

A rational double mutation in the serine acetyl xylo-oligosaccharide esterase from G. stearothermophilus T6 (Axe2) converts the enzyme from an octameric form to a dimeric form. The dimeric double mutant (Axe2-Y184F-W190P) has been crystallized in the orthorhombic space group P212121 and a complete X-ray diffraction data set has been measured for these crystals at 2.3 Å resolution for use in a full three-dimensional structural analysis of this form of the protein.
Keywords: Geobacillus stearothermophilus, xylan utilization, xylan esterase, serine esterase, double mutant, oligomerization
Abstract
Xylans are polymeric sugars constituting a significant part of the plant cell wall. They are usually substituted with acetyl side groups attached at positions 2 or 3 of the xylose backbone units. Acetylxylan esterases are part of the hemicellulolytic system of many microorganisms which utilize plant biomass for growth. These enzymes hydrolyze the ester linkages of the xylan acetyl groups and thus improve the accessibility of main-chain-hydrolyzing enzymes and their ability to break down the sugar backbone units. The acetylxylan esterases are therefore critically important for those microorganisms and as such could be used for a wide range of biotechnological applications. The structure of an acetylxylan esterase (Axe2) isolated from the thermophilic bacterium Geobacillus stearothermophilus T6 has been determined, and it has been demonstrated that the wild-type enzyme is present as a unique torus-shaped octamer in the crystal and in solution. In order to understand the functional origin of this unique oligomeric structure, a series of rational noncatalytic, site-specific mutations have been made on Axe2. Some of these mutations led to a different dimeric form of the protein, which showed a significant reduction in catalytic activity. One of these double mutants, Axe2-Y184F-W190P, has recently been overexpressed, purified and crystallized. The best crystals obtained belonged to the orthorhombic space group P212121, with unit-cell parameters a = 71.1, b = 106.0, c = 378.6 Å. A full diffraction data set to 2.3 Å resolution has been collected from a flash-cooled crystal of this type at 100 K using synchrotron radiation. This data set is currently being used for the three-dimensional structure analysis of the Axe2-Y184F-W190P mutant in its dimeric form.
1. Introduction
Acetylxylan esterases hydrolyze the ester linkages of the acetyl moieties of xylan, which is one of the main polysaccharides constituting the plant cell wall. In many cases, these xylan polymers are decorated with acetyl side groups, which are attached at positions 2 or 3 of the xylose backbone units. For example, in the 4-O-methyl-d-glucuronoxylan polymer, the main hardwood hemicellulose, about 70% of the xylose units are acetylated (Sjöström, 1993 ▶). These acetyl substituents are usually involved in the intermolecular interactions between adjacent xylan chains and between xylan and other hemicelluloses and celluloses. As such, these groups contribute to the overall stability of the hardwood infrastructure, especially against enzymatic degradation. Hence, by removing the acetyl groups of xylan, these polymers become more accessible to main-chain-hydrolyzing enzymes such as xylanases and xylosidases, which can then break down their backbone to smaller sugar units (Biely et al., 2011 ▶). Acetylxylan esterases are classified in the CAZy database and are found in eight of the 16 carbohydrate esterase (CE) families characterized to date (Cantarel et al., 2009 ▶). Families CE3 (acetylxylan esterases) and CE12 (pectin acetylesterases, rhamnogalacturonan acetylesterases and acetylxylan esterases) are also classified as part of the SGNH superfamily of hydrolytic enzymes (CL0264) in the Pfam database of protein families (Punta et al., 2012 ▶).
Geobacillus stearothermophilus T6 is a Gram-positive thermophilic bacterium that possesses an extensive hemicellulolytic system, which is encoded by more than 40 genes (Shulami et al., 1999 ▶, 2011 ▶; Tabachnikov & Shoham, 2013 ▶). For the degradation and utilization of xylan, the bacterium initially secretes an extracellular xylanase (Gat et al., 1994 ▶; Khasin et al., 1993 ▶; Lapidot et al., 1996 ▶; Teplitsky et al., 1997 ▶, 2004 ▶; Bar et al., 2004 ▶), which partially degrades xylan to short decorated xylo-oligomers. These oligomers are then transported into the cell via a specific ATP-binding cassette (ABC) sugar transport system (Shulami et al., 2007 ▶). Inside the cell, the decorated xylo-oligomers are further hydrolyzed by side-chain-cleaving enzymes, including two α-arabinofuranosidases (Shallom, Belakhov, Solomon, Gilead-Gropper et al., 2002 ▶; Shallom, Belakhov, Solomon, Shoham et al., 2002 ▶; Hövel et al., 2003 ▶), an α-glucuronidase (Teplitsky et al., 1999 ▶; Zaide et al., 2001 ▶; Golan, Shallom et al., 2004 ▶; Shallom et al., 2004 ▶) and two xylan esterases (Alalouf et al., 2011 ▶; Lansky, Alalouf et al., 2013 ▶, 2014 ▶). The final degradation of the resulting xylo-oligomers is performed by an intracellular xylanase (Teplitsky et al., 2000 ▶; Solomon et al., 2007 ▶) and three β-xylosidases (Bravman, Michaly et al., 2001 ▶; Bravman, Zolotnitsky et al., 2001 ▶; Bravman et al., 2003 ▶; Czjzek et al., 2005 ▶; Shallom et al., 2005 ▶; Brüx et al., 2006 ▶; Ben-David et al., 2007 ▶).
In the framework of a wider study of the hemicellulolytic system of G. stearothermophilus, we have recently cloned, overexpressed, purified and characterized one of the two xylan esterases mentioned above, Axe2, the axe2 gene product (GenBank accession No. ABI49953.1). Axe2 is an acetyl xylo-oligosaccharide serine esterase belonging to the lipase GDSL 2 family (UniProtKB/TrEMBL accession No. Q09LX1), which is made up of 219 amino acids and has a calculated molecular mass of 24 770 Da (Alalouf et al., 2011 ▶). Axe2 deacetylates completely xylobiose peracetate (fully acetylated) and is active on synthetic substrates such as 2-naphthyl acetate, 4-nitrophenyl acetate, 4-methylumbelliferyl acetate and phenyl acetate. Based on bioinformatic analysis, Axe2 and its homologues do not belong to any of the known families in the CAZy database and thus represent a new family of carbohydrate esterases (Alalouf et al., 2011 ▶). The crystal structures of native Axe2 (Axe2-WT) and one of its catalytic mutants (Axe2-S15A) have been resolved and analyzed at high resolution (Lansky, Alalouf et al., 2013 ▶, 2014 ▶), demonstrating that the enzyme has the general SGNH hydrolase fold consisting of five central parallel β-sheets flanked by two layers of helices. Interestingly, the crystal structure of Axe2 revealed a unique ‘doughnut-shaped’ homo-octamer quaternary structure, made of two staggered tetrameric rings, where the eight active sites face a wide internal cavity. Gel-filtration, TEM (transmission electron microscopy) and SAXS (small-angle X-ray scattering) experiments confirmed that the octameric torus structure of Axe2-WT is also the main form of the enzyme present in solution (Lansky, Alalouf et al., 2014 ▶).
Although several hypotheses have been proposed for the functional role of the octameric assembly of Axe2 (Lansky, Alalouf et al., 2014 ▶), none of them could be unequivocally supported by other experimental results. A dedicated study combining rational mutagenesis and structural analysis was therefore initiated in order to further examine the unique quaternary structure of the enzyme and its functional significance, if any. The oligomeric arrangement of Axe2 creates three specific types of monomer–monomer interfaces, each of which involves a number of intermolecular interactions. These specific interactions were rationally disrupted by a series of site-specific mutations, followed by the characterization of the resulting Axe2 mutant in terms of its oligomeric state and its catalytic activity. This study demonstrated that several mutations of this kind turn Axe2 from its native octameric form to a different dimeric form (based on gel-filtration and SAXS experiments), with a significant reduction in catalytic activity (Alalouf, 2013 ▶). In this report we describe the crystallization and preliminary crystallographic characterization of one of these dimeric double mutants, Axe2-Y184F-W190P. Two complete crystallographic diffraction data sets have been collected from crystals of this mutant to 2.86 Å resolution (using our in-house crystallographic setup) and to 2.30 Å resolution (using a synchrotron set-up). These data are currently being used for the full three-dimensional structure determination of this dimeric form of the mutated protein.
2. Experimental
2.1. Production and purification of the Axe2-Y184F-W190P mutant
Site-directed mutagenesis was performed using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, California, USA). The Axe2-Y184F-W190P double mutant was obtained by two sequential mutations. The mutagenic primers for the mutations were as follows (the mutated nucleotides are given in bold): Axe2-Y184F, 5′-GTA-TTA-AAG-ACG-CTT-TTT-CCG-GCT-GCG-CTC-GCT-3′; Axe2-W190P, 5′-G-GCT-GCG-CTC-GCT-CCG-GAT-CGG-GTT-CAC-C-3′. The mutated gene was sequenced to confirm that only the desired mutation has been inserted. The axe2 (Y184F, W190P) gene was cloned in the pET9d vector and overexpressed in Escherichia coli BL21(DE3) as described previously (Alalouf et al., 2011 ▶). In short, E. coli BL21 [pET9d-axe2(Y184F, W190P)] cultures were grown overnight (500 ml medium in 2 l baffled flasks at 310 K) in Terrific Broth medium containing kanamycin (25 µg ml−1). Following overnight growth (average OD of 600 nm of 14.5), the cells were harvested, resuspended in about 45 ml buffer solution (50 mM Tris–HCl pH 7.0, 100 mM NaCl, 0.02% NaN3), disrupted by two passages through an EmulsiFlex-C3 homogenizer (Avestin Inc., Ottawa, Canada) and centrifuged (14 000 rev min−1 for 30 min), to obtain a soluble extract. The soluble extract was treated with protamine sulfate (0.2% final concentration) to remove nucleic acids; following centrifugation (14 000 rev min−1 for 30 min) the protein was purified by gel filtration using a HiLoad 26/600 Superdex 200 pg column (GE Healthcare Life Sciences). The final protein yield was about 150 mg per litre of culture and the protein appeared to be more than 95% pure based on SDS–PAGE.
2.2. Crystallization experiments
Crystallization experiments were set up immediately after the last purification step of the Axe2-Y184F-W190P mutant. The purified protein was concentrated using Centricon centrifugal concentrators (Millipore, Massachusetts, USA) to approximately 3 mg ml−1 and this protein solution (containing 50 mM Tris–HCl pH 7.0, 100 mM NaCl, 0.02% NaN3) was directly used for the crystallization experiments. All initial crystallization experiments were performed by the hanging-drop vapour-diffusion method, using an extensive series of different factorial screens (Jancarik & Kim, 1991 ▶). The final Axe2-Y184F-W190P crystallization drops were prepared by mixing the concentrated protein solution with each of the specific screen conditions (in various protein solution:screen solution proportions) to a final drop volume of 5.0 µl. Because of the different protein/screen ratios, the protein concentration in the drop varied in the range of 30–70% of its initial concentration. Each of these protein drops was then suspended over a 0.50–1.0 ml reservoir of the screen solution in 4 × 6 VDX crystallization plates (Hampton Research, California, USA) for a period of about 1–10 d at a constant temperature of 293 K. In general, these initial conditions were based on commercially available sets of ‘ready-to-use’ screening solutions. Once positive results had been obtained (i.e. crystals or microcrystals), further refinement experiments of these crystallization conditions were performed with specially prepared solutions, optimizing parameters of the initial conditions, such as pH, ionic strength, protein concentration, temperature, precipitating additives, protein drop volume and drop-to-reservoir ratio (Almog et al., 1993 ▶, 1994 ▶; Teplitsky et al., 1997 ▶, 1999 ▶, 2000 ▶; Gilboa et al., 1998 ▶; Bar et al., 2004 ▶; Golan, Zharkov et al., 2004 ▶; Reiland et al., 2004 ▶; Solomon et al., 2013 ▶; Lansky, Alalouf et al., 2013 ▶; Lansky, Salama et al., 2013 ▶; Lansky, Zehavi et al., 2014 ▶).
These screening experiments produced potentially useful crystals from two different screen solutions: condition No. 46 of the Wizard Classic 1 Screen (10% PEG 8K, 0.1 M imidazole–HCl buffer pH 8, 200 mM calcium acetate) and condition No. 18 of the Wizard Classic 2 Screen (20% PEG 3K, 0.1 M Tris–HCl buffer pH 7, 200 mM calcium acetate) (Emerald Bio, Bainbridge Island, Washington, USA). These initial conditions were then optimized, varying the parameters listed above, to produce crystals suitable for data collection (see below).
Diffraction data measurements were performed initially at the Technion Center for Structural Biology (TCSB, Technion, Haifa, Israel) using our in-house X-ray source. A second round of diffraction measurements was performed on the BM14 beamline of the European Synchrotron Research Facility (ESRF, Grenoble, France; see below). Processing, reduction, indexing, integration and scaling of the diffraction data were conducted using the HKL-2000 (Otwinowski & Minor, 1997 ▶) and HKL-3000 (Minor et al., 2006 ▶) program suites.
3. Results and discussion
3.1. Crystal forms of Axe2-Y184F-W190P
Reasonable crystals of the Axe2-Y184F-W190P double mutant have been obtained from three different crystallization conditions after optimizing the two initial screening ‘hits’ mentioned above. Condition 1, refined from Wizard Classic 1 Screen solution No. 46, comprised a reservoir solution consisting of 7.5–10% PEG 8K, 0.1 M imidazole–HCl buffer pH 6.5–7.2, 200 mM calcium acetate. Condition 2, refined from Wizard Classic 2 Screen solution No. 18, comprised a reservoir solution consisting of 16–21% PEG 3 K, 0.1 M Tris–HCl buffer pH 7.5, 200 mM calcium acetate. Condition 3, refined from a combination of the two screen solutions mentioned above (Wizard Classic 1 Screen solution No. 46 and Wizard Classic 2 Screen solution No. 18), comprised a reservoir solution consisting of 16.5–18.5% PEG 8K, 0.1 M Tris–HCl buffer pH 8.0, 100 mM calcium acetate. The crystals obtained with the Condition 1 reservoir solution usually appeared as ‘rectangular rods’ (typical dimensions of 0.02 × 0.02 × 0.15 mm) and were hence categorized as the RR1 crystal habit. Very similar ‘rectangular rod’ crystals were obtained from the Condition 2 reservoir solution, which were categorized as the RR2 crystal habit (Fig. 1 ▶). The RR2 crystals seemed to be more variable in terms of appearance and overall dimensions. They could appear as single rods (Fig. 1 ▶ a, centre, and Fig. 1 ▶ b), as small loosely bound clusters of four to eight rods (Fig. 1 ▶ a, bottom right) or as a larger tightly bound cluster of ten to 20 rods (Fig. 1 ▶ c). Their dimensions varied in the range of 0.02–0.05 × 0.02–0.05 × 0.07–0.35 mm. The crystals obtained with the Condition 3 reservoir solution appeared usually as small rectangular bars (typical dimensions of 0.02 × 0.03 × 0.05 mm) and were categorized as the RB1 crystal habit.
Figure 1.

Typical crystals of Axe2-Y184F-W190P of the RR2 crystal habit. These crystals appeared as single rods (a, top right; b), as small loosely bound clusters of four to eight rods (a, bottom right), or as a larger tightly bound cluster of ten to 20 rods (c). Their dimensions varied in the range of 0.02–0.05 × 0.02–0.05 × 0.07–0.35 mm. The crystals observed in (b) were used for the measurement of the full diffraction data sets described here at 2.86 and 2.30 Å resolution. Scale bars (in µm) are presented at the bottom corner of each of the figures.
Several crystals of all three habits were used for a detailed crystallographic characterization and measurement of X-ray diffraction data under cryogenic conditions. Initial experiments were performed at the TCSB using the FR-X rotating anode (Rigaku, Japan) as the X-ray source (λ = 1.54 Å) coupled to an R-AXIS HTC system (Rigaku, Japan) as the detector, and an Oxford Cryosystem as the crystal-cooling system (100 K). The crystal-cooling procedure used for these experiments included a short soak of the target crystal (about 20–40 s) in a cryoprotecting solution consisting of the original crystallization mother liquor with 15%(v/v) additional glycerol. Each of these pre-soaked crystals was submitted to a flash-cooling procedure, first by immediate immersion in liquid nitrogen followed by rapid transfer into the centre of a cold nitrogen-gas stream (100 K), flowing around the crystal throughout the X-ray data collection.
The observed diffraction pattern for crystals of the RR1 habit was generally rather weak and usually did not show significant diffraction beyond 3.5 Å resolution. Nevertheless, some of these crystals diffracted to around 3.0 Å resolution and their diffraction pattern indicated that the crystals belonged to an orthorhombic crystal system (space group P212121), with average crystallographic unit-cell parameters a = 72.6, b = 109.6, c = 390.0 Å. The RR2 crystals appeared to be closely isomorphous to the RR1 crystals in both space group (P212121) and the cell dimensions (average crystallographic unit-cell parameters a = 71.2, b = 106.0, c = 378.3 Å), but their observed resolution limits seemed to be a little better (around 2.9 Å). Different crystals of this crystallization batch gave similar unit-cell dimensions with an overall deviation from these average values of less than 0.3%. The RB1 crystals did not show useful diffraction beyond 6 Å resolution and it was therefore decided not to include them in further crystallographic studies. Although the crystals of the RR1 and RR2 habits seemed to be very similar, RR2 crystals were obtained more readily, more systematically, and diffracted slightly better, and it was therefore decided to continue the structural studies with only this crystal form (see below).
3.2. X-ray diffraction data for Axe2-Y184F-W190P using in-house system
One of the RR2 crystals of the Axe2-Y184F-W190P double mutant (Fig. 1 ▶ b) was used for a full medium-resolution X-ray diffraction data measurement at 2.86 Å resolution (Fig. 2 ▶), performed with the TCSB crystallographic set-up (see above). This oscillation data set included 900 frames measured with Δϕ of 0.2°, 110 s exposure time per frame and a crystal-to-detector distance of 250 mm. The relatively small oscillation angle (0.2°) used for the data collection was chosen mainly because of the exceptional length of one of the unit-cell parameters (about 380 Å), which caused the diffraction pattern to be very dense and difficult to resolve at oscillation angles larger than 0.2°. The raw diffraction images were processed and integrated with the HKL-3000 program suite (Minor et al., 2006 ▶).
Figure 2.

X-ray diffraction pattern of Axe2-Y184F-W190P (RR2 crystal), obtained using the TCSB crystallographic set-up. The circle corresponds to a 2.9 Å resolution limit. The insets represent magnified views of the sections indicated by the corresponding squares (bottom left, low resolution; top right, medium resolution).
A total of 267 968 accepted reflections [F > 1.0σ(F)] were measured in the 50.0–2.86 Å resolution range and resulted in 68 346 independent reflections with 97.5% completeness to 2.86 Å resolution. The overall multiplicity in the full data set was 4.0, the overall mosaicity was 0.57°, the average 〈I/σ(I)〉 was 6.4 (2.5 for the highest resolution shell, 2.91–2.86 Å) and the final R merge for the whole data was 17.6% (53.9% for the highest resolution shell) (Table 1 ▶). The relatively high R merge and the relatively low completeness in the highest resolution shell (72.9%) originate probably from the rather weak diffraction pattern and its high degree of anisotropy. While reasonable diffraction could be measured to 2.86 Å resolution for some of the crystal orientations (Fig. 2 ▶), relatively weak diffraction was observed in other crystal orientations (especially at the outer diffraction shells). Moreover, significant radiation damage was noticed during the rather long data collection (about 30 h), a factor that could also lead to non-ideal values of R merge and completeness.
Table 1. Representative parameters from the crystallographic data measurement of the Axe2-Y184F-W190P double mutant measured at the TCSB facility and at the ESRF synchrotron facility (BM14 beamline).
Values in parentheses are for the outer shell.
| Medium resolution | High resolution | |
|---|---|---|
| Crystallographic set-up | TCSB | BM14, ESRF |
| Wavelength (Å) | 1.54 | 0.979 |
| Space group | P212121 | P212121 |
| Unit-cell parameters | ||
| a (Å) | 71.2 | 71.1 |
| b (Å) | 106.0 | 106.0 |
| c (Å) | 378.3 | 378.6 |
| Resolution (Å) | 50.00–2.86 (2.91–2.86) | 35.00–2.30 (2.34–2.30) |
| No. of reflections | ||
| Total | 267968 | 533604 |
| Unique | 68346 | 127473 |
| Multiplicity | 4.0 (3.2) | 4.2 (4.7) |
| 〈I〉/〈σ(I)〉 | 6.4 (2.5) | 9.7 (5.8) |
| Mosaicity (°) | 0.57 | 0.25 |
| Completeness (%) | 97.5 (72.9) | 98.9 (99.8) |
| R merge † (%) | 17.6 (53.9) | 7.1 (25.6) |
R
merge = 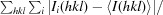
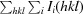 , where Ii(hkl) is the intensity of the ith observation of reflection hkl.
, where Ii(hkl) is the intensity of the ith observation of reflection hkl.
3.3. X-ray diffraction data for Axe2-Y184F-W190P using synchrotron radiation
Since the diffraction data described above appeared to be borderline in terms of quality and resolution, it was decided to collect another diffraction data set using synchrotron radiation, assuming that the higher intensity of the X-ray beam, the better optics and the more sensitive detector available at the synchrotron facility will allow a significantly better data set to be obtained.
This was indeed the case, as demonstrated by the high-resolution X-ray diffraction experiment conducted at the BM14 beamline of the ESRF. In this experiment, a single crystal of the Axe2-Y184F-W190P double mutant (RR2 form, Fig. 1b ▶) was subjected to a well collimated (50 µm) synchrotron X-ray beam (λ = 0.979 Å), and diffraction data were obtained using a CCD area detector (MAR 225, MAR Research Inc., USA), resulting in a full diffraction data set to 2.30 Å resolution (Fig. 3 ▶). Since the crystal showed some propensity to radiation damage (see above) and since the crystal was significantly long relative to the beam cross section (Fig. 1 ▶ b), it was decided to collect the data from three different sections along the long axis of the crystal. The oscillation data were measured with a Δϕ of 0.25°, 6 s exposure time per frame and a crystal-to-detector distance of 244 mm. The first data set (taken from the left side of the crystal) included 240 frames measured between ϕ = 0 and 60°. The second data set (taken from the centre of the crystal) included 240 additional frames measured between ϕ = 60 and 120°. The third data set (taken from the right side of the crystal) included 240 additional frames measured between ϕ = 120 and 180°. These three data sets were then processed and merged with the HKL-2000 program suite (Otwinowski & Minor, 1997 ▶).
Figure 3.

X-ray diffraction pattern of Axe2-Y184F-W190P (RR2 crystal), obtained using synchrotron radiation (BM14, ESRF). The circle corresponds to a 2.3 Å resolution limit. The insets represent magnified views of the sections indicated by the corresponding squares (bottom left, high resolution; top left, medium resolution; bottom right, low resolution).
The merged data set included a total of 533 604 accepted reflections [F > 1.0σ(F)] in the 35.0–2.30 Å resolution range and resulted in 127 473 independent reflections with an overall completeness of 98.9%. The overall multiplicity in the combined data set was 4.2, the overall mosaicity was 0.25°, the average 〈I/σ(I)〉 was 9.7 (5.8 for the highest resolution shell of 2.34–2.30 Å) and the final R merge for the whole data set was 7.1% (25.6% for the highest resolution shell). These parameters confirm that the data collected at BM14 of the ESRF represent a nearly complete diffraction data set of relatively high quality, with obvious improvements over the diffraction data collected earlier using our in-house crystallographic system (Table 1 ▶).
3.4. Content of the unit cell
A rough calculation of the specific ratio of volume/protein (V M) was performed in order to estimate the number of protein monomers per crystallographic asymmetric unit. The volume of the Axe2-Y184F-W190P crystallographic unit cell, as determined from the mean value of the unit-cell parameters at 100 K, is 2.85 × 106 Å3. Assuming that the V M and the overall water content values here are within the normal range observed for soluble proteins (Matthews, 1968 ▶; Kantardjieff & Rupp, 2003 ▶), there should be between eight and 16 Axe2-Y184F-W190P monomers (219 amino acids; molecular weight 24 770 Da; Alalouf et al., 2011 ▶) in the crystallographic asymmetric unit. With eight molecules in the P212121 asymmetric unit (32 in the unit cell), the calculated V M is 3.60 Å3 Da−1 and the corresponding solvent content in the crystals is 63.4%. With 16 molecules in the asymmetric unit (64 in the unit cell), the calculated V M is 1.80 Å3 Da−1 and the corresponding solvent content in the crystal is 31.7%. Both the high-limit and the low-limit V M values have been observed for soluble proteins, but it is expected that the true content of the asymmetric unit will be somewhere between these two extreme possibilities. The actual content of the Axe2-Y184F-W190P mutant asymmetric unit will be unequivocally resolved only when the crystallographic protein structure is fully determined.
The two full data sets described above (2.86 and 2.30 Å resolution) should now be used for the detailed three-dimensional structural analysis of the Axe2 double mutant in its dimeric form. As mentioned above, the complete high-resolution structure of Axe2-WT has recently been solved in our laboratory (Lansky, Alalouf et al., 2014 ▶), presenting a well defined homo-octameric structure. Although some differences in local conformations of the protein are definitely possible, we do not expect the overall structure of the Axe2 monomer to be dramatically different in different oligomeric forms of the enzyme. It would therefore be reasonable to use the available octameric structure as the basis for the structure determination of the current dimeric form of the Axe2 protein. We will thus use the coordinates of one of the monomers of the octameric structure of Axe2-WT (PDB entry 3w7v; Lansky, Alalouf et al., 2014 ▶) as a search model for molecular-replacement calculations, which are expected to solve the phase problem and provide a good starting model for the full structural analysis of the dimeric form of the Axe2-Y184F-W190P double mutant. Such analyses are currently in progress in our laboratory. Once completed, we hope that this structure will clarify, at least in part, the critical structural parameters involved in the octamer–dimer conversion of Axe2 and their relevance to the catalytic activity of the enzyme.
Acknowledgments
This work was supported by the Israel Science Foundation Grants 500/10 and 152/11, the I-CORE Program of the Planning and Budgeting Committee, the Ministry of Environmental Protection and the Grand Technion Energy Program (GTEP), and comprises part of The Leona M. and Harry B. Helmsley Charitable Trust reports on Alternative Energy series of the Technion, Israel Institute of Technology and the Weizmann Institute of Science. YS acknowledges partial support by the Russell Berrie Nanotechnology Institute and The Lorry I. Lokey Interdisciplinary Center for Life Science and Engineering, Technion. HD thanks the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement No. 330879- MC-CHOLESTRUCTURE for financial support. We thank the staff at the European Synchrotron Research Facility (ESRF, BM14 beamline) for their helpful support in the X-ray synchrotron data measurement and analysis. The synchrotron experiments at the ESRF were also supported by the ESRF internal funding program. YS holds the Erwin and Rosl Pollak Chair in Biotechnology at the Technion.
References
- Alalouf, O., Balazs, Y., Volkinshtein, M., Grimple, Y., Shoham, G. & Shoham, Y. (2011). J. Biol. Chem. 286, 41933–42001. [DOI] [PMC free article] [PubMed]
- Alalouf, O. (2013). PhD thesis. Technion, Haifa, Israel.
- Almog, O., Greenblatt, H. M., Spungin, A., Ben-Meir, D., Blumberg, S. & Shoham, G. (1993). J. Mol. Biol. 230, 342–344. [DOI] [PubMed]
- Almog, O., Klein, D., Braun, S. & Shoham, G. (1994). J. Mol. Biol. 235, 760–762. [DOI] [PubMed]
- Bar, M., Golan, G., Nechama, M., Zolotnitsky, G., Shoham, Y. & Shoham, G. (2004). Acta Cryst. D60, 545–549. [DOI] [PubMed]
- Ben-David, A., Bravman, T., Balazs, Y. S., Czjzek, M., Schomburg, D., Shoham, G. & Shoham, Y. (2007). Chembiochem, 8, 2145–2151. [DOI] [PubMed]
- Biely, P., Mastihubová, M., Tenkanen, M., Eyzaguirre, J., Li, X.-L. & Vršanská, M. (2011). J. Biotechnol. 151, 137–142. [DOI] [PubMed]
- Bravman, T., Michaly, A., Shulami, S., Belakhov, V., Baasov, T., Shoham, G. & Shoham, Y. (2001). FEBS Lett. 495, 115–119. [DOI] [PubMed]
- Bravman, T., Zolotnitsky, G., Belakhov, V., Shoham, G., Henrissat, B., Baasov, T. & Shoham, Y. (2003). Biochemistry, 42, 10528–10536. [DOI] [PubMed]
- Bravman, T., Zolotnitsky, G., Shulami, S., Belakhov, V., Solomon, D., Baasov, T., Shoham, G. & Shoham, Y. (2001). FEBS Lett. 495, 39–43. [DOI] [PubMed]
- Brüx, C., Ben-David, A., Shallom-Shezifi, D., Leon, M., Niefind, K., Shoham, G., Shoham, Y. & Schomburg, D. (2006). J. Mol. Biol. 359, 97–109. [DOI] [PubMed]
- Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V. & Henrissat, B. (2009). Nucleic Acids Res. 37, D233–D238. [DOI] [PMC free article] [PubMed]
- Czjzek, M., Ben David, A., Bravman, T., Shoham, G., Henrissat, B. & Shoham, Y. (2005). J. Mol. Biol. 353, 838–846. [DOI] [PubMed]
- Gat, O., Lapidot, A., Alchanati, I., Regueros, C. & Shoham, Y. (1994). Appl. Environ. Microbiol. 60, 1889–1896. [DOI] [PMC free article] [PubMed]
- Gilboa, R., Bauer, A. J. & Shoham, G. (1998). Acta Cryst. D54, 1467–1470. [DOI] [PubMed]
- Golan, G., Shallom, D., Teplitsky, A., Zaide, G., Shulami, S., Baasov, T., Stojanoff, V., Thompson, A., Shoham, Y. & Shoham, G. (2004). J. Biol. Chem. 279, 3014–3024. [DOI] [PubMed]
- Golan, G., Zharkov, D. O., Fernandes, A. S., Zaika, E., Kycia, J. H., Wawrzak, Z., Grollman, A. P. & Shoham, G. (2004). Acta Cryst. D60, 1476–1480. [DOI] [PubMed]
- Hövel, K., Shallom, D., Niefind, K., Belakhov, V., Shoham, G., Baasov, T., Shoham, Y. & Schomburg, D. (2003). EMBO J. 22, 4922–4932. [DOI] [PMC free article] [PubMed]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst. 24, 409–411.
- Kantardjieff, K. A. & Rupp, B. (2003). Protein Sci. 12, 1865–1871. [DOI] [PMC free article] [PubMed]
- Khasin, A., Alchanati, I. & Shoham, Y. (1993). Appl. Environ. Microbiol. 59, 1725–1730. [DOI] [PMC free article] [PubMed]
- Lansky, S., Alalouf, O., Solomon, V., Alhassid, A., Govada, L., Chayan, N. E., Belrhali, H., Shoham, Y. & Shoham, G. (2013). Acta Cryst. F69, 430–434. [DOI] [PMC free article] [PubMed]
- Lansky, S., Alalouf, O., Solomon, H. V., Alhassid, A., Govada, L., Chayen, N. E., Belrhali, H., Shoham, Y. & Shoham, G. (2014). Acta Cryst. D70, 261–278. [DOI] [PubMed]
- Lansky, S., Salama, R., Solomon, V. H., Belrhali, H., Shoham, Y. & Shoham, G. (2013). Acta Cryst. F69, 695–699. [DOI] [PMC free article] [PubMed]
- Lansky, S., Zehavi, A., Dann, R., Dvir, H., Belrhali, H., Shoham, Y. & Shoham, G. (2014). Acta Cryst. F70, 225–231. [DOI] [PMC free article] [PubMed]
- Lapidot, A., Mechaly, A. & Shoham, Y. (1996). J. Biotechnol. 51, 259–264. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. (2006). Acta Cryst. D62, 859–866. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Punta, M. et al. (2012). Nucleic Acids Res. 40, D290–D301. [DOI] [PMC free article] [PubMed]
- Reiland, V., Funano-Hershcovitz, Y., Golan, G., Gilboa, R., Shoham, Y. & Shoham, G. (2004). Acta Cryst. D60, 2371–2376. [DOI] [PubMed]
- Shallom, D., Belakhov, V., Solomon, D., Gilead-Gropper, S., Baasov, T., Shoham, G. & Shoham, Y. (2002). FEBS Lett. 514, 163–167. [DOI] [PubMed]
- Shallom, D., Belakhov, V., Solomon, D., Shoham, G., Baasov, T. & Shoham, Y. (2002). J. Biol. Chem. 277, 43667–43673. [DOI] [PubMed]
- Shallom, D., Golan, G., Shoham, G. & Shoham, Y. (2004). J. Bacteriol. 186, 6928–6937. [DOI] [PMC free article] [PubMed]
- Shallom, D., Leon, M., Bravman, T., Ben-David, A., Zaide, G., Belakhov, V., Shoham, G., Schomburg, D., Baasov, T. & Shoham, Y. (2005). Biochemistry, 44, 387–397. [DOI] [PubMed]
- Shulami, S., Gat, O., Sonenshein, A. L. & Shoham, Y. (1999). J. Bacteriol. 181, 3695–3704. [DOI] [PMC free article] [PubMed]
- Shulami, S., Raz-Pasteur, A., Tabachnikov, O., Gilead-Gropper, S., Shner, I. & Shoham, Y. (2011). J. Bacteriol. 193, 2838–2850. [DOI] [PMC free article] [PubMed]
- Shulami, S., Zaide, G., Zolotnitsky, G., Langut, Y., Feld, G., Sonenshein, A. L. & Shoham, Y. (2007). Appl. Environ. Microbiol. 73, 874–884. [DOI] [PMC free article] [PubMed]
- Sjöström, E. (1993). Wood Chemistry: Fundamentals and Applications, 2nd ed. London: Academic Press.
- Solomon, H. V., Tabachnikov, O., Feinberg, H., Govada, L., Chayen, N. E., Shoham, Y. & Shoham, G. (2013). Acta Cryst. F69, 1114–1119. [DOI] [PMC free article] [PubMed]
- Solomon, V., Teplitsky, A., Shulami, S., Zolotnitsky, G., Shoham, Y. & Shoham, G. (2007). Acta Cryst. D63, 845–859. [DOI] [PubMed]
- Tabachnikov, O. & Shoham, Y. (2013). FEBS J. 280, 950–964. [DOI] [PubMed]
- Teplitsky, A., Feinberg, H., Gilboa, R., Lapidot, A., Mechaly, A., Stojanoff, V., Capel, M., Shoham, Y. & Shoham, G. (1997). Acta Cryst. D53, 608–611. [DOI] [PubMed]
- Teplitsky, A., Mechaly, A., Stojanoff, V., Sainz, G., Golan, G., Feinberg, H., Gilboa, R., Reiland, V., Zolotnitsky, G., Shallom, D., Thompson, A., Shoham, Y. & Shoham, G. (2004). Acta Cryst. D60, 836–848. [DOI] [PubMed]
- Teplitsky, A., Shulami, S., Moryles, S., Shoham, Y. & Shoham, G. (2000). Acta Cryst. D56, 181–184. [DOI] [PubMed]
- Teplitsky, A., Shulami, S., Moryles, S., Zaide, G., Shoham, Y. & Shoham, G. (1999). Acta Cryst. D55, 869–872. [DOI] [PubMed]
- Zaide, G., Shallom, D., Shulami, S., Zolotnitsky, G., Golan, G., Baasov, T., Shoham, G. & Shoham, Y. (2001). Eur. J. Biochem. 268, 3006–3016. [DOI] [PubMed]