

Abstract
Chemoattraction of leukocytes into the brain after induction of middle cerebral artery occlusion (MCAO) increases the lesion size and worsens disease outcome. Our previous studies demonstrated that partial MHC class II constructs can reverse this process. However, the potential application of pMHC to human stroke is limited by the need to rapidly match recipient MHC class II with the β1 domain of the pMHC construct. We designed a novel recombinant protein comprised of the HLA-DRα1 domain linked to MOG-35-55 peptide but lacking the β1 domain found in pMHC and treated MCAO after 4 h reperfusion in humanized DR2 mice. Infarct volumes were quantified after 96 h reperfusion and immune cells from the periphery and CNS were evaluated for expression of CD74 and other cell surface, cytokine and pathway markers. This study demonstrates that four daily treatments with DRα1-MOG-35-55 reduced infarct size by 40 % in the cortex, striatum and hemisphere, inhibited the migration of activated CD11b+CD45high cells from the periphery to the brain and reversed splenic atrophy. Furthermore, DRα1-MOG-35-55 bound to CD74 on monocytes and blocked both binding and downstream signaling of macrophage migration inhibition factor (MIF) that may play a key role in infarct development. The novel DRα1-MOG-35-55 construct is highly therapeutic in experimental stroke and could be given to all patients at least 4 h after stroke onset without the need for tissue typing due to universal expression of DRα1 in humans.
Keywords: Stroke, Inflammation, Immunotherapy, Recombinant T-cell receptor Ligand, MHC class II invariant chain
Introduction
It has now been well established that experimental stroke triggers inflammation in brain as well as rapid activation of the peripheral immune system, resulting in migration of monocytes, neutrophils and T-cells across the blood–brain barrier into the growing infarct and further activation of microglial cells (Gee et al. 2007; Nilupul Perera et al. 2006; Wang et al. 1993). These infiltrating cells contribute to ischemic damage through localized inflammation. The magnitude of the inflammatory response is strongly associated with stroke outcome in patients (Emsley et al. 2005; Smith et al. 2004). Furthermore, the peripheral immune system is massively activated after cerebral ischemia. This vast activation is followed by immunosuppression that is marked by atrophy of the spleen and thymus (Offner et al. 2006a, b). Thus, immunotherapeutic approaches for treatment of ischemic stroke could reduce the inflammatory milieu, target specific mechanisms of the inflammatory pathway and maintain homeostasis of peripheral immunity.
Recombinant T-cell receptor ligand (RTL) molecules consist of the α1 and β1 domains of MHC class II molecules expressed as a single polypeptide with or without antigenic amino terminal extensions (Burrows et al. 1999; Vandenbark et al. 2003). We previously demonstrated that RTL could prevent and/or reverse clinical signs of experimental autoimmune encephalomyelitis (EAE) and subsequently showed that an RTL construct could effectively treat experimental stroke in mice (Akiyoshi et al. 2011; Burrows et al. 1998, 2001; Huan et al. 2004; Subramanian et al. 2009; Vandenbark et al. 2003). We reported previously that treatment with RTL1000 that is comprised of an HLA-DR2 moiety linked to human MOG-35-55 peptide in humanized DR2 mice reduced infarct size (Akiyoshi et al. 2011). Recently, we found that the RTL and HLA-DRα1 domain directly binds to and downregulates the cell surface expression of the MHC class II invariant chain (CD74) on CD11b+ monocytes, and the RTL inhibit binding of macrophage migration inhibitory factor (MIF) to CD74 and blocks downstream inflammatory effects in the CNS (Benedek et al. 2013; Vandenbark et al. 2013). We have further demonstrated that the potency of the DRα1 domain could be enhanced by addition of a peptide extension (MOG-35-55 peptide) (Meza-Romero et. al. manuscript in preparation). Moreover, because the DRα1 domain is present in all humans and thus would not be recognized as foreign, treatment using DRα1 constructs would not require HLA screening of potential recipients and could be used for treatment of CNS diseases.
We demonstrate herein that DRα1-MOG-35-55 strongly reduces infarct size and reverses splenic atrophy after stroke when administered at a clinically relevant time-point; 4 h after the onset of stroke, mediated in part by reduced expression of the CD74 MIF cell surface receptor and migration of CD11b+ monocytes into the ischemic brain.
Materials and methods
Ethics statement
The study was conducted in accordance with National Institutes of Health guidelines for the use of experimental animals, and the protocols were approved by the Animal Care and Use Committees at Oregon Health & Science University and the Portland Veteran Affairs Medical Center.
Animals
All experiments used age-matched, sexually mature (20 to 25 g) male HLA-DRB1*1502 (DR2-Tg) mice produced by Dr. Chella David (Gonzalez-Gay et al. 1996). The mice were housed and bred at the Veterans Affairs Medical Center, and studies were conducted at Oregon Health & Science University.
HLA-DRα1-MOG-35-55 cloning, production and purification
DRα1-MOG-35-55 domain cloning, production and purification have been described elsewhere (Meza-Romero et. al. manuscript in preparation). Briefly, DRα1-MOG-35-55 was constructed using the DRα1 construct as a template. The mouse MOG (35–55) peptide DNA encoding sequence was attached to the N-terminus of the DRα1 domain with a linker-thrombin-linker intervening element.
Treatment with DRα1-MOG-35-55
Mice were randomized to receive 0.1 ml (500 μg) DRα1-MOG-35-55 or 0.1 ml Vehicle (5 % dextrose in Tris–HCl, pH 8.5) by subcutaneous injection 4 h after the onset of reperfusion followed by similar doses at 24, 48, and 72 h of reperfusion for a total of 4 treatments each of DRα1-MOG-35-55 or Vehicle. Both DRα1-MOG-35-55 and Vehicle treated MCAO mice were euthanized at the 96 h time-point for evaluation of tissues and cells.
Transient middle cerebral artery occlusion
Transient focal cerebral ischemia was induced in male DR2-Tg mice for 1 h by reversible right MCAO under isoflurane anesthesia followed by 96 h of reperfusion, as described previously with slight modifications (Offner et al. 2006a). Head and body temperature were controlled at 36.5±0.5 °C throughout MCAO surgery with warm water pads and a heating lamp. Laser-Doppler flowmetry (LDF; Model DRT4, Moor Instruments Ltd., Oxford, England) was monitored throughout the ischemic period with a LDF probe affixed to the skull to ensure effective occlusion and reperfusion. The common carotid artery was exposed and the external carotid artery was ligated and cauterized. Unilateral MCAO was accomplished by inserting a 6–0 nylon monofilament surgical suture (ETHICON, Inc., Somerville, NJ, USA) with a heat-rounded and silicone-coated (Xantopren comfort light, Heraeus, Germany) tip into the internal carotid artery via the external carotid artery stump. Animals were excluded if mean intra-ischemic LDF was greater than 30 % pre-ischemic baseline. At 1 h of occlusion, the occluding filament was withdrawn to allow for reperfusion. Mice were then allowed to recover from anesthesia and survived for 96 h following initiation of reperfusion.
Determination of infarct size
The brains were harvested after 96 h of reperfusion and sliced into four 2-mm-thick coronal sections for staining with 1.2 % 2,3,5-triphenyltetrazolium chloride (TTC; Sigma, St. Louis, MO, USA) in saline as described previously (Hurn et al. 2007). The 2-mm brain sections were incubated in 1.2 % TTC for 15 min at 37 °C, and then fixed in 10 % formalin for 24 h. Infarct volume was measured using digital image analysis software (Systat, Inc., Point Richmond, CA, USA). To control for edema, infarct volume (cortex, striatum, and hemisphere) was calculated by subtraction of the ipsilateral non-infarcted regional volume from the contralateral regional volume. This value was then divided by the contralateral regional volume and multiplied by 100 to yield regional infarct volume as a percent of the contralateral region.
Leukocyte isolation from brain and spleen
Spleens from MCAO-treated mice were removed and a single-cell suspension was prepared by passing the tissue through a 100 μm nylon mesh (BD Falcon, Bedford, MA). The cells were washed using RPMI 1640 and the red cells lysed using 1× red cell lysis buffer (eBioscience, Inc., San Diego, CA) and incubated for 3 min. The cells were then washed twice with RPMI 1640, counted and resuspended in stimulation medium (RPMI, containing 2 % FBS, 1 % sodium pyruvate, 1 % L-glutamine, 0.4 % βME). The brain was divided into the ischemic (right) and nonischemic (left) hemispheres, digested for 60 min with 1 mg/ml Type IV collagenase (Sigma Aldrich, St. Louis, MO) and DNase I (50 mg/ml, Roche Diagnostics, Indianapolis, IN) at 37 °C with shaking at 200 rpm. Samples were mixed every 15 min. The suspension was washed 1× in RPMI, resuspended in 80 % Percoll overlayed with 40 % Percoll and centrifuged for 30 min at 1600 RPM. The cells were then washed twice with RPMI 1640, counted and resuspended in staining medium.
Flow cytometry
All antibodies were purchased (BD Biosciences, San Jose, CA or eBioscience, Inc., San Diego, CA) as published. Four-color (FITC, PE, APC and PerCP) fluorescence flow cytometry analyses were performed to determine the phenotypes of cells following standard antibody staining procedures. One million cells were washed with staining medium (PBS containing 0.1 % NaN3 and 1 % bovine serum albumin (Sigma, Illinois) and incubated with combinations of the following monoclonal antibodies: CD80 (16-10A1), HLA-DR (TU39), CD11b (MAC-1), CD74 (In-1), CD45 (Ly-5), CD62L (MEL14), ICAM-1 (3B2), and CD44 (IM7), CCR2 (475301), CD4 (GK1.5) for 20 min at 4 °C. Propidium iodide was added to identify dead cells. Data were collected with CELLQUEST (BD Biosciences, San Jose, CA) and FCS EXPRESS (De Novo Software, Los Angeles, CA) software on a FACSCalibur (BD Biosciences).
Intracellular staining for TNF-α
Splenocytes were resuspended (2×106 cells/ml) in stimulation media (RPMI 1640 media containing 2 % FCS, 1 mM pyruvate, 200 μg/ml penicillin, 200 U/ml streptomycin, 4 mM L-Glutamine, and 5×10−5 M 2-β-ME with PMA [50 ng/ml], ionomycin [500 ng/ml], and Brefeldin A [10 μg/ml] [all reagents from BD Bioscience]) for 4 h. Fc receptors were blocked with mouse Fc receptor-specific mAb (2.3 G2; BD PharMingen) before cell-surface staining and then fixed and permeabilized using a Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer’s instructions. Permeabilized cells were washed with 1×Permeabilization Buffer (BD Bioscience) and stained with either PE-conjugated TNF-α (MP6-XT22) or Isotype matched mAb that served as a negative control. Data were collected with CELLQUEST (BD Biosciences, San Jose, CA) and FCS EXPRESS (De Novo Software, Los Angeles, CA) software on a FACSCalibur (BD Biosciences).
Real time PCR
Splenocytes or brain cells were isolated from DR*1502-Tg mice. Total RNA was isolated from cells using an RNeasy cultured cell kit according to the manufacturer’s instructions. (Qiagen, Valencia, CA, USA). Quantitative real time PCR was performed using the ABI7000 sequence detection system with gene-on-demand assay products (Applied Biosystems) for TaqMan array mouse immune response or for IL-4 (Assay ID: Mm00445259_m1), ACE (Assay ID: Mm00802048_m1) and CCL3 (Assay ID: Mm00441249_g1). GAPDH housekeeping gene was amplified as an endogenous control. Primers were used according to manufacturer’s instructions.
Statistical analysis
Data are presented as mean ± SEM. Statistical differences in cortical, striatal, and total (hemispheric) infarct volume, as well as spleen and brain cell counts and percentages of cellular subtypes from FACS analyses were determined by Student’s t-test. Statistical significance was p <0.05.
Results
DRα1-MOG-35-55 treatment significantly reduces infarct size after MCAO in DR2-Tg mice
Evaluation of brain infarcts 96 h after MCAO demonstrated that DRα1-MOG-35-55-treated male DR2-Tg mice had significantly reduced infarct volumes compared with the Vehicle-treated group as shown in Fig. 1a. Cortical infarct volume was 25.9±4.67 % in DRα1-MOG-35-55-treated compared to 47.0±2.5 % in vehicle-treated mice (p <0.01). Similarly, striatal infarct volume was 40.8±5.4 % vs. 64.5±2.0 % respectively in DRα1-MOG-35-55-treated vs. Vehicle-treated mice (p <0.01), whereas the total hemispheric infarct volume was 19.4±3.6 % vs. 31.1±1.71 %, respectively (p <0.01). Quantitative assay of TTC stained cerebral sections after 96 h of reperfusion illustrated the smaller infarct area in DRα1-MOG-35-55-treated mice compared with Vehicle-treated mice (Fig. 1b). There were no significant differences in laser-Doppler perfusion before, during or immediately after MCAO between DRα1-MOG-35-55-treated and Vehicle-treated groups (data not shown).
Fig. 1.

DRα1-MOG-35-55 treatment decreased infarct size compared to vehicle 96 h after MCAO. a Infarct volumes were significantly decreased in DRα1-MOG-35-55 (n =10) versus Vehicle-treated (n =10) mice after 60 min MCAO and 96 h of reperfusion in cortex (CTX) and caudateputamen (CP), as well as in total hemisphere (HMSPHR) (*p ≤0.05). B). Representative 2,3,5-triphenyltetrazolium chloride stained cerebral sections after 96 h of reperfusion following 1 h of MCAO
DRα1-MOG-35-55 reduces the number of activated microglia and infiltrating monocytes and their CD74 cell surface expression in the ischemic brain
We sought to determine if the reduced infract size after DRα1-MOG-35-55 treatment was due to diminished number of infiltrating inflammatory cells in the brain after MCAO. DRα1-MOG-35-55 treatment significantly reduced the absolute number of mononuclear cells in the right ischemic brain compared with Vehicle-treated mice (13.14×104 vs. 21.33×104 respectively, p <0.05) (Fig. 2a). This difference is attributed mainly to the reduction in the number of activated CD11b+CD45high monocytic cells (4.7× 104 vs. 11.85×104 respectively, p <0.01). There were no significant differences in the absolute numbers of the CD4+ T cells or the resting microglia (CD11b+CD45low) (Fig. 2b). In the non-ischemic left brain there were no differences in the total absolute number of mononuclear cells or in any cell type (Supplemental Fig. 1).
Fig. 2.

DRα1-MOG-35-55 treatment reduces number of activated CD11b+ cells and their CD74 surface expression in the ischemic brain 96 h after MCAO. a Absolute lymphocytes numbers in the non-ischemic left and ischemic right brain from DRα1-MOG-35-55-treated (n =7) versus Vehicle-treated mice (n =9). b Absolute numbers of CD11b+CD45low, CD11b+CD45high and CD4+ cells in the right brain. c CD74 cell surface expression on CD11b+CD45high cells in the right brain. Data are presented as mean ± SEM. *p <0.05, **p < 0.01, ***p <0.001 Student’s t-test
Previously, it was demonstrated that DRα1 treatment of EAE also leads to reduction of activated CD11b+ cells in the Central Nervous System (CNS) and that DRα1 reduces the cell surface expression of the MIF receptor, CD74, on the activated CD11b+ cells. In order to determine if DRα1-MOG-35-55 treatment has the same effect in MCAO, expression of CD74 cell surface levels was evaluated on CD11b+CD45high cells. As shown in Fig. 2c, there was a significant reduction in the level of CD74 expression as measured by the Mean Fluorescent Intensity (MFI) in the DRα1-MOG-35-55-treated mice compared with the Vehicle-treated mice (p <0.01). Taken together, these results demonstrate that DRα1-MOG-35-55 can reduce the number of activated microglia and infiltrating monocytes in the ischemic brain and affect the expression of CD74 on the surface of these cells.
DRα1-MOG-35-55 treatment affects the immune gene expression profile in the ischemic brain after MCAO
In order to assess the DRα1-MOG-35-55 treatment effect on the expression of immune related genes in brain, mRNA was isolated from the ischemic brains of 3 Vehicle-treated mice and 3 DRα1-MOG-35-55-treated mice. A real-time PCR assay was performed on pooled cDNA samples and expression levels of the DRα1-MOG-35-55-treated sample was analyzed relative to the Vehicle-treated sample (Fig. 3). Validation of 3 genes: IL-4, CCL3 and ACE, using individual samples, demonstrated that the expression trends were the same as in the immune array, although not all of the genes showed a significant difference (Supplemental Fig. 2). The immune array data demonstrate that there was a decrease in the expression of monocyte-related genes such as CCL3, CCL2 and increases in Th1 and Th2 related genes such as IL-12, Tbx21, IL-4 and IL-13. It is important to note that several genes that were associated previously with cerebrovacular function and ischemic brain injury, including ACE and EDN1, were down regulated after DRα1-MOG-35-55 treatment relative to Vehicle treatment.
Fig. 3.
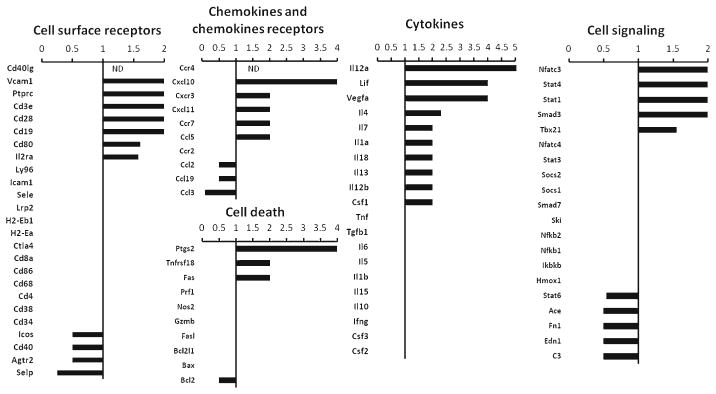
Gene expression profile of right ischemic brain from DRα1-MOG-35-55 treated relative to Vehicle treated mice 96 h after MCAO. Relative expression of mRNA of inflammatory genes was analyzed by real-time PCR from pooled ischemic brain samples of DRα1-MOG-35-55-treated (n =3) relative to Vehicle-treated (n =3) mice. Values <1, 1, >1 represent down-regulated, unchanged or up-regulated genes, respectively, of samples from DRα1-MOG-35-55-treated mice relative to Vehicle-treated mice. ND, not detected
DRα1-MOG-35-55 treatment reverses MCAO-Induced splenic atrophy
To evaluate the effects of DRα1-MOG-35-55 treatment on stroke-induced splenic atrophy, cell numbers in the spleen were counted in post-ischemic Vehicle- and DRα1-MOG-35-55-treated mice. As expected, MCAO induced a significant decrease in spleen numbers in the Vehicle-treated group (Fig. 4a). Interestingly, viable cell counts were significantly increased in spleens of DRα1-MOG-35-55-treated versus Vehicle-treated mice (64.67×106 vs. 32.11×106 respectively, p <0.01) 96 h after reperfusion. The increase in spleen cell numbers was reflected in the absolute numbers of CD11b+ cells (p <0.01) and CD4+ cells (p <0.05) (Fig. 4b). Although DRα1-MOG-35-55 treatment increased the cell numbers in the spleen, it reduced the frequency of CD4+ cells and did not change the frequency of the CD11b+ cells in the spleen (Fig. 4c).
Fig. 4.

DRα1-MOG-35-55 treatment reverses MCAO-induced splenic atrophy. a Absolute cell numbers in the spleen from DRα1-MOG-35-55-treated (n =10) versus Vehicle-treated mice (n =12). b Absolute numbers of CD11b+ and CD4+ cells in spleen c. Frequency of CD11b+ and CD4+ cells in spleen. Data are presented as mean ± SEM. * p <0.05, **p <0.01 Student’s t-test
DRα1-MOG-35-55 treatment increases the frequency of activated CD4+ cells but does not change the activation state of CD11b+ cells in the spleen after MCAO
We further sought to assess the activation stated of CD11b+ and CD4+ cells in the spleen 96 h after reperfusion. As shown in Fig. 5a there were no differences in the level of expression of CD80, ICAM-1, HLA-DR and CCR2 on CD11b+ cells between DRα1-MOG-35-55-versus Vehicle-treated mice. Evaluation of CD4+ activation by the expression of CD44 and CD62L revealed that the frequency of both CD62L low and high activated CD4+ cells in the spleen were increased after treatment with DRα1-MOG-35-55 compared with Vehicle (p <0.05 for both). In addition, spleen cells from DRα1-MOG-35-55- or Vehicle-treated mice were stimulated with PMA/Ionomycine for 4 h and evaluated by flow cytometry for intracellular staining of TNF-α. There was no difference in production of TNF-α by CD4+ or CD11b+ cells in spleens from DRα1-MOG-35-55- or Vehicle-treated mice (Supplemental Fig. 3).
Fig. 5.

DRα1-MOG-35-55 treatment increases the frequency of activated CD4+ cells but does not change the activation state of CD11b+ cells in the spleen 96 h after MCAO. a CD11b+ cells in the spleen of DRα1-MOG-35-55-treated (n =10) versus Vehicle-treated mice (n =12) were analyzed for the expression of CD80, HLA MHC class II, ICAM-1 and CCR2. b CD4+ cells in spleen were analyzed for the expression of CD44 and CD62L. Data are presented as mean ± SEM. * p <0.05, Student’s t-test
DRα1-MOG-35-55 treatment affects the immune gene expression profile in the spleen after MCAO
In order to assess the DRα1-MOG-35-55 treatment effect on the expression of immune related genes in the spleen, mRNA was isolated from spleens of 6 Vehicle-treated mice and 6 DRα1-MOG-35-55-treated mice. A real-time PCR assay was performed on pooled cDNA samples and expression levels from the DRα1-MOG-35-55-treated mice were analyzed relative to the Vehicle-treated mice (Fig. 6). The expression levels of 3 genes: IL-4, CCL3 and ACE were validated using individual samples (Supplemental Fig. 4). Interestingly, the expression of several of the genes in the spleen, such as CCL3, IL-4, Stat6, ACE, FN1 and C3) had an opposite trend compared with their expression in the brain after DRα1-MOG-35-55 treatment. In addition, the expression of monocyte related genes, such as CD68, were increased in the spleen of DRα1-MOG-35-55-treated mice relative to Vehicle-treated mice. These expression results are in correlation to the absolute numbers of cells in the spleen.
Fig. 6.
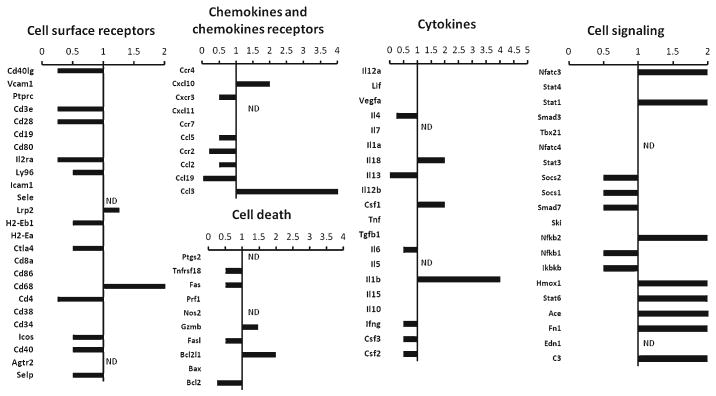
Gene expression profile of spleens from DRα1-MOG-35-55 treated relative to Vehicle treated mice 96 h after MCAO. Relative expression of mRNA of inflammatory genes was analyzed by real-time PCR from pooled spleen samples of DRα1-MOG-35-55-treated (n =6) relative to Vehicle-treated (n =6) mice. Values <1, 1, >1 represent down-regulated, unchanged or up-regulated genes, respectively, of samples from DRα1-MOG-35-55-treated mice relative to Vehicle-treated mice. ND, not detected
Discussion
Our study demonstrates for the first time that treatment with DRα1-MOG-35-55 after onset of MCAO reduces infract size, inhibits infiltration of activated monocytes into the ischemic brain and reverses splenic atrophy, which is typically induced after MCAO. We have previously demonstrated that partial MHC class II constructs which are comprised of the linked β1α1 domains tethered to antigenic peptides, could treat CNS diseases such as experimental stroke and EAE (Akiyoshi et al. 2011; Sinha et al. 2009, 2007; Subramanian et al. 2009). However, the novel DRα1-MOG-35-55 construct represents an improved immunotherapy which would not require HLA screening prior to use. In contrast to RTL1000, in which the MOG peptide is linked to the DR2β1 domain and thus can be given only to HLA-DR2 matched subjects, the MOG peptide in the new construct is linked to the DRα1 domain. Hence, the lack of the polymorphic β1 domain and the fact that the DRα1 domain is invariable and thus universally tolerogenic enables its use without pre-screening for HLA genotype.
We have recently shown that DRα1-MOG-35-55 binds to and down-regulates CD74 on CD11b+ cells. This binding has a bi-functional inhibitory effect on MIF/CD74 interactions and blocks downstream MIF effects in the CNS during EAE (Meza-Romero et. al. manuscript in preparation). In addition, we have demonstrated the RTL can inhibit MIF binding to CD74 (Benedek et al. 2013). However, it is not yet clear whether the treatment effect of DRα1-MOG-35-55 in MCAO is MIF dependent, MIF independent or both. In the literature, the contribution of MIF to the outcome of MCAO is controversial and one report even indicates that deletion of MIF worsens MCAO in females (Inacio et al. 2011a, b, c; Turtzo et al. 2013), suggesting a protective effect. It is thus important to note that the parent RTL1000 construct can treat MCAO in both male and female mice ((Akiyoshi et al. 2011) and HO unpublished data). Assuming similar mechanisms with RTL1000, the DRα1-MOG-35-55 treatment might thus have a MIF independent effect, at least in female mice, either via the tethered MOG-35-55 peptide or by reduction of CD74 surface expression on activated CD11b+ cells. Consistent with this line of reasoning, Fan et. al. demonstrated that macrophages from CD74 KO mice showed increased random migration compared with WT macrophages, while the random migration levels of MIF KO and WT macrophages did not differ (Fan et al. 2011). This result would suggest a MIF-independent role of CD74 on macrophage migration that might also be affected by DRα1-MOG-35-55.
The infiltration of inflammatory cells into the brain after MCAO was reported in detail previously and it was shown that of the activated cells in the brain the predominant population is macrophages/activated microglial cells (both cell types are CD11b+CD45high in inflamed CNS) (Stevens et al. 2002; Subramanian et al. 2009). Our results clearly demonstrate that DRα1-MOG-35-55 treatment reduces the infarct volume and the number and frequency of CD11b+CD45high cells in the ischemic brain. This result is very similar to the previously described effect of RTLs in blocking activation of resident microglia and cellular infiltration into the CNS during EAE and MCAO (Benedek et al. 2013; Subramanian et al. 2009).
The gene expression profile of immune related genes in the brain of DRα1-MOG-35-55-treated relative to Vehicle-treated mice was concurrent with the reduction of activated CD11b+ cells in the ischemic brain 96 h after MCAO. Interestingly, several genes that were shown to be involved in the induction and outcome of stroke, such as Endothelin 1 (EDN1) (Ziv et al. 1992) and Angiotensin-converting enzyme (ACE) were down regulated (Yusuf et al. 2000) at this time point. It is also important to note that up-regulated genes such as CXCL10, CXCR3 and CCR7 in DRα1-MOG-35-55-treated vs. Vehicle MCAO mice could reflect a lesser degree of MCAO-induced immunosuppression (similar to sham-treated vs. untreated MCAO mice) at 96 h(Offner et al. 2006b). Thus, treatment with DRα1-MOG-35-55 is directly affecting the number of infiltrating cells, mainly monocytes, into the brain and in addition is restoring immune homeostasis to the ischemic brain.
The effects of cerebral damage from stroke on the peripheral immune system are profound (Offner et al. 2006a, b; Seifert et al. 2012). This drastic loss of cell numbers in the spleen 96 h after MCAO induction was largely reversed after DRα1-MOG-35-55 treatment. The absolute numbers of CD11b+ and CD4+ cells as well as total splenocytes were significantly increased. We also observed that DRα1-MOG-35-55 treatment did not alter the frequency of CD11b+ cells or their activation state. As for the CD4+ cells, although the frequency of these cells was reduced in the spleen after DRα1-MOG-35-55 treatment, the frequency of the activated CD4+ cells was significantly higher suggesting that the activated cells are retained in the spleen.
The gene expression profile of the spleen from DRα1-MOG-35-55-treated mice relative to Vehicle-treated mice was almost a mirror image to the expression in the ischemic brain. Taken together, these data suggest that the DRα1-MOG-35-55 mechanism of action in stroke involves alteration of the migration patterns of activated cells in the periphery. Down regulation of CD74 on activated CD11b+ cells by DRα1-MOG-35-55 could induce random migration or inhibit chemotactic migration of these cells, as was shown in CD74 deficient macrophages or by down regulation of CD74 cell surface expression by RTLs.
In summary, our study demonstrates that four daily treatments with DRα1-MOG-35-555 beginning 4 h after MCAO reduces the migration of activated cells from spleen to brain, thus contributing to a reduction in infarct size. This immunomodulatory agent might thus represent the next generation of partial MHC class II constructs with therapeutic activity for CNS diseases. Further studies are underway to determine how long after MCAO DRα1-MOG-35-55 is effective and what is the minimal effective dose that can be given.
Supplementary Material
Acknowledgments
The authors wish to thank Melissa S. Barber for assistance with manuscript submission. This work was supported by NIH Grants #NS076013 (STTR), and NS047661 and by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development. The contents do not represent the views of the Department of Veterans Affairs or the United States Government.
Abbreviations
- MCAO
Middle cerebral artery occlusion
- RTL
Recombinant T-cell receptor ligand
- MHC
Major histocompatibility complex
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s11011-013-9440-0) contains supplementary material, which is available to authorized users.
Conflict of interest No Conflicts of Interest.
Contributor Information
Gil Benedek, Neuroimmunology Research, R&D-31, Portland Veterans Affairs Medical Center, 3710 SW US Veterans Hospital Rd, Portland, OR 97239, USA. Department of Neurology, Oregon Health & Science University, Portland, OR, USA.
Wenbin Zhu, Department of Anesthesiology and Perioperative Medicine, Oregon Health & Science University, Portland, OR, USA.
Nicole Libal, Department of Anesthesiology and Perioperative Medicine, Oregon Health & Science University, Portland, OR, USA.
Amanda Casper, Department of Anesthesiology and Perioperative Medicine, Oregon Health & Science University, Portland, OR, USA.
Xiaolin Yu, Neuroimmunology Research, R&D-31, Portland Veterans Affairs Medical Center, 3710 SW US Veterans Hospital Rd, Portland, OR 97239, USA. Department of Neurology, Oregon Health & Science University, Portland, OR, USA.
Roberto Meza-Romero, Neuroimmunology Research, R&D-31, Portland Veterans Affairs Medical Center, 3710 SW US Veterans Hospital Rd, Portland, OR 97239, USA. Department of Neurology, Oregon Health & Science University, Portland, OR, USA.
Arthur A. Vandenbark, Neuroimmunology Research, R&D-31, Portland Veterans Affairs Medical Center, 3710 SW US Veterans Hospital Rd, Portland, OR 97239, USA. Department of Neurology, Oregon Health & Science University, Portland, OR, USA. Department of Molecular Microbiology & Immunology, Oregon Health & Science University, Portland, OR, USA
Nabil J. Alkayed, Department of Anesthesiology and Perioperative Medicine, Oregon Health & Science University, Portland, OR, USA
Halina Offner, Email: offnerva@ohsu.edu, Neuroimmunology Research, R&D-31, Portland Veterans Affairs Medical Center, 3710 SW US Veterans Hospital Rd, Portland, OR 97239, USA. Department of Anesthesiology and Perioperative Medicine, Oregon Health & Science University, Portland, OR, USA. Department of Neurology, Oregon Health & Science University, Portland, OR, USA.
References
- Akiyoshi K, Dziennis S, Palmateer J, Ren X, Vandenbark AA, Offner H, Herson PS, Hurn PD. Recombinant T cell receptor ligands improve outcome after experimental cerebral ischemia. Transl Stroke Res. 2011;2(3):404–410. doi: 10.1007/s12975-011-0085-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedek G, Meza-Romero R, Andrew S, Leng L, Burrows GG, Bourdette D, Offner H, Bucala R, Vandenbark AA. Partial MHC class II constructs inhibit MIF/CD74 binding and downstream effects. Eur J Immunol. 2013;43(5):1309–1321. doi: 10.1002/eji.201243162. 10.1002/eji. 201243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows GG, Bebo BF, Jr, Adlard KL, Vandenbark AA, Offner H. Two-domain MHC class II molecules form stable complexes with myelin basic protein 69–89 peptide that detect and inhibit rat encephalitogenic T cells and treat experimental autoimmune encephalomyelitis. J Immunol. 1998;161(11):5987–5996. [PubMed] [Google Scholar]
- Burrows GG, Chang JW, Bachinger HP, Bourdette DN, Offner H, Vandenbark AA. Design, engineering and production of functional single-chain T cell receptor ligands. Protein Eng. 1999;12(9):771–778. doi: 10.1093/protein/12.9.771. [DOI] [PubMed] [Google Scholar]
- Burrows GG, Chou YK, Wang C, Chang JW, Finn TP, Culbertson NE, Kim J, Bourdette DN, Lewinsohn DA, Lewinsohn DM, Ikeda M, Yoshioka T, Allen CN, Offner H, Vandenbark AA. Rudimentary TCR signaling triggers default IL-10 secretion by human Th1 cells. J Immunol. 2001;167(8):4386–4395. doi: 10.4049/jimmunol.167.8.4386. [DOI] [PubMed] [Google Scholar]
- Emsley HC, Smith CJ, Georgiou RF, Vail A, Tyrrell PJ, Barberan EM, Rothwell NJ, Hopkins SJ. Correlation of systemic inflammatory response with infarct volume in acute ischemic stroke patients. Stroke. 2005;36(2):228–229. doi: 10.1161/01.STR.0000155197.88944.ac. author reply 228–229. [DOI] [PubMed] [Google Scholar]
- Fan H, Hall P, Santos LL, Gregory JL, Fingerle-Rowson G, Bucala R, Morand EF, Hickey MJ. Macrophage migration inhibitory factor and CD74 regulate macrophage chemotactic responses via MAPK and Rho GTPase. J Immunol. 2011;186(8):4915–4924. doi: 10.4049/jimmunol.1003713. 10. 4049/jimmunol.1003713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee JM, Kalil A, Shea C, Becker KJ. Lymphocytes: potential mediators of postischemic injury and neuroprotection. Stroke. 2007;38(2 Suppl):783–788. doi: 10.1161/01.STR.0000248425.59176.7b. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Gay MA, Zanelli E, Khare SD, Krco CJ, Zhou P, Inoko H, Griffiths MM, Luthra HS, David CS. Human leukocyte antigen-DRB1*1502 (DR2Dw12) transgene reduces incidence and severity of arthritis in mice. Hum Immunol. 1996;50(1):54–60. doi: 10.1016/0198-8859(96)00123-1. [DOI] [PubMed] [Google Scholar]
- Huan J, Subramanian S, Jones R, Rich C, Link J, Mooney J, Bourdette DN, Vandenbark AA, Burrows GG, Offner H. Monomeric recombinant TCR ligand reduces relapse rate and severity of experimental autoimmune encephalomyelitis in SJL/J mice through cytokine switch. J Immunol. 2004;172(7):4556–4566. doi: 10.4049/jimmunol.172.7.4556. [DOI] [PubMed] [Google Scholar]
- Hurn PD, Subramanian S, Parker SM, Afentoulis ME, Kaler LJ, Vandenbark AA, Offner H. T- and B-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab. 2007;27(11):1798–1805. doi: 10.1038/sj.jcbfm.9600482. 10.1038/sj.jcbfm. 9600482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inacio AR, Bucala R, Deierborg T. Lack of macrophage migration inhibitory factor in mice does not affect hallmarks of the inflammatory/immune response during the first week after stroke. J Neuroinflammation. 2011a;8:75. doi: 10.1186/1742-2094-8-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inacio AR, Ruscher K, Leng L, Bucala R, Deierborg T. Macrophage migration inhibitory factor promotes cell death and aggravates neurologic deficits after experimental stroke. J Cereb Blood Flow Metab. 2011b;31(4):1093–1106. doi: 10.1038/jcbfm.2010.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inacio AR, Ruscher K, Wieloch T. Enriched environment downregulates macrophage migration inhibitory factor and increases parvalbumin in the brain following experimental stroke. Neurobiol Dis. 2011c;41(2):270–278. doi: 10.1016/j.nbd.2010.09.015. [DOI] [PubMed] [Google Scholar]
- Nilupul Perera M, Ma HK, Arakawa S, Howells DW, Markus R, Rowe CC, Donnan GA. Inflammation following stroke. J Clin Neurosci. 2006;13(1):1–8. doi: 10.1016/j.jocn.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Parker SM, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab. 2006a;26(5):654–665. doi: 10.1038/sj.jcbfm.9600217. [DOI] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Parker SM, Wang C, Afentoulis ME, Lewis A, Vandenbark AA, Hurn PD. Splenic atrophy in experimental stroke is accompanied by increased regulatory Tcells and circulating macrophages. J Immunol. 2006b;176(11):6523–6531. doi: 10.4049/jimmunol.176.11.6523. [DOI] [PubMed] [Google Scholar]
- Seifert HA, Hall AA, Chapman CB, Collier LA, Willing AE, Pennypacker KR. A transient decrease in spleen size following stroke corresponds to splenocyte release into systemic circulation. J Neuroimmune Pharmacol. 2012;7(4):1017–1024. doi: 10.1007/s11481-012-9406-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Subramanian S, Miller L, Proctor TM, Roberts C, Burrows GG, Vandenbark AA, Offner H. Cytokine switch and bystander suppression of autoimmune responses to multiple antigens in experimental autoimmune encephalomyelitis by a single recombinant T-cell receptor ligand. J Neurosci. 2009;29(12):3816–3823. doi: 10.1523/JNEUROSCI.5812-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Subramanian S, Proctor TM, Kaler LJ, Grafe M, Dahan R, Huan J, Vandenbark AA, Burrows GG, Offner H. A promising therapeutic approach for multiple sclerosis: recombinant T-cell receptor ligands modulate experimental autoimmune encephalomyelitis by reducing interleukin-17 production and inhibiting migration of encephalitogenic cells into the CNS. J Neurosci. 2007;27(46):12531–12539. doi: 10.1523/JNEUROSCI.3599-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJ, Emsley HC, Gavin CM, Georgiou RF, Vail A, Barberan EM, del Zoppo GJ, Hallenbeck JM, Rothwell NJ, Hopkins SJ, Tyrrell PJ. Peak plasma interleukin-6 and other peripheral markers of inflammation in the first week of ischaemic stroke correlate with brain infarct volume, stroke severity and long-term outcome. BMC Neurol. 2004;4:2. doi: 10.1186/1471-2377-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens SL, Bao J, Hollis J, Lessov NS, Clark WM, Stenzel-Poore MP. The use of flow cytometry to evaluate temporal changes in inflammatory cells following focal cerebral ischemia in mice. Brain Res. 2002;932(1–2):110–119. doi: 10.1016/s0006-8993(02)02292-8. [DOI] [PubMed] [Google Scholar]
- Subramanian S, Zhang B, Kosaka Y, Burrows GG, Grafe MR, Vandenbark AA, Hurn PD, Offner H. Recombinant T cell receptor ligand treats experimental stroke. Stroke. 2009;40(7):2539–2545. doi: 10.1161/STROKEAHA.108.543991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turtzo LC, Li J, Persky R, Benashski S, Weston G, Bucala R, Venna VR, McCullough LD. Deletion of macrophage migration inhibitory factor worsens stroke outcome in female mice. Neurobiol Dis. 2013;54:421–431. doi: 10.1016/j.nbd.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbark AA, Meza-Romero R, Benedek G, Andrew S, Huan J, Chou YK, Buenafe AC, Dahan R, Reiter Y, Mooney JL, Offner H, Burrows GG. A novel regulatory pathway for autoimmune disease: binding of partial MHC class II constructs to monocytes reduces CD74 expression and induces both specific and bystander T-cell tolerance. J Autoimmun. 2013;40:96–110. doi: 10.1016/j.jaut.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbark AA, Rich C, Mooney J, Zamora A, Wang C, Huan J, Fugger L, Offner H, Jones R, Burrows GG. Recombinant TCR ligand induces tolerance to myelin oligodendrocyte glycoprotein 35–55 peptide and reverses clinical and histological signs of chronic experimental autoimmune encephalomyelitis in HLA-DR2 transgenic mice. J Immunol. 2003;171(1):127–133. doi: 10.4049/jimmunol.171.1.127. [DOI] [PubMed] [Google Scholar]
- Wang PY, Kao CH, Mui MY, Wang SJ. Leukocyte infiltration in acute hemispheric ischemic stroke. Stroke. 1993;24(2):236–240. doi: 10.1161/01.str.24.2.236. [DOI] [PubMed] [Google Scholar]
- Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342(3):145–153. doi: 10.1056/NEJM200001203420301. [DOI] [PubMed] [Google Scholar]
- Ziv I, Fleminger G, Djaldetti R, Achiron A, Melamed E, Sokolovsky M. Increased plasma endothelin-1 in acute ischemic stroke. Stroke. 1992;23(7):1014–1016. doi: 10.1161/01.str.23.7.1014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.