

Abstract
Purpose
To determine how often loss of ATM protein expression occurs in primary pancreatic ductal adenocarcinomas and to determine its prognostic significance.
Experimental Design
The expression of ATM and TP53 was determined by immunohistochemistry in 397 surgically-resected pancreatic ductal adenocarcinomas (Hopkins), a second set of 159 cases (Emory) and 21 cancers after neoadjuvant chemoradiotherapy. Expression was correlated with the clinicopathologic parameters, including survival.
Results
Tumoral ATM loss was observed in one cancer known to have bi-allelic inactivation of ATM and 50 of the first 396 (12.8%) cases, significantly more often in patients with a family history of pancreatic cancer (12/49; 24.5%) than in those without (38/347; 11.0%) (p=0.019). In the Hopkins series, ATM loss was associated with a significantly decreased overall survival in patients whose cancers had normal TP53 expression (p=0.019) and was a significant independent predictor of decreased overall survival (p=0.014). Seventeen (10.7%) of 159 Emory cases had tumoral ATM loss and tumoral ATM loss/normal TP53 was associated with poorer overall survival (p=0.1). Multivariate analysis of the combined Hopkins/Emory cases found tumoral ATM loss/normal TP53 was an independent predictor of decreased overall survival (HR 2.61, CI1.27–5.37, p=0.009). Of 21 cancers examined after neoadjuvant chemoradiotherapy one had tumoral loss of ATM; it had no histological evidence of tumor response.
Conclusions
Tumoral loss of ATM protein was detected more often in patients with a family history of pancreatic cancer than in those without. Patients whose pancreatic cancers had loss of ATM but normal TP53 had worse overall survival after pancreatic resection.
Keywords: pancreatic cancer, ATM, p53
INTRODUCTION
Pancreatic ductal adenocarcinoma is the fourth leading cause of cancer-related mortality in the United States (1). Pancreatic cancers are rarely detected at an early stage, with ~80% of cases being diagnosed after regional invasion or distant metastasis, and pancreatic cancer-related mortality remains poor (1). Even among patients who present with surgically resectable disease, at least 80% of these patients will subsequently develop local recurrences or distant metastases within 2 years of surgery (2). Mortality from pancreatic cancer requires better early detection strategies and a better understanding of the molecular alterations that predict outcome. Efforts to improve the early detection of pancreatic cancer have relied on identifying individuals with an inherited susceptibility to pancreatic cancer and to evaluate the utility of screening these individuals to identify pancreatic cancer precursors and early invasive pancreatic cancers that could be cured with appropriate therapeutic intervention (3, 4).The genetic alterations responsible for the familial aggregation of pancreatic cancer are still under investigation. The genes with germline mutations that have been identified as being associated with an increased pancreatic cancer susceptibility include BRCA2, PALB2, p16, STK11, PRSS1 and ATM (5). BRCA2 has been the gene most commonly identified as predisposing to familial pancreatic cancer when mutated in the germline (5), with germline mutations in PALB2, ATM, p16, PRSS1 and STK11 together accounting for less than 10% of the familial clustering of pancreatic cancer (6–8).
The ataxia-telangiectasia-mutated (ATM) gene is a tumor suppressor gene of approximately 150kb-length which is located on 11q22–23. It encodes the ATM protein, which upon activation phosphorylates downstream targets involved in DNA damage repair and cell cycle checkpoint activation, such as p53 and Chk2, in response to DNA damage and thus maintains genomic integrity (9–11). The population prevalence of germline ATM gene mutation carriers has been estimated to be approximately 0.5% to 1% (10, 12), and germline biallelic inactivation of the ATM gene results in ataxia-telangiectasia, a rare autosomal recessive disease which is characterized by various clinical manifestations including cerebellar ataxia, oculo-cutaneous telangiectasia, immunodeficiency, radiosensitivity and increased risk of malignancies, especially hematolymphoid malignancies (12).
Germline mutations in the ATM gene have also been associated with increased risks of certain solid cancers, particularly familial breast cancers that do not harbor BRCA mutations (13). Recently, deleterious ATM mutations have been recognized in the germline of families with familial pancreatic cancer, suggesting that ATM is a susceptibility gene in pancreatic cancer (14). Truncating somatic ATM mutations in pancreatic ductal adenocarcinomas have also been recently demonstrated (15).
In this study, we sought to identify the prevalence of alterations of ATM protein expression in pancreatic ductal adenocarcinomas, including both sporadic and familial cancers, and to analyze the prognostic significance of ATM protein expression status in these tumors.
MATERIALS AND METHODS
Case selection and tissue microarray construction
We reviewed the clinicopathological records of three hundred and ninety-seven patients who had a pancreatic cancer resected at Johns Hopkins Medical Institutions between January 1998 and June 2006. These patients were included because we had sufficient formalin-fixed paraffin-embedded pancreatic ductal adenocarcinoma tissues available for ATM immunohistochemical analysis. The clinicopathologic findings of each case were carefully reviewed from the regularly updated clinical database and the hematoxylin-eosin slides, including tumor size, histologic differentiation, presence of vascular or perineural invasion, tumor stage, the presence of a family history of pancreatic cancer, treatment modality and follow up data. Vascular invasion was defined by the presence of neoplastic cells inside the lumen of vascular structures containing smooth muscle layers, in the peritumoral pancreas, intra-tumoral tissue, or in fibroadipose tissues. Only conventional ductal adenocarcinomas were enrolled in the study; variants such as adenosquamous carcinomas, medullary or undifferentiated carcinomas were excluded. Pathologic tumor staging was performed according to the TNM classification of the American Joint Committee on Cancer (AJCC) Staging Manual, 7th edition. The time of relapse (local recurrence or distant metastasis) and time of death were obtained from the Johns Hopkins Pathology Database, patient records and radiology reports. This study was conducted under the approval of the Johns Hopkins Hospital institutional review board.
Tissue microarrays (TMAs) were constructed from archival formalin-fixed paraffin-embedded tissue blocks as previously described (16). Four 1.5 mm-sized cores were punched from each patient’s tumor and non-neoplastic pancreatic tissue donor blocks and harvested into recipient blocks. These TMAs were designed to obtain uniform immunohistochemical labeling of pancreatic tissues and limit intra-assay variation, and normal tissues from various other organs, including tonsil, colon, testis and liver, were included in each TMA for quality control.
To obtain an independent estimate of the prevalence of tumoral ATM loss in primary pancreatic ductal adenocarcinomas and further examine its prognostic significance, we performed immunohistochemical analysis of ATM and TP53 on tissue microarrays from an additional 159 surgically resected pancreatic cancer cases treated at Emory University Hospital between 1978 and 2012 and examined clinicopathological associations as described above.
Immunohistochemistry
Immunohistochemical labeling was performed with ATM rabbit monoclonal antibody (1:50, ab32420, Abcam, Cambridge, MA) and TP53 mouse monoclonal antibody (ready-to-use, clone DO-7, DakoCytomation, Glostrup, Denmark). Briefly, 4μm-thick sections from the TMA paraffin blocks were deparaffinized in xylene, rehydrated in graded alcohol and quenched in 3% H2O2. Antigen retrieval was performed in Tris-HCl buffer (pH 7.5) (ATM) or citrate buffer (pH 6.0) (TP53) in a steam pressure cooker for 15 minutes and the TMA slides were incubated with primary antibodies in a humidified chamber for 1 hour at room temperature. After rinsing, the slides were incubated with a secondary antibody (PowerVision poly-HRP anti-rabbit IgG, Leica Biosystems, Newcastle-upon-Tyne, UK) for 30 minutes. 3,3-diaminobenzidine was used as a chromogen, and the sections were counterstained with hematoxylin. Some tissues did not immunolabel successfully due to loss of tissue cores during the sectioning or labeling process. The immunohistochemical scoring was performed blinded to any other patient data including outcome. Nuclear labeling with the ATM antibody was regarded as positive, and the percentage of immunolabeled nuclei were calculated for each TMA core. “Loss of ATM expression” was defined as labeling of less than 10% of neoplastic cell nuclei. “Abnormal TP53 expression” was defined as either positive labeling in 50% or more of tumor cell nuclei or total absence of nuclear labeling as described previously (17). To examine the specificity of the ATM antibody, we obtained primary pancreatic cancer tissue from a patient known to have a germline mutation in ATM and loss of the wild-type ATM allele in their cancer (14).
ATM Mutation Analysis
In order to identify germline mutations in the ATM gene, Sanger sequencing was performed as previously described using matched non-neoplastic duodenal or pancreatic tissues from cases with loss of ATM protein expression (18). Sixty-three primer sets which covered 62 exons of ATM were used for sequencing. Information regarding the ATM primer sequence is summarized in Supplementary Table 1. As a positive control, one case with a previously described germline ATM mutation was included in the sequencing analyses (14).
ATM and TP53 Expression Status in Post-neoadjuvant Treatment Pancreatic Cancers
Twenty-two additional pancreatic cancer cases which had been treated by chemoradiotherapy prior to surgical resection at Johns Hopkins Medical Institutions were analyzed for ATM and TP53 expression by immunohistochemistry. Adequate labeling could not be obtained for one of these cases. Tumor response to chemoradiotherapy was evaluated histologically, focusing on the extent of fibrosis, necrosis, tumor cellularity, and the presence of cytopathic effects (19).
Statistical Analysis
Data were analyzed using SPSS software, version 19.0 (SPSS Inc, Chicago, IL, USA) and Stata, version 11 (StataCorp, College Station, TX). The differences in the continuous and categorical clinicopathologic variables according to ATM expression status were analyzed using the student t-test, chi-square test and Fisher-exact test, as deemed appropriate. At the time we initiated the study we did not have much data to refer to regarding the prevalence of ATM loss in pancreatic cancer or its prognostic significance, so we estimated our study power assuming ATM had a prevalence of 11% and a similar prognostic magnitude of other genes inactivated in pancreatic cancer such as SMAD4 (20–22). We estimated our power to detect a survival difference with our sample size. Assuming an overall median survival of ~21 months in our study population, with 396 cases in the Hopkins series of which 44 had ATM loss, we would have a 71% probability of detecting a difference in survival with ATM expressing vs. non-expressing cases assuming a survival of 24 months in ATM-expressing cases and 16 months in ATM loss cases with a standard Type I error of 0.05.
The overall survival, local recurrence-free survival and distant metastasis-free survival were analyzed for patients with pancreatic adenocarcinoma; overall survival was defined as the time from primary surgical resection to death or the date of last follow-up and local recurrence-free survival or distant metastasis-free survival as the time from primary resection to first local recurrence or distant metastasis, respectively, or the date of last follow-up. Survival rates were calculated using the Kaplan-Meier method and statistical significance was evaluated using the log-rank test. The clinical charts, imaging study reports and the pathology database were carefully reviewed in order to identify patients with relapse. Clinicopathological variables with p<0.1 on univariate analysis were checked for the proportional hazards assumption by examining the Schoenfeld residuals, and the variables satisfying the proportional hazards assumption were included in the multivariate Cox regression analysis. Univariate and multivariate analysis was performed for each site and then for the overall study population. The combined multivariate analysis also controlled for institution. In the Emory series, seven patients were lost to follow-up, and regarded as censored from the overall survival analysis. For the relapse-free survival analysis of Hopkins cases, only patients with complete follow-up data at the time of analysis were included (n=345). Treatment periods were defined as 5-year intervals as follows: before 1985, 1986~1990, 1991~1995, 1996~2000, 2001~2005, 2006~2010, and 2011 and later. Statistical significance was defined as p<0.05.
RESULTS
Clinicopathologic correlation and survival analysis (Hopkins series)
The clinicopathologic features of the 397 patients and their pancreatic ductal adenocarcinomas from Johns Hopkins Hospital who underwent pancreatic resection without prior therapy are summarized in Table 1.
Table 1.
Summary of clinicopathologic characteristics of 556 pancreatic ductal adenocarcinomas from two institutions*
| Clinicopathologic variable | Hopkins (n=397) | Emory (n=159) |
|---|---|---|
| Age (years; mean±s.d.) | 66.4±10.8 | 64.8±11.4 |
| ≥65 years | 230 (57.9) | 86 (55.1) |
| Gender | ||
| Male | 209 (52.6) | 67 (42.7) |
| Female | 188 (47.4) | 90 (57.3) |
| T stage | ||
| T1 | 9 (2.3) | 12 (10.2) |
| T2 | 12 (3.0) | 3 (1.7) |
| T3 | 358 (90.2) | 101 (85.6) |
| T4 | 15 (3.8) | 2 (1.7) |
| N stage | ||
| N0 | 60 (15.1) | 40 (25.8) |
| N1 | 337 (84.9) | 115 (74.2) |
| Tumor size (cm; mean±s.d.) | 3.2±1.4 | 3.5±1.6 |
| Differentiation | ||
| Well differentiated | 12 (3.0) | 28 (17.8)) |
| Moderately differentiated | 214 (53.9) | 89 (56.7) |
| Poorly differentiated | 169 (42.6) | 40 (25.5) |
| Vascular invasion | 192 (48.4) | 77 (77.0) |
| Perineural invasion | 372 (93.7) | 138 (95.2) |
| Positive surgical resection margin | 133 (33.6) | 44 (28.8) |
| Follow up [months; median (range)] | 13.7 (0.1–165.8) | 12.8 (0–114.0) |
| Death | 338 (85.1) | 112 (71.3) |
| Local recurrence | 35 (10.2) | |
| Distant metastasis | 88 (25.5) | |
Number of cases (%), unless otherwise specified; s.d. standard deviation
In non-neoplastic pancreata, the antibody to ATM labeled the normal nuclei of ductal and acinar cells (Figure 1). Complete loss of tumoral ATM expression was evident in one pancreatic cancer ascertained from a patient known to carry a deleterious germline ATM mutation and loss of the wild-type ATM allele in their cancer (14). ATM loss was observed in 50 out of 396 (12.8%) remaining pancreatic cancers. When the sporadic (n=347) and familial (n=49), pancreatic cancers were analyzed separately (excluding the positive control case with a known germline ATM mutation), ATM loss was observed significantly more frequently in familial cases (12/49; 24.5%, 95% confidence interval: 14.6–38.1%) compared to sporadic cases (38/347; 11.0%, 95% C.I. 8.8–14.7%) (p=0.019).
Figure 1.
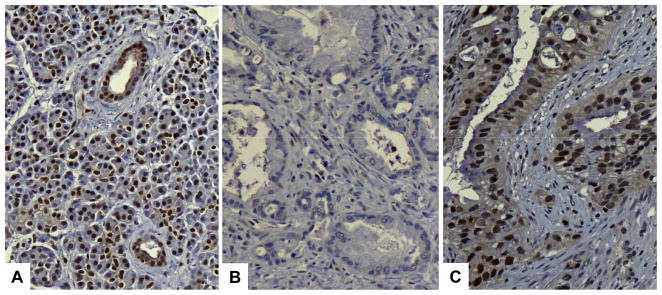
Immunohistochemical labeling for ATM protein. Nuclear ATM expression is seen in most nuclei of ducts and acini in the normal pancreas (A). Pancreatic ductal adenocarcinoma with loss of ATM expression (immunolabeling in less than 10% of tumor cells) (B). Pancreatic ductal adenocarcinoma with nuclear ATM expression (“intact ATM”) (C).
Sanger sequencing was performed using matched non-neoplastic duodenal or pancreatic tissues from 26 cases with loss of ATM expression. Of these 26 cases, 6 cases were associated with a family history of pancreatic cancer (including the positive control case). No deleterious germline ATM mutations were found apart from the positive control case (c.8266A>AT; p.K2756X), which was reported previously (14).
There were no significant associations between loss of ATM expression and other clinicopathologic variables, including age, tumor size, perineural invasion and histologic differentiation. Pancreatic cancers with ATM loss tended to have more vascular invasion (63.3%) and lymph node metastasis (92.2%) compared with cases without ATM loss (49.4% and 84.1%, respectively) (p=0.070 and p=0.129, respectively).
Clinicopathological factors associated with decreased overall survival included older age at diagnosis (>67 years, p=0.030), higher T stage (p=0.052), lymph node metastasis (p=0.001), presence of vascular invasion (p=0.071), poor differentiation (p<0.001), larger tumor size (>3cm, p=0.005) and positive surgical resection margins (p<0.001). Among patients with available data on their metastasis-free survival patients whose pancreatic cancers had ATM loss (p=0.004), poor differentiation (p=0.022) and those who had lymph node metastasis in their resection specimen (p=0.035) demonstrated significantly reduced distant metastasis-free survival by univariate analysis. After multivariate analysis, ATM loss remained the only significant independent predictor of decreased distant metastasis-free survival (HR=1.90, 95% C.I., 1.08–3.33, p=0.025). Lymph node metastasis (p=0.003), poor differentiation (p=0.015) and positive resection margins (p<0.001) were independent predictors for poor overall survival (Table 2). There was no significant difference in overall survival or local recurrence-free survival according to ATM expression status. Since TP53 status is known to influence cellular responses to ATM deficiency we evaluated ATM stratified by TP53 status.
Table 2.
Significant independent predictors of decreased overall and distant metastasis-free survival on multivariate analysis
| p-value | Hazard ratio (95% C.I.) | |
|---|---|---|
| Overall survival (n=397) | ||
| Lymph node metastasis | p=0.003 | 1.70 (1.19–2.44) |
| Differentiation (WD/MD vs. PD) | p=0.015 | 2.49 (1.20–5.19) |
| Positive surgical resection margin | p<0.001 | 1.75 (1.37–2.24) |
| No abnormal TP53 and ATM loss | p=0.014 | 2.63 (1.22–5.67) |
| Distant metastasis-free survival (n=345) | ||
| ATM loss | p=0.025 | 1.90 (1.08–3.33) |
WD: well-differentiated, MD: moderately differentiated, PD: poorly differentiated
Patient outcome by ATM and TP53 expression status
Abnormal TP53 expression was seen in 293 (73.8%) of the Hopkins cases. There was no difference in the prevalence of ATM expression loss in pancreatic cancers by TP53 protein expression status (42/293, 14.3%) in pancreatic cancers with abnormal TP53 expression compared to (9/104, 8.7%) with normal TP53 expression (p=0.172). ATM loss was associated with a significantly decreased overall survival in patients whose pancreatic adenocarcinomas had normal TP53 expression (p=0.019), but not in patients whose pancreatic cancers had abnormal TP53 expression (Table 3, Figure 2). Nine cases which demonstrated both ATM loss and normal TP53 expression had a significantly reduced overall survival (p=0.010) compared to the other 388 pancreatic cancers, and after multivariate analysis, combined ATM loss and normal TP53 expression remained a significant independent predictor of decreased overall survival (HR=2.63, 95% C.I., 1.22–5.67, p=0.014).
Table 3.
Overall survival in pancreatic cancer patients according to TP53 and ATM expression status (Hopkins series)
| Median survival (months) | 95% C.I. (months) | p-value | |
|---|---|---|---|
| Abnormal TP53 expression (n=293) | p=0.762 | ||
| Intact ATM | 15.4 | 12.8–18.0. | |
| ATM loss | 15.4 | 10.8–20.1 | |
| No abnormal TP53 expression (n=104) | p=0.019 | ||
| Intact ATM | 16.6 | 11.9–21.3 | |
| ATM loss | 9.8 | 4.8–14.7 |
Figure 2.
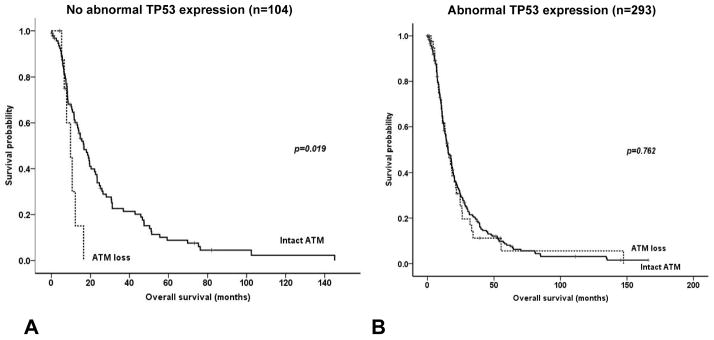
Kaplan-Meier survival curves according to ATM expression status in pancreatic ductal adenocarcinomas with and without abnormal TP53 expression. ATM loss is associated with a significantly decreased overall survival in tumors without abnormal TP53 expression (A), while there is no survival difference according to ATM expression status in pancreatic cancers with abnormal TP53 expression (B).
Clinicopathologic correlation and survival analysis (Emory series)
We evaluated the pancreatic cancers of 159 patients who underwent pancreatic resection at Emory University Hospital. The clinicopathologic characteristics are summarized in Table 1. Loss of ATM expression was demonstrated in 17 out of 159 (10.7%) cases, and was significantly associated with decreased overall survival on univariate analysis (p=0.005). There were no significant relationships between ATM or TP53 expression status and other clinicopathologic variables, such as patient age, gender, tumor size, vascular invasion, perineural invasion, histologic differentiation, or lymph node metastasis (family history information was not available). Abnormal TP53 expression was seen in 107/159 (67.3%) cases. There was no significant difference in the prevalence of ATM loss according to TP53 expression status and there were no significant differences in overall survival according to TP53 expression status. Survival data was available for 152 of the 159 pancreatic cancers. A trend towards decreased overall survival was noted for Emory patients with pancreatic cancers having both ATM loss and normal TP53 expression (n=5) compared to other pancreatic cancers (n=147) (p=0.10). Perineural invasion (p=0.010), lymphatic invasion (p=0.006) and the presence of lymph node metastasis (p=0.046) were also associated with decreased overall survival. After multivariate analysis ATM loss remained the only significant independent predictor of decreased overall survival (HR=3.199, 95% C.I., 1.341–7.627, p=0.009).
Combined analysis of the Hopkins/Emory series
Tumoral ATM loss was detected in 67/555 cases (12%). Clinicopathological data and tumoral ATM and TP53 analysis were examined in the 549 patients with information on overall survival. This analysis was performed just as for the prior analysis but we also included treating institution and treatment period (by 5-year interval) in the model. Factors that were significant in the univariate model and included in the multivariate model included patient age, tumor size (>3cm), T stage, margin status, lymph node status, differentiation (well/moderate vs. poor), tumoral ATM loss, tumoral ATM loss with normal TP53, perineural and vascular invasion. Since ATM loss cases with normal TP53 were a subset of cases with tumoral ATM loss, we ran a separate analysis for these two predictors. In the multivariate models, tumoral ATM loss was not an independent predictor of outcome, but tumoral ATM loss with normal TP53 was an independent predictor of poorer overall survival (HR=2.61, C.I., 1.27–5.37, p=0.009). Other statistically significant factors associated with poorer overall survival included age >65 years, positive tumor margin, poor tumor differentiation, positive lymph nodes, as well as institution and treatment period (survival improved in recent years compared to earlier years) (Table 4).
Table 4.
Joint analysis of Hopkins/Emory cases for significant independent predictors of decreased overall survival on multivariate analysis
| p-value | Hazard ratio (95% C.I.) | |
|---|---|---|
| Overall survival (n=549) | ||
| Tumoral ATM loss without abnormal TP53 | p=0.009 | 2.61 (1.27–5.37) |
| Poor Differentiation (WD/MD vs. PD) | p<0.001 | 1.81 (0.99–3.29) |
| Positive surgical resection margin | p<0.001 | 1.55 (1.23–1.94) |
| Lymph node metastasis | p<0.001 | 1.97 (1.41–2.76) |
| Age (≥65 years) | p=0.024 | 1.28 (1.03–1.59) |
| Treating institution | p=0.042 | 0.62 (0.40–0.98) |
| Treatment period | p=0.003 | 0.06 (0.01–0.44) |
WD: well-differentiated, MD: moderately differentiated, PD: poorly differentiated
Tumoral ATM and TP53 expression in patients treated with neoadjuvant therapy
Of 22 resected pancreatic ductal adenocarcinoma tissues available for analysis ATM immunohistochemistry was evaluable for 21 cases. One of these 21 cases (4.8%) had loss of tumoral ATM expression in the resected specimen. This case also had abnormal TP53 expression. Interestingly, there was no evidence of histologic response to treatment in this case: tumor cellularity was abundant and there was minimal fibrosis or necrosis.
DISCUSSION
In this study we found loss of tumoral ATM expression in 12.6% of our cohort of resected pancreatic cancers indicating that ATM frequently undergoes bi-allelic inactivation during pancreatic tumor development (14). Our results are consistent with recent genetic evidence that ATM functions as a tumor suppressor gene in pancreatic ductal adenocarcinomas. The International Cancer Genome Consortium (ICGC) pancreatic cancer exome project identified somatic mutations in ATM in ~5% of sporadic pancreatic cancers (15), similar to our finding of loss of ATM expression in 11% of sporadic pancreatic cancers in the Hopkins series and in the Emory series. We also found that loss of tumoral ATM expression was significantly more common in pancreatic cancers from individuals with a family history of pancreatic cancer. Notably, we excluded any cases with known germline or somatic ATM mutations from our estimates of the prevalence of ATM loss in pancreatic cancers. Although we examined the germline DNA of patients with loss of tumoral ATM expression we did not identify new deleterious germline mutations, perhaps because the majority of these patients had sporadic pancreatic cancer. It should be noted that we did not have germline DNA available from all cases with tumoral loss of ATM expression so we cannot exclude the possibility that some patients in our series had a germline ATM mutation.
In our prior report, we found germline ATM mutations in only ~2.4% of patients with familial pancreatic cancer and these familial cases were enriched for those with more than 2 affected first-degree relatives (14). This low prevalence of germline ATM mutations even in patients with familial pancreatic cancer likely means that the tumoral loss of ATM in both familial and sporadic pancreatic cancers arises in most cases as a somatic event. Somatic ATM inactivation is generally by intragenic mutation according to the literature, although other mechanisms are possible (23). Recent studies have identified other mechanisms of ATM downregulation via microRNAs and the mTOR pathway (24). In most cases, ATM mutations are nonsense mutations, giving rise to truncated ATM protein, although other types of mutations have been reported, including missense mutations or in-frame deletions producing catalytically inactive ATM proteins (9). The higher prevalence of tumoral ATM loss in cases with a family history indicates that there may be differences in the prevalence of cancer-associated mutations in familial compared to sporadic pancreatic cancers. We know from initial studies that the same genes are mutated in pancreatic cancers from familial and sporadic cases (25), but more data regarding the mutational spectrum of familial vs. sporadic pancreatic cancers is needed, not only to confirm if ATM inactivation is more common in familial pancreatic cancers but also if there are differences in the activation or inactivation of other major driver genes of pancreatic cancer. Patients with familial pancreatic cancers may not only differ from sporadic cases with respect to inherited susceptibility, they may differ in the prevalence of other factors that contribute to pancreatic cancer development (e.g. smoking)(26), factors that can influence the pattern of genes and pathways targeted for mutation in a cancer.
The observation that biallelic inactivation of ATM is not unusual in pancreatic cancers could have important therapeutic implications. ATM is a critical member of the DNA repair pathway with complex protein-protein interactions facilitating recognition of double-stranded DNA breaks, recruitment of DNA repair proteins and activation of cell cycle check points to enable repair to occur. ATM activation involves interaction with the MRE11-RAD50-NBS1 complex to trigger downstream cell cycle check point activation through phosphorylation of Chk2 and other important substrates including p53 (27). ATM is required to repair DNA after treatment with poly (ADP-ribose) polymerase inhibition (PARP)(28, 29) and ATM inactivation is thought to predispose cancers to PARP inhibition (30). Cancers with inactivation of BRCA2 are also characterized by a remarkable sensitivity to PARP inhibition (31) and there is evidence that PALB2 inactivation renders cancers similarly sensitive (32). In contrast, cells lacking intact members of the Fanconi anemia complex upstream of PALB2 and BRCA2 rely on ATM for cell viability indicating the complementary nature of these pathways (33).
It is not known if pancreatic cancers with loss of ATM expression are more sensitive to radiotherapy or certain chemotherapies. In other cancer types, the chemosensitivity of ATM deficient cells depends on p53 status. In a recent study by Jiang et al., suppression of ATM or its downstream target Chk2 in the presence of functional (wild-type) p53 promoted genotoxic drug resistance of the tumor cells by preventing efficient execution of a p53-dependent apoptotic response, while ATM suppression sensitized the tumors to such chemotherapy in the absence of wild-type p53 (34). They also found that decreased or absent ATM immunolabeling in the presence of wild-type p53 levels correlated with a significantly decreased overall survival rate in breast cancers compared to those with intact ATM expression consistent with our results. Further investigation is needed to determine if pancreatic cancers with ATM loss and p53 inactivation are sensitive to chemotherapies that induce DNA strand breaks.
In summary, loss of ATM protein expression was observed in 12% of primary pancreatic ductal adenocarcinomas, was more common in patients with a family history of pancreatic cancer than in those without, and is not generally associated with germline mutations in the ATM gene. Loss of tumoral ATM expression is significantly associated with poorer overall survival among patients whose tumors retained normal p53 protein status.
Supplementary Material
Statement of Translational Relevance.
Pancreatic cancer is the fourth leading cause of cancer death in the United States and is characterized by resistance to most therapeutic treatments. Identifying molecular subsets is an important step towards identifying more effective therapies for this disease. The gene ataxia-telangiectasia-mutated (ATM) has been recently identified as a pancreatic cancer susceptibility gene and to undergo somatic mutation in some pancreatic ductal adenocarcinomas. Tumoral inactivation of ATM influences chemosensitivity responses. Herein, we analyzed resection specimens from almost 400 patients with pancreatic cancer for ATM protein expression and found that loss of ATM protein expression occurs in over 10% of cases; significantly more often in patients with a family history of pancreatic cancer, and it portended a poor prognosis among patients whose tumors retained TP53 expression. The relatively high prevalence and prognostic significance of ATM loss suggests tumoral ATM testing of pancreatic cancers should be considered for patients on clinical trials.
Acknowledgments
Dr. Goggins is supported by the National Cancer Institute grants (CA62924, RC2148346), the family of Susan Wojcicki and Dennis Troper, the Alan Graff Memorial Foundation and the Michael Rolfe Foundation.
Footnotes
Conflicts of Interest: None
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol. 2010;7:163–72. doi: 10.1038/nrclinonc.2009.236. [DOI] [PubMed] [Google Scholar]
- 3.Canto MI, Harinck F, Hruban RH, Offerhaus GJ, Poley JW, Kamel I, et al. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut. 2012 doi: 10.1136/gutjnl-2012-303108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Canto MI, Hruban RH, Fishman EK, Kamel IR, Schulick R, Zhang Z, et al. Frequent detection of pancreatic lesions in asymptomatic high-risk individuals. Gastroenterology. 2012;142:796–804. doi: 10.1053/j.gastro.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartsch DK, Gress TM, Langer P. Familial pancreatic cancer--current knowledge. Nat Rev Gastroenterol Hepatol. 2012;9:445–53. doi: 10.1038/nrgastro.2012.111. [DOI] [PubMed] [Google Scholar]
- 6.Slater EP, Langer P, Niemczyk E, Strauch K, Butler J, Habbe N, et al. PALB2 mutations in European familial pancreatic cancer families. Clin Genet. 2010;78:490–4. doi: 10.1111/j.1399-0004.2010.01425.x. [DOI] [PubMed] [Google Scholar]
- 7.Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klein AP. Identifying people at a high risk of developing pancreatic cancer. Nat Rev Cancer. 2013;13:66–74. doi: 10.1038/nrc3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–68. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 10.Taylor AM, Byrd PJ. Molecular pathology of ataxia telangiectasia. J Clin Pathol. 2005;58:1009–15. doi: 10.1136/jcp.2005.026062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nature reviews Molecular cell biology. 2008;9:759–69. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 12.Swift M, Sholman L, Perry M, Chase C. Malignant neoplasms in the families of patients with ataxia-telangiectasia. Cancer Res. 1976;36:209–15. [PubMed] [Google Scholar]
- 13.Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38:873–5. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 14.Roberts NJ, Jiao Y, Yu J, Kopelovich L, Petersen GM, Bondy ML, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012;2:41–6. doi: 10.1158/2159-8290.CD-11-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong SM, Li A, Olino K, Wolfgang CL, Herman JM, Schulick RD, et al. Loss of E-cadherin expression and outcome among patients with resectable pancreatic adenocarcinomas. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2011;24:1237–47. doi: 10.1038/modpathol.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yemelyanova A, Vang R, Kshirsagar M, Lu D, Marks MA, Shih Ie M, et al. Immunohistochemical staining patterns of p53 can serve as a surrogate marker for TP53 mutations in ovarian carcinoma: an immunohistochemical and nucleotide sequencing analysis. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2011;24:1248–53. doi: 10.1038/modpathol.2011.85. [DOI] [PubMed] [Google Scholar]
- 18.Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 19.Hartman DJ, Krasinskas AM. Assessing treatment effect in pancreatic cancer. Archives of pathology & laboratory medicine. 2012;136:100–9. doi: 10.5858/arpa.2011-0144-RA. [DOI] [PubMed] [Google Scholar]
- 20.Iacobuzio-Donahue CA, Fu B, Yachida S, Luo M, Abe H, Henderson CM, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol. 2009;27:1806–13. doi: 10.1200/JCO.2008.17.7188. Epub 2009 Mar 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tascilar M, Skinner HG, Rosty C, Sohn T, Wilentz RE, Offerhaus GJ, et al. The SMAD4 protein and prognosis of pancreatic ductal adenocarcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2001;7:4115–21. [PubMed] [Google Scholar]
- 22.Blackford A, Serrano OK, Wolfgang CL, Parmigiani G, Jones S, Zhang X, et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin Cancer Res. 2009;15:4674–9. doi: 10.1158/1078-0432.CCR-09-0227. Epub 2009 Jul 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bose S, Starczynski J, Chukwuma M, Baumforth K, Wei W, Morgan S, et al. Down-regulation of ATM protein in HRS cells of nodular sclerosis Hodgkin’s lymphoma in children occurs in the absence of ATM gene inactivation. The Journal of pathology. 2007;213:329–36. doi: 10.1002/path.2232. [DOI] [PubMed] [Google Scholar]
- 24.Shen C, Houghton PJ. The mTOR pathway negatively controls ATM by up-regulating miRNAs. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:11869–74. doi: 10.1073/pnas.1220898110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brune K, Hong SM, Li A, Yachida S, Abe T, Griffith M, et al. Genetic and epigenetic alterations of familial pancreatic cancers. Cancer Epidemiol Biomarkers Prev. 2008;17:3536–42. doi: 10.1158/1055-9965.EPI-08-0630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsubayashi H, Maeda A, Kanemoto H, Uesaka K, Yamazaki K, Hironaka S, et al. Risk Factors of Familial Pancreatic Cancer in Japan: Current Smoking and Recent Onset of Diabetes. Pancreas. 2011 doi: 10.1097/MPA.0b013e3182156e1b. [DOI] [PubMed] [Google Scholar]
- 27.D’Amours D, Jackson SP. The Mre11 complex: at the crossroads of dna repair and checkpoint signalling. Nature reviews Molecular cell biology. 2002;3:317–27. doi: 10.1038/nrm805. [DOI] [PubMed] [Google Scholar]
- 28.Bryant HE, Helleday T. Inhibition of poly (ADP-ribose) polymerase activates ATM which is required for subsequent homologous recombination repair. Nucleic Acids Res. 2006;34:1685–91. doi: 10.1093/nar/gkl108. Print 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–15. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 30.Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJ, et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood. 2010;116:4578–87. doi: 10.1182/blood-2010-01-265769. [DOI] [PubMed] [Google Scholar]
- 31.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 32.Buisson R, Dion-Cote AM, Coulombe Y, Launay H, Cai H, Stasiak AZ, et al. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol. 2010;17:1247–54. doi: 10.1038/nsmb.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kennedy RD, Chen CC, Stuckert P, Archila EM, De la Vega MA, Moreau LA, et al. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J Clin Invest. 2007;117:1440–9. doi: 10.1172/JCI31245. Epub 2007 Apr 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang H, Reinhardt HC, Bartkova J, Tommiska J, Blomqvist C, Nevanlinna H, et al. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 2009;23:1895–909. doi: 10.1101/gad.1815309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.