

Abstract
We report the design and implementation of the first phase 3 trial of CoenzymeQ10 (CoQ10) in children with genetic mitochondrial diseases. A novel, rigorous set of eligibility criteria was established. The trial, which remains open to recruitment, continues to address multiple challenges to the recruitment of patients, including widely condoned empiric use of CoQ10 by individuals with proven or suspected mitochondrial disease and skepticism among professional and lay mitochondrial disease communities about participating in placebo-controlled trials. These attitudes represent significant barriers to the ethical and scientific evaluation–and ultimate approval–of nutritional and pharmacological therapies for patients with life-threatening inborn errors of energy metabolism.
Keywords: Coenzyme Q10, Mitochondrial disease, Respiratory chain, Clinical trials
1.1 Introduction
1.1.1
Treatment of mitochondrial diseases has been generally disappointing and has usually been approached in an uncontrolled manner (Kerr, 2010; Vockley J and Vockley CM, 2010). Despite the routine use of specialized diets, nutritional supplements or investigational drugs, there is no objectively proven human therapy for any genetic mitochondrial disease that directly impacts cellular energy metabolism. However, coenzyme Q10 (CoQ10) has garnered enthusiasm as a potential treatment for specific deficiencies of the respiratory chain, including coenzyme Q10, deficiency because of its apparent safety, its integral role in the processes of electron transport, and its antioxidant properties. Results from open label and randomized clinical trials of CoQ10 in mitochondrial diseases have been reviewed recently (Haas, 2007; Stacpoole, 2011a). None of these trials evaluated prospectively the chronic safety and efficacy of CoQ10 in a pediatric population. Consequently, evidence-based guidelines for the use of CoQ10 in children with mitochondrial disorders are lacking.
1.1.2
We now report the design, implementation and eligibility criteria of the first phase 3 trial of CoQ10 in children with confirmed primary mitochondrial diseases. The term “primary” refers to disease caused by a biochemically or genetically proven disorder of mitochondrial respiratory metabolism (oxidative phosphorylation), in contrast to an acquired condition. The trial’s central hypothesis is that oral CoQ10 is a safe and effective treatment for children with primary disorders of mitochondrial respiratory metabolism, and that this beneficial action is reflected in improved motor function and quality of life.
1.2 Methods and Results
1.2.1 Study Organization
The organization and implementation of the trial was funded by a grant from the Orphan Products Division of the U.S. Food and Drug Administration (FDA) and is carried out under the auspices of IND 60,902 issued to Tischon Corporation that provides liquid CoQ10 and placebo gratis to the trial participants. The trial includes 4 clinical centers in which patients are evaluated and treated, diagnostic biochemistry cores, a clinical pharmacology core and a data coordinating center (DCC) (Table 1). Patients are evaluated and treated at the clinical sites. The biochemistry cores are based in the Center for Inherited Disorders of Energy Metabolism at Case Western Reserve University and at Sick Children’s Hospital at the University of Toronto. They undertake enzymological measurement of respiratory chain enzymes (complexes I–V) and, when necessary, additional biochemical determinations (see Eligibility Criteria). A pharmacology core at the University of Cincinnati measures plasma CoQ10 concentrations and a data coordinating core at the University of Florida oversees patient randomization, data management and statistical analysis. A Steering and Planning Committee (SPC) comprised of the clinical site investigators and heads of the various cores meets bi-weekly by teleconference to review progress and discuss the possible eligibility of prospective patients. During the trial’s initial years, the Hospital for Sick Children at the University of Toronto served as a clinical and diagnostic venue, but changed in late 2011 when the lead clinician moved to the University of California, San Diego. A data and safety monitoring board (DSMB) is comprised of experts in inborn errors of metabolism and biostatistics and includes a lay representative. The DSMB meets every 6 months by teleconference.
Table 1.
Operational aspects of trial.
| Institution | Role in project |
|---|---|
| University of Florida1 | Clinical center; data coordinating center |
| Case Western Reserve University | Clinical center; diagnostic core |
| University of Cincinnati | Clinical center; pharmacology core |
| University of Toronto2 | Clinical center; diagnostic core |
Lead Institution
Replaced by University of California, San Diego, as a Clinical center in Fall, 2011.
1.2.2 Study Design
The trial is a multinational randomized, double-blind comparison of CoQ10 (as ubiquinol) and placebo each administered for 6 months, in a crossover design. The two primary outcome measures are a validated, standardized test of motor function and quality of life. All patients are randomly assigned first to group A or B, stratified by age and gender (Fig. 1). Stratifying by diagnosis is neither practical for this small trial nor scientifically meaningful, because of the clinical heterogeneity among individuals who harbor the same biochemical or even molecular genetic defect. The randomized groups are: Group 1: Arm A→Arm B (25 patients) or Group 2: Arm B→Arm A (25 patients), where the arms are defined as: Arm A: placebo for six months (25 cases) and Arm B: CoQ10 10 mg/kg/day, max. 400 mg/day, for six months (25 cases).
Fig. 1.
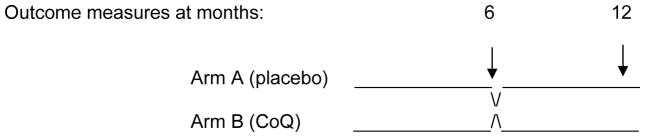
J Schematic of crossover trial. N=25 per treatment arm.
1.2.2.1
We provide two liquid formulations to patients: liquid ubiquinol (LiQNOL; Tishcon) and placebo, which are formulated to be identical in labeling, appearance, consistency and taste. CoQ10, as ubiquinol, or placebo is administered once daily with (or after) the evening meal. The randomization code and product strength code are kept by the DCC, which informs a Clinical Center as to what supply of coded product a newly enrolled patient will be given. CoQ10 is lipid soluble and susceptible to degradation on prolonged exposure to light and high ambient temperatures. Therefore, the liquid products are provided patients in opaque, screw-capped containers and are accompanied by instructions for home storage. CoQ10 and placebo containers appear identical. Patients return their unused product, in sealed containers, to the Clinical Center at each visit for volume measurement by the Research Coordinator, as a compliance check. Lots are replaced every 3 months by a new lot in which the appropriate formulation strength has been verified.
1.2.2.2
We chose this dose of CoQ10 for our trial based on a review of both open label and randomized controlled trials of CoQ10 in adults and children (Haas, 2007; Kerr 2010), indicating that most patients received doses of ≤ 20 mg/kg/d without evidence of harm. Furthermore, in a study of children with suspected mitochondrial diseases (Miles et al, 2008) using the liquid ubiquinol (LiQNOL) formulation by Tishcon (Miles et al, 2006), demonstrated excellent bioequivalence (Miles et al, 2002) and bioavailability (Evans et al, 2009) and a CoQ10 dosage of 1 mg/kg/d provided plasma CoQ10 levels generally within or slightly above the pediatric reference range (0.48–1.80 μmol/l) (Duncan et al, 2005). Their data also showed that a LiQNOL dosage of 5 mg/kg/d would provide an approximately 4-fold to 5-fold increase in baseline plasma concentration that was similar to the concentration range reported in a study of adults with Parkinson disease who received 1200 mg/d (approximately 4 to 8 μmol/d) (Shults et al, 2002).
1.2.2.3
No previous studies have correlated CoQ10 doses with blood and/or muscle concentration in patients with respiratory chain deficiencies. However, a recent multicenter evaluation (Sacconi et al, 2010) of muscle CoQ10 status in 76 patients with molecular genetic and/or clinical evidence of a primary mitochondrial disorder disclosed a relatively high proportion with mild (21% – 68% below the control mean; N=25) or severe (≤ 20% below control; N=3) CoQ10 deficiency. In at least nine of these cases, the low muscle CoQ10 level was secondary to a pathological nDNA or mtDNA mutation in a gene not involved in the CoQ10 biosynthetic pathway. Response to CoQ10 supplementation was not investigated in that study.
1.2.2.4
An almost universal, but completely empiric, practice among physicians and patients with mitochondrial diseases, especially those with respiratory chain (RC) defects, is to administer various ‘cocktails’ of nutritional supplements, as standard treatment. Typical ingredients include supra-physiological doses of ascorbate, B vitamins, CoQ10 and carnitine, in various admixtures. We recognized both the emotional import of these generally innocuous (but questionably effective) cocktails for patients, and their potential as confounding variables in a clinical trial. Therefore, all patients who enter the CoQ10 trial are asked to discontinue their current supplements, and instead receive a capsulated nutritional supplement of the following composition: vitamin C 40 mg, riboflavin 10 mg, thiamine 10 mg, and carnitine 40 mg. The number of capsules each subject receives is based on the child’s body weight as follows:
| Up to 4 kg | 1 capsule daily |
| 4.1 to 8 kg | 2 capsules daily |
| 8.1 to 12 kg | 3 capsules daily |
| 12.1 to 16 kg | 4 capsules daily |
1.2.2.5
Every additional 4 kg weight = 1 additional supplement capsule up to a maximum of 10 capsules daily. The supplement is prepared by Tishcon Corp. and provided without charge. Patients also receive a standard, RDA-compliant multivitamin and mineral preparation (equivalent to Centrum Kids Complete) that is chewable or that can be administered by mouth or feeding tube in pulverized form. Families are encouraged to continue participation in the study regardless of their child’s compliance with the nutritive cocktail mixture.
1.2.3 Clinical endpoints
Valuable predictive biomarkers of disease progression for most mitochondrial diseases, are lacking. Patients with RC defects who die during the neonatal period typically present with fulminant lactic acidosis and presumably have a profound deficiency in the activity of the affected enzyme, although this may be variably expressed in cultured fibroblasts or lymphocytes. In less severely affected patients, however, the magnitude of the enzyme defect measured in cultured cells correlates rather poorly with other biochemical and clinical manifestations of the disease (Munnich at el, 2001). This “fact of life” about mitochondrial diseases is frustrating, but predictable, because of the variable expression of the biochemical defect and the difficulty in biopsying the most relevant tissues. There is limited value of such biomarkers of disease or disease progression as lactate concentration, neuroimaging studies, or heteroplasmic load in relatively unaffected cells, such as lymphocytes or fibroblasts. Consequently, alternative functional measures are warranted to quantify the presentation and course of mitochondrial diseases among individuals with diverse genotypes and phenotypes. Potential clinical tools include assessment of motor function, validated quality of life (QOL) questionnaires and neurobehavioral testing.
1.2.3.1
The CoQ10 trial’s primary outcome measures are 1) motor function and 2) quality of life. Quantitation of motor function employs the complete version of the Gross Motor Function Measure (GMFM-88) by trained personnel. The test was originally designed to evaluate change in gross motor function in children with cerebral palsy and has been used subsequently in children with other neuromuscular developmental disorders (Lundkvist et al, 2009; Russell et al, 1989). Additional quantitative muscle strength testing is performed in any patient attaining a maximum score on the GMFM-88. A standard six minute walk test is also conducted in patients capable of undertaking this procedure. Home quality of life is measured by a questionnaire developed at the University of Florida and applied in an earlier randomized clinical trial (RCT) in children with heterogeneous forms of congenital lactic acidosis (Stacpoole et al, 2006).
1.2.4 Data and Safety Monitoring
The trial’s Data Safety and Monitoring Board (DSMB) meets every six months by email, teleconferences or in person since the beginning of the trial to review its progress and reviews, on an ad hoc basis, any Serious Adverse Events (SAEs). There have been 70 AEs reported since the start of the trial; only one was considered an SAE and involved a teenage female with MELAS who developed post-ictal mental status changes. No AE was considered to be related to the study medication or to procedures by the investigators or by the DSMB but, rather, were judged to be secondary to the underlying disease (e.g., seizure or death) process or to intercurrent, transient, illnesses (e.g., cold, diarrhea). Those in which such a relationship could not be ruled out included symptoms of generalized fatigue or muscle weakness, increased levels of serum transaminases (1 case) or loose stools (3 cases). In general, these findings are consistent with those of others in the chronic administration of LiQNOL to children with mitochondrial disease or Down syndrome (6). AEs and DSMB reports are forwarded to the trial’s sponsor, Tishcon Corporation that, in turn, files the reports with the FDA.
1.2.5 Eligibility and recruitment (Table 2)
Table 2.
Eligibility criteria for the trial.
Inclusion criteria:
|
Exclusion criteria:
|
Eligible patients must have well-defined deficiencies of RC complexes I, II, III, IV, or V, or combinations of these, or have defined mtDNA or nDNA mutations affecting subunits or assembly of these complexes that are associated with known clinical/pathological features, such as Chronic Progressive External Ophthalmoplegia (CPEO), Kearns-Sayre, Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-like Episodes (MELAS), Mitochondrial Encephalopathy and Ragged Red Fibers (MERRF), Neuropathy, Ataxia and Retinitis Pigmentosa (NARP) or Leigh syndrome. We consider “well-defined” deficiencies of an RC complex to have a reduction in enzymological activity of at least one complex to <25% of the mean or two or more complexes <35% of the mean, provided that at least 3 other mitochondrial enzyme activities are ≥60% of the mean, to assure sample integrity and that the measured activity is specific to the affected complex(s), rather than an artifact of the assay or general deterioration of the sample. Furthermore, patients with pathological mtDNA or nDNA mutations associated with disruption of the RC or oxidative phosphorylation (OXPHOS) (e.g., SURF1, SUCLA2, ATP6) must have recognizable clinical/pathological manifestations to be eligible for enrollment. Thus, if a patient (or family member) carried a mtDNA mutation but was not affected in some recognizable way as a clinical manifestation of that mutation, we would assume their level of whole body heteroplasmy was below the clinical threshold and they would be precluded from entering the study.
1.2.5.1
Virtually all children enrolled in this trial have neuromuscular complications as the dominant clinical expressions of disease. At baseline, these include some degree of cognitive delay and hypotonia in most patients, i.e., clinical manifestations that may benefit from CoQ10 treatment and that will be quantitated prospectively by validated, objective tools described below. At entry, the inclusion criteria listed in Table 2 must have already been met. Determination of diagnoses of specific deficiencies of mitochondrial disorders of electron chain complexes I, II, III, IV or V, or mtDNA will depend on standard, well-established procedures for evaluation of muscle biopsies at the diagnostic laboratory, including pathological examination, enzymatic assays, and mutational analyses of mtDNA (and nDNA in selected cases, e.g., SURF mutations). In some cases, some or all of this diagnostic information will have been obtained at other laboratories but reviewed by the SPC is to determine consistency of the findings and eligibility for randomization. The specific biochemical and diagnostic criteria we employ, including quality control measures, are summarized in Table 3.
Table 3.
Detailed Diagnostic Criteria.
|
1.2.5.2
Once a potential subject is identified, the patient is invited to participate in the study. A parent or legal guardian is asked to give consent for the child’s medical records to be reviewed by the SPC to determine if the child has met all inclusion/exclusion criteria.
1.2.5.3
A prospective patient may be self-referred to one of the participating institutions. At the time of initial referral to a center, a subject may or may not have had a diagnosis of a primary mitochondrial disease that, on review, is considered definitive. Further diagnostic studies may be performed in (and may require) more than one cell or tissue type (e.g., skin, muscle, blood cells). Primary diagnostic testing must be completed before entering the study and is part of the pre-recruitment clinical service. The expenses for these studies are borne by the patient or by third parties, as is currently required by the diagnostic laboratories. Supplementary or confirmatory testing may be performed by one of the diagnostic core laboratories, if recommended following the eligibility review.
1.2.6 Current status of the trial
We screened 107 potential patients through March, 2012 (Fig. 2). Note that “possibly eligible” subject data are not cumulative, whereas the other categories in Fig. 2 do reflect cumulative data. Potentially eligible patients are actively moved to the eligible or ineligible category as additional diagnostic information becomes available. Clinical and laboratory information on each subject is discussed at biweekly conference calls. Five new subjects have been declared eligible since January, 2012. To date, 9 patients have dropped out because of family decisions regarding clinical status, logistical hardships associated with travel to the treatment site or, in 2 patients, because of dissatisfaction with the taste of the blinded product formulation (CoQ10 or placebo). All randomized subjects have provided blood specimens during both the drug and placebo phases of our study for CoQ10 measurement. In 17 patients in whom complete data are currently available, the mean ± SD plasma total CoQ10 level was 5.44 ± 2.95 μg/ml during drug administration and 0.64 ± 0.25 μg/ml during placebo administration. These values are similar to those reported by Glover et al. (Glover et al, 2010) in adults treated with ~20 mg/kg/d CoQ10 (as ubiquinol).
Fig. 2.
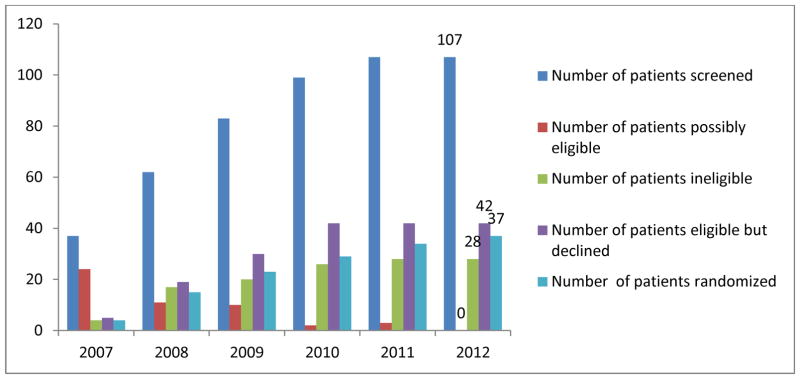
Subject screening and enrollment, 2007-March, 2012.
1.2.7 Recruitment challenges
A few eligible families (~3) have declined to participate in this trial in part or solely because of the logistical challenges associated with travel to a treatment site. However, the principal challenge to recruitment relates to the intervention itself and to deeply ingrained attitudes by many in the professional and lay mitochondrial disease communities regarding placebo-controlled trials (Stacpoole, 2011a; Vockley et al, 2010). Virtually all subjects we have screened for this study have already been receiving a commercial CoQ10 preparation as part of a nutritional cocktail prescribed by their primary caregiver and/or through their family’s efforts. We anticipated this high frequency of CoQ10 exposure and designed the trial so that treatment comparisons would occur after 6 months each of CoQ10 and placebo exposure, i.e., at months 6 and 12. However, the major reason why a disproportionately low number of eligible patients have entered the trial is reluctance of families and practitioners to engage in a placebo-controlled trial that, in their perception, could possibly jeopardize the child’s clinical status by temporarily removing an ingredient from a nutritional concoction of presumed efficacy and safety. We continue to address this difficult challenge in publications (2, 4) and at national meetings (Stacpoole, 2011b; Stacpoole 2011c).
1.2.7.1
Our approach to patient recruitment is based on the following activities: 1) word of mouth among colleagues in pediatrics, neurology and genetics; 2) annual talks given by some of us to families at annual meetings of the UMDF; 3) an informative booth about this trial for families and physicians at UMDF meetings; 4) twice-annual announcements about the trial posted on UMDF, NORD and Exceptional Parent websites. UMDF meetings and electronic advertisements have typically generated a spurt of interest and have contributed to a substantial number of enrolled subjects; 5) All CoQ10 trial sites are part of the new North American Mitochondrial Disease Consortium (NAMDC), which is part of the NIH’s Rare Disease Clinical Research Network. NAMDC publicizes the trial in its continued communications with its member institutions and is developing a patient registry of patients with mitochondrial diseases to facilitate clinical trial participation; 6) Webinars and articles by trial investigators sponsored by the UMDF and Exceptional Parent, the latter reaching a worldwide audience; and 7) contacting families of patients who have had diagnostic RC assays performed through the Center for Inherited Disorders of Energy Metabolism (CIDEM) and whose results match our biochemical eligibility criteria. Families are contacted prospectively with the expectation that they will be more motivated to consider the CoQ10 trial soon after diagnosis. This approach is based on sending referring physicians a cover letter and IRB-approved information about the trial, with a copy of the diagnostic report and additional information for families and how they can contact us, if interested. CIDEM-affiliated investigators then follow-up with a phone call to the referring physician.
1.3 Discussion
1.3.1
CoQ10 supplementation may be expected to benefit children with disorders of the mitochondrial respiratory chain by several mechanisms that are not mutually exclusive. The first would be CoQ10 (ubiquinone) deficiency, detected by low muscle CoQ10, which may be primary or secondary, and is therefore clinically heterogeneous (Ogasahara et al, 1989; Musumeci et al, 2001; Lamperti et al, 2003). These relatively infrequent cases may have been identified and already treated prior to entering into the proposed study. Some of these subjects have been treated with infant doses as high as 30 mg/kg/d or adult doses of 3000 mg/d, well above the higher dose planned for this trial (Musumeci et al, 2001; Rahman et al, 2001). The second mechanism could be facilitating electron transport by circumventing a block in the electron transport chain. For example, if the subject had a block in complex III, supplemental CoQ10 could accept electrons from either the normal ubiquinone binding site or the FeS site of complex I (or both), and donate them to complex IV via ascorbate. This mechanism was originally demonstrated in an individual with complex III deficiency treated with vitamin K3 (menadione) and ascorbate (Arogov et al, 1986). In the proposed study, CoQ10 would serve as a preferred electron acceptor, replacing menadione, which is potentially toxic for infants (Arogov et al, 1986). Presumably, the higher dose of CoQ10 would be more effective in this situation. The third mechanism is that CoQ10 can serve as an antioxidant, accepting electrons from disrupted electron transport and reducing the risk of formation of reactive oxygen species that might cause damage to mitochondrial membranes, proteins, lipids, and DNA (Geromel et al, 2002; Beal, 2003; Turunen et al, 2004). This is the most general mechanism, potentially applicable to any defect of electron transport, and presumably the implicit rationale for widespread current use of CoQ10 in management of children with mitochondrial disorders.
1.3.1.1
However, it could be argued that the rationale for conducting a randomized controlled trial of CoQ10 in the targeted population is rendered moot by the disappointing results of other CoQ10 trials in adults with mitochondrial (Haas, 2007) or other neurodegenerative diseases (Huntington Study Group, 2001; Mancuso et al, 2010; Shults et al, 2002; Stamelou et al, 2008). Furthermore, mixtures containing CoQ10 and other nutritional supplements (Rodriguez et al, 2007) or newly developed CoQ10 analogs (Bergamini et al, 2012; Enns et al, 2012) have been advocated as being potentially superior because of potency and/or enhanced bioavailability to ubiquinol alone in mitigating the clinical complications of primary mitochondrial disease. However, such claims remain based, for the most part, on anecdotal reports and early phase clinical trials in pediatric subjects or in controlled trials conducted in adults. Thus, there remains an unmet need for a scientifically and ethically rigorous examination of CoQ10 per se in the treatment of young children with OXPHOS disorders who, on theoretical biochemical grounds, may be the most likely to benefit from supplementation with this naturally occurring molecule.
1.3.1.2
The use of CoQ10 as sole interventional therapy in this trial must also account for the almost universal tendency of patients with possible or proven mitochondrial disease to be prescribed or to self-administer supplemental “cocktails” of naturally occurring substances. Clinical trialists are thus faced with the dilemma of deciding to adopt a “purist” approach by prohibiting any such mixtures – a decision almost certainly dooming effective patient recruitment – or of adjusting to this reality and minimizing its impact as a confounding variable. Therefore, we chose to first, assure potential subjects and their families that nutritional supplements would be continued throughout the trial and, second, sought to standardize the cocktail’s composition to reduce confounding. Although the dose of most cocktail ingredients is adjusted according to body weight, the relatively short duration of the trial has so far caused only trivial changes in the actual amount of any supplement a given subject consumes.
1.3.1.3
One of the most critical requirements of the design of any phase 3 trial is to develop a set of rigorous eligibility criteria that ensure a high confidence level in determining specificity in both the pathological diagnosis and the enrollment of appropriate patients. Two diagnostic schemes (Bernier et al, 2002; Wolf and Smeitink, 2002) for children with suspected mitochondrial disease were published well before the CoQ10 trial was organized, so why did we chose not to adopt them for our study? The more widely used Bernier criteria originally evolved from an earlier classification of adults with presumed RC disorders and a retrospective review of patient records at a single hospital. Analysis of these databases led to the designation of “definite, probable, possible or unlikely” diagnostic categories, based on clinical, histological, enzymological, functional and molecular information. Accordingly, such data were further categorized into so-called “major” or “minor” diagnostic criteria, in which a definite diagnosis of a RC disorder could be made on the basis of two major criteria or one major plus two minor criteria, providing other possible metabolic or non-metabolic disorders had already been excluded by appropriate tests.
1.3.1.4
The Bernier criteria have been employed in non-interventional studies of children with RC diseases (e.g., Diogo et al, 2010; Kang et al, 2007). However, to our knowledge, neither it nor the diagnostic approach developed by Wolf and Smeitnik (Wolf and Smeitink, 2002) have been used in randomized controlled trials. As pointed out by Haas et al (Haas, 2007), both diagnostic approaches require extensive laboratory testing, based on each group’s own quality control criteria, which are notoriously variable among laboratories engaged in the diagnosis of mitochondrial diseases. Moreover, neither approach incorporates criteria for the diagnosis of pathological mutations that do not directly involve an RC subunit or complex, such as POLG mutations, both of which are part of the CoQ10 trial inclusion criteria (Table 3). In contrast to the Bernier and Wolf and Smeitnik algorithms, we excluded as diagnostic criteria muscle histopathology, such as cytochrome oxidase or ragged red fiber staining or mitochondrial morphology on electron microscopy because of their lack of specificity in terms of identifying a particular enzyme defect or pathological mutation. Consequently, only subjects who had clinical signs or symptoms consistent with a mitochondrial disease and who demonstrated 1) unequivocal deficiency of CoQ10 measured in biopsied skeletal muscle; 2) markedly decreased enzyme activity; and/or 3) a proven pathological nDNA or mtDNA mutation were considered eligible for our trial.
1.3.1.5
We recruit patients from throughout North America and many subjects are referred on the basis of enzymological tests conducted by laboratories using variable criteria for tissue integrity and disease diagnosis. Thus, we often conduct additional biochemical assays when residual tissue is available and/or refer patients for molecular genetic testing before making definitive decisions regarding diagnosis and eligibility. Such procedures frequently lead to exclusion of potential subjects and to weeks-months delays in the final acceptance of eligible patients. Despite this limitation in the speed of patient recruitment, such rigor greatly enhances confidence in rendering a diagnosis. Therefore, creating and adhering to stringent diagnostic guidelines are time-consuming, but essential features of randomized controlled trials that must be taken into account of by investigators, review groups and funding agencies in developing and adjudicating such studies.
1.3.1.6
This trial is conducted at four North American sites that were chosen because of past experience in participating in randomized controlled trials of inborn errors of metabolism and because of their track record as referral centers for the diagnosis and treatment of primary mitochondrial disorders. Funding limitations prevented incorporating additional centers to the trial, which became a significant impediment to recruiting patients from geographically distant locations, despite invaluable assistance from Mercy Medical Airlift. Those involved in the design and funding of future controlled trials for mitochondrial and other rare diseases must be mindful of this caveat and seek creative strategies for ensuring a broad geographic distribution of participating institutions while adhering to tight fiscal constraints.
1.3.1.7
Finally, ingrained lay and even professional misunderstanding and skepticism about subjecting children with life-threatening mitochondrial diseases to placebo-controlled trials continues to be the major barrier to their timely and efficient conduct and, consequently, a major cause of why no proven therapies for mitochondrial diseases exist. Thus, we encourage patient advocacy groups and related scientific organizations to assume a prominent and continuous responsibility for educating lay and professional communities about the necessity of participation in rigorously controlled trials. Indeed, this will be essential if the current therapeutic void is ever to be overcome with safe, effective, approved and affordable alternatives treatments for mitochondrial disorders.
Table 4.
Demographics of Randomized Patients.
| Diagnosis | Male (age at entry 2 – 17 yrs) | Female (age at entry 2 – 17 yrs) |
|---|---|---|
| Complex I Deficiency | 6 | 6 |
| Complex II Deficiency | 0 | 2 |
| Complex III Deficiency | 2 | 2 |
| Complex IV Deficiency | 0 | 1 |
| Multiple Complex Deficiencies | 5 | 4 |
| Homoplasmic m.5814T>C (in the tRNA Cys) Mutation | 0 | 1 |
| MELAS: ⋄ Mutation A3243G |
2 | 2 |
| Leigh Syndrome: ⋄ Mutation T8993G in the ATPase 6 gene |
||
| Homoplasmic 961 T>G in the 12S rRNA gene | 1 | 0 |
| POLG1 Mutation | 1 | 1 |
| Total | 17 | 20 |
Through March, 2012
Highlights.
First phase 3 trial of CoQ10 in children with mitochondrial diseases.
Rigorous, laboratory-based diagnostic and eligibility criteria applicable to future trials.
Novel and extensive patient recruitment strategies applicable to future trials.
Inclusion of qualitative and quantitative clinical endpoints important to patient home functionality.
Acknowledgments
This work was supported in part by a grant from the Food and Drug Administration R01 FD003032, the NIH/NCATS Clinical and Translational Science Award to the University of Florida UL1 TR000064 and Tishcon Corporation. We thank the United Mitochondrial Disease Foundation for publicizing the trial, Mercy Medical Airlife for patient transportation and C. Caputo for editorial assistance.
Abbreviations
- AE
Adverse Event
- ATP6
Adenosine triphosphate synthase subunit 6
- CIDEM
Center for Inherited Disorders of Energy Metabolism
- CoQ10
Coenzyme Q10
- CPEO
Chronic Progressive External Ophthalmoplegia
- DCC
Data Coordinating Center
- DSMB
Data Safety Monitoring Board
- FDA
Food and Drug Administration
- GMFM88
Gross Motor Function Measure, version 88
- LiQNOL®
Liquid ubiquinol
- MELAS
Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-like Episodes
- MERFF
Mitochondrial Encephalopathy and Ragged Red Fibers
- NAMDC
North American Mitochondrial Disease Consortium
- NARP
Neuropathy, Ataxia and Retinitis Pigmentosa
- nDNA
Nuclear Deoxyribonucleic Acid
- NORD
National Organization for Rare Disorders
- mtDNA
Mitochondrial Deoxyribonucleic Acid
- OXPHOS
Oxidative Phosphorylation
- POLG
polymerase gamma
- RC
Respiratory chain
- RCT
Randomized Clinical Trial
- SAE
Serious Adverse Event
- SUCLA2
Succinyl-CoA Synthetase Ligase
- SURF1
Surfeit gene 1
- SPC
Steering and Planning Committee
- UMDF
United Mitochondrial Disease Foundation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arogov Z, Bank WJ, Maris J, et al. Treatment of mitochondrial myopathy due to complex III deficiency with vitamins K3 and C: A 31P-NMR follow-up study. Ann Neurol. 1986;19:598. doi: 10.1002/ana.410190615. [DOI] [PubMed] [Google Scholar]
- Beal MF. Mitochondria, oxidative damage, and inflammation in Parkinson’s disease. Ann NY Acad Sci. 2003;991:120–31. doi: 10.1111/j.1749-6632.2003.tb07470.x. [DOI] [PubMed] [Google Scholar]
- Bergamini C, Moruzzi N, Sblendido A, Lenaz G, Fato R. A water soluble CoQ10 formulation improves intravellular distribution and promotes mitochondrial respiration in cultured cells. PLoS One. 2012;7:e33712. doi: 10.1371/journal.pone.0033712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier FP, Boneh A, Dennett X, Chow CW, Cleary MA, Thorburn DR. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology. 2002;12:1406–11. doi: 10.1212/01.wnl.0000033795.17156.00. [DOI] [PubMed] [Google Scholar]
- Diogo L, Cordeiro M, Garcia P, Fineza I, Moura C, Oliveira CR, Veiga M, Garcia T, Grazina M. Value of brain magnetic resonance imaging in mitochondrial respiratory chain disorders. Pediatr Neurol. 2010;42:196–200. doi: 10.1016/j.pediatrneurol.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Duncan AJ, Heales SJR, Mills K, Eaton S, Land JM, Hargreaves IP. Determination of coenzyme Q10 status in blood mononuclear cells, skeletal muscle, and plasma by HPLC with di-propoxy-coenzyme Q10 as an internal standard. Clin Chem. 2005;51:2380–2382. doi: 10.1373/clinchem.2005.054643. [DOI] [PubMed] [Google Scholar]
- Enns GM, Kinsman SL, Perlman SL, Spicer KM, Abdenur JE, Cohen BH, Arnagala A, Barnes A, Kheifets V, Shrader WD, Thoolen M, Blankenberg F, Miller G. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol Genet Metab. 2012;105:91–102. doi: 10.1016/j.ymgme.2011.10.009. [DOI] [PubMed] [Google Scholar]
- Evans M, Baisley J, Barss S, Guthrie N. A randomized, double-blind trial on the bioavailability of two CoQ10 formulations. J Functional Foods. 2009;1:65–73. [Google Scholar]
- Geromel V, Darin N, Chretien D, et al. Coenzyme Q10 and idebenone in the therapy of respiratory chain disorders: rationale and comparative benefits. Molecular Genetics and Metabolism. 2002;77:21–30. doi: 10.1016/s1096-7192(02)00145-2. [DOI] [PubMed] [Google Scholar]
- Glover EI, Martin J, Maher A, Thornhill RE, Moran GR. A randomized trial of coenzyme Q10 in mitochondrial disorders. Muscle Nerve. 2010;42:739–748. doi: 10.1002/mus.21758. [DOI] [PubMed] [Google Scholar]
- Haas RH. The evidence basis for coenzyme Q therapy in oxidative phosphorylation disease. Mitochondrion. 2007;7S:S136–S145. doi: 10.1016/j.mito.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Huntington Study Group. A randomized, placebo-controlled trial of coenzyme Q10 and remacemide in Huntington’s disease. Neurology. 2001;57:397–404. doi: 10.1212/wnl.57.3.397. [DOI] [PubMed] [Google Scholar]
- Kang HC, Kwon JW, Lee YM, Kim HD, Lee HJ, Hahn SH. Nonspecific mitochondrial disease with epilepsy in children: diagnostic approaches and epileptic phenotypes. Childs Nerv Syst. 2007;23:1301–7. doi: 10.1007/s00381-007-0369-7. [DOI] [PubMed] [Google Scholar]
- Kerr DS. Treatment of mitochondrial electron transport chain disorders, 2010. A review of clinical trials over the past decade. Mol Genet Metab. 99(3):246–255. doi: 10.1016/j.ymgme.2009.11.005. [DOI] [PubMed] [Google Scholar]
- Lamperti C, Naini A, Hirano M, et al. Cerebellar ataxia and coenzyme Q10 deficiency. Neurology. 2003l;60:1206–08. doi: 10.1212/01.wnl.0000055089.39373.fc. [DOI] [PubMed] [Google Scholar]
- Lundkvist JA, Jarnlo GB, Gummesson C, Nordmark E. Longitudinal construct validity of the GMFM-88 total score and goal total score and the GMGM-66 score in a 5-year follow-up study. Phys Ther. 2009;89:342–40. doi: 10.2522/ptj.20080037. [DOI] [PubMed] [Google Scholar]
- Mancuso M, Orsucci D, Volpi L, Calsolaro V, Siciliano G. Coenzyme Q10 in neuromuscular and neurodegenerative disorders. Curr Drug Targets. 2010;11(1):111–121. doi: 10.2174/138945010790031018. [DOI] [PubMed] [Google Scholar]
- Miles MC, Horn P, Miles L, Tang P, Steele P, DeGrauw T. Bioequivalence of coenzyme Q10 from OTC supplements. Nutrition Research. 2002;22(8):919–929. [Google Scholar]
- Miles MV, Miles L, Tang PH, Horn PS, Steele PE, DeGrauw AJ, Wong BL, Bove KE. Systematic evaluation of muscle coenzyme Q10 content in children with mitochondrial respiratory chain enzyme deficiencies. Mitochondrion. 2008;8:170–180. doi: 10.1016/j.mito.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Miles MV, Patterson BJ, Schapiro MB, Hickey FJ, Chalfonte-Evans M, Horn PS, Hotze SL. Coenzyme Q10 Absorpotion and Tolerance in Children With Down Syndrome: A Dose-Ranging Trial. Pediatr Neurol. 2006;35:30–37. doi: 10.1016/j.pediatrneurol.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Munnich A, Rotig A, Cormier V, Rustin P. Clinical presentation of respiratory chain deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, et al., editors. Metabolic and Molecular Bases of Inherited Disease. Mc-Graw-Hill; New York: 2001. pp. 2261–2271. [Google Scholar]
- Musumeci O, Naini A, Slonim AE, et al. Familial cerebellar ataxia with muscle Q10 deficiency. Neurology. 2001;56:849–55. doi: 10.1212/wnl.56.7.849. [DOI] [PubMed] [Google Scholar]
- Ogasahara S, Engel AG, Frens D, Mack D. Muscle coenzyme Q deficiency in familial mitochondrial encephalopathy. Proc Natl Acad Sci USA. 1989;86:2379–82. doi: 10.1073/pnas.86.7.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S, Hargreaves I, Clayton P, Heales S. Neonatal presentation of coenzyme Q10 deficiency. J Pediatr. 2001;139:456–8. doi: 10.1067/mpd.2001.117575. [DOI] [PubMed] [Google Scholar]
- Rodriguez MC, MacDonald JR, Mahoney DJ, Parise G, Beal MF, Tamopolsky MA. Beneficial effects of creatine, CoQ10 and lipoic acid in mitochondrial disorders. Muscle Nerve. 2007;35:235–242. doi: 10.1002/mus.20688. [DOI] [PubMed] [Google Scholar]
- Russell DJ, Rosenbaum PL, Cadman DT, Gowland C, Hardy S, Jarvis S. The Gross Motor Function Measure: a means to evaluate the effects of physical therapy. Dev Med Child Neurol. 1989;31:341–352. doi: 10.1111/j.1469-8749.1989.tb04003.x. [DOI] [PubMed] [Google Scholar]
- Sacconi S, Trevisson E, Salviati L, Aymé, Rigal O, Redondo AG, Mancuso M, Siciliano G, Tonin P, Angelini C, Auré K, Lombès, Desnuelle C. Coenzyme Q10 is frequently reduced in muscle of patients with mitochondrial myopathy. Neuromuscular Disorders. 2010;20:44–48. doi: 10.1016/j.nmd.2009.10.014. [DOI] [PubMed] [Google Scholar]
- Shults CW, Oakes D, Kieburtz K, Beal F, Haas R, Plumb S, Juncos JL, Nutt J, Shoulson I, Carter J, Kompoliti K, Perimutter JS, Reich S, Stern M, Watts RL, Kurlan R, Molho E, Harrison M, Lew M Parkinson Study Group. Effects of coenzyme Q10 in early Parkinson disease. Evidence of slowing of the functional decline. Arch Neurol. 2002;59:1541–1523. doi: 10.1001/archneur.59.10.1541. [DOI] [PubMed] [Google Scholar]
- Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, Felitsyn NM, Gilmore Rl, Greer M, Henderson GH, Hutson AD, Neiberger RE, O’Brien RG, Perkins LA, Quisling RG, Shroads AL, Shuster JJ, Silverstein JH, Theriaque DW, Valenstein E. A controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics. 2006;117:1519–1531. doi: 10.1542/peds.2005-1226. [DOI] [PubMed] [Google Scholar]
- Stacpoole PW. Why are there no proven therapies for genetic mitochondrial diseases? Mitochondrion. 2011a;11(5):679–85. doi: 10.1016/j.mito.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacpoole PW. Why are there no proven therapies for genetic mitochondrial diseases/Society for Inherited Metabolic Disorders Annual Meeting; Asilomar, CA. February 28-March 3; 2011b. (Invited Symposium speaker.) [Google Scholar]
- Stacpoole PW. Forward Together! Joining researchers and community in advancing clinical trials for mitochondrial diseases. United Mitochondrial Disease Foundation Annual Symposium; Chicago, IL. June 15–19; 2011c. (Symposium Chairman) [Google Scholar]
- Stamelou M, Reuss A, Pilatus U, Magerkurth J, Niklowitz P, Eggert KM, Krisp A, Menke T, Schade-Brittinger C, Oertel WH, Höglinger GU. Short-term effects of coenzyme Q10 in progressive supranuclear palsy: a randomized, placebo-controlled trial. Movement Disorders. 2008;23(7):942–949. doi: 10.1002/mds.22023. [DOI] [PubMed] [Google Scholar]
- Turunen M, Olsson, Dallner G. Metabolism and function of coenzyme Q. Biochim Biophys Acta. 2004;1660:171–99. doi: 10.1016/j.bbamem.2003.11.012. [DOI] [PubMed] [Google Scholar]
- Vockley J, Vockley CM. Clinical trials: curing a critical deficiency in metabolic medicine. Mol Genet Metab. 2010;99(3):244–245. doi: 10.1016/j.ymgme.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Wolf NI, Smeitink JAM. Mitochondrial disorders: A proposal for consensus diagnostic criteria in infants and children. Neurology. 2002;59:1402–1405. doi: 10.1212/01.wnl.0000031795.91814.d8. [DOI] [PubMed] [Google Scholar]