPK-induced cell lysis with autophagy. However a similar association was not seen for breast cancer cells.
ΔPK infection induces LC3-II accumulation but does not alter Atg5 expression in melanoma spheroids
To further examine the relationship between ΔPK and autophagy, we asked whether infection induces autophagy markers. We focused on the membrane-bound phosphatidyl-ethanolamine conjugated form of microtubule-associated protein 1 light chain 3 (LC3II), which binds autophagosome membranes, and Atg5, which conjugates with Atg12 to generate an E3 ubiquitin ligase-like enzyme required for autophagy. Both of these are widely used markers to identify autophagy 38-40. In a first series of experiments protein extracts from A2058 and A375 spheroids mock-infected with PBS or infected with ΔPK (moi = 1) were immunoblotted with LC3 antibody and examined for the conversion of LC3I to LC3II. The stripped blots were probed with antibody to actin (loading control) and the results were quantified by densitometric scanning. ΔPK caused a significant increase (34-fold) in the LC3-II/LC3-I ratio relative to that seen in the mock-infected cultures in both melanoma cultures. LC3-II increase was seen as early as 1 hr p.i. and it was still present at 24 hrs p.i. (Fig. 5a). In the second series of experiments, A2058 spheroids were mock-infected or infected with ΔPK (moi = 1; 24 hrs) in the absence or presence of CQ (10μM) and protein extracts were immunoblotted with antibody to Atg5. Atg5 was strongly expressed in the mock-infected cultures and ΔPK did not alter its levels, which were similar to those seen in mock-infected cultures and in cultures infected with ΔPK in the presence of CQ (Fig. 5b). Collectively, the data indicate that ΔPK modulates some (viz. LC3II), but not other (viz. Atg5) markers of autophagy.
Figure 5. ΔPK induces LC3-II accumulation and calpain-dependent clearance of p62/SQSTM1.

(a) A2058 and A375 spheroid cultures were dissociated into single cell suspensions and mock- or ΔPK-infected (moi=1) for 1,4 or 24 hrs and protein extracts were immunoblotted with antibodies to LC3 followed by actin (loading control). Data were quantified by densitometric scanning and the results obtained for three replicate experiments are expressed as LC3-II/LC3-I ratio. (b) A2058 spheroid cultures dissociated into single cell suspensions were mock- or ΔPK-infected (moi=1; 24hrs) in the absence or presence of PD150606 (100 μM) or CQ (10μM) and protein extracts were immunoblotted with antibodies to Atg5, p62/SQSTM1, the calpain p28 regulatory subunit, AIF, or GAPDH (loading control). The blots were stripped between antibodies and representatives of three replicate experiments are shown. Similar results were obtained in A375 spheroids. Data were quantified by densitometric scanning and the results obtained for three replicate experiments are expressed as densitometric units ± SD for Atg5, p62/SQSTM1 and p28 (calpain).
p62/SQSTM1 is cleared from ΔPK-infected melanoma spheroids
p62/SQSTM1 is a stress-inducible protein that interacts with LC3. It has a crucial role as an assembly factor for ubiquitinated proteins and organelles and is ultimately degraded by autophagy 41. Having seen that ΔPK modulates some but not other autophagy markers we wanted to better understand its effect on p62/SQSTM1, which also functions as a signaling hub in life and death pathways and is implicated in tumorigenesis 41-43. A2058 and A375 spheroids were mock-infected or infected with ΔPK in the absence or presence of CQ (10 μM) and immunoblotted with antibody to p62/SQSTM1. The blots were stripped and re-blotted with antibody to GAPDH and the levels of p62/SQSTM1 determined by densitometric scanning and analyzed relative to GAPDH. As shown in Fig. 5b for A2058 cells, p62/SQSTM1 was expressed in the mock, but not ΔPK-infected cultures, indicating that ΔPK induces p62/SQSTM1 clearance. Significantly, however, expression was not restored by treatment of the infected cells with the autophagy inhibitor CQ (Fig. 5b,c), suggesting that the ΔPK-mediated clearance of p62/ SQSTM1 is through a mechanism other than autophagy. Similar results were obtained for A375 spheroids. This is particularly relevant from the standpoint of virus oncolysis because in addition to being implicated in tumorigenesis43, high levels of p62/ SQSTM1 were also associated with significantly worse prognosis and tumor progression, at least in lung and breast cancer44,45
ΔPK-mediated p62/SQSTM1 clearance is calpain-dependent
To better understand the mechanism responsible for the clearance of p62/SQSTM1, duplicate spheroid cultures were infected with ΔPK (moi=1; 24h) in the absence or presence of PD150606 (100μM) and protein extracts were immunoblotted with antibody to p62/SQSTM1 followed by immunoblotting of the sequentially stripped blots with antibodies to the calpain regulatory subunit p28, the loss of which documents calpain activation, or apoptosis-inducing factor (AIF), which is an apoptosis-associated calpain target 46. p62/SQSTM1 expression was fully restored in the PD150606-treated ΔPK-infected spheroid cultures, indicating that its clearance is calpain-dependent (Fig. 5b,c). Indeed, ΔPK induced calpain activation as evidenced by: (i) loss of the p28 regulatory subunit in ΔPK-infected cells, and (ii) restored expression in infected cells treated with the calpain inhibitor PD150606 (Fig. 5b,c). The levels of AIF were similar under all conditions (Fig. 5b,c) indicating that calpain does not function at this level. Collectively, the data indicate that calpain-mediated p62/SQSTM1 degradation is a key contributor to the ΔPK oncolytic activity in CSC-enriched 3D melanoma cultures.
DISCUSSION
The salient feature of the data presented in this report is the finding that ΔPK has strong oncolytic activity in CSC-enriched melanoma and breast cancer 3D cultures that involves calpain activation and the clearance of p62/SQSTM1 in the melanoma cultures. The following comments seem pertinent with respect to these findings.
Direct studies of CSC lysis are relatively scant. oHSV, including those engineered to increase tumor penetration and virus spread, were shown to kill glioblastoma, neuroblastoma, and rhabdomyosarcoma cells with CSC-like properties 2,10-17. However, tumors appear to harbor multiple phenotypically and/or genetically distinct CSCs 5,6,31 , underscoring the therapeutic advantage of a virus that elicits PCD pathways tailored to distinct cell types. ΔPK differs from most oHSV in that it is based on HSV-2 and is deleted in ICP10PK, which is not conserved in HSV-1, but it retains the replication-associated ICP34.5 and ribonucleotide reductase activities 2,7. ICP10PK activates Ras signaling pathways and contributes to virus growth, such that its deletion enables tumor selectivity, reduces replicative potential and induces multiple PCD pathways 18,21,22,24,35. The ICP10PK-deleted virus, ΔPK, was well tolerated in phase I/II human clinical trials25 and it has strong in vivo oncolytic activity in melanoma, with xenograft-bearing animals remaining tumor-free for at least 1 year after treatment18. While this implies that CSC are not resistant to ΔPK, their lysis remained to be documented.
We studied 3D melanoma and breast cancer cultures that are enriched in the widely distributed CSC phenotypic markers CD44+CD24-/low (breast cancer) and CD271 (melanoma)5,31-33,47,48. These cultures are enriched for slow cycling pluripotent with enhanced tumorigenicity and drug and mimic in vivo tumors in their microenvironment, pH and oxygen gradients, growth factors distribution and interaction with the extracellular matrix 27-30. They are known to resist oncolytic virotherapy due to reduced virus penetration and spread 13-15. Our data indicate that ΔPK effectively penetrates 3D spheroid cultures even when given at the very low titer of 0.1 pfu/cell and it lyses most of the cells by 10 days p.i. The few cells that survived this treatment were equally susceptible to lysis by these low virus titers and the entire cultures were completely eradicated through 4 cycles of infection, indicative of a fundamental lack of resistance to ΔPK lysis. Resistance development is a major therapeutic challenge2,34 and its absence is clinically relevant, because multiple injections are typically required during a treatment regimen. While these and previous 18 findings suggest that ΔPK has effective tumor penetration, the potential contribution of density, hypoxic regions, intratumoral heterogeneity, stromal tissue barriers and blood flow remains to be determined and studies of cancer-initiating cells from actual tumors are still needed.
In addition to lysing CSC-enriched cultures, ΔPK inhibited the 3D growth potential of both melanoma and breast cancer cells, as measured by spheroid and agarose colony formation. Growth was restored by treatment with PD150606, which is a cell-permeable, non-competitive and selective inhibitor of both calpain-1 and -2 that is directed towards the calpain Ca2+ binding sites. The calpain specificity of PD150606 is fully established and it was used at independently established doses 18,46,49-51. Growth was not restored by PD145305, which is an inactive analog of PD150606 (data not shown). Together with the findings that: (i) ΔPK caused the loss of the p28 calpain regulatory subunit, and (ii) PD150606 restored its expression, the data indicate that calpain activation contributes to the oncolytic activity of ΔPK in the 3D melanoma and breast cancer cultures. Calpains are Ca2+ activated nonlysosomal cysteine proteases that have paradoxical functions in the perturbed cancer apoptotic pathways. Calpain activity was implicated in the pro-survival activities of the tumor suppressor protein p53 and nuclear factor-κB, but it was also shown to cause cell death by inducing the collapse of the mitochondrial membrane potential (ΔΨm) and the induction of both caspase-dependent and independent (AIF-dependent) death pathways 46,51. Interestingly, the pancaspase inhibitor ZVAD-fmk did not restore 3D spheroid/agarose growth, although caspase activation is involved in the oncolytic activity of ΔPK in 2D melanoma cultures18, potentially reflecting increased CSC expression of anti-apoptotic proteins (viz. Bcl-2 and survivin) 52.
Autophagy has been implicated in maintaining normal physiological processes, particularly in CSC, where it enables survival in the tumor microenvironment 26 , and it contributes to breast cancer tumorigeniciy 53. However, a growing body of evidence supports the contribution of bona fide autophagic cell death to various pathological conditions, with various factors governing the cellular decision to die by autophagy 54. In cancer, autophagy can be pro- or anti-tumorigenic depending on the specific tissue and tumor stage55. Currently available data for melanoma support both protective and cytotoxic activities, albeit by still poorly understood mechanisms 56. We found that CQ, used at a dose at which it is known to inhibit autophagy, restored the 3D growth of ΔPK-infected melanoma, but not breast cancer cultures, providing a measure of specificity. In addition, the ΔPK-infected melanoma spheroids had increased levels of LC3-II accompanied by p62/SQSTM1 clearance, an established signature of autophagy 41,42. However, the relationship between ΔPK and autophagy is confounded by the finding that ΔPK did not alter the expression of Atg5, which has a critical role in autophagy activation by participating in two essential pathways, that is, Atg12-Atg5 conjugation and LC3 lipidation 38-40. Moreover, the expression of p62/SQSTM1 was not restored by CQ, but rather by the calpain inhibitor PD150606. A possible interpretation of our findings is that the observed LC3-II accumulation reflects a residual effort by the cells to survive ΔPK-induced cell death. However, implicit in this interpretation is the conclusion that the ability of CQ to restore 3D growth is through a pro-survival function other than autophagy that is melanoma specific. While such a function remains elusive, and discounting the possible contribution of dose, it may be important to point out that CQ (used at a dose 5-fold higher than ours), was recently shown to have melanoma-specific BH3-protein dependent activity.57 Further studies are needed in order to test this interpretation for ΔPK and to better elucidate the contribution of autophagy, if any, to its oncolytic activity.
Notwithstanding, to the extent of our knowledge, ours is the first report that calpain activation induces anti-tumor activity through p62/SQSTM1 degradation. Indeed, p62/SQSTM1 was previously implicated in Ras-induced tumorigenesis and its loss inhibited Ras-controlled transformation, apparently through ROS-induced cell death58. Moreover, high levels of p62/SQSTM1 were associated with worse prognosis and tumor progression in breast and non-small-cell lung cancer 43-45. While, the potential contribution of other death pathways and inflammatory signals 18 to the ability of ΔPK to lyse breast cancer spheroids remains unclear, our data underscore the importance of the tumor cell type in the selection of an effective oncolytic platform and highlight the versatility of ΔPK.
MATERIALS AND METHODS
Cells and virus
A2058 and A375 melanoma cells were obtained from the American Type Culture Collection (Manassas, VA). HS578T breast cancer cells were the gift of Dr. Angela Brodie (University of Maryland, Baltimore). They were grown in adherent monolayers cultures in Dulbeco modified Eagle's medium (DMEM, Invitrogen, Carlsbad, CA, USA) supplemented with L-glutamine (4 mM) and 10% fetal bovine serum (FBS, Gemini Bioproducts, Calabasos, CA). WI-38 cells (normal human embryonic lung fibroblasts) are an expansion from passage 9 and have a limited lifespan of 50 population doublings. They were cultured in minimal essential medium with Earle's salts, 10% FBS, 1 mm sodium pyruvate and 0.1 mm nonessential amino acids. The generation and properties of the HSV-2 mutant ΔPK that is deleted in the kinase domain of large subunit of ribonucleotide reductase (R1, also known as ICP10) were previously described 18,21-24,46.
Antibodies, pharmacological inhibitors and chemical reagents
The generation and specificity of the rabbit polyclonal antibody to ICP10, which recognizes an epitope that is retained by both ICP10 and the PK deleted p95 protein were previously described 18,21-24,46. The following antibodies were purchased and used according to manufacturer's instructions. FITC-conjugated anti-CD20 and APC-conjugated anti-CD133 antibodies were from Mylteni Biotech (Auburn, CA). FITC-conjugated anti-CD24, APC-conjugated anti-CD44, APC-conjugated anti-ABCG2, FITC-conjugated IgG1 isotype, APC-conjugated IgG2b isotype and APC-conjugated anti-mouse antibodies were from BD Biosciences (San Jose, CA). Antibodies to AIF, LC3, calpain and GAPDH were from Santa Cruz Biotechnologies (Santa Cruz, CA), antibodies to p62 and ATG5 from Cell Signaling Technologies (Danvers, MA) and antibody to VP5 from Virusys (Taneytown, MD). Alexafluor 594 conjugated anti-mouse, Alexafluor 488-conjugated anti-rabbit antibody and SlowFade Gold (with DAPI) mounting medium were from Invitrogen. Rapamycin was purchased from Cell Signaling Technologies, the calpain inhibitor PD150606 from Calbiochem (La Jolla, CA) and the pancaspase inhibitor benzyloxcarbonyl-Val-Ala-Asp-fluormethyl ketone (z-VAD-fmk) from Promega (Madison, WI). CQ, PI, low melting point agarose and Tween 20 were from Sigma Aldrich (Carlsbad, CA) and Accutase from Innovative Cell Technologies, Inc (San Diego, CA). All the inhibitors were selected for their target specificity and used at doses recommended by the manufacturer and confirmed in the literature 18,47,49-51.
Anchorage-independent growth
Anchorage independent growth was assessed by both spheroid and soft agar growth assays. For spheroid growth, cells were suspended in serum free DMEM supplemented with 20ng/ml basic fibroblast growth factor (bFGF, R&D Systems) and 20ng/ml epidermal growth factor (EGF, R&D Systems), plated (1×104 /well) onto ultra-low attachment plates (Corning, Corning, NY) and grown at 37°C for 7 days. The assay was performed in triplicate, spheroids (≥ 500μm in diameter) were counted and the results are expressed as No. of spheroids/104 cells ± SD. Soft agar colonies were grown as previously described 59. Briefly, cells (500 or 5000/well) were suspended in 0.3% low melting temperature agarose in 1×DMEM and overlaid onto 0.6% agarose in 1× DMEM in 24-well plates. The solidified agarose-cell mixture was overlaid with 1× DMEM, the plates were incubated at 37°C for 14 days, and colonies (defined as ≥ 50μm diameter) were counted. The assay was performed in triplicate and the results are expressed as No. of colonies/104 cells ± SD.
Virus infection of spheroid cultures
Spheroid cultures were either infected as intact 3-D cultures or after they were dissociated into single cell suspensions by treatment with accutase (37°C, 5 min). To estimate the multiplicity of infection (moi = pfu/cell) for the intact spheroid cultures, duplicate representative cultures were dissociated before cell counting. For all cultures, virus was adsorbed in adsorption medium (PBS with 0.2% glucose and 0.2% bovine serum albumin) for 1 h at 4°C (synchronized adsorption) and removed by centrifugation (800-1500 rpm; 5-10 min) prior to re-incubation at 37°C.
Cell death
Staining with propidium iodide (PI), a cell impermeant red fluorescent nuclear dye used to determine cell death was done as per manufacturer's instructions and visualized by microscopy using a Nikon E4100 fluorescent microscope that utilizes Brightfield and a Texas Red (540-580nm) cube. Stained cells were counted in five randomly selected 3mm2 fields (≥250 cells each), and the % positive cells was calculated relative to total number of cells imaged by phase contrast microscopy23, 24,46.
Immunofluorescence and Flow Cytometry (FCM)
Immunofluorescent staining was as previously described 23,24,46. Briefly, cells were fixed with 4% paraformaldehyde overnight at 4°C, blocked with 5% normal goat serum and 5% BSA (30 min RT) and incubated with primary antibody overnight at 4°C. They were washed in PBS with 0.1% Tween 20, exposed to fluorochrome-labeled secondary antibodies (37°C, 1hr) and mounted in Vectashield with DAPI (Vector Laboratories, Burlingame, CA, USA). Slides were visualized with an Olympus BX50 fluorescence microscope utilizing FITC (330–380 nm), UV (for DAPI) (465–495nm), and Texas red (540–580nm) cubes. Stained cells were counted in five randomly selected 3mm2 fields (≥250 cells each) and the percentage of positive cells was calculated relative to total number of cells imaged by DAPI. FCM was done as previously described 60. Briefly, washed cells were re-suspended (106 cells/tube) in FCM buffer (2% FBS in 1× PBS) and incubated (10 min.) at room temperature (RT) with human Fc-binding inhibitor (eBiosciences; San Diego, CA). This was followed by incubation (20 min. 4°C in the dark) with primary antibodies followed by the corresponding secondary antibodies (20 min., RT) and the washed cells were fixed with 1% paraformaldehyde. Isotype-matched APC, and FITC conjugated antibodies were used as controls. Data were acquired using a LSRII benchtop flow cytometer (BD Biosciences) and analyzed with the FlowJo software package (Tree Star, Ashland, OR, USA).
Immunoblotting
Preparation of protein extracts and immunoblotting were as previously described18,21-24,46. Briefly, cultured cells were lysed with radioimmuno-precipitation buffer [RIPA; 20 mM Tris-HCl (pH 7.4), 0.15 mM NaCl, 1% Nonidet P-40, 0.1% sodium dodecyl sulfate (SDS), 0.5% sodium deoxycholate] supplemented with protease and phosphatase inhibitor cocktails (Sigma-Aldrich) and sonicated twice for 30 seconds at 25% output power with a Sonicator ultrasonic processor (Misonix, Inc., Farmingdale, NY). Protein concentrations were determined by the bicinchoninic assay (Pierce, Rockford, IL) and 100 mg protein samples were resolved by SDS-polyacrylamide gel elecrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride membranes. The membranes were blocked (1hr RT) in 5% nonfat milk in TN-T buffer (0.01 M Tris–HCl pH 7.4, 0.15 M NaCl, 0.05% Tween-20), exposed (1hr) to primary antibodies, washed in TN-T buffer and incubated (1 hr) in HRP-conjugated secondary antibodies. Detection was with ECL reagents (Amersham, Pittsburg, PA) and high performance chemiluminescence film (Hyperfilm ECL, Amersham). Quantitation was by densitometric scanning with the Bio-Rad GS-700 imaging densitometer (Bio-Rad, Hercules, CA) and pixel density was determined using Multi-Analyst software (Bio-Rad, Hercules, CA). Results of three independent experiments are expressed as the mean GAPDH-adjusted densitometric units ± SD.
LC3-GFP transduction
The Premo™ Autophagy Sensor (LC3-GFP) BacMam 2.0 expression system (Invitrogen/Molecular Probes) was used according to the manufacturer's directions. Briefly, A2058 cells were plated onto glass coverslips at ~70% confluency, transduced with the LC3-GFP vector (moi=30) and incubated (24h; 37°C) in complete growth medium.
Statistical Analysis
Analysis of variance (ANOVA) was performed with SigmaStat version 3.1 for Windows (Systat Software, Point Richmond, CA).
Supplementary Material
Acknowledgments
The studies were supported by Public Health Service grant AR053512 from NIAMS, NIH. AC was supported by grant ES07263 from NIEHS, NIH.
Footnotes
Supplementary information is available at Gene Therapy's website
Conflict of Interest Statement:
The authors declare no conflict of interest.
REFERENCES
- 1.Aghi M, Martuza RL. Oncolytic viral therapies - the clinical experience. Oncogene. 2005;24:7802–16. doi: 10.1038/sj.onc.1209037. [DOI] [PubMed] [Google Scholar]
- 2.Wakimoto H, Kesari S, Farrell CJ, Curry WT, Jr, Zaupa C, Aghi M, et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009;69(8):3472–81. doi: 10.1158/0008-5472.CAN-08-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeyaullah M, Patro M, Ahmad I, Ibraheem K, Sultan P, Nehal M, et al. Oncolytic viruses in the treatment of cancer: a review of current strategies. Pathol Oncol Res. 2012;18:771–81. doi: 10.1007/s12253-012-9548-2. [DOI] [PubMed] [Google Scholar]
- 4.Fukunaga-Kalabis M, Roesch A, Herlyn M. From cancer stem cells to tumor maintenance in melanoma. J Invest Dermatol. 2011;131:1600–1604. doi: 10.1038/jid.2011.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quintana E, Shackleton M, Foster HR, Fullen DR, Sabel MS, Johnson TM, et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell. 2010;18:510–23. doi: 10.1016/j.ccr.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10:717–728. doi: 10.1016/j.stem.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 7.Kanai R, Zaupa C, Sgubin D, Antoszczyk SJ, Martuza RL, Wakimoto H, et al. Effect of γ34.5 deletions on oncolytic herpes simplex virus activity in brain tumors. J Virol. 2012;86:4420–4431. doi: 10.1128/JVI.00017-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Todo T. Active immunotherapy: oncolytic virus therapy using HSV-1. Adv Exp Med Biol. 2012;746:178–86. doi: 10.1007/978-1-4614-3146-6_14. [DOI] [PubMed] [Google Scholar]
- 9.Senzer NN, Kaufman HL, Amatruda T, Neumunaitis M, Daniels G, Gonzalez R, et al. Phase II clinical trial of a granulocyte-macrophage Colony- stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J. Clin. Oncol. 2009;27:5763–5771. doi: 10.1200/JCO.2009.24.3675. [DOI] [PubMed] [Google Scholar]
- 10.Cheema TA, Wakimoto H, Fecci PE, Ning J, Kuroda T, Jeyaretna DS, et al. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc Natl Acad Sci U S A. 2013;110:12006–12011. doi: 10.1073/pnas.1307935110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahller YY, Williams IP, Baird WH, et al. Neuroblastoma cell lines contain pluripotent tumor initiating cells that are susceptible to a targeted oncolytic virus. PLoS One. 2009;4:e4235. doi: 10.1371/journal.pone.0004235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pressey JG, Haas MC, Pressey CS, Kelly VM, Parker JN, Gillespie GY, et al. CD133 marks a myogenically primitive subpopulation in rhabdomyosarcoma cell lines that are relatively chemoresistant but sensitive to mutant HSV. Pediatr Blood Cancer. 2013;60:45–52. doi: 10.1002/pbc.24117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hong CS, Fellows W, Niranjan A, Alber S, Watkins S, Cohen JB, et al. Ectopic matrix metalloproteinase-9 expression in human brain tumor cells enhances oncolytic HSV vector infection. Gene Ther. 2010;17:1200–1205. doi: 10.1038/gt.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu G, Su W, Jin G, et al. Glioma stem cells targeted by oncolytic virus carrying endostatin-angiostatin fusion gene and the expression of its exogenous gene in vitro. Brain Res. 2011;1390:59–69. doi: 10.1016/j.brainres.2011.03.050. [DOI] [PubMed] [Google Scholar]
- 15.Dmitrieva N, Yu L, Viapiano M, et al. Chondroitinase ABC I-mediated enhancement of oncolytic virus spread and antitumor efficacy. Clin Cancer Res. 2011;17:1362–1372. doi: 10.1158/1078-0432.CCR-10-2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanai R, Wakimoto H, Martuza RL, Rabkin SD. A novel oncolytic herpes simplex virus that synergizes with phosphoinositide 3-kinase/Akt pathway inhibitors to target glioblastoma stem cells. Clin Cancer Res. 2011;17(11):3686–96. doi: 10.1158/1078-0432.CCR-10-3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhuang X, Zhang W, Chen Y, Han X, Li J, Zhang, et al. Doxorubicin-enriched, ALDH(br) mouse breast cancer stem cells are treatable to oncolytic herpes simplex virus type 1. BMC Cancer. 2012;12:549. doi: 10.1186/1471-2407-12-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colunga AG, Laing JM, Aurelian L. The HSV-2 mutant DeltaPK induces melanoma oncolysis through nonredundant death programs and associated with autophagy and pyroptosis proteins. Gene Ther. 2010;17:315–327. doi: 10.1038/gt.2009.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smalley KS, Haass NK, Brafford PA, Lioni M, Flaherty KT, Herlyn M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol Cancer Ther. 2006;5:1136–1144. doi: 10.1158/1535-7163.MCT-06-0084. [DOI] [PubMed] [Google Scholar]
- 20.Nagano S, Perentes JY, Jain RK, Boucher Y. Cancer cell death enhances the penetration and efficacy of oncolytic herpes simplex virus in tumors. Cancer Res. 2008;68:3795–3802. doi: 10.1158/0008-5472.CAN-07-6193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith CC, Peng T, Kulka M, Aurelian L. The PK domain of the large subunit of herpes simplex virus type 2 ribonucleotide reductase (ICP10) is required for immediate-early gene expression and virus growth. J Virol. 1998;72:9131–9141. doi: 10.1128/jvi.72.11.9131-9141.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith CC, Nelson J, Aurelian L, Gober M, Goswami BB. Ras-GAP binding and phosphorylation by herpes simplex virus type 2 RR1 PK (ICP10) and activation of the Ras/MEK/MAPK mitogenic pathway are required for timely onset of virus growth. J Virol. 2000;74:10417–10429. doi: 10.1128/jvi.74.22.10417-10429.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laing JM, Gober MD, Golembewski EK, Thompson SM, Gyure KA, Yarowsky PJ, Aurelian L. Intranasal administration of the growth-compromised HSV-2 vector DeltaRR prevents kainate-induced seizures and neuronal loss in rats and mice. Mol Ther. 2006;13(5):870–81. doi: 10.1016/j.ymthe.2005.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Golembewski EK, Wales SQ, Aurelian L, Yarowsky PJ. The HSV-2 protein ICP10PK prevents neuronal apoptosis and loss of function in an in vivo model of neurodegeneration associated with glutamate excitotoxicity. Exp Neurol. 2007;203(2):381–93. doi: 10.1016/j.expneurol.2006.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aurelian L. Herpes simplex virus type 2 vaccines: new ground for optimism? Clin Diagn Lab Immunol. 2004;11:437–445. doi: 10.1128/CDLI.11.3.437-445.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rausch V, Liu L, Apel A, Rettig T, Gladkich J, Labsch S, et al. Autophagy mediates survival of pancreatic tumour-initiating cells in a hypoxic microenvironment. J Pathol. 2012;227(3):325–35. doi: 10.1002/path.3994. [DOI] [PubMed] [Google Scholar]
- 27.Dufau I, Frongia C, Sicard F, Dedieu L, Cordelier P, Ausseil F, et al. Multicellular tumor spheroid model to evaluate spatio-temporal dynamics effect of chemotherapeutics: application to the gemcitabine/CHK1 inhibitor combination in pancreatic cancer. BMC Cancer. 2012;12:15. doi: 10.1186/1471-2407-12-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ludwig K, Tse ES, Wang JY. Colon cancer cells adopt an invasive phenotype without mesenchymal transition in 3-D but not 2-D culture upon combined stimulation with EGF and crypt growth factors. BMC Cancer. 2013;13(1):221. doi: 10.1186/1471-2407-13-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loessner D, Flegg JA, Byrne HM, Clements JA, Hutmacher DW. Growth of confined cancer spheroids: a combined experimental and mathematical modelling approach. Integr Biol (Camb) 2013;5(3):597–605. doi: 10.1039/c3ib20252f. [DOI] [PubMed] [Google Scholar]
- 30.Grimshaw MJ, Cooper L, Papazisis K, Coleman JA, Bohnenkamp HR, Chiapero-Stanke L, et al. Mammosphere culture of metastatic breast cancer cells enriches for tumorigenic breast cancer cells. Breast Cancer Res. 2008;10:R52. doi: 10.1186/bcr2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140:62–73. doi: 10.1016/j.cell.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 32.Perego M, Tortoreto M, Tragni G, Mariani L, Deho P, Carbone A, et al. Heterogeneous phenotype of human melanoma cells with in vitro and in vivo features of tumor-initiating cells. J Invest Dermatol. 2010;130:1877–1886. doi: 10.1038/jid.2010.69. [DOI] [PubMed] [Google Scholar]
- 33.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Valyi-Nagy K, Dosa S, Kovacs SK, Bacsa S, Voros A, Shukla D, et al. Identification of virus resistant tumor cell subpopulations in three-dimensional uveal melanoma cultures. Cancer Gene Ther. 2010;17:223–234. doi: 10.1038/cgt.2009.73. [DOI] [PubMed] [Google Scholar]
- 35.Perkins D, Pereira EF, Gober M, Yarowsky PJ, Aurelian L. The herpes simplex virus type 2 R1 protein kinase (ICP10 PK) blocks apoptosis in hippocampal neurons, involving activation of the MEK/MAPK survival pathway. J Virol. 2002;76(3):1435–39. doi: 10.1128/JVI.76.3.1435-1449.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoon YH, Cho KS, Hwang JJ, Lee SJ, Choi JA, Koh JY. Induction of lysosomal dilation, arrested autophagy, and cell death by chloroquine in cultured ARPE-19 cells. Invest Opthalmol Vis Sci. 2010;51(11):6030–7. doi: 10.1167/iovs.10-5278. [DOI] [PubMed] [Google Scholar]
- 37.Egger ME, Huang JS, Yin W, McMasters KM, McNally LR. Inhibition of autophagy with chloroquine is effective in melanoma. J Surg Res. 2013;184(1):274–81. doi: 10.1016/j.jss.2013.04.055. [DOI] [PubMed] [Google Scholar]
- 38.Mehrpour M, Esclatine A, Beau I, Codogno P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010;20:748–762. doi: 10.1038/cr.2010.82. [DOI] [PubMed] [Google Scholar]
- 39.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pyo JO, Yoo SM, Ahn HH, Nah J, Hong SH, Kam TI, et al. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun. 2013;4:2300. doi: 10.1038/ncomms3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Komatsu M, Kageyama S, Ichimura Y. p62/SQSTM1/A170: physiology and pathology. Pharmacol Res. 2012 Dec;66(6):457–62. doi: 10.1016/j.phrs.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 42.Moscat J, Diaz-Meco MT. p62: a versatile multitasker takes on cancer. Trends Biochem Sci. 2012;37(6):230–6. doi: 10.1016/j.tibs.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parkhitko A, Myachina F, Morrison TA, Hindi KM, Auricchio N, Karbowniczek M, et al. Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)-dependent. Proc Natl Acad Sci U S A. 2011;108(30):12455–60. doi: 10.1073/pnas.1104361108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Inoue D, Suzuki T, Mitsuishi Y, Miki Y, Suzuki S, Sugawara S, et al. Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci. 2012;103:760–766. doi: 10.1111/j.1349-7006.2012.02216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rolland P, Madjd Z, Durrant L, Ellis IO, Layfield R, Spendlove I. The ubiquitin-binding protein p62 is expressed in breast cancers showing features of aggressive disease. Endocr Relat Cancer. 2007;14:73–80. doi: 10.1677/erc.1.01312. [DOI] [PubMed] [Google Scholar]
- 46.Wales SQ, Laing JM, Chen L, Aurelian L. ICP10PK inhibits calpain-dependent release of apoptosis-inducing factor and programmed cell death in response to the toxin MPP+. Gene Ther. 2008;15:1397–1409. doi: 10.1038/gt.2008.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP, et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature. 2010;466:133–137. doi: 10.1038/nature09161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jaggupilli A, Elkord E. Significance of CD44 and CD24 as Cancer Stem Cell Markers: An Enduring Ambiguity. Clin Dev Immunol. 2012:708036. doi: 10.1155/2012/708036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ali MA, Stepanko A, Fan X, Holt A, Schulz R. Calpain inhibitors exhibit matrix metalloproteinase-2 inhibitory activity. Biochem Biophys Res Commun. 2012;423(1):1–5. doi: 10.1016/j.bbrc.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 50.Darnell GA, Schroder WA, Antalis TM, Lambley E, Major L, Gardner J, et al. Human papillomavirus E7 requires the protease calpain to degrade the retinoblastoma protein. J Biol Chem. 2007;282:37492–500. doi: 10.1074/jbc.M706860200. [DOI] [PubMed] [Google Scholar]
- 51.Storr SJ, Carragher NO, Frame MC, Parr T, Martin SG. The calpain system and cancer. Nat Rev Cancer. 2011;11(5):364–74. doi: 10.1038/nrc3050. [DOI] [PubMed] [Google Scholar]
- 52.Kim JW, Ho WJ, Wu BM. The role of the 3D environment in hypoxia-induced drug and apoptosis resistance. Anticancer Res. 2011;31:3237–3245. [PubMed] [Google Scholar]
- 53.Gong C, Bauvy C, Tonelli G, Yue W, Deloménie C, Nicolas V, et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene. 2012;32(18):2261–72. doi: 10.1038/onc.2012.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clarke PG, Puyal J. Autophagic cell death exists. Autophagy. 2012;8:867–869. doi: 10.4161/auto.20380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosenfeldt MT, Ryan KM. The multiple roles of autophagy in cancer. Carcinogenesis. 2011;32(7):955–63. doi: 10.1093/carcin/bgr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu H, He Z, Simon HU. Targeting autophagy as a potential therapeutic approach for melanoma therapy. Semin Cancer Biol. 2013;23(5):352–60. doi: 10.1016/j.semcancer.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 57.Lakhter AJ, Sahu RP, Sun Y, Kaufmann WK, Androphy EJ, Travers JB, et al. Chloroquine promotes apoptosis in melanoma cells by inhibiting BH3 domain-mediated PUMA degradation. J Invest Dermatol. 2013;133(9):2247–54. doi: 10.1038/jid.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13(4):343–54. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 59.Jariwalla RJ, Aurelian L, Ts'o PO. Immortalization and neoplastic transformation of normal diploid cells by defined cloned DNA fragments of herpes simplex virus type 2. Proc Natl Acad Sci U S A. 1983;19:5902–6. doi: 10.1073/pnas.80.19.5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ono F, Sharma BK, Smith CC, Burnett JW, Aurelian L. CD34+ cells in the peripheral blood transport herpes simplex virus DNA fragments to the skin of patients with erythema multiforme (HAEM). J Invest Dermatol. 2005;124(6):1215–24. doi: 10.1111/j.0022-202X.2005.23712.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.