

Abstract
Background
We have previously reported stromal upregulation of the endogenous angiogenesis inhibitor thrombospondin-2 (TSP-2) during multistep carcinogenesis, and we found accelerated and enhanced skin angiogenesis and carcinogenesis in TSP-2 deficient mice.
Goals
To investigate whether enhanced levels of TSP-2 might protect from skin cancer development.
Methods
We established transgenic mice with targeted overexpression of TSP-2 in the skin and subjected hemizygous TSP-2 transgenic mice and their wild-type littermates to a chemical skin carcinogenesis regimen.
Results
TSP-2 transgenic mice showed a significantly delayed onset of tumor formation compared to wild-type mice, whereas the ratio of malignant conversion to squamous cell carcinomas was comparable in both genotypes. Computer-assisted morphometric analysis of blood vessels revealed pronounced tumor angiogenesis already in the early stages of carcinogenesis in wild type mice. TSP-2 overexpression significantly reduced tumor blood vessel density in transgenic mice but had no overt effect on LYVE-1 positive lymphatic vessels. The percentage of desmin surrounded, mature tumor-associated blood vessels and the degree of epithelial differentiation remained unaffected. The antiangiogenic effect of transgenic TSP-2 was accompanied by a significantly increased number of apoptotic tumor cells in transgenic mice.
Conclusion
Our results demonstrate that enhanced levels of TSP-2 in the skin result in reduced susceptibility to chemically-induced skin carcinogenesis and identify TSP-2 as a new target for the prevention of skin cancer.
Introduction
In contrast to the plethora of reports on tumor angiogenesis factors, much less is known about the biological role of endogenous inhibitors of angiogenesis during tumor development, in particular during the early stages of tumor promotion. Several endogenous inhibitors of tumor angiogenesis have been identified including thrombospondin-(TSP)-1 [1], TSP-2 [2], angiostatin [3], endostatin [4], vasostatin [5] and tumstatin [6]. Although TSP-1 and TSP-2 are members of the same family of glycoproteins with considerable structural similarities [7, 8], the expression of TSP-2 differs spatially and temporally from TSP-1 during embryonic development [9] [10]. Moreover, the regulation of TSP-2 gene expression by growth factors is distinct from TSP-1 [7]. Previously, TSP-2 was shown to diminish the angiogenic activity of basic fibroblast growth factor [11] and the formation of focal adhesions in aortic endothelial cells [12], indicating its role in controlling angiogenesis. TSP-2 deficient mice are characterized by increased vascular density in several tissues including the skin [13] and display accelerated healing of excisional wounds by virtue of their highly vascularized granulation tissue [14].
Previously, we identified stromal up-regulation of TSP-2 expression as a potential host anti-tumor mechanism during multistep skin carcinogenesis [15], and we found that stable overexpression of TSP-2 in human squamous cell carcinoma xenotransplants inhibited tumor growth and vascularization even more potently than TSP-1 [2]. Furthermore, systemic treatment with an N-terminal 80 kDa recombinant fragment of TSP-2 inhibited angiogenesis and tumor growth in squamous cell carcinoma (SCC) bearing mice [16].
However, human cancers arise through a multistep progression pathway, and the role of TSP-2 in the early stages of cancer development has remained unknown. Based on our previous observation that TSP-2 deficient mice display enhanced skin carcinogenesis and angiogenesis, we hypothesized that TSP-2 might play a role in the control of early tumorigenesis. To directly evaluate the biological effects of TSP-2 in multistep epithelial tumor development, we established transgenic mice with targeted overexpression of TSP-2 in epidermal keratinocytes of the skin. Hemizygous TSP-2 transgenic mice and their wild-type littermates were subjected to a standard two-step skin carcinogenesis regimen, using topical application of 7,12-dimethylbenz (α) anthracene (DMBA) for tumor initiation and phorbol 12-myristate 13-acetate (PMA) for tumor promotion. This established model of skin carcinogenesis [17] allows detailed insights into the premalignant as well as the malignant stages of skin cancer development.
We found that targeted overexpression of TSP-2 in the skin of transgenic mice reduced the incidence of early, premalignant stages of tumor development as well as the formation of squamous cell carcinomas. Tumor angiogenesis was significantly inhibited in all stages of skin carcinogenesis in TSP-2 transgenic mice, but lymphangiogenesis remained unaffected. Moreover, the number of apoptotic tumor cells in TSP-2 transgenic mice was significantly increased over wild-type controls, identifying TSP-2 as a potential factor for the prevention of skin cancer.
Materials and Methods
Generation of TSP-2 transgenic mice
We cloned a 4.1-kb mouse TSP-2 cDNA sequence, comprising the full TSP-2 coding sequence, into a pGEM-3Z vector containing the human keratin 14 (K14) promoter (kindly provided by Dr. Elaine Fuchs, Chicago) that targets transgene expression to epidermal keratinocytes of the skin [18]. We have previously used the identical expression vector to establish transgenic mice overexpressing the mouse VEGF164 gene [19] and the human TSP-1 gene [20]. The correct sequence and orientation of the TSP-2 insert were verified by restriction mapping and direct sequencing using the Sanger dideoxy method.
After digestion of the complete construct with the restriction enzymes KpnI and HindIII, the 8.3-kb expression vector (Fig. 1A) was used to generate transgenic mice as described [19]. Transgenic founders were detected by Southern blot analysis of BamHI digested genomic tail DNA obtained 2 weeks after birth. For rapid identification, genomic tail DNA was subjected to PCR using an 18-mer primer and a 21-mer primer that bind, respectively, to positions 321–338 and 650–630 of the human growth hormone gene in the transgene construct, leading to selective amplification of a 330-bp fragment when the transgene construct was incorporated into the genome. Transgenic lines were established in the FVB/J genetic background. Transgene expression was confirmed by Northern hybridization of total RNA extracted from the skin of mice (Fig. 1B) Northern blot analysis confirmed transgenic TSP-2 mRNA expression in the skin of TSP-2 transgenic mice whereas TSP-2 mRNA was not significantly increased above baseline in the liver of transgenic mice demonstrating selective transgenic expression in the skin. This was further confirmed by in situ hybridization demonstrating strong TSP-2 overexpression in basal epidermal keratinocytes and in outer root sheath keratinocytes of hair follicles (Fig. 3B, D).
Figure 1. Schematic representation of the K14-TSP2 transgene construct.

A 4100 bp mouse TSP-2 BamHI cDNA fragment was ligated into the BamHI restriction site of the keratin 14 promoter expression cassette (A). Overexpression of TSP-2 mRNA in the skin of 3-week-old K14-TSP-2 transgenic mice was confirmed by northern blotting. TSP-2 mRNA was not increased above wild-type (wt) levels in the liver of TSP-2 transgenic mice. Hybridization with a murine b-actin probe served as control for equal loading (B).
Figure 3. Differential TSP-2 expression in normal skin, SCC and tumor stroma in wild-type and transgenic mice.
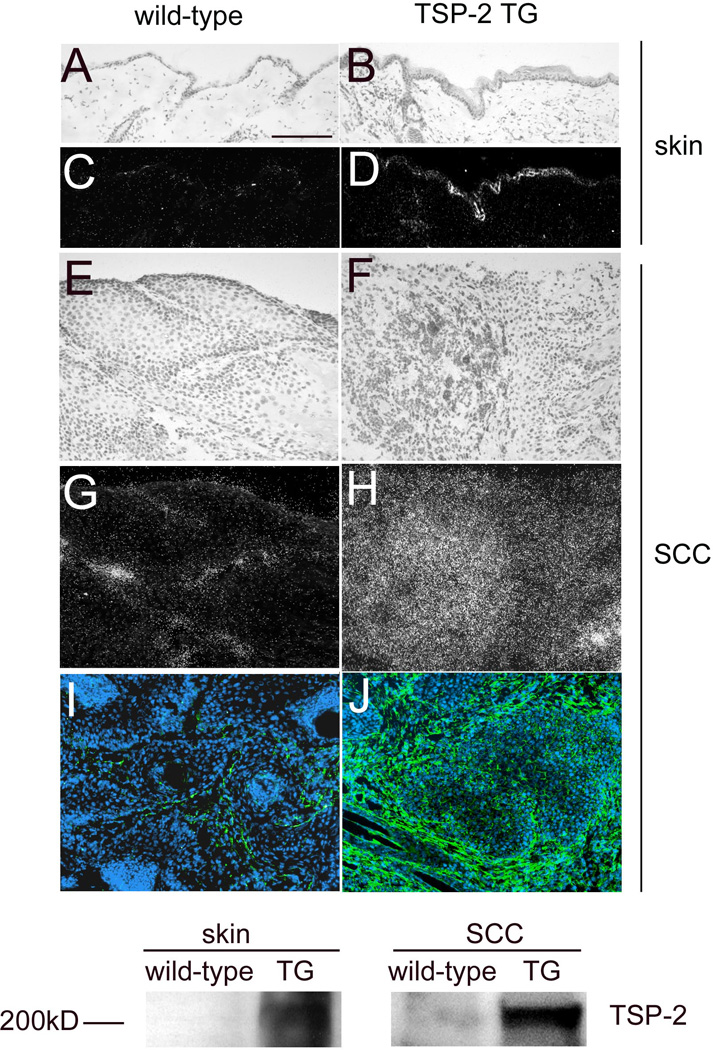
In situ hybridization confirmed moderately enhanced TSP-2 mRNA expression in the normal skin (B,D) of TSP-2 transgenic mice and a strong TSP-2 expression in the SCC of TSP-2 transgenic mice (F, H), whereas little or no TSP-2 mRNA was detectable in the normal skin (A, C) or the SCC of wild-type mice (E, G). Immunofluorescence stains for TSP-2 (green) demonstrate pronounced deposition of TSP-2 protein predominantly in the tumor stroma in TSP-2 transgenic SCC (J), as compared to weak TSP-2 protein expression in wild-type SCC (I). Bar = 100 µm. Western blot demonstrates high levels of TSP-2 expression in the skin and in tumors of TSP-2 transgenic mice in contrast to low protein levels in wild-type mice (K).
Western blot analysis
For western blot analyses, murine skin and squamous cell carcinomas (SCCs) were obtained from four different wild-type and hemizygous TSP-2 transgenic mice and were homogenized as described [20]. Protein concentrations were determined using the Bio-Rad protein assay (Bio-Rad, Hercules, CA). Samples were boiled in denaturating Laemmli sample buffer (Bio-Rad) with β-mercaptoethanol. Thirty microliters of each sample were electrophoresed on 12% SDS polyacrylamide gels and blotted onto nitrocellulose membranes (Bio-Rad). Membranes were incubated overnight in phosphate-buffered saline (PBS) containing 5% skim milk to block nonspecific binding and were then incubated with a goat anti-mouse VEGF-A antibody (R&D Systems, Minneapolis, MN) or a polyclonal rabbit anti mouse TSP-2 antibody (kindly provided by Paul Bornstein, University of Washington, Seattle, WA) After incubation with horseradish peroxidase-conjugated anti-goat IgG or anti-rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, PA), immunoreactive proteins were visualized using a chemiluminescence detection system (ECL; Amersham). To confirm equal loading, the same membranes were stained with Naphthol Blue Black solution (Sigma, St. Louis, MO).
Chemical skin carcinogenesis regimen
For tumor initiation, 50 µg of DMBA (Sigma), dissolved in 200 µl acetone, were topically applied to the shaved back skin of 8-weeks-old female hemizygous TSP-2 transgenic mice (n=25) and their wild-type littermates (n=25), followed by weekly topical application of 5 µg of the tumor promoter PMA (Sigma) over 20 weeks, as described [21] [22] In addition, 5 wild-type and 5 TSP-2 transgenic mice each were treated either with PMA alone or with a single application of DMBA. Raised lesions of a minimum diameter of 1 mm which had been present for at least one week were recorded as tumors. Mice were sacrificed after 32 weeks or earlier if tumors reached 10 mm in diameter or became ulcerated. The ratio of malignant conversion was calculated as the total number of squamous cell carcinomas (SCC) divided by the total number of papillomas, expressed as a percentage. The two-sided unpaired Student’s t-test was used to analyze differences in the number of tumors per mouse between the two genotypes. In vivo metastasis data were evaluated using the Mann-Whitney test and the Alternate Welch test. All animal studies were approved by the Massachusetts General Hospital Subcommittee on Research Animal Care.
Histology and immunohistochemistry
Immunofluorescence stains were performed on 6 µm frozen sections of biopsies obtained from normal untreated skin (n=9 for each genotype), small papillomas (1–3 mm in diameter; n=11), large papillomas (>3 mm in diameter; n=11), and SCCs (n=10), using antibodies against murine TSP-2 (P. Bornstein), mouse CD31 (Pharmingen, San Diego, CA), mouse LYVE-1 (kindly provided by Dr. David Jackson, Oxford, UK) or desmin (Chemicon, Temecula, CA) and with corresponding secondary antibodies labeled with Alexa Fluor 488 or 594 (Molecular Probes, Eugene, Oregon). For immunohistochemical stains, frozen sections were incubated with antibodies to mouse keratin 6, 10, 14 or loricrin (Babco, Richmond, CA), followed by biotin-conjugated secondary antibodies. Labeling was visualized using the ABC kit (Vector Laboratories, Burlingame, CA) and DAB (Zymed Laboratories Inc., San Francisco, CA) according to the manufacturer’s instructions. Papillomas and SCC were diagnosed histologically on hematoxylin/eosin stained tissue sections. Representative micrographs of a small papilloma, a large papilloma and a SCC are shown in Figure 2 G–I.
Figure 2. Delayed and reduced skin carcinogenesis in TSP-2 transgenic mice.

Delayed and diminished incidence of papillomas (small papillomas, SP, and large papillomas, LP) in TSP-2 transgenic mice (n=25, filled circles), as compared with wildtype mice (n=25, open squares). The incidence of papillomas is expressed as percentage of mice with detectable tumor formation >1mm (A). Significantly decreased frequency of papilloma formation, expressed as the average number of papillomas (SP and LP) per mouse, in TSP-2 transgenic mice (B). Markedly reduced incidence of large papillomas (LP, >3mm) in TSP-2 transgenic mice (C). Decreased average number of LP per mouse (D). Delayed and decreased incidence of squamous cell carcinomas (SCC) in TSP-2 transgenic mice (E). Decreased average number of SCCs per mouse in TSP-2 transgenic mice (F). H&E staining of representative small papilloma (G), large papilloma (H) and squamous cell carcinoma (I). The inserts in panel I reveal severe dysplastic cells and invasion of tumor cells through the basement membrane.
In situ hybridization
In situ hybridization was performed on 6 µm paraffin sections obtained from untreated skin (n=8 for each genotype), small papillomas (n=10), large papillomas (n=8) and SCCs (n=8) of wild-type and TSP-2 transgenic mice as described [23, 24]. An RNA probe to mouse TSP-2 was transcribed from a pBluescript II KS+ vector containing a 350-bp fragment of the coding region that is specific for mouse TSP-2 but not TSP-1 [15]. Antisense and sense single-stranded 35S RNA probes for VEGF-A were prepared from a 393 bp PCR rat VGF-A cDNA fragment, cloned into pGEM-3Zf(+) [19]. Transcription reactions were carried out using the Riboprobe Gemini II kit (Promega) in the presence of (α-35S) UTP. Anti-sense and control sense probes were evaluated on alternate sections.
Morphometric vessel analyses
Computer-assisted vessel analysis was performed on CD31/LYVE-1 double-stained frozen sections, obtained from biopsies of normal untreated skin (n=9 per genotype), small papillomas (n=11), large papillomas (n=11), and SCCs (n=10) as described [20], using the IPLab software (Scanalytics, Billerica, MA). The extent of pericyte coverage of tumor vessels was determined on CD31/desmin double-stained sections of small papillomas and SCC (n=3 for each type of tumor and each genotype). In 5 to 10 representative areas of each specimen, the number of CD31-positive vessels that were associated with desmin-positive pericytes was determined and expressed as a percentage of CD31-positive vessels. Values were expressed as means±SEM. Statistical analysis was performed using the two-sided unpaired Student’s t-test.
Cell proliferation and apoptosis assays
Two hours prior to sacrifice, mice were injected intraperitoneally with 40 mM bromodeoxyuridine (BrdU, Sigma) in 200 µl of 0.95% NaCl solution. Papillomas were obtained from TSP-2 transgenic (n=6) and wild-type (n=6) mice. Proliferating tumor cells were visualized by double immunofluorescence stains with an FITC-conjugated anti-BrdU antibody (Pharmingen) and a rabbit antibody to mouse keratin K14, and were counted in an area overlying a total length of 43.7 mm of basement membrane in TSP-2 transgenic mice and of 55.8 mm in wild-type mice as described [15]. Apoptotic cells were identified by TUNEL staining using the Fluorescein-FragEL DNA fragmentation kit (Oncogene, Cambridge, MA) according to the manufacturer's instructions. Nuclei were stained with propidium iodide in PBS (2 µg/ml). Labeled tumor cells were counted in an area overlying a total length of 14.7 mm of basement membrane in TSP-2 transgenic mice and of 19.5 mm in wild-type mice. Results are expressed as the mean ± SEM of BrdU- or TUNEL-positive nuclei per mm basement membrane. The two-sided unpaired Student’s t-test was used to analyze differences of proliferation and apoptosis rates.
Results
Delayed and reduced skin carcinogenesis in TSP-2 transgenic mice
We subjected hemizygous TSP-2 transgenic mice and their wild-type littermates to a standard two-step skin carcinogenesis protocol by using topical application of a single dose of DMBA for tumor initiation, followed by 20 weekly topical applications of PMA for tumor promotion. TSP-2 transgenic mice showed delayed onset of pre-malignant skin papillomas, with an average latency period of 18 weeks after the first PMA application, as compared with 13.5 weeks in wild-type mice (Fig. 2A). The first small papillomas were visible after 7 weeks in wildtype mice and after 8 weeks in TSP-2 transgenic mice (Fig. 2A). At 18 weeks, 100% of the wild-type mice but only 52% of the TSP-2 transgenic mice showed visible tumor formation. The number of epithelial tumors per mouse was significantly decreased in TSP-2 transgenic mice, as compared with wild-type mice. After 20 weeks of PMA promotion, TSP-2 transgenic mice had developed less than 3 papillomas per mouse, as compared with more than 6 papillomas in wild-type mice (P<0.001, Fig. 2B). The evaluation of larger papillomas (>3 mm) revealed that the first large papilloma developed after 14 weeks of PMA promotion in TSP-2 transgenic mice, 3 weeks later than in wild-type mice (Fig. 2C). Following 20 weeks of PMA promotion, the incidence of large papillomas was 73% in wild-type mice, as compared with only 39% in TSP-2 transgenic mice (Fig. 2C), with a more than 3-fold increased average number of large papillomas in wild-type mice (2.04 large papillomas per mouse) compared with TSP-2 transgenic littermates (0.68 large papillomas per mouse, Fig. 2C), indicating that papilloma growth was significantly delayed in the skin of TSP-2 transgenic mice. Malignant conversion of papillomas to SCC was first detected at 17 weeks after TPA promotion in wild-type mice and after 23 weeks in TSP-2 transgenic mice (Fig. 2E). After 32 weeks, 41% of TSP-2 transgenic mice had developed SCC as compared with 55% of wild-type mice. The average number of SCC per mouse was significantly decreased in TSP-2 transgenic mice (0.45+0.15 at week 32; P<0.01), as compared with wild-type mice (0.86+0.19; Fig. 2F). However, the ratio of malignant conversion of papillomas to SCC was comparable in wild-type (13.9%) and TSP-2 transgenic mice (15.6%; Fig. 2G). No tumors were observed in wild-type or TSP-2 transgenic mice treated with DMBA or with PMA alone (data not shown). Representative micrographs of H&E stained sections of a small papilloma, a large papilloma and a SCC are shown in Fig. 2 G–I.
Pronounced TSP-2 expression is maintained in tumors of TSP-2 transgenic mice
In situ hybridization revealed high levels of TSP-2 mRNA expression in the untreated skin of TSP-2 transgenic mice (Fig. 3B, D) compared with low or undetectable TSP-2 mRNA expression in the skin of wild-type littermates (Fig. 3A, C). High levels of TSP-2 mRNA expression were maintained in SCC of TSP-2 transgenic mice (Fig.3F, H). In contrast, little or no TSP-2 mRNA expression was found in SCC of wild-type mice (Fig. 3E, G). Immunofluorescence stains for TSP-2 demonstrated pronounced deposition of TSP-2 protein in the stroma of TSP-2 transgenic SCC (Fig. 3J) as compared with scarce protein detection in wild-type SCC (Fig. 3I). In accordance, Western blot analysis revealed high levels of TSP-2 in the normal skin and SCCs of TSP-2 transgenic mice, whereas only moderate expression of TSP-2 was detected in the tumors of wild-type mice (Fig. 3 K).
Decreased tumor angiogenesis in TSP-2 transgenic mice
Computer-assisted morphometric analyses of skin sections stained for the panendothelial marker CD31 and the lymphatic marker LYVE-1 showed comparable vessel density in the untreated skin of wild-type (Fig.4A) and TSP-2 transgenic mice (Fig. 4B). Evaluation of papillomas revealed that the ‘angiogenic switch’ from vascular quiescence to induction of angiogenesis was already detected at the early stages of epithelial tumorigenesis, as evidenced by the increased vessel density in pre-malignant, small papillomas. As shown by comparative analysis of CD31-positive vessels in early-stage papillomas, the extent of tumor angiogenesis was less pronounced in TSP-2 transgenic mice (Fig. 4D) compared to wild-type mice (Fig. 4C). TSP-2 transgenic mice also showed reduced numbers of large angiogenic vessels. Morphometric image analysis revealed that both the vessel density and the average vessel size were decreased in early papillomas in TSP-2-transgenic mice, as compared with wild-type mice, resulting in a significant reduction of the relative area occupied by tumor blood vessels (11.7 + 0.46 in wild-type mice, 8.6 + 0.55 in TSP-2 transgenic mice, p< 0.001, Fig. 4G).
Figure 4. Diminished tumor angiogenesis in TSP-2 transgenic mice.

Differential immunofluorescence analyses of untreated skin for CD31 (red) and LYVE-1 (green) demonstrate comparable vascularization in wild-type (A) and TSP-2 transgenic skin (B). Increased density of CD31+ vessels in small papillomas (SP; C, D) and squamous cell carcinomas (SCC; E, F) in both genotypes is accompanied by insignificant alterations of LYVE-1+ vessels. Tumor angiogenesis was less pronounced in TSP-2 transgenic mice with a prominent reduction of enlarged angiogenic vessels. Bar = 100 µm. Quantitative image analysis of CD31-stained vessels revealed a significant decrease of the average vascular area in TSP-2 transgenic mice (filled bars) already at early stages of papilloma formation and throughout the progressive stages of skin carcinogenesis, as compared with wild-type mice (open bars, G). Quantitative image analysis of LYVE-1+ lymphatic vessels revealed a comparable average lymphatic vascular area in untreated skin and throughout the development of skin tumors in wild type (open bars) and TSP-2 transgenic mice (filled bars, H). skin; untreated skin (n=9), small papilloma (SP <3mm, n=11), large papilloma (LP >3mm, n=11), squamous cell carcinoma (SCC, n=11). Data are expressed as mean values+SEM., ***p<0.001.
Whereas the relative area occupied by tumor blood vessels continuously increased during tumor progression in wild-type mice (14.7+0.76 in large papillomas and 16.8+0.9 in SCC, Fig. 4G), the angiogenic response remained at significantly (p<0.001) lower levels in TSP-2 transgenic mice (relative area occupied by tumor blood vessels 7.9+0.6 in large papillomas and 9.4s+0.4 in SCC, Fig. 4G), indicating that the anti-angiogenic effect of TSP-2 was sustained during tumor progression. Computer-assisted morphometric analysis of lymphatic vessels revealed a comparable density of LYVE-1 positive vessels in tumors of TSP-2 transgenic and wild-type mice (Fig.4 H).
Comparable VEGF expression in tumors of both genotypes
To investigate whether TSP-2 overexpression might have resulted in reduced levels of VEGF-A expression, we next performed in situ hybridization analyses of VEGF-A mRNA expression. In situ hybridization revealed comparable VEGF mRNA expression levels in SCCs of wild-type (Fig. 5A,C) and of TSP-2 transgenic mice (Fig. 5 B, D). Moreover, Western blot analyses of SCCs did not reveal any major differences in VEGF protein levels between wild-type and TSP-2 transgenic mice. Equal protein loading was confirmed by blotting for actin (Fig. 5E).
Figure 5. Comparable expression of VEGF-A in wild-type and TSP-2 transgenic mice.

In situ hybridization revealed comparable VEGF mRNA expression by SCCs of wild-type (A, C) and TSP-2 transgenic mice (B, D). Bright-field (A, B) and dark-field (C, D) micrographs, Bar = 100 µm. Western Blot confirmed comparable VEGF-A protein expression in SCCs of wild-type and TSP-2 transgenic mice (E). Equal protein loading was confirmed by staining with Naphthol Blue Black solution (data not shown).
Increased tumor cell apoptosis in TSP-2 transgenic mice
In agreement with our previous results [24], decreased angiogenesis resulted in significantly increased numbers of apoptotic tumor cells in TSP-2 transgenic as compared to wild-type mice. Tunel assays revealed a more than 2.8-fold increase of apoptotic keratinocyte nuclei/mm basement membrane in TSP-2 transgenic papillomas (66+6.9; Fig. 6A), as compared with wild-type tumors (23+2.8, p<0.001; Fig. 6 B,C). No major differences of tumor cell proliferation were detected in wild-type (Fig. 6D) and TSP-2 transgenic mice (Fig. 6E, F).
Figure 6. Increased number of apoptotic cells in TSP-2 transgenic tumors.

Tunnel assays demonstrated a significant increase in the number of apoptotic epithelial cells per mm basement membrane in TSP-2 transgenic papillomas (filled bar, B), as compared with wild-type tumors (open bar, A). No significant difference was found in the number of BrdU labeled proliferating tumor cells in wild-type (D) and TSP-2 transgenic mice (E, F). Data are expressed as mean values+SEM. ***P<0.001. Tunnel assay (n=11), proliferation assay (n=12).
Comparable pattern of epithelial differentiation in tumors of wild-type and TSP-2 transgenic mice
We next evaluated the expression of epidermal differentiation markers including keratin 6, 10, and loricrin in papillomas and SCC of wild-type and TSP-2 transgenic mice. Hyperproliferation-associated keratin 6 was abundantly expressed in all tumors of wild-type and TSP-2 transgenic mice (Fig. 7 A–D). The expression of the early differentiation marker keratin 10 and the late differentiation marker loricrin was pronounced in papillomas of both genotypes (Fig. 7 E, F, I, J), whereas in SCC an equally reduced expression of these markers was observed in wild-type (Fig. 7 G, H) and TSP-2 transgenic mice (Fig. 7 K, L).
Figure 7. Comparable expression of epidermal differentiation markers in wild-type and transgenic mice.

Immunohistochemical stains demonstrated equally strong expression of the proliferation-associated cytokeratin 6 (K6) in all stages of skin carcinogenesis in wild-type (A, C) and TSP-2 transgenic mice (B, D). The early differentiation marker K10 and the late differentiation marker loricrin were abundantly expressed in early, small papillomas of both genotypes (E, F, I, J) and were comparably decreased in SCCs of wild-type and transgenic mice (G, H, K, L). Bar = 100 µm.
Transgenic TSP-2 overexpression does not enhance vessel maturation during tumor progression
To explore whether transgenic TSP-2 might have effects on tumor vessel maturation, we evaluated the percentage of blood vessels enveloped by pericytes. Immunohistochemical double labeling for CD31 and for the pericyte marker desmin confirmed a pronounced reduction of tumor vessels in papillomas and SCC of TSP-2 overexpressing mice (Fig. 8 B, D) compared to wild-type littermates (Fig. 8 A, C). Importantly, in both genotypes, more than 90% of vessels were associated with pericytes in papillomas (95.7%±1 in wildtype, 91.3±1.9 in TSP-2 transgenic mice) and SCCs (Fig. 8 E).
Figure 8. Comparable expression of desmin associated CD31-positive vessels both in wild-type and transgenic mice.

Differential immunofluorescence stains for CD31 and the pericyte marker desmin showed that the majority of vessels were surrounded by pericytes in papillomas (A, B) and SCC (C, D) of both genotypes. Bar = 50 µm. Quantification of desmin-associated CD31-positive vessels demonstrated a comparable percentage of desmin-positive vessels (E). Values expressed as mean+SEM; n.s.: not significant).
Discussion
It is well established that tumor growth and progression critically depend on angiogenesis, i.e. the formation of new tumor associated blood vessels. In recent years, several proteins have been demonstrated to promote or inhibit angiogenic processes in vitro and in vivo [25, 26]. Previously, VEGF has been identified as the major tumor angiogenesis factor in the vast majority of human malignancies and in experimental tumor models [27, 28]
However, there is increasing evidence that tumors acquire the ability to secrete additional angiogenic factors which orchestrate the growth of newly formed vessels during tumor progression. Accordingly, therapies aiming to block VEGF activity have been found to reduce tumor progression in experimental models [29, 30], but clinical studies with anti-VEGF therapies have shown relatively modest efficiency in human malignancies [31]. Endogenous inhibitors of angiogenesis are thought to outweigh the pro-angiogenic stimulators and maintain the vascular quiescence by directly blocking tumor angiogenesis. One of the first proteins shown to inhibit angiogenesis was thrombospondin, later designated as TSP-1 upon the discovery of additional members of the TSP family [32]. Of the five proteins that comprise the family, only TSP-1 and -2 have been shown to exert influence on vascular development and angiogenesis. TSP-1 and -2 suppress the angiogenic response by antagonizing VEGF-induced cell survival and by inducing apoptotic pathways in blood vessels [8, 33]. An inhibitory role of TSP-2 in angiogenesis has been demonstrated by its effects on sprouting blood vessels in the chick chorioallantoic membrane assay [11]. Targeted disruption of the TSP-2 gene resulted in increased density of blood vessels in several organs of TSP-2 deficient mice [34], in enhanced angiogenesis induced by foreign body implantation [13], and in increased vascularization of full thickness wounds [14]. Moreover, TSP-2 deficient mice displayed pronounced inflammatory response in experimental DTH reactions [35].
There is increasing evidence for the importance of TSP-2 in tumor-associated angiogenesis, as demonstrated in several in vivo models. Circulating levels of TSP-2 have been increased by implanting biodegradable polymers containing fibroblasts that were engineered to overexpress TSP-2. These increased TSP-2 serum levels persisted for up to 5 weeks and were sufficient to inhibit experimental squamous cell carcinomas, Lewis lung carcinomas and melanomas [36]. Moreover, systemic treatment of mice with a recombinant protein that contains the N-terminal 80 kDa fragment of human TSP-2 inhibited experimental squamous cell carcinoma [16] and breast cancer growth [37] . Moreover, we previously found a profound upregulation of TSP-2 during multistep carcinogenesis and an enhanced susceptibility to skin carcinogenesis in TSP-2 deficient mice, accompanied by accelerated and increased tumor formation [15].
Our findings reveal that TSP-2 overexpressing mice show a significantly delayed tumor development and a dramatically reduced incidence of papillomas and squamous cell carcinomas compared to wild-type mice. The extent of tumor formation in wild-type mice was comparable to previously reported results in the FVB/J genetic background [15, 21]. Our data reveal that the transgenic overexpression of TSP-2 leads to the delayed onset of early premaligant tumors in skin carcinogenesis. However, taking into account the higher number of papillomas in the wild-type mice, the malignant conversion rate of benign lesions to SCC was almost identical in TSP-2 tansgenic and wild-type mice. This result indicates that the abundance of an antiangiogenic factor in the skin is not sufficient to prevent the genetic changes involved in the progression from premalignant to malignant neoplasia.
Our findings indicate that TSP-2 leads to a delayed growth of premalignant tumors but does not abolish the emergence of malignant cutaneous tumors. In line with the observation that TSP-2 inhibits early stages of carcinogenesis but not invasive tumors TGF-β (transforming growth factor beta) was shown to stimulate the synthesis of TSP-2 [38]. It is known to be a pro-apoptotic and anti-inflammatory agent in early stages of carcinogenesis. In case of late malignant events, TGF-β may promote transformation, tumor aggressiveness and metastatic capability [39]. Therefore, one underlying mechanism of the restricted inhibitory role of TSP-2 in early tumor development might be a mediated by the TGF-β signaling pathway. Beside endothelial cells, the microenvironment is composed of fibroblasts, pericytes, leukocytes, and extra-cellular matrix which might provide additional pro-neoplastic effects [40].
Keratin 14 expression is correlated with the proliferative activity of keratinocytes - there is a much stronger K14 promoter activity in hyperplastic epidermis, whereas the transgene expression, driven by K14, is rather low in normal skin. Therefore, the differential expression of TSP-2 in TSP-2 transgenic versus wildtype skin is much more pronounced in the hyperproliferative epidermis of papillomas and SCC than in normal skin. This explains the potent inhibitory effect of TSP-2 on tumor angiogenesis in K14/TSP-2 transgenic mice, in the absence of major alterations of the cutaneous vasculature in untreated skin. Our data are in agreement with previous findings that specific overexpression of TSP-1 in the epidermis preferentially interfered with wound healing associated angiogenesis, rather than with the angiogenesis associated with normal development and skin homeostasis [24]. Importantly, we found that the induction of angiogenesis occurred already in small papillomas in both genotypes, indicating that the angiogenic switch is turned on already in early stages of premalignant neoplastic development. However, TSP-2 overexpression led to a profound reduction of tumor-associated blood vessels in early premalignant small papillomas as well as in large papillomas and SCC. Our data demonstrate that despite reduced angiogenesis, tumor cell proliferation was comparable in wild-type and TSP-2 overexpressing SCCs. The correlation of diminished angiogenesis and retarded onset of papillomas in our study is in accordance with findings by others, showing that angiogenesis is a rate-limiting step in multistage carcinogenesis [42]. The significantly enhanced apoptosis in the tumors of TSP-2 transgenic mice is in line with the observation that therapeutic antiangiogenesis induces apoptosis [43] and supports the concept that tumor angiogenesis may act as a paracrine regulator of tumor apoptosis [25].
Our results demonstrate that the reduced vascularization in tumors of TSP-2 transgenic mice was not attributable to an interference of transgenic TSP-2 with VEGF, since in situ hybridization and western blotting revealed comparable VEGF mRNA and protein expression levels in both genotypes. These results are consistent with previous findings that overexpression of TSP-2 in SCC xenotransplants maintained VEGF expression at high levels [2]. It is of interest that transgenic overxpression of TSP-2 did not inhibit tumor-associated lymphangiogenesis as evidenced by morphometric analysis of immunostains for the lymphatic-specific marker LYVE-1. These observations are in accordance with previously published results [20] [16], indicating that thrombospondins have no overt effect on lymphatic vessels. This is most likely attributable to the scant expression of the TSP receptor CD36 on lymphatic endothelium.
Pericyte coverage of tumor endothelial cells is an indicator of vessel maturation [44, 45]. Because mature vessels with periendothelial support are a critical determinant for blood flow in the microvasculature, we addressed the issue of vessel maturation by analyzing periendothelial cell coverage of tumor endothelial cells. Double immunofluorescence stains for CD31 and for the pericyte marker desmin revealed a comparable percentage of CD31-positive blood vessels surrounded by desmin positive pericytes in tumors of both wild-type and TSP-2 transgenic mice. Our data are in accordance with recently published results showing a similar amount of desmin decorated blood vessels in the tumors of a murine melanoma model [46]. They also indicate that transgenic overexpression of TSP-2 in the skin exerts anti-angiogenic effects without overt influence on vessel maturation. Taken together, our data demonstrate that transgenic overexpression of TSP-2 in the skin exerts anti-angiogenic effects leading to the prevention of premalignant skin tumors but has no significant effect on the conversion into SCCs. With regard to cutaneous malignancies, angiogenesis inhibition thus appears to be more suitable in the treatment of premalignant lesions or as an adjuvant treatment in patients with low tumor load. Therefore, anti-angiogenic treatment likely should be tailored to specific tumor stages and be combined with other targeted treatment approaches.
Acknowledgements
This work was supported by National Institutes of Health grant CA69184, Swiss National Foundation grants 3100A0-108207 and 31003A-130627, Advanced European Research Council Grant LYVICAM, Krebsliga Schweiz and Krebsliga Zurich (to M.D.), the Jubilaeumsfonds of the Austrian Federal Reserve Bank grant 12747 (to R.K.) and Deutsche Forschungsgemeinschaft grants HA 2898 4-1 and 4-2 (to T.H.). The authors wish to thank Dr. Martin Mihm and Dr. Sam Dadras for critically evaluating H&E stained sections of skin tumors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have no conflict of interest to declare.
References
- 1.Weinstat-Saslow DL, Zabrenetzky VS, VanHoutte K, Frazier WA, Roberts DD, Steeg PS. Transfection of thrombospondin 1 complementary DNA into a human breast carcinoma cell line reduces primary tumor growth, metastatic potential, and angiogenesis. Cancer Res. 1994;54:6504–6511. [PubMed] [Google Scholar]
- 2.Streit M, Riccardi L, Velasco P, Brown LF, Hawighorst T, Bornstein P, et al. Thrombospondin-2: a potent endogenous inhibitor of tumor growth and angiogenesis. Proc Natl Acad Sci U S A. 1999;96:14888–14893. doi: 10.1073/pnas.96.26.14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O'Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, et al. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994;79:315–328. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 4.O'Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 5.Pike SE, Yao L, Jones KD, Cherney B, Appella E, Sakaguchi K, et al. Vasostatin, a calreticulin fragment, inhibits angiogenesis and suppresses tumor growth. J Exp Med. 1998;188:2349–2356. doi: 10.1084/jem.188.12.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maeshima Y, Sudhakar A, Lively JC, Ueki K, Kharbanda S, Kahn CR, et al. Tumstatin, an endothelial cell-specific inhibitor of protein synthesis. Science. 2002;295:140–143. doi: 10.1126/science.1065298. [DOI] [PubMed] [Google Scholar]
- 7.Bornstein P, Devarayalu S, Li P, Disteche CM, Framson P. A second thrombospondin gene in the mouse is similar in organization to thrombospondin 1 but does not respond to serum. Proc Natl Acad Sci U S A. 1991;88:8636–8640. doi: 10.1073/pnas.88.19.8636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bornstein P. Thrombospondins function as regulators of angiogenesis. J Cell Commun Signal. 2009;3:189–200. doi: 10.1007/s12079-009-0060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kyriakides TR, Zhu YH, Yang Z, Bornstein P. The distribution of the matricellular protein thrombospondin 2 in tissues of embryonic and adult mice. J Histochem Cytochem. 1998;46:1007–1015. doi: 10.1177/002215549804600904. [DOI] [PubMed] [Google Scholar]
- 10.Iruela-Arispe ML, Liska DJ, Sage EH, Bornstein P. Differential expression of thrombospondin 1, 2, and 3 during murine development. Dev Dyn. 1993;197:40–56. doi: 10.1002/aja.1001970105. [DOI] [PubMed] [Google Scholar]
- 11.Volpert OV, Tolsma SS, Pellerin S, Feige JJ, Chen H, Mosher DF, et al. Inhibition of angiogenesis by thrombospondin-2. Biochem Biophys Res Commun. 1995;217:326–332. doi: 10.1006/bbrc.1995.2780. [DOI] [PubMed] [Google Scholar]
- 12.Murphy-Ullrich JE, Gurusiddappa S, Frazier WA, Hook M. Heparin-binding peptides from thrombospondins 1 and 2 contain focal adhesion-labilizing activity. J Biol Chem. 1993;268:26784–26789. [PubMed] [Google Scholar]
- 13.Kyriakides TR, Leach KJ, Hoffman AS, Ratner BD, Bornstein P. Mice that lack the angiogenesis inhibitor, thrombospondin 2, mount an altered foreign body reaction characterized by increased vascularity. Proc Natl Acad Sci USA. 1999;96:4449–4454. doi: 10.1073/pnas.96.8.4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kyriakides TR, Tam JW, Bornstein P. Accelerated wound healing in mice with a disruption of the thrombospondin 2 gene. J Invest Dermatol. 1999;113:782–787. doi: 10.1046/j.1523-1747.1999.00755.x. [DOI] [PubMed] [Google Scholar]
- 15.Hawighorst T, Velasco P, Streit M, Kyriakides TR, Brown LF, Bornstein P, et al. Thrombospondin-2 plays a protective role in multistep carcinogenesis: a novel host antitumor defense mechanism. EMBO J. 2001;20:2631–2640. doi: 10.1093/emboj/20.11.2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noh YH, Matsuda K, Hong YK, Kunstfeld R, Riccardi L, Koch M, et al. An N-terminal 80 kDa recombinant fragment of human thrombospondin-2 inhibits vascular endothelial growth factor induced endothelial cell migration in vitro and tumor growth and angiogenesis in vivo. J Invest Dermatol. 2003;121:1536–1543. doi: 10.1046/j.1523-1747.2003.12643.x. [DOI] [PubMed] [Google Scholar]
- 17.Ito N, Hasegawa R, Imaida K, Hirose M, Asamoto M, Shirai T. Concepts in multistage carcinogenesis. Crit Rev Oncol Hematol. 1995;21:105–133. doi: 10.1016/1040-8428(94)00169-3. [DOI] [PubMed] [Google Scholar]
- 18.Vassar R, Rosenberg M, Ross S, Tyner A, Fuchs E. Tissue-specific and differentiation-specific expression of a human K14 keratin gene in transgenic mice. Proc Natl Acad Sci U S A. 1989;86:1563–1567. doi: 10.1073/pnas.86.5.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Detmar M, Brown LF, Schön MP, Elicker BM, Velasco P, Richard L, et al. Increased microvascular density and enhanced leukocyte rolling and adhesion in the skin of VEGF transgenic mice. J Invest Dermatol. 1998;111:1–6. doi: 10.1046/j.1523-1747.1998.00262.x. [DOI] [PubMed] [Google Scholar]
- 20.Hawighorst T, Oura H, Streit M, Janes L, Nguyen L, Brown LF, et al. Thrombospondin-1 selectively inhibits early-stage carcinogenesis and angiogenesis but not tumor lymphangiogenesis and lymphatic metastasis in transgenic mice. Oncogene. 2002;21:7945–7956. doi: 10.1038/sj.onc.1205956. [DOI] [PubMed] [Google Scholar]
- 21.Hirakawa S, Kodama S, Kunstfeld R, Kajiya K, Brown LF, Detmar M. VEGF-A induces tumor and sentinel lymph node lymphangiogenesis and promotes lymphatic metastasis. J Exp Med. 2005;201:1089–1099. doi: 10.1084/jem.20041896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirakawa S, Brown LF, Kodama S, Paavonen K, Alitalo K, Detmar M. VEGF-Cinduced lymphangiogenesis in sentinel lymph nodes promotes tumor metastasis to distant sites. Blood. 2007;109:1010–1017. doi: 10.1182/blood-2006-05-021758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Detmar M, Brown LF, Claffey KP, Yeo KT, Kocher O, Jackman RW, et al. Overexpression of vascular permeability factor/vascular endothelial growth factor and its receptors in psoriasis. J Exp Med. 1994;180:1141–1146. doi: 10.1084/jem.180.3.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Streit M, Velasco P, Riccardi L, Spencer L, Brown LF, Janes L, et al. Thrombospondin-1 suppresses wound healing and granulation tissue formation in the skin of transgenic mice. EMBO J. 2000;19:3272–3282. doi: 10.1093/emboj/19.13.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 26.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 27.Giatromanolaki A, Koukourakis MI, Sivridis E, Chlouverakis G, Vourvouhaki E, Turley H, et al. Activated VEGFR2/KDR pathway in tumour cells and tumour associated vessels of colorectal cancer. Eur J Clin Invest. 2007;37:878–886. doi: 10.1111/j.1365-2362.2007.01866.x. [DOI] [PubMed] [Google Scholar]
- 28.Weigand M, Hantel P, Kreienberg R, Waltenberger J. Autocrine vascular endothelial growth factor signalling in breast cancer. Evidence from cell lines and primary breast cancer cultures in vitro. Angiogenesis. 2005;8:197–204. doi: 10.1007/s10456-005-9010-0. [DOI] [PubMed] [Google Scholar]
- 29.Drevs J, Hofmann I, Hugenschmidt H, Wittig C, Madjar H, Muller M, et al. Effects of PTK787/ZK 222584, a specific inhibitor of vascular endothelial growth factor receptor tyrosine kinases, on primary tumor, metastasis, vessel density, and blood flow in a murine renal cell carcinoma model. Cancer Res. 2000;60:4819–4824. [PubMed] [Google Scholar]
- 30.Shaheen RM, Tseng WW, Vellagas R, Liu W, Ahmad SA, Jung YD, et al. Effects of an antibody to vascular endothelial growth factor receptor-2 on survival, tumor vascularity, and apoptosis in a murine model of colon carcinomatosis. Int J Oncol. 2001;18:221–226. [PubMed] [Google Scholar]
- 31.Mita MM, Rowinsky EK, Forero L, Eckhart SG, Izbicka E, Weiss GR, et al. A phase II, pharmacokinetic, and biologic study of semaxanib and thalidomide in patients with metastatic melanoma. Cancer Chemother Pharmacol. 2007;59:165–174. doi: 10.1007/s00280-006-0255-0. [DOI] [PubMed] [Google Scholar]
- 32.Lawler JW, Slayter HS, Coligan JE. Isolation and characterization of a high molecular weight glycoprotein from human blood platelets. J Biol Chem. 1978;253:8609–8616. [PubMed] [Google Scholar]
- 33.Lawler J, Detmar M. Tumor progression: the effects of thrombospondin-1 and-2. Int J Biochem Cell Biol. 2004;36:1038–1045. doi: 10.1016/j.biocel.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 34.Kyriakides TR, Zhu YH, Smith LT, Bain SD, Yang Z, Lin MT, et al. Mice that lack thrombospondin 2 display connective tissue abnormalities that are associated with disordered collagen fibrillogenesis, an increased vascular density, and a bleeding diathesis. J Cell Biol. 1998;140:419–430. doi: 10.1083/jcb.140.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lange-Asschenfeldt B, Weninger W, Velasco P, Kyriakides TR, von Andrian UH, Bornstein P, et al. Increased and prolonged inflammation and angiogenesis in delayedtype hypersensitivity reactions elicited in the skin of thrombospondin-2--deficient mice. Blood. 2002;99:538–545. doi: 10.1182/blood.v99.2.538. [DOI] [PubMed] [Google Scholar]
- 36.Streit M, Stephen AE, Hawighorst T, Matsuda K, Lange-Asschenfeldt B, Brown LF, et al. Systemic inhibition of tumor growth and angiogenesis by thrombospondin-2 using cell-based antiangiogenic gene therapy. Cancer Res. 2002;62:2004–2012. [PubMed] [Google Scholar]
- 37.Koch M, Hussein F, Woeste A, Grundker C, Frontzek K, Emons G, et al. CD36-mediated activation of endothelial cell apoptosis by an N-terminal recombinant fragment of thrombospondin-2 inhibits breast cancer growth and metastasis in vivo. Breast Cancer Res Treat. 2011;128:337–346. doi: 10.1007/s10549-010-1085-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Negoescu A, Lafeuillade B, Pellerin S, Chambaz EM, Feige JJ. Transforming growth factors beta stimulate both thrombospondin-1 and CISP/thrombospondin-2 synthesis by bovine adrenocortical cells. Exp Cell Res. 1995;217:404–409. doi: 10.1006/excr.1995.1103. [DOI] [PubMed] [Google Scholar]
- 39.Gotzmann J, Huber H, Thallinger C, Wolschek M, Jansen B, Schulte-Hermann R, et al. Hepatocytes convert to a fibroblastoid phenotype through the cooperation of TGFbeta1 and Ha-Ras: steps towards invasiveness. J Cell Sci. 2002;115:1189–1202. doi: 10.1242/jcs.115.6.1189. [DOI] [PubMed] [Google Scholar]
- 40.Pietras K, Ostman A. Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res. 2010;316:1324–1331. doi: 10.1016/j.yexcr.2010.02.045. [DOI] [PubMed] [Google Scholar]
- 41.Franovic A, Holterman CE, Payette J, Lee S. Human cancers converge at the HIF-2alpha oncogenic axis. Proc Natl Acad Sci U S A. 2009;106:21306–21311. doi: 10.1073/pnas.0906432106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bergers G, Javaherian K, Lo KM, Folkman J, Hanahan D. Effects of angiogenesis inhibitors on multistage carcinogenesis in mice. Science. 1999;284:808–812. doi: 10.1126/science.284.5415.808. [DOI] [PubMed] [Google Scholar]
- 43.Holmgren L, O'Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med. 1995;1:149–153. doi: 10.1038/nm0295-149. [DOI] [PubMed] [Google Scholar]
- 44.Benjamin LE, Hemo I, Keshet E. A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development. 1998;125:1591–1598. doi: 10.1242/dev.125.9.1591. [DOI] [PubMed] [Google Scholar]
- 45.Abramsson A, Berlin O, Papayan H, Paulin D, Shani M, Betsholtz C. Analysis of mural cell recruitment to tumor vessels. Circulation. 2002;105:112–117. doi: 10.1161/hc0102.101437. [DOI] [PubMed] [Google Scholar]
- 46.Helfrich I, Scheffrahn I, Bartling S, Weis J, von Felbert V, Middleton M, et al. Resistance to antiangiogenic therapy is directed by vascular phenotype, vessel stabilization, and maturation in malignant melanoma. J Exp Med. 2010;207:491–503. doi: 10.1084/jem.20091846. [DOI] [PMC free article] [PubMed] [Google Scholar]