

Abstract
Diabetic complications encompass macrovascular events, mainly the result of accelerated atherosclerosis, and microvascular events that strike the eye (retinopathy), kidney (nephropathy), and nervous system (neuropathy). The traditional view is that hyperglycemia-induced dysregulated biochemical pathways cause injury and death of cells intrinsic to the organs affected. There is emerging evidence that diabetes compromises the function of the bone marrow (BM), producing a stem cell niche-dependent defect in hematopoietic stem cell mobilization. Furthermore, dysfunctional BM-derived hematopoietic cells contribute to diabetic complications. Thus, BM cells are not only a victim but also an accomplice in diabetes and diabetic complications. Understanding the underlying molecular mechanisms may lead to the development of new therapies to prevent and/or treat diabetic complications by specifically targeting these perpetrators.
Keywords: diabetes mellitus, complications, diabetic nephropathy, diabetic retinopathy, diabetic neuropathy, bone marrow, hematopoietic cells
Hyperglycemia induced organ dysfunctions
The discovery of insulin about ninety years ago along with other advances in medical therapy have markedly improved the quality of life and life expectancy of people with type 1 and type 2 diabetes. Nowadays, diabetic patients rarely die of the acute complications of ketoacidosis. With a much longer lifespan, however, the majority of people with diabetes will develop chronic diabetic complications, the cause of much morbidity and mortality.
Hyperglycemia is the ultimate cause of diabetic complications. Hyperglycemia induces a number of biochemical processes with important pathogenic implications, such as rendering cells more vulnerable to oxidative stress, increased production of advanced glycation end-products (AGEs) that alter the function of intracellular proteins and extracellular matrix, increased protein kinase C activity that causes blood flow abnormalities, vascular permeability, and microvascular matrix protein accumulation, and post-translation modification of transcription factors that results in altered gene expression [1–3]. Cells in various tissues and organs, such as endothelial cells, pericytes (capillary support cells), and Müller cells in the retina, endothelial cells, mesangial cells, and podocytes in the kidney glomerulus, and neurons and Schwann cells as well as endothelial cells of the vasa nervorum in the peripheral nerves, are susceptible to hyperglycemia-induced damage. Since target organs are constantly in contact with circulating hematopoietic cells and known downstream effects of hyperglycemia encompass inflammatory signaling which can recruit hematopoietic cells, these cells likely play crucial roles in diabetic complications.
Until recently, the bone marrow (BM) was not generally considered as a target organ for chronic diabetic complications. However, in the last decade, Fadini et al. have observed decreased circulating endothelial progenitor cells (EPCs) and hematopoietic stem/progenitor cells (HSPCs) in diabetic rodents and humans (see Glossary) [4–6]. In addition, microangiopathy (small vessel disease) [7, 8] and niche dysfunction [9] were reported in the diabetic BM. HSPC dysfunction [7, 8, 10] and altered hematopoiesis with increases in inflammatory monocytes [11–14] have also been observed. It is now clear that the BM is another target organ afflicted by diabetic complication and BM-derived hematopoietic cells play an active role in the development of diabetic microvascular complications in multiple organs [15–19]. This review will discuss how the diabetic milieu modulates the distribution, abundance and function of hematopoietic cells and their pathogenic role in the development of chronic diabetic complications. Of note, HSPC is used to denote both hematopoietic stem cells (HSCs) and hematopoietic progenitor cells (HPCs) (see Glossary). ‘Hematopoietic cells’ is used collectively to refer to HSCs, HPCs, fully differentiated cells, and other progenitor/stem cells of hematopoietic origin. We will designate cells with a specific set of surface markers when it helps understanding. Notably, there is inconsistency over the use of the acronym EPC in the literature. Instead of a true progenitor cell of endothelial cell lineage, i.e., a non-hematopoietic cell, the name EPC has been applied by some to a myeloid lineage cell, i.e., a hematopoietic cell with vascular repair and angiogenesis capacity. Some of the discrepancies in different reports seem to have resulted from the relatively lax use of the term [20]. We will summarize in the following section the dysfunctional EPCs in diabetes. Fadini et al. have published several recent review articles on this aspect of BM-derived cells (BMDCs) in diabetes [20–22].
Diabetes induced changes in the BM
Impaired EPC and HSPC Mobilization in Diabetes
Asahara et al. first identified circulating EPCs by showing that human CD34+ cells (a HSPC marker) or mouse VEGFR-2+ cells (Flk1+ in mice and KDR in humans, an endothelial marker) from the peripheral blood can develop endothelial-like phenotypes in vitro and contribute to neovascularization in response to ischemia [23]. EPCs, typically identified in humans as peripheral mononuclear cells that are positive for the CD34 and VEGFR-2, and/or CD133 stem cell markers, are involved in vascular repair through re-endothelialization and neovascularization [24] and have been used as a biologic marker for vascular health [25]. It was believed that EPCs repair vascular damage by differentiating into endothelial cells; in addition, paracrine actions of EPCs have also been recognized, which may represent a more dominant functional mechanism than endothelialization per se [26–27]. As noted above, EPCs represent heterogeneous groups of cells ranging from mostly proangiogenic hematopoietic cells to subsets of HSPCs and other progenitor/stem cells [20, 28].
A decrease in circulating EPCs was first recognized as a cardiometabolic risk factor in people about a decade ago [25]. Subsequently, many studies have shown diabetes-associated changes in EPCs, which include defects in proliferation and vascular tubal formation, in vitro, in type 1 [29] and in type 2 diabetes [30]. Indeed, both type 1 and type 2 diabetic individuals have a reduced number of circulating EPCs [4, 31, 32], a phenotype also associated with diabetic complications [33]. Furthermore, CD34+ cells are reduced in the peripheral blood as well as in BM aspirates [6] and their response to granulocyte-colony-stimulating factor (G-CSF) is impaired in diabetic people [9, 34, 35]. Observations in diabetic animals reveal similar findings. Rodents with streptozocin (STZ)-induced diabetes have decreased circulating EPCs and impaired mobilization in response to limb ischemia [5] or wound injury [36]. Mechanistically, diabetic animals exhibit decreased release of a chemoattractant signaling molecule, C-X-C motif chemokine 12 (CXCL12, also called SDF-1α) from local tissues as well as decreased activation of a mobilization enzymatic pathway, endothelial nitric oxide synthase (eNOS), in the BM. Mice with STZ-induced diabetes also show poor HSPC mobilization in response to G-CSF [9]. These studies strongly implicate defective BM and impaired BM function in diabetes and highlight possible structural and functional changes in the BM induced by diabetes.
Diabetic BM Microangiopathy and Niche Dysfunction
The concept of diabetic BM microangiopathy has evolved over the last few years (Box 1 and Figure 1). Busik et al. reported adrenergic denervation as a cause of impaired EPC mobilization in BBZDR/Wor rats, a model of type 2 diabetes [37]. Another group examined the functional and structural changes in the BM of mice with long-term (27–30 weeks) STZ-induced diabetes, and found that these animals have microvascular rarefaction with poor perfusion, decreased hematopoietic fraction, and increased fat accumulation in the BM. Lineage-negative (Lin)−stem-cell antigen 1 (SCA1)+KIT+ (LSK) stem cells, a source of HSCs with both long- (LT-HSCs) and short-term (ST-HSCs) renewal capacity, are reduced especially at hypoperfused locations. There is reduced colony formation of multipotent progenitor cells, but not lineage committed progenitor cells [7]. These structural changes were, however, not observed in another study using mice with STZ induced diabetes followed up to 20 weeks, though decrease in LSK stem cells was again found with reduced repopulation capacity on competitive engraftment [10]. Ferraro et al. examined the BM niche function to further dissect the mechanism that underlies impaired HSPC mobilization in mice with STZ diabetes of a shorter duration of 5–8 week [9]. They found in the BM an increased number of LSK cells with intact repopulating potential. BM transplant (BMT) experiments in mice showed that diabetic recipients exhibit impairment in mobilization, whereas nondiabetic recipients that have received diabetic BM do not display such defects. They further observed a substantial impairment of adrenergic stimulation-mediated down-regulation of CXCL12 in the nestin+ mesenchymal stem cells (MSCs, stromal cells that are found exclusively in the perivascular space) resulting in the retention of HSPCs in the BM despite a two-fold increase in sympathetic nerve terminals; blockade of CXCR4 (CXCL12 receptor) alleviates mobilization defects in diabetic mice, consistent with BM niche dysfunction in diabetes. These findings show that the BM undergoes structural and functional changes in diabetes associated with quantitative and qualitative changes in HSPCs and their niche in the BM. Discrepancies in the degree of the response of different BM components and the repopulating potential of HSCs may be related to the duration of diabetes and the particular model used (Box 1 and Figure 1).
BOX 1. Diabetic Bone Marrow Dysfunction.
The impaired mobilization of EPCs in diabetes suggests that the bone marrow (BM) also bears the brunt of diabetes-induced organ damage. There is a rich network of nerve fibers in the BM. Katayama et al. showed that the sympathetic nervous system (SNS) is needed to mobilize HSPCs and that G-CSF induces mobilization of HSPCs, in part through the SNS-mediated suppression of osteoblast activity and downregulation of CXCL12 in the bone [93]. Méndez-Ferrer et al. found that circulating HSPCs exhibit circadian fluctuations mediated by circadian secretion of norepinephrine by the SNS into the BM, which down-regulates CXCL12 via β3 adrenergic receptor on nestin+ MSCs [94, 95]. In this regard, Busik et al. studied sympathetic nerve terminals in the BM in association with EPC release in Bio-Breeding Zucker diabetic (BBZDR/Wor) rats, a model of type 2 diabetes [37]. These rats start showing a decrease in sympathetic nerve terminals two months after the onset of diabetes. Another two months later, there is decreased circadian release of EPCs, which is associated with dysregulated circadian plasma norepinephrine levels. EPCs are increased in the BM but exhibit decreased rate of proliferation. Subsequent detailed analysis of the diabetic BM in mice 27–30 weeks after STZ induced diabetes by Oikawa et al. revealed microvascular rarefaction and thus decreased perfusion, endothelial barrier dysfunction, decreased hematopoietic fraction with depleted hematopoietic stem cells (HSC) predominantly at the poorly perfused endosteal surface, increased fat accumulation, and osteopenia. The endothelial cells in the BM do not respond to chemo-attractant signals such as CXCL 12 and VEGF-A, and fail to form an endothelial network in vitro. The authors labeled the diabetes-associated BM pathology “bone marrow microangiopathy” [7]. Ferraro et al. found that diabetic patients do not mobilize CD34+ cells well after G-CSF injection. Using rodent models, they also demonstrated that mice with relatively short duration of diabetes (db/db mice younger than 12 weeks or 5–8 weeks after STZ injection) have similarly impaired HSPC mobilization in response to G-CSF, a defect that is correlated with blood glucose level. HSC numbers increase in the BM and their repopulation capacity is maintained in STZ diabetes. Impaired mobilization is not cell-autonomous but microenvironment-dependent, i.e., mobilization defects occur in nondiabetic-to-diabetic BMT but not in diabetic-to-nondiabetic BMT. Further examination of niche function revealed decreased number of osteoblasts (which contributes to mobilization defects), increased number of sympathetic nerve terminals, no changes in the number of nestin+ MSCs, and elevated baseline sympathetic activation but blunted suppression of CXCL12 in nestin+ MSCs by sympathetic stimulation [9]. In summary, diabetes causes stem cell niche dysfunction, and in rodents, that is also reflected by structural and functional changes in the BM (Figure 1).
Figure 1. Diabetic BM microangiopathy and niche dysfunction.
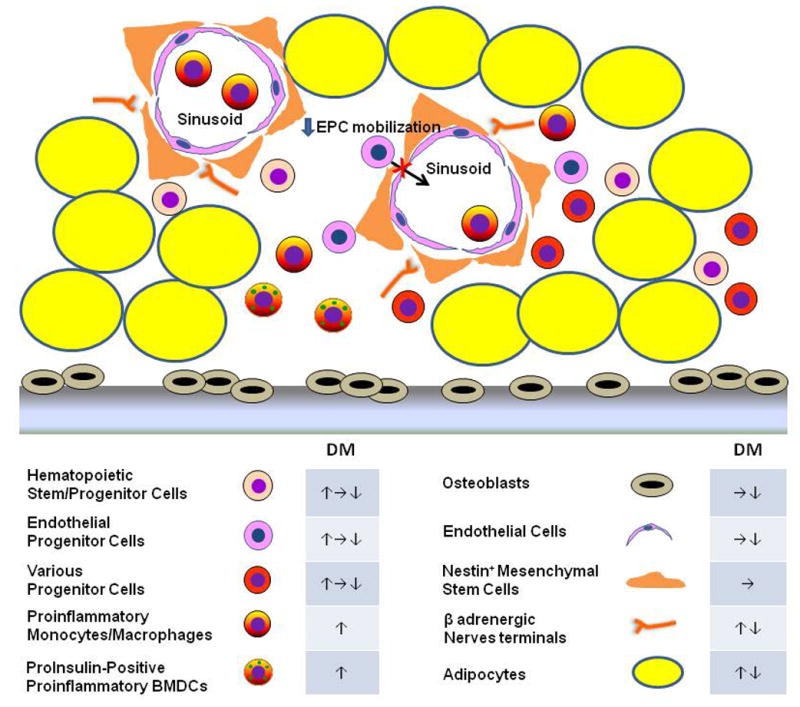
Hyperglycemia induces structural and functional changes in the cellular composition of the BM, including the ones depicted in this figure. These include: unchanged capillary [10] or capillary rarefaction (↓endothelial cells) [7]; decreased [7, 10, 11], unchanged [36] or increased [9] fractions of hematopoietic and other progenitor cells; unchanged [10] or decreased [9] osteoblasts; decreased [11] or increased [7, 37] fat accumulation; and decreased [37] or increased [9] sympathetic nerve fibers. Functional defects, i.e. niche dysfunction, accompany the structural changes. Different laboratories have observed impaired mobilization of EPCs in response to ischemic injury [5] and of HSPCs in response to G-CSF [9] associated with β adrenergic dysfunction. There is down-regulated proliferative potential of multipotent progenitor cells [7], but increased production of pro-inflammatory monocytes [11, 38]. In addition, PI-BMDCs appear in the diabetic BM [42]. PI-BMDCs share many of the characteristics of monocytes/macrophages. They are proinflammatory in nature and are actively involved in the pathogenesis of diabetic complications (see Box 2)
Abbreviations: DM, abnormalities in diabetic mellitus BM; CSF, colony stimulating factor; EPC, endothelial progenitor cell; HSPC, hematopoietic stem and progenitor cell.
Altered Hematopoiesis in Diabetes
The changes in the BM microenvironment in diabetes are also accompanied by altered hematopoiesis. A number of studies revealed an impairment in the colony-forming capability of the BM, while the number of Ly6Chi (proinflammatory) monocytes in the peripheral blood is increased in mice with long standing STZ diabetes and in diabetic mice with an insulin mutation (Akita) [11, 38]. Mechanistically, hyperglycemia induces secretion of non-AGE ligands for AGE receptors (RAGE) from neutrophils and interaction of these molecules with RAGE on common myeloid progenitor cells results in enhanced myelopoiesis [38].
Hyperglycemia not only affects hematopoiesis, it also alters the functional characteristics of BM-derived hematopoietic cells. For example, incubation of BM cells in high glucose in vitro was found to induce insulin gene transcription [39, 40] and TNF-α expression [40, 41]. Kojima et al. found the appearance of pro-insulin (PI) positive cells in multiple extrapancreatic tissues in rodents with hyperglycemia (STZ injection, leptin deficiency, high fat diet, and glucose injections alone) [42]. These cells are of BM origin and have been called PI-positive bone marrow derived cells (PI-BMDCs) (Box 2). PI production is very low and serves only as a biomarker as it identifies a hyperglycemia-induced inflammatory (TNF-α-expressing) hematopoietic cell population [40, 43–45]. PI-BMDCs have been found to be involved in the pathogenesis of diabetic complications because of their proinflammatory nature and fusogenicity [46] (discussed below).
BOX 2. Discovery of PI-BMDCs and their Role in Diabetic Complications.
PI-producing bone marrow-derived cells (PI-BMDCs) were discovered by serendipity during a gene therapy experiment. A research team delivered the gene of the islet transcription factor NeuroD [96] or Neurogenin3 [97] (required for normal pancreatic islet development) to the liver of STZ-treated diabetic mice. The maneuver robustly induced PI-positive cells in the liver of these animals. While such cells were not observed in untreated nondiabetic controls, unexpectedly, small numbers of PI-producing cells were found in the liver of sham-treated diabetic controls. Subsequent experiments revealed that the transcription factor-induced PI-positive cells were islet-like cells derived from hepatic progenitor cells [97], whereas PI-positive cells in sham-treated diabetic mice were derived from the BM, i.e., PI-BMDCs [96]. PI-BMDCs were found in the liver, adipose depots, and BM of type 1 (STZ) and type 2 (high fat diet and ob/ob) diabetic mice. In addition, incubation of rodent BM cells in vitro induces insulin mRNA expression [39]. Thus PI-BMDCs might potentially be used as a form of cell therapy to treat insulin-deficient diabetes. However, Kojima et al. found that these PI-BMDCs do not secrete any measurable insulin or proinsulin in vitro and have no therapeutic potential [96].
PI-BMDCs are morphologically similar to macrophage and are F4/80+. They appear in mice and rats within 1–3 days of intermittent hyperglycemia [42]. Terashima et al. examined mice and rats 8–12 weeks after STZ-induced diabetes and found PI-positive neurons (sciatic nerves and DRG neurons) and Schwann cells. BMT experiments using genetically marked donor and recipient animals showed that the PI-positive cells are fusion cells formed between neurons (or Schwann cells) and PI-BMDCs. Furthermore, all PI-positive cells were polyploid whereas all PI-negative neurons were diploid, again indicating that these morphologically neuron-like PI-positive cells are actually neuron-PI-BMDC fusion cells. Compared with unfused neurons, the fusion cells are much more prone to apoptosis. Histochemical analysis revealed that apoptotic neurons are not detectable, i.e., exceedingly rare, in the DRGs of nondiabetic animals but comprise ~0.4% of the neurons in the DRGs of diabetic mice and rats; all apoptotic cells are also PI-positive, i.e. fusion cells [40, 43]. The fusion of PI-BMDCs with neurons in hyperglycemic states enhances their pathogenicity. The fused cells, fixed in the DRG, provide a stable source of TNF-α, which diffuses to neighboring neurons and causes them to malfunction.
Therefore, the health status of the BM cells is as important as the health status of the neurons in the natural history of diabetic neuropathy development.
BM dysfunction in diabetic patients
There is very limited published data on BM dysfunction in diabetic patients. One study showed reduced hematopoietic tissue, increased fat deposition, and microvascular rarefaction in diabetic individuals, more so in patients with vascular complications, consistent with the presence of diabetic microangiopathy. These BM changes were associated with decrease in HSPCs [8]. In addition to mobilization defects of EPCs and HSPCs, diabetic people tend to have increased number of peripheral monocytes [12] and enhanced inflammatory monocyte profile [13, 14]. Therefore, both preclinical and clinical studies indicate that diabetes induces BM dysfunction.
Hematopoietic cells in common chronic diabetic complications
Hyperglycemia-induced BM microangiopathy and niche dysfunction, inflammatory gene expression, modulation of stem/progenitor cells, and altered myelopoiesis appear to be important etiologic factors for chronic diabetic complications. In addition, diabetes-associated cellular malfunctions have tissue- and organ-specific consequences in individual complications. The role of macrophages in atherosclerosis is well established [3]. Hyperglycemia may accelerate this process by increasing myelopoiesis and monocyte recruitment to atherosclerotic lesions [38]. The role of EPCs in the process has also been studied extensively [20, 22, 47]. Additionally, the lipid abnormalities that commonly occur in type 2 diabetes provide fertile soil for macrovascular disease and complications. The following sections will focus on the emerging roles of hematopoietic cells in diabetic microvascular complications.
Diabetic Nephropathy
Diabetic nephropathy is the most common cause of end-stage kidney disease. It is characterized by glomerular hyperfiltration followed by progressive decline of kidney function in association with proteinuria, glomerulosclerosis, and tubulointerstitial damage. Hyperglycemia induces mesangial cell proliferation and hypertrophy with excessive extracelullar matrix accumulation and thickening of glomerular basement membrane, loss of podocytes, tubular hypertrophy followed by thickening of tubular basement membrane and interstitial fibrosis, and arteriolar hyalinization [48].
BMT experiments in rodents showed that the BM-derived stem/progenitor cells, including HSCs, EPCs, and MSCs, have the capacity to regenerate tubular epithelial cells, glomerular endothelial cells, podocytes, and mesangial cells [49]. The interpretation of these experiments is, however, limited by the necessary manipulation of the donor cells and the effect of irradiation. Inverse relationship of peripheral CD34+ and CD34+/CD133+ cells and microalbuminuria in diabetic patients suggests defective replacement of damaged renal cells as a possible pathogenic mechanism in diabetic nephropathy [20]. In mouse experiments, BMT from diabetic donors into non-diabetic recipients causes albuminuria; it is interesting that donor BM-derived mesangial cells are found in the glomerular lesion of the recipient mice [50]. Transfer of BM MSCs into mice with STZ-induced diabetes prevents nephropathy; it is also associated with the incorporation of the donor cells into the recipient kidneys [51]. Nonetheless, there is minimal suggestive evidence to date that BM-derived progenitor/stem cells contribute to the development of diabetic nephropathy.
Like other chronic renal diseases [52], the role of inflammatory hematopoietic cells, mostly macrophages, in diabetic nephropathy is generally well established [15]. Macrophages infiltrate into the glomerulus and contribute to extracellular matrix accumulation and development of fibrosis in rats with STZ-induced diabetes [53]. Macrophage infiltration also occurs in the glomerulus and the tubulointerstitium in fafa rats and db/db mice, both models of type 2 diabetes [54, 55]. The degree of macrophage accumulation correlates with blood glucose and hemoglobin A1c level, degree of proteinuria, as well as hypertrophy, and fibrosis in the glomerulus and interstitium. It is noteworthy that the severity of kidney dysfunction is best correlated with the interstitial macrophages [55].
Macrophages play an important pathogenic role. Glycemic control with insulin administration or macrophage depletion by irradiation or mycophenolate prevents macrophage infiltration and the associated downstream pathology in the glomerulus [53, 56, 57]. Furthermore, deficiency of intracellular adhesion molecule-1 (ICAM-1), a key cell adhesion protein on the endothelium, and chemokine (C-C motif) ligand 2 (CCL2, also called MCP1) or its receptor CCR2 (a key chemokine signaling molecule in inflammatory monocyte recruitment) markedly suppresses macrophage infiltration and prevents diabetic nephropathy [58–62]. Intervention studies showed that CCR2 antagonists can prevent diabetic nephropathy in STZ-treated or db/db mice [62–64]. In addition, blockade of colony-stimulating factor-1 (CSF-1) receptors, which promote macrophage accumulation, activation, and survival, also attenuates macrophage infiltration and subsequent renal damage [65]. Moreover, adoptive transfer of M2 polarized macrophages prevents diabetic nephropathy, though interpretation is clouded by the concomitant improvement in hyperglycemia and hemoglobin A1c levels [66]. Recently, proinflammatory TNF-α-producing PI-BMDCs were observed in the kidney of type 1 (STZ) and type 2 (high fat diet-fed) diabetic mouse models. Moreover, these PI-BMDCs were found to form fusion cells with tubular epithelial cells, a pathological process that may have contributed to the diabetic kidney disease in these animals [45].
In summary, rodent experiments indicate that monocyte/macrophages contribute to the development of diabetic nephropathy. This is further supported by the presence of macrophages infiltrating the glomerulus [67] and the interstitial compartment [68] in people with diabetic glomerulosclerosis. In addition, glomerular macrophage infiltration correlates mainly with baseline kidney function, while interstitial macrophage infiltration is associated with baseline kidney function, proteinuria, interstitial fibrosis, and the rate of progression of nephropathy in diabetic individuals [69]. There are ongoing clinical trials that study the efficacy of blocking CCR2 in diabetic nephropathy. The role of other cells including progenitor/stem cells and lymphoid cells, however, is less clear.
Diabetic Retinopathy
Diabetic retinopathy is a disease of the retinal microvasculature. Retinal capillary vasoregression occurs early, which is followed by retinal neovascularization in response to ischemia. Hyperglycemia induces biochemical changes in different retinal cells including endothelial cells, pericytes, Müller cells, microglia, and neurons, often leading to secretion of vasoregulatory factors from Müller cells and loss of endothelial cells and pericytes, producing an acellular capillary without perfusion [16, 70]. It also up-regulates inflammatory cytokines and mediators which play key roles in retinopathy development. Consequently, new but aberrant blood vessels proliferate and may hemorrhage. The paradoxical involvement of EPC in proliferative retinopathy was recently reviewed elsewhere [33, 71]. The role of hematopoietic cells in the development of vasoregression, especially via leukostasis [17, 18], is further discussed below (Figure 2).
Figure 2. Role of hematopoietic cells in diabetic retinopathy.
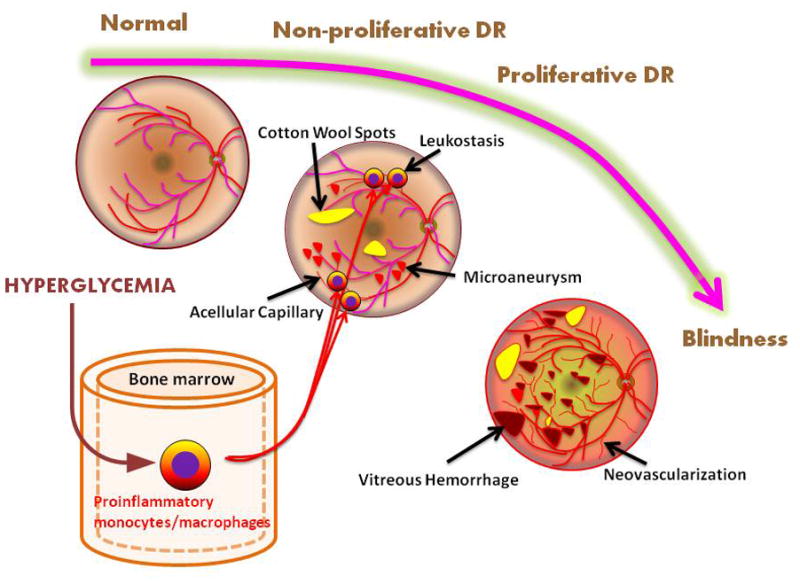
Vasoregression is an early event in diabetic retinopathy associated with hyperglycemia-induced leukostasis, causing loss of capillary endothelial cells and pericytes and thus leading to acellular capillaries without perfusion [16]. Ischemia secondary to vasoregression manifests as cotton wool spots (nerve fiber layer infarcts) and microaneurysms (aborted attempts at neovascularization). Progressive ischemia results in fragile neovascularization that is prone to preretinal and vitreous hemorrhage. Fibrotic changes from hemorrhages and fibrovascular proliferation can eventually lead to retinal detachment and loss of vision. Macular edema can occur at any stage of diabetic retinopathy and is another cause of loss of vision.
Retinal leukostasis was first reported in rats with alloxan-induced diabetes. Increased numbers of intracapillary monocytes and granulocytes were observed in the retina with accompanying capillary occlusions and endothelial damages [72]. Diabetic retinal vascular leakage and non-perfusion are temporally and spatially associated with leukostasis in rats with STZ diabetes. Leukostasis is related to the expression of ICAM-1; blockade of ICAM-1 prevents leukostasis and vascular leakage [73]. Deficiency of either ICAM-1 or CD18 (integrin beta2, a ICAM-1 ligand on leukocytes) reduces adherent leukocytes in the retinal capillary and attenuates loss of endothelial cells and pericytes, reducing acellular capillaries and vascular leakage in STZ diabetic mice [74]. Importantly, BMT of diabetic BM cells produces retinal leukostasis in non-diabetic recipients though leukostasis is worst when both donors and recipients are diabetic [75].
Inducible nitric oxide synthase (iNOS) and poly ADP-ribose polymerase-1 (PARP-1) are major mediators of inflammation in response to hyperglycemia; mice that receive BMT from iNOS- or PARP-1-deficient BM donors (as compared to wild-type donors) are resistant to the development of capillary degeneration, superoxide production, proinflammatory gene expression, and leukostasis associated with STZ-induced diabetes [76]. Lipid mediators, such as leukotrienes and prostanoids, released locally by leukocytes, may also contribute to retina capillary degeneration [77]. Therefore, BM-derived hematopoietic cells play a central role in the pathogenesis of early diabetic retinopathy.
Leukostasis is mainly from an increase in monocytes (CD11b) but not granulocytes (Gr1) or lymphocytes (CD3) [75]. There is evidence, however, that neutrophils are also important in retinopathy. For example, both monocytes and neutrophils can promote leukocyte-mediated endothelial death in vitro. Furthermore, granulocytopenia from transplantation of G-CSF receptor-deficient BM prevents leukocyte-induced endothelial death and capillary degeneration in STZ injected mice [76].
The role of inflammatory hematopoietic cells in diabetic retinopathy is further supported by a postmortem study in humans that also revealed increased expression of ICAM-1 and neutrophils in the retina of diabetic subjects [78]. Unlike diabetic nephropathy, the role of CCL2-CCR2 in the development of retinopathy is not as well defined [75, 79]. The therapeutic implications of these findings are unclear.
Diabetic Neuropathy
Diabetic peripheral neuropathy (DPN) is the most common diabetic neuropathic syndrome that occurs in over half of all diabetic individuals. DPN is a symmetric, axon length-dependent sensorimotor polyneuropathy, which typically starts in the toes and gradually moves proximally. The cause of diabetic neuropathy is complex but, like other complications, hyperglycemia fuels its development. Hyperglycemia damages neurons and Schwann cells as well as endothelial cells of the vasa nervorum in the peripheral nerves. In addition, ER stress, inflammation, and dyslipidemia, especially in type 2 diabetes, are important causative factors [80].
Recent studies have uncovered the important role of hematopoietic cells in the pathogenesis of DPN. In animal models, injection of BM derived stem/progenitor cells such as EPCs and MSCs ameliorates diabetic neuropathy through their neurotrophic and angiogenic activities [81]. It seems that BM-derived stem/progenitor cells may protect against damages inflicted by hyperglycemia [20], though their reparative role in the development of diabetic neuropathy is unclear. On the other hand, inflammatory changes in the peripheral nerves induced by hematopoietic cells are well recognized in diabetic neuropathy. Increased macrophage infiltration was found in the sciatic nerve of rats with STZ diabetes [82]. Transplantation of iNOS or PARP-1-deficient BM was shown to prevent tactile allodynia, a manifestation of diabetic neuropathy [76]. As with other microvascular complications, RAGE plays a central role in diabetic neuropathy [2], and RAGE-deficient mice are protected from the development of diabetic neuropathy [83]. RAGE deficiency also improves axonal regeneration in superimposed acute peripheral nerve crash in mice with STZ-induced diabetes. Using BMT experiment, Juranek et al. discovered that absence of RAGE in the BM only was sufficient to recapitulate the beneficial effects of global RAGE deficiency on axonal regeneration in STZ diabetic mice [84].
As noted earlier, TNF-α-expressing PI-BMDCs are detected in multiple tissues and organs of type 1 and type 2 diabetic rodents [42]. Intriguingly, Terashima et al. subsequently observed PI-expressing neurons and Schwann cells, including the sciatic nerve as well as neurons in the dorsal root ganglion (DRG) in mice and rats with diabetic neuropathy [40, 43]. These PI-positive cells in the DRG turned out to be PI-BMDC-neuron fusion cells. Importantly, the fusion cells express TNF-α and are much more prone to apoptosis than the neighboring unfused DRG neurons (Box 2 and Figure 3). Furthermore, while PARP-1-deficient BMT protects against diabetic neuropathy, PARP-1-expressing (wild-type) BM cells confer susceptibility to diabetic neuropathy when they are transplanted to PARP-1-deficient mice, which are normally resistant to neuropathy [40]. In sum, PI-BMDCs play an active pathogenic role in the development of diabetic neuropathy.
Figure 3. Role of hematopoietic cells, especially PI-BMDCs, in diabetic neuropathy.
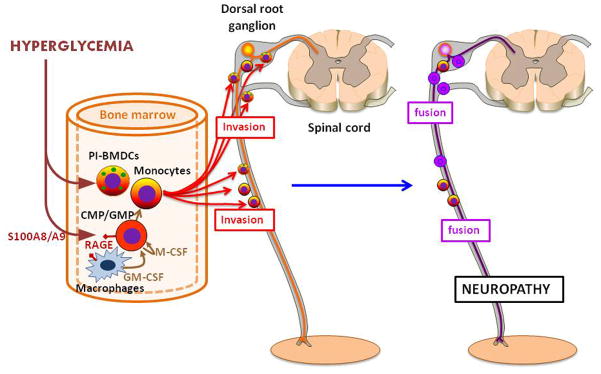
Hyperglycemia increases myelopoiesis through the activation of RAGE on the macrophages and common myeloid progenitors (CMPs) in the bone marrow [38]. Stimulated macrophages and CMPs secrete granulocyte-macrophage colony stimulating factor (GM-CSF) and macrophage-colony stimulating factor (M-CSF) leading to proliferation of granulocyte macrophage progenitors (GMPs) and CMPs/GMPs, respectively. RAGE+ BM-derived cells exhibit impaired axon regeneration after injury in diabetic mice [84]. Hyperglycemia also induces the emergence of proinsulin positive and TNF-α producing inflammatory cell in the BM [42]. These PI-BMDCs invade the peripheral nervous system, where they fuse with neurons and Schwann cells in the peripheral nerves, e.g., sciatic nerve, and with neurons in the dorsal root ganglia (DRG). The fusion damages the neurons and causes premature apoptosis. Furthermore, the PI-BMDC-neuron fusion cells continue to be a source of TNF-α, which amplifies the ill effects of PI-BMDCs [40, 43] (see Box 2).
So far, there is no clinical data to support the role of hematopoietic cells in diabetic neuropathy. There is ongoing investigation on the role of CCL2-CCR2. Pharmacologic blockade of the CCL2-CCR2 axis was reported to ameliorate nondiabetic neuropathic pain by interfering with pain signaling within the peripheral nervous system rather than recruitment of inflammatory cells [85]. However, recent phase 2 trials of a CCR2 antagonist in patients with painful diabetic neuropathy and posttraumatic neuralgia failed to improve symptoms [86, 87].
Concluding Remarks
Recent improvements in life expectancy have increased paradoxically the burden of chronic diabetic complications in people with diabetes. According to traditional wisdom, dysregulated biochemical pathways downstream of hyperglycemia directly harm cells in multiple organ systems, causing them to fail and to self-destruct. This appears to be true for all organs affected by the classic diabetic complications. However, it is only half the story. We now know that BM-derived hematopoietic cells are as culpable as the local cells in pathogenesis.
The realization that hematopoietic cells play an active role in the pathogenesis of diabetic complications is only the first chapter of an exciting story. It is common knowledge that hematopoietic cells infiltrate tissues afflicted by microvascular complications; traditionally, they were thought to be the clean-up crew that removes the debris from tissue damage. We now know that they actively participate in tissue damage. While the local cells undergo metabolic stress and express chemoattractant molecules as a result of inflammatory signaling, hematopoietic cells are primed for pro-inflammatory gene expression. Activation of RAGE in hematopoietic cells as well as the local cells may be a common mechanism of hyperglycemia-induced dysfunction [2].
RAGE and PARP-1 expression/activation are known to drive diabetic complications for over a decade. Recent BMT experiments in rodents show that blocking their expression on hematopoietic cells only is sufficient to ward off diabetic neuropathy. This suggests that the development of diabetic neuropathy is dependent on priming of hematopoietic cells. In addition to such conditioning of the BMDCs, the vulnerability of different organs to diabetic complications seems to also require additional factors, as not all organs develop overt complications, and those that do may do it at different rates.
One important question is how soon hyperglycemia is able to induce dysfunctional changes in hematopoietic cells versus the local cells, and how soon euglycemia can reverse cell dysfunction. Surprisingly, PI-BMDCs appear as short as three days after intermittent glucose injections. Acute hyperglycemia-induced inflammatory priming of hematopoietic cells may be a mechanism for the poor outcome in patients who develop hyperglycemia in acute coronary syndrome and ischemic stroke [88]. On the flipside, hyperglycemia-induced changes may linger on even after restoration of euglycemia, as exemplified by the appearance of leukostasis in the retina of non-diabetic recipients given BM cells from diabetic donors [75]. The persistent cellular dysfunction is likely the result of epigenetic changes in BM-derived cells [89] and may account for the development of neuropathy in prediabetic patients with mild or intermittent hyperglycemia [90].
A full knowledge of the interactions between hematopoietic cells and the local cells in diabetes will not be forthcoming until there is full characterization of the hyperglycemia-primed proinflammatory hematopoietic cells. Experiments using genetic or pharmacologic blockade models have provided clues on the key roles of RAGE, PARP-1, and CCR-2 in these cells. The very low level expression of PI is not thought to have any biological effect. It happens to be a useful biomarker that identifies a population of hyperglycemia-induced proinflammatory hematopoietic cells. Future studies can take advantage of such a marker to further characterize the hyperglycemia-induced changes in hematopoietic cells. It is noteworthy that PI is normally expressed in an exceedingly low level in myeloid cells [91] and the level goes up during hyperglycemia coincident with the appearance of TNF-α expression [40, 43–45].
From a therapeutic standpoint, maintaining stable euglycemia is the best and primary way of preventing diabetic complications. Unfortunately, it is difficult to achieve tight glycemic control without occasional hypoglycemia. Before we can achieve perfect euglycemia, targeting the proinflammatory or other detrimental characteristics, e.g., RAGE, PARP-1, or TNFα expressed by these hyperglycemia-associated hematopoietic cells would be effective, if not target cell-specific, forms of treatment. Such therapies have been shown to be effective in diabetic neuropathy rodent models [40, 84, 92]. Clinical trials are needed to test their safety and therapeutic efficacy in diabetic patients.
Finally, some BM subpopulations are actually known to have reparative function [20, 49, 81]. One treatment approach is actually the use of BM-derived stem/progenitor cells. Multifactorial diabetic complications such as diabetic foot ulcers may benefit from the BM reparative cells, which could induce simultaneously improvements in circulation, tissue regeneration, and peripheral nerve function. It is interesting that some forms of BM cells contribute to chronic diabetic complications, whereas other sets of BM cells may possess protective function. Despite much recent progress, this irony underlines how little we know about the various BM subpopulations and their role in diabetic complications.
In conclusion, there are emerging roles of dysfunctional hematopoietic cells in diabetic complications. Details of the molecular pathways mediated by these cells in different diabetic complications are yet to be deciphered. It is a fertile area for investigation; not only will we learn of new pathways in pathogenesis, we may also identify new treatments targeting these cells to slow down or reverse diabetic complications.
Highlights.
Hyperglycemia directly compromises the function of bone marrow (BM) cells.
Hyperglycemia produces a microenvironment (stem cell niche)-dependent defect in hematopoietic stem cell mobilization.
Hyperglycemia-induced dysregulated hematopoietic cells travel to organs affected by diabetes.
The dysregulated hematopoietic cells cause inflammation, cellular dysfunction, and accelerated apoptosis.
Acknowledgments
We thank E. Buras, L. Yang, M. Katagi, Y. Nakae, T. Terashima, J. Okano, and Yu-Chih Ku for valuable assistance and discussions. The research in the authors’ laboratories that was discussed in this review was supported by the US National Institutes of Health (P30-DK079638 for a Diabetes Endocrinology Research Center at Baylor College of Medicine, T32-HL066991, and R01-HL051586 to L.C.), and by the Ministry of Education, Culture, Sports, Science and Technology, Japan (Grant-in-Aid #23590378 to H.K.), and the President’s Discretionary Fund from Shiga University of Medical Science (#1515503I to H.K.). L.C. was also supported by the Betty Rutherford Chair for Diabetes Research from St. Luke’s Medical Center, Houston, Texas.
Glossary
- CC chemokine ligand 2 (CCL2)
also known as MCP1, is expressed by most nucleated cells in response to proinflammatory cytokines or stimulation of innate immune receptors. CCL2 binds to its receptor CCR2 (CCR2 also binds to CCL7 or MCP3) expressed by monocytes, HSCs and a subset of natural killer cells, and mediates recruitment of these cells to inflammatory foci
- CXC chemokine ligand 12 (CXCL 12)
also known as SDF1-α, is expressed by stromal cells including nestin+ MSCs in part under the control of the sympathetic nervous system. CXCL 12 binds to its receptor CXCR4, also known as CD184, expressed on hematopoietic cells. The CXCL 12/CXCR4 complex is involved in regulating and retaining HSPCs in the BM
- Endothelial progenitor cells (EPCs)
commonly defined as mononuclear cells positive for both immature cell and endothelial markers such as CD34 and VEGFR-2 and/or CD133 in humans. They exist in the peripheral blood and the BM and enhance vascular repair through re-endothelialization and neovascularization. EPCs are decreased in various vascular disorders. They also have received attention for their potential utility in cell therapy
- Hematopoietic stem cells (HSCs)
a group of self-renewing cells capable of producing daughter cells that proliferate and mature to provide all adult blood cells in erythroid, myeloid, and lymphoid lineages
- Hematopoietic stem and progenitor cells (HSPCs)
a term used to describe both hematopoietic stem cells (HSC) and progenitor cells (HPC). HSCs differentiate and become multipotent progenitor cells. Multipotent progenitor cells further differentiate into more committed oligopotent progenitor cells that eventually mature to individual lineages of hematopoietic cells. HPCs have little to no self-renewal capacity. HSPC is frequently used when distinction between HSC and HPC is unclear or unnecessary
- Leukostasis
an acute syndrome characterized by abnormal intravascular leukocyte aggregation and clumping. Inflammatory hematopoietic cells adhere to capillary endothelial cell occluding blood flow and damaging endothelial cells
- LSK cells
a lineage-negative (Lin)−stem-cell antigen 1 (SCA1)+KIT+ (LSK) population that is used to isolate HSCs in mice. The subset of LSK cells is heterogeneous in terms of self-renewal potential and contains long-term reconstituting HSCs (LT-HSCs) as well as short-term reconstituting HSCs (ST-HSCs). LT-HSCs maintain potential for self-renewal and multi-lineage differentiation throughout life and are the bona fide stem cells of hematopoiesis, whereas ST-HSCs, which derive from LT-HSCs, are multipotent but limited in self-renewal potential. ST-HSCs subsequently produce multi- and oligo-potent progenitors. Lineage means a collection of cell surface markers for all terminally differentiated populations
- Stem cell niche
the physical, molecular, and cellular microenvironment that regulates stem cell function in harmony with stem cell autonomous mechanisms, maintaining the balance between quiescence, self-renewal, differentiation, and mobilization of stem cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brownlee M. The pathobiology of diabetic complications a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 2.Manigrasso MB, et al. Unlocking the biology of RAGE in diabetic microvascular complications. Trends Endocrinol Metab. 2013 doi: 10.1016/j.tem.2013.08.002. http://dx.doi.org/10.1016/j.tem.2013.08.002. [DOI] [PMC free article] [PubMed]
- 3.Rask-Madsen C, King GL. Vascular complications of diabetes: mechanisms of injury and protective factors. Cell Metab. 2013;17:20–33. doi: 10.1016/j.cmet.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fadini GP, et al. Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J Am Coll Cardiol. 2005;45:1449–1457. doi: 10.1016/j.jacc.2004.11.067. [DOI] [PubMed] [Google Scholar]
- 5.Fadini GP, et al. Diabetes impairs progenitor cell mobilisation after hindlimb ischaemia-reperfusion injury in rats. Diabetologia. 2006;49:3075–3084. doi: 10.1007/s00125-006-0401-6. [DOI] [PubMed] [Google Scholar]
- 6.Fadini GP, et al. Time course and mechanisms of circulating progenitor cell reduction in the natural history of type 2 diabetes. Diabetes Care. 2010;33:1097–1102. doi: 10.2337/dc09-1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oikawa A, et al. Diabetes mellitus induces bone marrow microangiopathy. Arterioscler Thromb Vasc Biol. 2010;30:498–508. doi: 10.1161/ATVBAHA.109.200154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spinetti G, et al. Global remodeling of the vascular stem cell niche in bone marrow of diabetic patients: implication of the microRNA-155/FOXO3a signaling pathway. Circ Res. 2013;112:510–522. doi: 10.1161/CIRCRESAHA.112.300598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferraro F, et al. Diabetes impairs hematopoietic stem cell mobilization by altering niche function. Sci Transl Med. 2011;3:104ra101. doi: 10.1126/scitranslmed.3002191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orlandi A, et al. Long-term diabetes impairs repopulation of hematopoietic progenitor cells and dysregulates the cytokine expression in the bone marrow microenvironment in mice. Basic Res Cardiol. 2010;105:703–712. doi: 10.1007/s00395-010-0109-0. [DOI] [PubMed] [Google Scholar]
- 11.Hazra S, et al. Long-term type 1 diabetes influences haematopoietic stem cells by reducing vascular repair potential and increasing inflammatory monocyte generation in a murine model. Diabetologia. 2013;56:644–653. doi: 10.1007/s00125-012-2781-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Min D, et al. Alterations in monocyte CD16 in association with diabetes complications. Mediators Inflamm. 2012;2012:649083. doi: 10.1155/2012/649083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fadini GP, et al. An unbalanced monocyte polarisation in peripheral blood and bone marrow of patients with type 2 diabetes has an impact on microangiopathy. Diabetologia. 2013;56:1856–1866. doi: 10.1007/s00125-013-2918-9. [DOI] [PubMed] [Google Scholar]
- 14.Fadini GP, et al. Monocyte-macrophage polarization balance in pre-diabetic individuals. Acta Diabetol. 2013;50:977–982. doi: 10.1007/s00592-013-0517-3. [DOI] [PubMed] [Google Scholar]
- 15.Navarro-González JF, et al. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol. 2011;7:327–340. doi: 10.1038/nrneph.2011.51. [DOI] [PubMed] [Google Scholar]
- 16.Hammes HP, et al. Diabetic retinopathy: targeting vasoregression. Diabetes. 2011;60:9–16. doi: 10.2337/db10-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adamis AP, Berman AJ. Immunological mechanisms in the pathogenesis of diabetic retinopathy. Semin Immunopathol. 2008;30:65–84. doi: 10.1007/s00281-008-0111-x. [DOI] [PubMed] [Google Scholar]
- 18.Kern TS. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp Diabetes Res. 2007;2007:95103. doi: 10.1155/2007/95103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan L, et al. Pathogenesis of diabetic neuropathy: bad to the bone. Ann N Y Acad Sci. 2011;1240:70–76. doi: 10.1111/j.1749-6632.2011.06309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fadini GP. A reappraisal of the role of circulating (progenitor) cells in the pathobiology of diabetic complications. Diabetologia. 2014;57:4–15. doi: 10.1007/s00125-013-3087-6. [DOI] [PubMed] [Google Scholar]
- 21.Fadini GP, Avogaro A. It is all in the blood: the multifaceted contribution of circulating progenitor cells in diabetic complications. Exp Diabetes Res. 2012;2012:742976. doi: 10.1155/2012/742976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Menegazzo L, et al. Endothelial progenitor cells in diabetes mellitus. Biofactors. 2012;38:194–202. doi: 10.1002/biof.1016. [DOI] [PubMed] [Google Scholar]
- 23.Asahara T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–966. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 24.Urbich C, Dimmeler S. Endothelial progenitor cells characterization and role in vascular biology. Circ Res. 2004;95:343–353. doi: 10.1161/01.RES.0000137877.89448.78. [DOI] [PubMed] [Google Scholar]
- 25.Hill JM, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 26.Asahara T, et al. Concise review: circulating endothelial progenitor cells for vascular medicine. Stem Cells. 2011;29:1650–1655. doi: 10.1002/stem.745. [DOI] [PubMed] [Google Scholar]
- 27.Hagensen MK, et al. Circulating endothelial progenitor cells do not contribute to regeneration of endothelium after murine arterial injury. Cardiovasc Res. 2012;93:223–231. doi: 10.1093/cvr/cvr278. [DOI] [PubMed] [Google Scholar]
- 28.Yoder MC. Human endothelial progenitor cells. Cold Spring Harb Perspect Med. 2012;2:a006692. doi: 10.1101/cshperspect.a006692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loomans CJM, et al. Endothelial progenitor cell dysfunction a novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes. 2004;53:195–199. doi: 10.2337/diabetes.53.1.195. [DOI] [PubMed] [Google Scholar]
- 30.Tepper OM, et al. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002;106:2781–2786. doi: 10.1161/01.cir.0000039526.42991.93. [DOI] [PubMed] [Google Scholar]
- 31.Sibal L, et al. Circulating endothelial progenitor cells, endothelial function, carotid intima-media thickness and circulating markers of endothelial dysfunction in people with type 1 diabetes without macrovascular disease or microalbuminuria. Diabetologia. 2009;52:1464–1473. doi: 10.1007/s00125-009-1401-0. [DOI] [PubMed] [Google Scholar]
- 32.Egan CG, et al. Generalised reduction of putative endothelial progenitors and CXCR4-positive peripheral blood cells in type 2 diabetes. Diabetologia. 2008;51:1296–1305. doi: 10.1007/s00125-008-0939-6. [DOI] [PubMed] [Google Scholar]
- 33.Avogaro A, et al. Endothelial dysfunction in diabetes the role of reparatory mechanisms. Diabetes Care. 2011;34:S285–S290. doi: 10.2337/dc11-s239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fadini GP, et al. Diabetes impairs stem cell and proangiogenic cell mobilization in humans. Diabetes care. 2013;36:943–949. doi: 10.2337/dc12-1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fadini GP, Avogaro A. Diabetes impairs mobilization of stem cells for the treatment of cardiovascular disease: A meta-regression analysis. Int J Cardiol. 2013;168:892–897. doi: 10.1016/j.ijcard.2012.10.089. [DOI] [PubMed] [Google Scholar]
- 36.Gallagher KA, et al. Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1α. J Clin Invest. 2007;117:1249–1259. doi: 10.1172/JCI29710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Busik JV, et al. Diabetic retinopathy is associated with bone marrow neuropathy and a depressed peripheral clock. J Exp Med. 2009;206:2897–2906. doi: 10.1084/jem.20090889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagareddy PR, et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013;17:695–708. doi: 10.1016/j.cmet.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oh SH, et al. Adult bone marrow-derived cells trans-differentiating into insulin-producing cells for the treatment of type I diabetes. Lab Invest. 2004;84:607–617. doi: 10.1038/labinvest.3700074. [DOI] [PubMed] [Google Scholar]
- 40.Terashima T, et al. Bone marrow expression of poly (ADP-ribose) polymerase underlies diabetic neuropathy via hematopoietic-neuronal cell fusion. FASEB J. 2012;26:295–308. doi: 10.1096/fj.11-186262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.González F, et al. In vitro evidence that hyperglycemia stimulates tumor necrosis factor-alpha release in obese women with polycystic ovary syndrome. J Endocrinol. 2006;188:521–529. doi: 10.1677/joe.1.06579. [DOI] [PubMed] [Google Scholar]
- 42.Kojima H, et al. Extrapancreatic insulin-producing cells in multiple organs in diabetes. Proc Natl Acad Sci. 2004;101:2458–2463. doi: 10.1073/pnas.0308690100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Terashima T, et al. The fusion of bone-marrow-derived proinsulin-expressing cells with nerve cells underlies diabetic neuropathy. Proc Natl Acad Sci. 2005;102:12525–12530. doi: 10.1073/pnas.0505717102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fujimiya M, et al. Fusion of proinsulin-producing bone marrow-derived cells with hepatocytes in diabetes. Proc Natl Acad Sci. 2007;104:4030–4035. doi: 10.1073/pnas.0700220104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamashita T, et al. Fusion of bone marrow-derived cells with renal tubules contributes to renal dysfunction in diabetic nephropathy. FASEB J. 2012;26:1559–1568. doi: 10.1096/fj.11-183194. [DOI] [PubMed] [Google Scholar]
- 46.Chan L, et al. Chronic diabetic complications: the body’s adaptive response to hyperglycemia gone awry? Trans Am Clin Climatol Assoc. 2006;117:341–352. [PMC free article] [PubMed] [Google Scholar]
- 47.Shantsila E, et al. Endothelial progenitor cells in cardiovascular disorders. J Am Coll Cardiol. 2007;49:741–752. doi: 10.1016/j.jacc.2006.09.050. [DOI] [PubMed] [Google Scholar]
- 48.Kanwar YS, et al. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol. 2011;6:395–423. doi: 10.1146/annurev.pathol.4.110807.092150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo JK, Cantley LG. Cellular maintenance and repair of the kidney. Annu Rev Physiol. 2010;72:357–376. doi: 10.1146/annurev.physiol.010908.163245. [DOI] [PubMed] [Google Scholar]
- 50.Zheng F, et al. Development of Albuminuria and Glomerular Lesions in Normoglycemic B6 Recipients of db/db Mice Bone Marrow The Role of Mesangial Cell Progenitors. Diabetes. 2004;53:2420–2427. doi: 10.2337/diabetes.53.9.2420. [DOI] [PubMed] [Google Scholar]
- 51.Ezquer F, et al. Endovenous administration of bone marrow-derived multipotent mesenchymal stromal cells prevents renal failure in diabetic mice. Biol Blood Marrow Transplant. 2009;15:1354–1365. doi: 10.1016/j.bbmt.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 52.Wang Y, Harris CH. Macrophages in renal disease. J Am Soc Nephrol. 2011;22:21–27. doi: 10.1681/ASN.2010030269. [DOI] [PubMed] [Google Scholar]
- 53.Sassy-Prigent C, et al. Early glomerular macrophage recruitment in streptozotocin-induced diabetic rats. Diabetes. 2000;49:466–475. doi: 10.2337/diabetes.49.3.466. [DOI] [PubMed] [Google Scholar]
- 54.Coimbra TM, et al. Early events leading to renal injury in obese Zucker (fatty) rats with type II diabetes. Kidney Int. 2000;57:167–182. doi: 10.1046/j.1523-1755.2000.00836.x. [DOI] [PubMed] [Google Scholar]
- 55.Chow F, et al. Macrophages in mouse type 2 diabetic nephropathy: correlation with diabetic state and progressive renal injury. Kidney Int. 2004;65:116–128. doi: 10.1111/j.1523-1755.2004.00367.x. [DOI] [PubMed] [Google Scholar]
- 56.Young BA, et al. Cellular events in the evolution of experimental diabetic nephropathy. Kidney Int. 1995;47:935–944. doi: 10.1038/ki.1995.139. [DOI] [PubMed] [Google Scholar]
- 57.Utimura R, et al. Mycophenolate mofetil prevents the development of glomerular injury in experimental diabetes1. Kidney Int. 2003;63:209–216. doi: 10.1046/j.1523-1755.2003.00736.x. [DOI] [PubMed] [Google Scholar]
- 58.Okada S, et al. Intercellular adhesion molecule-1-deficient mice are resistant against renal injury after induction of diabetes. Diabetes. 2003;52:2586–2593. doi: 10.2337/diabetes.52.10.2586. [DOI] [PubMed] [Google Scholar]
- 59.Chow FY, et al. Intercellular adhesion molecule-1 deficiency is protective against nephropathy in type 2 diabetic db/db mice. J Am Soc Nephrol. 2005;16:1711–1722. doi: 10.1681/ASN.2004070612. [DOI] [PubMed] [Google Scholar]
- 60.Chow FY, et al. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 2006;69:73–80. doi: 10.1038/sj.ki.5000014. [DOI] [PubMed] [Google Scholar]
- 61.Chow FY, et al. Monocyte chemoattractant protein-1-induced tissue inflammation is critical for the development of renal injury but not type 2 diabetes in obese db/db mice. Diabetologia. 2007;50:471–480. doi: 10.1007/s00125-006-0497-8. [DOI] [PubMed] [Google Scholar]
- 62.Awad AS, et al. Monocyte/macrophage chemokine receptor CCR2 mediates diabetic renal injury. Am J Physiol Renal Physiol. 2011;301:F1358–F1366. doi: 10.1152/ajprenal.00332.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sayyed SG, et al. An orally active chemokine receptor CCR2 antagonist prevents glomerulosclerosis and renal failure in type 2 diabetes. Kidney Int. 2011;80:68–78. doi: 10.1038/ki.2011.102. [DOI] [PubMed] [Google Scholar]
- 64.Seok SJ, et al. Blockade of CCL2/CCR2 signalling ameliorates diabetic nephropathy in db/db mice. Nephrol Dial Transplant. 2013;28:1700–1710. doi: 10.1093/ndt/gfs555. [DOI] [PubMed] [Google Scholar]
- 65.Lim AKH, et al. Antibody blockade of c-fms suppresses the progression of inflammation and injury in early diabetic nephropathy in obese db/db mice. Diabetologia. 2009;52:1669–1679. doi: 10.1007/s00125-009-1399-3. [DOI] [PubMed] [Google Scholar]
- 66.Zheng D, et al. Transfused macrophages ameliorate pancreatic and renal injury in murine diabetes mellitus. Nephron Exp Nephrol. 2011;118:e87–e99. doi: 10.1159/000321034. [DOI] [PubMed] [Google Scholar]
- 67.Furuta T, et al. The role of macrophages in diabetic glomerulosclerosis. Am J Kidney Dis. 1993;21:480–485. doi: 10.1016/s0272-6386(12)80393-3. [DOI] [PubMed] [Google Scholar]
- 68.Bohle A, et al. The pathogenesis of chronic renal failure in diabetic nephropathy: investigation of 488 cases of diabetic glomerulosclerosis. Pathol Res Pract. 1991;187:251–259. doi: 10.1016/s0344-0338(11)80780-6. [DOI] [PubMed] [Google Scholar]
- 69.Nguyen D, et al. Macrophage accumulation in human progressive diabetic nephropathy. Nephrology (Carlton) 2006;11:226–231. doi: 10.1111/j.1440-1797.2006.00576.x. [DOI] [PubMed] [Google Scholar]
- 70.Simó R, et al. Neurodegeneration is an early event in diabetic retinopathy: therapeutic implications. Br J Ophthalmol. 2012;96:1285–1290. doi: 10.1136/bjophthalmol-2012-302005. [DOI] [PubMed] [Google Scholar]
- 71.Anand-Apte B. Studies on Retinal and Choroidal Disorders. Springer; 2012. Dysfunction of Circulating Endothelial Progenitor Cells in Diabetic Retinopathy; pp. 517–528. [Google Scholar]
- 72.Schröder S, et al. Activated monocytes and granulocytes, capillary nonperfusion, and neovascularization in diabetic retinopathy. Am J Pathol. 1991;139:81–100. [PMC free article] [PubMed] [Google Scholar]
- 73.Miyamoto K, et al. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci. 1999;96:10836–10841. doi: 10.1073/pnas.96.19.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Joussen AM, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004;18:1450–1452. doi: 10.1096/fj.03-1476fje. [DOI] [PubMed] [Google Scholar]
- 75.Serra AM, et al. CD11b+ bone marrow-derived monocytes are the major leukocyte subset responsible for retinal capillary leukostasis in experimental diabetes in mouse and express high levels of CCR5 in the circulation. Am J Pathol. 2012;181:719–727. doi: 10.1016/j.ajpath.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 76.Li G, et al. Marrow-derived cells regulate the development of early diabetic retinopathy and tactile allodynia in mice. Diabetes. 2012;61:3294–3303. doi: 10.2337/db11-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Talahalli R, et al. Leukocytes regulate retinal capillary degeneration in the diabetic mouse via generation of leukotrienes. J Leukoc Biol. 2013;93:135–143. doi: 10.1189/jlb.0112025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McLeod DS, et al. Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am J Pathol. 1995;147:642–653. [PMC free article] [PubMed] [Google Scholar]
- 79.Dong N, et al. Upregulation of retinal neuronal MCP-1 in the rodent model of diabetic retinopathy and its function in vitro. Invest Ophthalmol Vis Sci. 2012;53:7567–7575. doi: 10.1167/iovs.12-9446. [DOI] [PubMed] [Google Scholar]
- 80.Vincent AM, et al. Diabetic neuropathy: cellular mechanisms as therapeutic targets. Nat Rev Neruol. 2011;7:573–583. doi: 10.1038/nrneurol.2011.137. [DOI] [PubMed] [Google Scholar]
- 81.Han JW, et al. Cell Therapy for Diabetic Neuropathy Using Adult Stem or Progenitor Cells. Diabetes & metabolism journal. 2013;37:91–105. doi: 10.4093/dmj.2013.37.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nukada H, et al. Increased susceptibility to ischemia and macrophage activation in STZ-diabetic rat nerve. Brain Res. 2011;1373:172–182. doi: 10.1016/j.brainres.2010.11.084. [DOI] [PubMed] [Google Scholar]
- 83.Toth C, et al. Receptor for advanced glycation end products (RAGEs) and experimental diabetic neuropathy. Diabetes. 2008;57:1002–1017. doi: 10.2337/db07-0339. [DOI] [PubMed] [Google Scholar]
- 84.Juranek JK, et al. RAGE deficiency improves postinjury sciatic nerve regeneration in type 1 diabetic mice. Diabetes. 2013;62:931–943. doi: 10.2337/db12-0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jung H, et al. Visualization of chemokine receptor activation in transgenic mice reveals peripheral activation of CCR2 receptors in states of neuropathic pain. J Neurosci. 2009;29:8051–8062. doi: 10.1523/JNEUROSCI.0485-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kalliomäki J, et al. A randomized, double-blind, placebo-controlled trial of a chemokine receptor 2 (CCR2)-antagonist in posttraumatic neuralgia. Pain. 2013;154:761–767. doi: 10.1016/j.pain.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 87.Kalliomäki J, et al. Evaluation of a novel chemokine receptor 2 (CCR2)-antagonist in painful diabetic polyneuropathy. Scand J Pain. 2013;4:77–83. doi: 10.1016/j.sjpain.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 88.Balasanthiran A, et al. Hyperglycaemia in the acute care setting. Clinical Medicine. 2012;12:272–275. doi: 10.7861/clinmedicine.12-3-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.El-Osta A, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. 2008;205:2409–2417. doi: 10.1084/jem.20081188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Papanas N, et al. Neuropathy in prediabetes: does the clock start ticking early? Nat Rev Endocrinol. 2011;7:682–690. doi: 10.1038/nrendo.2011.113. [DOI] [PubMed] [Google Scholar]
- 91.Narendran P, et al. Proinsulin is encoded by an RNA splice variant in human blood myeloid cells. Proc Natl Acad Sci. 2006;103:16430–16435. doi: 10.1073/pnas.0607380103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yamakawa I, et al. Inactivation of TNF-α ameliorates diabetic neuropathy in mice. Am J Physiol Endocrinol Metab. 2011;301:E844–E852. doi: 10.1152/ajpendo.00029.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Katayama Y, et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124:407–421. doi: 10.1016/j.cell.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 94.Méndez-Ferrer S, et al. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452:442–447. doi: 10.1038/nature06685. [DOI] [PubMed] [Google Scholar]
- 95.Méndez-Ferrer S, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kojima H, et al. NeuroD-betacellulin gene therapy induces islet neogenesis in the liver and reverses diabetes in mice. Nat Med. 2003;9:596–603. doi: 10.1038/nm867. [DOI] [PubMed] [Google Scholar]
- 97.Yechoor V, et al. Neurogenin3 is sufficient for transdetermination of hepatic progenitor cells into neo-islets in vivo but not transdifferentiation of hepatocytes. Dev Cell. 2009;16:358–373. doi: 10.1016/j.devcel.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]