

Abstract
Purpose
EphA2 is an attractive therapeutic target due to its diverse roles in cancer growth and progression. Dasatinib is a multi-kinase inhibitor that targets EphA2 and other kinases. However, reliable predictive markers and a better understanding of the mechanisms of response to this agent are needed.
Experimental design
The effects of dasatinib on human uterine cancer cell lines were examined using a series of in vitro experiments, including MTT, Western blot, and plasmid transfection. In vivo, an orthotopic mouse model of uterine cancer was utilized to identify the biological effects of dasatinib. Molecular markers for response prediction and the mechanisms relevant to response to dasatinib were identified by using RPPA, immunoprecipitation, and double immunofluorescence staining.
Results
We show that high levels of CAV-1, EphA2 phosphorylation at S897 and the status of PTEN are key determinants of dasatinib response in uterine carcinoma. A set of markers essential for dasatinib response was also identified and includes CRaf, pCRafS338, pMAPKT202/Y204 (MAPK pathway), pS6S240/244, p70S6kT389 (mTOR pathway) and pAKTS473. A novel mechanism for response was discovered whereby high expression level of CAV-1 at the plasma membrane disrupts the BRaf/CRaf heterodimer and thus inhibits the activation of MAPK pathway during dasatinib treatment.
Conclusions
Our in vitro and in vivo results provide a new understanding of EphA2 targeting by dasatinib and identify key predictors of therapeutic response. These findings have implications for ongoing dasatinib-based clinical trials.
Keywords: Dasatinib, EphA2, Caveolin-1 (CAV-1), Uterine Cancer
Introduction
EphA2 is a receptor tyrosine kinase involved in many processes crucial to malignant progression (1–3). Overexpression of EphA2, which has been reported for many human cancers, including breast, melanoma, prostate, lung, ovarian (4, 5) and uterine cancers (6, 7), is often associated with poor prognostic features (8). For these reasons, EphA2 is considered an important therapeutic target. Various therapeutic strategies targeting EphA2 have been developed, including monoclonal antibodies, immunoconjugates, small-molecule tyrosine kinase inhibitors, vaccines, and RNA interference (9, 10). Among these, dasatinib is the farthest along with regard to clinical development, even though it was primarily developed as an oral dual inhibitor of Bcr/Abl and Src family kinases. Genomic and proteomic profiling have shown EphA2 to be a direct target of dasatinib (11–13). Dasatinib has shown activity in a fraction of patients with solid tumors (14–17); however, reliable predictors of response to this agent are not known.
Here, we sought to identify a set of markers in uterine carcinoma that are essential for dasatinib response and to investigate the underlying mechanisms of response based on EphA2 function. We discovered that caveolin-1 (CAV-1)-mediated cross-talk between EphA2 and BRaf is a key determinant of dasatinib response in uterine cancer cells. Moreover, EphA2 phosphorylation at S897 and the status of PTEN are required for response to dasatinib.
Materials and Methods
Cell Cultures
We selected uterine cancer cell lines HEC1-A, Ishikawa, SKUT-2, RL95-2 and KLE (all EphA2 positive) and SPEC-2, AN3CA and HEC-265 (all EphA2 negative). Cells were purchased from the MD Anderson Characterized Cell Line Core Facility (Houston, Texas), which supplies authenticated cell lines. The cell lines were routinely tested to confirm the absence of mycoplasma, and all experiments were performed with cell lines at 60%–80% confluence. Cells were maintained in specific culture medium as described previously (6). The molecular features of cell lines (EphA2 status, CAV-1 status, RAS mutation, and PTEN mutation) are listed in Fig 1a (left).
Figure 1. In vitro effects of dasatinib in uterine cancer cell lines.

(a) Expression level of EphA2 and CAV-1 and expression of wild-type or mutant RAS and PTEN for each cell line. −, No expression; +, low expression; +++, high expression (left). Median inhibitory concentration (IC50) after treatment with dasatinib (middle). Cell viability after treatment with dasatinib at 0, 10, 50, 100, 150, 1,000, 1,500, or 10,000 nM for 72 hours (right). Data represent means of triplicate measurements with error bars to represent SEM. (b) Immunoblot analysis of proteins associated with the Src/FAK/EphA2 pathway in cells treated with (+) or without (−) dasatinib at 100 nM for 16 hours (left). Quantification of band intensity relative to actin intensity is shown graphically. Black bars, no treatment. White bars, treatment (right). (c) Immunoprecipitation (IP) and Western blot(WB) analysis of tyrosine-phosphorylated EphA2 in HEC1-A, Ishikawa, and SKUT-2 cells treated with (+) or without (−) dasatinib at 100 nM for 16 hours. (d) Effect of wild-type (WT) EphA2 and pEphA2S897 status on the sensitivity of SKUT-2 cells to dasatinib at 0, 10, 50, 100, 150, 1,000, 1,500, or 10,000 nM for 72 hours. Cell viability assay was performed with SKUT-2 cells stably transfected with Myc-DDK-tagged plasmid with WT EphA2, inactivated pEphA2S897, or constitutively activated pEphA2S897 (left). Median inhibitory concentration (IC50) after treatment with dasatinib(right, top) at 100 nM for 16 hours. Western blotresults showing status of EphA2 and pEphA2S897 in SKUT-2 cells after transfection. Anti-Myc antibody was used as a marker for efficiency of transfection. β-Actin was used as a loading control(right, bottom). EV, empty vector.
MTT Assay
Inhibition of cell growth by dasatinib was detected using the MTT cell proliferation assay, a tetrazolium-based colorimetric assay performed in quadruplicate. Additional details regarding the treatment and MTT assay are provided in the Supplementary Methods (available online).
Western Blot Analysis and IP Analyses of Dasatinib Targets In Vitro
The expression levels of pEphA2S897, pSrcY416, pFAKY397, pFAKY925, pPaxillinY118, pAKTS473, pp130CasY410, CAV-1, and pS6S240/244 in the uterine cells lines were determined using Western blot analysis. Additional details regarding the Western blot analysis are provided in the Supplementary Methods (available online). To test the interaction between EphA2, BRaf, and CAV-1, whole-cell lysates of related cells before and after dasatinib treatment were immunoprecipitated with antibodies against EphA2 (Millipore, Billerica, MA and Santa Cruz Biotechnology, Santa Cruz, CA), BRaf, or CAV-1 (CST, Danvers, MA and Santa Cruz Biotechnology), and blots were detected with antibodies against them. Additional details regarding the IP assays are provided in the Supplementary Methods (available online).
Site-Directed Mutagenesis of EphA2 at Serine 897
We designed two types of point mutations at S897 of EphA2, one for inactivated pEphA2S897 (serine-897 to alanine) (5′-GCGTGTCTATCCGGCTCCCCGCCACGAGCGGCTCGGAGGGGGTG-3′ and 5′-CACCCCCTCCGAGCCGCTCGTGGCGGGGAGCCGGATAGACACGC-3′) and one for constitutively activated pEphA2S897 (serine-897 to glutamine) (5′-GCGTGTCTATCCGGCTCCCCGAAACGAGCGGCTCGGAGGGGGTG-3′ and 5′-CACCCCCTCCGAGCCGCTCGTTTCGGGGAGCCGGATAGACACGC-3′). The human cDNA open reading frame Myc-DDK-tagged clone of EPHA2 (NM_004431, Origene, Rockville, MD) was amplified using these primers containing the desired mutation by polymerase chain reaction. The product was digested with DPN1 for 1 hour to eliminate methylated DNA. The final mutant EphA2 was confirmed by sequencing (SeqWright, Houston, TX).
Double Immunofluorescence Staining
To test the dynamic changes in the localization of EphA2, CAV-1, and BRaf in SKUT-2 cells treated with dasatinib at 100 nM for 16 hours, double staining was carried out using the rabbit monoclonal antibody against CAV-1 (red) and mouse monoclonal antibody for BRaf (green) or EphA2 (red or green). The stained cells were visualized by confocal microscopy at ×200 magnification.
In Situ Proximity Ligation Assay
In situ PLA was performed according to manufacturer’s instructions (Olink Bioscience). Briefly, after incubation with primary antibodies, the cells were incubated with a combination of corresponding PLA probes, secondary antibodies conjugated to oligonucleotides (mouse MINUS and rabbit PLUS). Subsequently, ligase was added forming circular DNA strands when PLA probes were bound in close proximity, along with polymerase and oligonucleotides to allow rolling circle amplification. Fluoroscently labeled probes complementary in sequence to the rolling circle amplification product was hybridized to the rolling circle amplification product (Duolink Detection Kit 563; Olink Bioscience). Thus, each individual pair of proteins generated a spot (blob) that could be visualized using fluorescent microscopy at ×200 magnification.
Transfection of siRNA
SiRNA was purchased from Sigma-Aldrich (Woodlands, TX). A non-silencing siRNA that did not share sequence homology with any known human mRNA based on a BLAST search was used as control for target siRNA. For in vitro delivery, siRNA (5 μg) was incubated with 30 μL RNAiFect transfection reagent (Qiagen) for 10 minutes at room temperature and added to cells in culture at 80% confluence in 35 mm culture plates. The medium was changed 6 hours later, and cells collected after 48 hours as lysate for Western blot analysis.
Detection of Multiple Signaling Pathways by RPPA
SKUT-2, SPEC-2, HEC1-A, and Ishikawa cells were treated with 1 μM dasatinib and 5 nM paclitaxel for 16 hours. Samples were probed with 176 validated primary antibodies by RPPA at the M.D. Anderson Cancer Center RPPA Core Facility (see Supplementary Methods (available online) for additional details.
Orthotopic In Vivo Model of Uterine Cancer and Tissue Processing
All animal studies were approved and supervised by the MD Anderson Institutional Animal Care and Use Committee. Two uterine cancer cell lines, SPEC-2 (no EphA2 expression) and SKUT-2 (high EphA2 expression) were used for in vivo experiments as previously described (6). For in vivo therapy experiments, 10 mice were randomly allocated into four treatment groups: control, dasatinib (15 mg/kg oral, daily), paclitaxel (100 μg in 200 μL of PBS intraperitoneally, weekly), or dasatinib plus paclitaxel. Therapy was initiated 2 weeks after cell injection. Additional details are provided in the Supplementary Methods (available online).
Immunohistochemical Staining in the Mouse Uterine Tumor Samples
Paraffin-embedded tissues were used to detect cell proliferation (with Ki67) and apoptosis (with cleaved caspase 3). The sections were incubated with the Ki67 antibody (1:400; Dako) and monoclonal mouse antibody against cleaved caspase 3 (1:100; Biocare Medical, Concord, CA). CD31 staining with rat monoclonal anti-mouse CD31 (1:800, PharMingen, San Diego, CA) was performed on frozen sections. Additional details regarding IHC method are provided in the Supplementary Methods (available online).
Immunohistochemical Staining of Human Uterine Cancer Specimens
After approval by the MD Anderson Institutional Review Board, slides from forty cases of uterine cancer were obtained from the surgical pathology files of MD Anderson. For human uterine cancer samples, immunohistochemical analysis for CAV-1 (1:200 dilution; CST), pAKTS473 (1:100 dilution; CST), and pEphA2Y594 and pEphA2S897 (1:200 dilution; Cell Applications) was performed as described previously (18). See Suplementary Methods (available online) for additional details.
Statistical Analysis
For in vivo therapy experiments, 10 mice were used in each group, which provided the power to detect a 50% reduction in tumor size (β error = 0.2). Continuous variables were compared using Student’s t test (two groups) or analysis of variance (all groups) if the data were normally distributed. For non-parametric distributions, the Mann-Whitney U or Kruskal-Wallis test (all groups) was used. A value of p<0.05 with two-tailed testing was deemed statistically significant.
Results
In Vitro Effect of Dasatinib on EphA2-Positive or -Negative Uterine Cancer Cell Lines
We first assessed the effect of dasatinib on a panel of six uterine cancer cell lines with known EphA2 expression levels (Fig 1a, left). The median inhibitory concentration (IC50) of these cell lines ranged from 0.03 to 17.9 μM (Fig 1a, middle). Among these, EphA2-positive SKUT-2 cells were the most sensitive to dasatinib, whereas EphA2-negative SPEC-2 cells were the most resistant (Fig 1a, right), suggesting that EphA2 status is a potential determinant of dasatinib sensitivity. The exception to this pattern was the EphA2-positive HEC1-A cell line (harbors a RAS mutation), with a higher IC50 value than other EphA2-negative cells. These observations were further supported by EphA2-negative HEC-265 and EphA2-positive KLE uterine cancer cells, and by ectopic expression of EphA2 in the EphA2-negative A2780 ovarian cancer cells (Supplementary Figs S1a and S1b).
Wild-Type PTEN Increased Sensitivity to Dasatinib in EphA2 Positive Cell Lines In Vitro
We found that dasatinib exhibited less growth inhibition in EphA2 positive RL95-2 cells, which harbors high basal level of p-AKTS473. Therefore, next we addressed whether PTEN, which is frequently altered in uterine carcinoma (19), influences response to dasatinib through its effects on p-AKTS473. AKT activation and PTEN mutation status in a panel of uterine cancer cell lines is shown in Figs 1a and 1b. Next, we transfected wild-type or mutant PTEN into ishikawa cells carrying mutant PTEN. The dasatinib-sensitivity of ishikawa cells transfected with wild type PTEN was significantly enhanced by 5-fold, compared to that with parental cells carrying PTEN mutation (Supplementary Fig S2), suggesting that wild type PTEN is also a determinant of dasatinib sensitivity in uterine cancer cells.
In Vitro Effects of Dasatinib on Src/FAK/EphA2 Signaling Pathway
Next, we examined signaling events in the Src/FAK/EphA2 pathway known to be perturbed by dasatinib. The expression level of the traditional targets of dasatinib, including pAKTS473, pSrcY416, pFAKY925, pPaxillinY118, and pp130CasY410, were reduced in all six uterine cancer cell lines after dasatinib treatment, whereas pFAKY397 expression level was not notably affected in any of the cell lines (Fig 1b). These results support the known effects of dasatinib, but they do not explain the sensitivity of specific uterine cancer cell lines to it.
pEphA2S897 Status is an Important Determinant of Response to dasatinib in Uterine Cancer Cells
Given the differential expression of EphA2 in dasatinib-sensitive versus -resistant cell lines and the role of ligand-independent pEphA2S897 in many oncogenic functions (20), we next examined the effects of dasatinib on pEphA2S897. The expression of pEphA2S897 was significantly decreased in SKUT-2 cells, but moderately so in HEC1-A and Ishikawa cells (Fig 1b and Suplementary Fig S4). Furthermore, because the expression levels of pEphA2S897 and pEphA2Y594 are inversely related (20), EphA2 tyrosine phosphorylation was decreased in HEC1-A and Ishikawa cells, but increased in SKUT-2 cells, after dasatinib treatment (Fig 1c). The extent of inhibition was directly related to dasatinib response. To further test whether phosphorylation at S897 is critical for dasatinib response, we performed site-directed mutagenesis of pEphA2 at S897 and transfected SKUT-2 cells with plasmids for wild-type EphA2, inactivated pEphA2S897, or constitutively activated pEphA2S897. The cells with constitutively activated pEphA2S897 had reduced sensitivity to dasatinib, whereas the cells with inactivated pEphA2S897 remained highly sensitive to dasatinib (Fig 1d). These results confirmed that pEphA2S897 status is an important determinant of response to dasatinib in uterine cancer cells (Table S1).
Identification of Predictive Molecular Markers
To explore other potential markers of response to dasatinib, we used RPPA to quantify protein expression of genes involved in cell cycle, apoptosis, angiogenesis, and adhesion that are modulated by dasatinib in the SPEC-2, SKUT-2, HEC1-A, and Ishikawa cell lines (Fig 2, Supplementary Figs S3, S4 and Tables S3-S10). The expression levels of total CRaf, pCRafS338, pS6S240/244, p70S6kT389, pAKTS473, total EphA2, pEphA2S897, pMAPKT202/Y204, mTOR, and pMEKS217/221 were significantly decreased in the dasatinib-sensitive SKUT-2 cells but not in the dasatinib-resistant SPEC-2, HEC1-A, or Ishikawa cells. Moreover, KRAS and PI3K expression levels did not change significantly in SKUT-2 cells. Interestingly, a high basal level of CAV-1 was detected in SKUT-2 cells compared with the three other cell lines. We next carried out a series of experiments to identify potential mechanistic explanations for response to dasatinib-based therapy. In SKUT-2 cells, Western blot analysis validated the changes in the levels of pS6S240/244 and CRaf following dasatinib therapy; a high basal level of CAV-1 was detected only in SKUT-2 cells and CAV-1 and BRaf had no notable changes (Figs 3a and 3b). Next, we carried out immunoprecipitation (IP) studies with antibodies against EphA2, CAV-1, and BRaf. IP analysis showed direct binding between EphA2 and CAV-1, EphA2 and BRaf, and CAV-1 and BRaf in SKUT-2 cells treated with dasatinib (Fig 3c), but not in HEC1-A and Ishikawa cells (data not shown), or in the absence of dasatinib. Meanwhile, our IP analysis showed that dasatinib induced the heterodimer of BRaf/CRaf in Ishikawa cells, but not in SKUT-2 cells (Fig 3d), indicating that co-localization between BRaf and CAV-1 induced by dasatinib in SKUT-2 cells interferes with the formation of the BRaf/CRaf heterodimer and therefore can inactivate signaling downstream of BRaf/CRaf (MAPK pathway). The heterodimer of BRaf/CRaf was found in HEC1-A cells treated with or without dasatinib (Fig 3d), likely due to the fact that HEC1-A bears a RAS mutation and is more resistant to dasatinib treatment (21). To further explore the association between EphA2, CAV-1, and BRaf, we immunostained SKUT-2 cells for these proteins with or without dasatinib treatment. Immunofluorescence staining analysis showed that BRaf was translocated from the cytoplasm to the plasma membrane and co-localized with CAV-1 after dasatinib treatment (Figs 4a, left, and 4b, left). EphA2 was also translocated and co-localized with CAV-1 after dasatinib treatment (Fig 4a, right, top), and EphA2 and BRaf showed co-localization as well (Fig 4a, right, bottom). No co-localization between BRaf and CAV-1 was observed following dasatinib treatment in SPEC-2 cells (Fig 4b, right) or in HEC1-A and Ishikawa cells (data not shown). Furthermore, following CAV-1 gene silencing, the growth-inhibitory effects of dasatinib on SKUT-2 cells were attenuated (Fig 4c). There was no co-localization between BRaf and CAV-1 (Fig 4b, right) or between BRaf and EphA2 (data not shown) in SKUT-2 cells following CAV-1 silencing, indicating that a high level of CAV-1 is important for the response to dasatinib in tumor cells and required for recruiting BRaf in response to dasatinib therapy. EphA2 silencing resulted in partially increased sensitivity to dasatinib in SKUT-2 cells, but it did not alter the response to dasatinib in HEC1-A cells (Supplementary Fig S5), which suggests that EphA2 is also important for the response to dasatinib in tumor cells.
Figure 2. Expression of multiple proteins associated With Src/FAK/EphA2 and RAS/RAF/MAPK signaling pathways as detected with reverse-phase protein array.

(a) Heatmap of molecules whose expression significantly (p<0.05) differed before and after treatment with dasatinib (100 nM), paclitaxel (5 nM), or both in SPEC-2 cells (top) and SKUT-2 cells (bottom) cells for 16 hours. (b and c) Normalized expression levels in SPEC-2 cells (b) and SKUT-2 cells (c) treated with (red) or without (blue) dasatinib. Data represent means of triplicate measurements.
Figure 3. Effect of casatinib treatment on protein expression of pS6S240/244, CAV-1, BRaf and CRaf in uterine cancer cells.

(a and b) Western blot (WB) analysis of the expression levels of pS6S240/244 and CAV-1 (a) and BRaf and CRaf (b) in uterine cancer cells treated with (+) or without (−) dasatinib at 100 nM for 16 hours (top). Densitometry was performed to objectively assess potential differences (bottom). (c) Immunoprecipitation (IP) and WB analysis of the interaction of CAV-1, BRaf and EphA2 in SKUT-2 cells treated with (+) or without (−) dasatinib for 8 hours. (d) Immunoprecipitation and WB analysis of the interaction of CRaf and BRaf in Ishikawa, SKUT-2, and HEC1-A cells treated with (+) or without (−) dasatinib for 8 hours.
Figure 4. Effect of dasatinib on direct binding between EphA2 and CAV-1, EphA2 and BRaf, or CAV-1 and BRaf in uterine cancer cells.

(a) Double Immunofluorescence (IF) staining analysis of distribution of CAV-1 (red)/BRaf (green) (left), CAV-1 (red)/EphA2 (green) and of EphA2 (red)/BRaf (green) in SKUT-2 cells (right), before and after dasatinib treatment for 8 hours. Nucleus staining by Sytox (blue). (b) Dynamic effect of dasatinib on distribution of BRaf (green) and CAV-1 (red) in SKUT-2 cells for the indicated time periods (left). IF staining analysis of the effect of CAV-1 silencing on distribution of BRaf (green) and CAV-1 (red) in SKUT-2 (right panel, left) or SPEC2 cells treated with or without dasatinib for 8 hours (right panel, right). Nucleus staining by Sytox (blue). (c) MTT analysis of dasatinib on SKUT-2 cells transfected with CAV-1 siRNA or control siRNA (bottom). Western blotresults showing the effect of CAV-1 silencing with CAV-1 siRNA in SKUT-2 cells (top). (d) PLA analysis of distribution of CAV-1 and BRaf (top, left) or EphA2 (bottom, left) in SKUT-2 and HEC1-A cells grown on small chambers, before and after dasatinib treatment for 8 hours. Each individual blob (red) represents CAV-1 in close proximity with BRaf or EphA2. Statitical analysis of the signals using the Blobfinder V3.2 software (right).
To further investigate whether BRaf or EphA2 have the capability to interact with CAV-1 we used in situ proximity ligation assay (PLA), which is a unique method developed to visualize subcellular localization and protein-protein interactions in situ (22). SKUT-2 and HEC1-A treated with or without dasatinib for 8 hours were incubated overnight with primary antibody pair of different species directed to BRaf or EphA2 (mouse monoclonal anibody) and to CAV-1 (rabbit monoclonal antibody), respectively. The secondary antibodies were modified by addition of complementary oligonucleotides capable of interacting when in close proximity, an event that was detected by PCR amplification using a fluorochrome-based detection method. We observed that the colocalization of CAV-1 with BRaf (Fig 4d, top) and EphA2 (Fig 4d, bottom) was increased substantially in SKUT-2 cells after dasatinib treatment, and that the majority of binding interactions were localized on the plasma membrane. No significant increase in the co-localization of CAV-1 with BRaf and EphA2 was observed following dasatinib treatment in HEC1-A cells. Notably, CAV-1 gene silencing by siRNA significantly inhibited the colocalization of CAV-1 with BRaf and EphA2 (Fig 4d), indicating that high expression of CAV-1 in the cells is required for CAV-1 binding to BRaf or EphA2.
In Vivo Effects of Dasatinib in Orthotopic Models of Uterine Cancers
Next, we tested the effects of dasatinib-based therapy in orthotopic mouse models of EphA2-positive and -negative uterine carcinoma. In the EphA2-positive SKUT-2 model, dasatinib treatment resulted in significant anti-tumor activity (79% [p<0.01] and 63% [p<0.01], respectively; Fig 5a). In contrast, in the EphA2-negative SPEC-2 model, dasatinib demonstrated only minimal anti-tumor activity compared with the untreated (control) group (15% reduction of tumor weight vs. control [p>0.05] and 10% decrease in number tumor nodules vs. control [p>0.05]; Fig 5b). Given the role of paclitaxel in combination with biologically targeted agents, we also tested dasatinib with paclitaxel. The addition of paclitaxel resulted in a large reduction in the SKUT-2 model (95% [p<0.001] and 68% [p<0.01], respectively; Fig 5a), but minimal anti-tumor activity in the SPEC-2 model (37% [p<0.05] and 30% [p>0.05], respectively; Fig 5b). No obvious toxicity was observed in the various groups in that the mean body weight was similar in all groups (Supplementary Figs S6a and S6b).
Figure 5. In vivo effects of therapy with dasatinib and paclitaxel in uterine cancer models.

(a and b) In vivo effect of dasatinib (15 mg/kg oral, daily), paclitaxel (100 μg in 200 μL of PBS intraperitoneally, weekly), or both in an EphA2-positive model (SKUT-2) (a) and an EphA2-negative model (SPEC-2) (b). Error bars indicate SEM. *p<0.05, **p<0.01, and ***p<0.001. (c and d) Immunohistochemical staining showing the effect of dasatinib, paclitaxel, or both on uterine cancer cells angiogenesis (CD31), proliferation (Ki67), and apoptosis (caspase 3) in the SKUT-2 model (c) and the SPEC-2 model (d). Immunohistochemical staining of pEphA2S897 expression in SKUT-2 cells (c, bottom). *p<0.05, **p<0.01, and ***p<0.001 compared with control group. Original magnification ×100 or ×200.
To examine the biological effects of dasatinib-based therapy, we examined tumors from the SKUT-2 and SPEC-2 models for markers of tumor cell proliferation (Ki67), angiogenesis (CD31), and apoptosis (cleaved caspase 3). Changes in these markers mirrored the anti-tumor activity in response to dasatinib. Specifically, significant reductions in proliferation and microvessel density and increased apoptosis were noted in the SKUT-2 model (Fig 5c), whereas more modest changes were noted with the SPEC-2 model (Fig 5d). In addition, the expression level of pEphA2S897 was significantly decreased in the SKUT-2 model(79% [p<0.01]) (Fig 5c, bottom).
Expression of Predictive Markers in Human Samples
We next examined the expression of the putative dasatinib response markers (CAV-1, pEphA2Y594, pEphA2S897, and pAKTS473) in 30 human uterine cancer samples and 10 normal uterine samples by immunohistochemistry. Representative pictures of immunohistochemical staining are presented in Fig 6a. CAV-1 was not highly expressed in the normal uterine samples and was highly expressed in only 6.6% of tumor samples, but varying expression levels of CAV-1 were found in the stroma around the tumor cells (Table S2). pEphA2S897 was overexpressed in none of the normal samples but 70% of tumors, whereas high pEphA2Y594 expression was detected in 90% of normal samples and 10% of tumor samples.
Figure 6. Expression of predictive markers in uterine cancer patients.
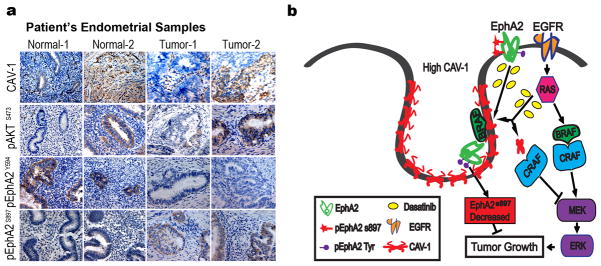
(a) Representative pictures of immunohistochemical staining of CAV-1, pAKTS473, pEphA2Y594, and pEphA2S897 expression in normal ueterine samples and uterine cancer samples. Original magnification ×200. (b) Cartoon for EphA2 signaling pathway and its interaction with CAV-1 and BRaf in dasatinib-sensitive uterine cancer cells. Dasatinib drives BRaf and EphA2 to high level CAV-1 at the plasma membrane, resulting in disruption of the BRaf/CRaf heterodimer and thus downregulating MAPK signaling.
Discussion
The focus of this work is on the multi-targeted tyrosine kinase dasatinib, which is used in the clinic for several diseases. Unfortunately, predictive markers guiding its use and subsequent response are largely absent resulting in “hit or miss” clinical application. Herein, we present evidence from in vitro and in vivo experiments for a set of markers essential for dasatinib response. Among these, CAV-1, EphA2 phosphorylation at S897 and the status of PTEN were key determinants of dasatinib response. In addition, CAV-1-mediated cross-talk between EphA2 and BRaf is required for response to dasatinib whereby dasatinib drives BRaf and EphA2 to CAV-1 at the plasma membrane, disrupting the BRaf/CRaf heterodimer and thus down-modulation of the mitogen-activated protein kinase (MAPK) pathway in dasatinib-sensitive cells, but not in dasatinib-resistant cells (Fig 6b).
The biological functions of CAV-1 on cancer cells have been controversial. CAV-1 is known to directly interact via its scaffolding domain with multiple signaling proteins and function as pre-organized signalosomes by sequestering and regulating proteins localized in caveolae, including receptor tyrosine kinases and their signaling effectors (23). In general, caveolins bind to and inactivate signaling molecules (24), including those of the MAPK signaling pathway (e.g., Ras, RAF-1, and MAPK) (25). Loss of CAV-1 expression confers a significant growth advantage that is associated with constitutive hyperactivation of the p42/44 MAPK pathway (26). In addition, interaction between Eph families, including EphA2 and CAV-1, has been reported (27). It has been reported that the Eph feedback loop in the activation of the HRAS/RAF/ERK pathway also increases EphA2 expression (28). We found that dasatinib inhibited EphA2 signalling, but unexpectedly stimulated Braf recruitment to the cell membrane, potentially promoting changes in the phosphorylation status and triggering its kinase activity (29). This resulted in increased heterodimerization with CRaf, which resulted in increased MEK/ERK activation (30). Further, we demonstrated the mechanistic basis for high CAV-1 at the plasma membrane holding BRaf, resulting in disruption of the BRAF/CRAF heterodimer and inhibiting MAPK activation following dasatinib treatment. Importantly, this finding could provide new approaches to enhance and extend the activity of dasatinib (e.g., CAV-1 peptide and RAF or MEK Inhibitors). The role of estrogen (ER) and progesterone (PR) receptor status could also be relevant for therapeutic considerations. For example, previously we have reported that tumors with EphA2 overexpression are more likely to be ER/PR negative. Thus, our findings may have therapeutic implications for patients with poorly differentiated endometrial cancers that are steroid hormone receptor deficient (8).
While our findings may have clinical implications for ongoing dasatinib-based clinical trials, some potenitial limitations should be considered. Whether the identified markers are indeed predictive of dasatinib sensitivity in human clinical trials is not known. A pilot and translational study (NSC#732517) of dasatinib, paclitaxel, and carboplatin for women with advanced-stage and recurrent uterine cancer is ongoing. This trial is evaluating pharmacodynamic variances of EphA2 signaling in response to both dasatinib alone (in the lead-in phase) and in combination with chemotherapy. Validation of these biomarkers will be important for the next generation of clinical investigation involving treatment allocation based on specific biomarkers (31). The diversity of biological characterization that represents “uterine cancer” is only just being described (32) and highlights the need for target prevalence description and prospective determination of expression to effectively test our portfolio of targeted therapeutics (33).
Supplementary Material
Translational Relevance.
EphA2 is considered an important therapeutic target as it is involved in many processes crucial to malignant progression. Among the various therapeutic strategies targeting EphA2, dasatinib is the farthest along with regard to clinical development. However, reliable predictors of response and the mechanisms relevant to dasatinib response have been largely lacking. We have identified a set of molecular predictors of dasatinib sensitivity in uterine cancer that may have significant implications for ongoing dasatinib-based clinical trials. Our key findings are that CAV-1, EphA2 phosphorylation at S897 and the status of PTEN are key determinants of dasatinib response. Moreover, we have identified a previously unknown mechanism involving the association between CAV-1, EphA2 and the heterodimer BRAF/CRAF, which has implications for designing new therapeutic combinations.
Acknowledgments
Financial support was provided by the National Institutes of Health (P50 CA098258, CA 109298, P50 CA083639, U54 CA151668, M. D. Anderson’s Cancer Center Support Grant CA016672), the Ovarian Cancer Research Fund, Inc. (Program Project Development Grant), the U.S. Department of Defense (OC073399, OC093146, BC085265), the Ann Rife Cox Chair in Gynecology, the Zarrow Foundation, the Marcus Foundation, the Betty Anne Asche Murray Distinguished Professorship, the RGK Foundation, and the Gilder Foundation. JBM and HJD are supported by an NCI-DHHS-NIH T32 Training Grant (T32 CA101642). None of the authors has any conflicts of interest to disclose. We thank Elizabeth L. Hess of the MD Anderson Department of Scientific Publications for helpful editing.
Footnotes
Disclosure: There was no conflict of interest in all authors.
References
- 1.Landen CN, Kinch MS, Sood AK. EphA2 as a target for ovarian cancer therapy. Expert Opin Ther Targets. 2005;9:1179–87. doi: 10.1517/14728222.9.6.1179. [DOI] [PubMed] [Google Scholar]
- 2.Nakamoto M, Bergemann AD. Diverse roles for the Eph family of receptor tyrosine kinases in carcinogenesis. Microsc Res Tech. 2002;59:58–67. doi: 10.1002/jemt.10177. [DOI] [PubMed] [Google Scholar]
- 3.Menges CW, McCance DJ. Constitutive activation of the Raf-MAPK pathway causes negative feedback inhibition of Ras-PI3K-AKT and cellular arrest through the EphA2 receptor. Oncogene. 2008;27:2934–40. doi: 10.1038/sj.onc.1210957. [DOI] [PubMed] [Google Scholar]
- 4.Landen CN, Jr, Lu C, Han LY, Coffman KT, Bruckheimer E, Halder J, et al. Efficacy and antivascular effects of EphA2 reduction with an agonistic antibody in ovarian cancer. J Natl Cancer Inst. 2006;98:1558–70. doi: 10.1093/jnci/djj414. [DOI] [PubMed] [Google Scholar]
- 5.Lin YG, Han LY, Kamat AA, Merritt WM, Landen CN, Deavers MT, et al. EphA2 overexpression is associated with angiogenesis in ovarian cancer. Cancer. 2007;109:332–40. doi: 10.1002/cncr.22415. [DOI] [PubMed] [Google Scholar]
- 6.Lee JW, Stone RL, Lee SJ, Nam EJ, Roh JW, Nick AM, et al. EphA2 targeted chemotherapy using an antibody drug conjugate in endometrial carcinoma. Clin Cancer Res. 2010;16:2562–70. doi: 10.1158/1078-0432.CCR-10-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merritt WM, Kamat AA, Hwang JY, Bottsford-Miller J, Lu C, Lin YG, et al. Clinical and biological impact of EphA2 overexpression and angiogenesis in endometrial cancer. Cancer Biol Ther. 2011;10:1306–14. doi: 10.4161/cbt.10.12.13582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kamat AA, Coffey D, Merritt WM, Nugent E, Urbauer D, Lin YG, et al. EphA2 overexpression is associated with lack of hormone receptor expression and poor outcome in endometrial cancer. Cancer. 2009;115:2684–92. doi: 10.1002/cncr.24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sierra JR, Cepero V, Giordano S. Molecular mechanisms of acquired resistance to tyrosine kinase targeted therapy. Mol Cancer. 2010;9:75. doi: 10.1186/1476-4598-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tandon M, Vemula SV, Mittal SK. Emerging strategies for EphA2 receptor targeting for cancer therapeutics. Expert Opin Ther Targets. 2011;15:31–51. doi: 10.1517/14728222.2011.538682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol. 2007;25:1035–44. doi: 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- 12.Chang Q, Jorgensen C, Pawson T, Hedley DW. Effects of dasatinib on EphA2 receptor tyrosine kinase activity and downstream signalling in pancreatic cancer. Br J Cancer. 2008;99:1074–82. doi: 10.1038/sj.bjc.6604676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang XD, Reeves K, Luo FR, Xu LA, Lee F, Clark E, et al. Identification of candidate predictive and surrogate molecular markers for dasatinib in prostate cancer: rationale for patient selection and efficacy monitoring. Genome Biol. 2007;8:R255. doi: 10.1186/gb-2007-8-11-r255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puls LN, Eadens M, Messersmith W. Current status of SRC inhibitors in solid tumor malignancies. Oncologist. 2011;16:566–78. doi: 10.1634/theoncologist.2010-0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schilder RJ, Brady WE, Lankes HA, Fiorica JV, Shahin MS, Zhou XC, et al. Phase II evaluation of dasatinib in the treatment of recurrent or persistent epithelial ovarian or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol. 2012;127:70–4. doi: 10.1016/j.ygyno.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dudek AZ, Pang H, Kratzke RA, Otterson GA, Hodgson L, Vokes EE, et al. Phase II study of dasatinib in patients with previously treated malignant mesothelioma (cancer and leukemia group B 30601): a brief report. J Thorac Oncol. 2012;7:755–9. doi: 10.1097/JTO.0b013e318248242c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson FM, Bekele BN, Feng L, Wistuba I, Tang XM, Tran HT, et al. Phase II study of dasatinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2010;28:4609–15. doi: 10.1200/JCO.2010.30.5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ali-Fehmi R, Morris RT, Bandyopadhyay S, Che M, Schimp V, Malone JM, Jr, et al. Expression of cyclooxygenase-2 in advanced stage ovarian serous carcinoma: correlation with tumor cell proliferation, apoptosis, angiogenesis, and survival. Am J Obstet Gynecol. 2005;192:819–25. doi: 10.1016/j.ajog.2004.10.587. [DOI] [PubMed] [Google Scholar]
- 19.Hanrahan AJ, Schultz N, Westfal ML, Sakr RA, Giri DD, Scarperi S, et al. Genomic complexity and AKT dependence in serous ovarian cancer. Cancer Discov. 2012;2:56–67. doi: 10.1158/2159-8290.CD-11-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miao H, Li DQ, Mukherjee A, Guo H, Petty A, Cutter J, et al. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell. 2009;16:9–20. doi: 10.1016/j.ccr.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Packer LM, Rana S, Hayward R, O’Hare T, Eide CA, Rebocho A, et al. Nilotinib and MEK inhibitors induce synthetic lethality through paradoxical activation of RAF in drug-resistant chronic myeloid leukemia. Cancer Cell. 2011;20:715–27. doi: 10.1016/j.ccr.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 23.Gosens R, Dueck G, Gerthoffer WT, Unruh H, Zaagsma J, Meurs H, et al. p42/p44 MAP kinase activation is localized to caveolae-free membrane domains in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1163–72. doi: 10.1152/ajplung.00471.2006. [DOI] [PubMed] [Google Scholar]
- 24.Goetz JG, Lajoie P, Wiseman SM, Nabi IR. Caveolin-1 in tumor progression: the good, the bad and the ugly. Cancer Metastasis Rev. 2008;27:715–35. doi: 10.1007/s10555-008-9160-9. [DOI] [PubMed] [Google Scholar]
- 25.Smart EJ, Graf GA, McNiven MA, Sessa WC, Engelman JA, Scherer PE, et al. Caveolins, liquid-ordered domains, and signal transduction. Mol Cell Biol. 1999;19:7289–304. doi: 10.1128/mcb.19.11.7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams TM, Lee H, Cheung MW, Cohen AW, Razani B, Iyengar P, et al. Combined loss of INK4a and caveolin-1 synergistically enhances cell proliferation and oncogene-induced tumorigenesis: role of INK4a/CAV-1 in mammary epithelial cell hyperplasia. J Biol Chem. 2004;279:24745–56. doi: 10.1074/jbc.M402064200. [DOI] [PubMed] [Google Scholar]
- 27.Vihanto MM, Vindis C, Djonov V, Cerretti DP, Huynh-Do U. Caveolin-1 is required for signaling and membrane targeting of EphB1 receptor tyrosine kinase. J Cell Sci. 2006;119:2299–309. doi: 10.1242/jcs.02946. [DOI] [PubMed] [Google Scholar]
- 28.Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer. 2010;10:165–80. doi: 10.1038/nrc2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cantwell-Dorris ER, O’Leary JJ, Sheils OM. BRAFV600E: implications for carcinogenesis and molecular therapy. Mol Cancer Ther. 2011;10:385–94. doi: 10.1158/1535-7163.MCT-10-0799. [DOI] [PubMed] [Google Scholar]
- 30.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–30. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferguson SE, Olshen AB, Levine DA, Viale A, Barakat RR, Boyd J. Molecular profiling of endometrial cancers from African-American and Caucasian women. Gynecol Oncol. 2006;101:209–13. doi: 10.1016/j.ygyno.2005.11.028. [DOI] [PubMed] [Google Scholar]
- 32.Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santin AD, Zhan F, Cane S, Bellone S, Palmieri M, Thomas M, et al. Gene expression fingerprint of uterine serous papillary carcinoma: identification of novel molecular markers for uterine serous cancer diagnosis and therapy. Br J Cancer. 2005;92:1561–73. doi: 10.1038/sj.bjc.6602480. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.