

The crystallization of PBPD2 from L. monocytogenes is described.
Keywords: penicillin-binding proteins, PBPD2, Listeria monocytogenes
Abstract
Penicillin-binding proteins (PBPs), which mediate the peptidoglycan biosynthetic pathway in the bacterial cell wall, have been intensively investigated as a target for the design of antibiotics. In this study, PBPD2, a low-molecular-weight PBP encoded by lmo2812 from Listeria monocytogenes, was overexpressed in Escherichia coli, purified and crystallized at 295 K using the sitting-drop vapour-diffusion method. The crystal belonged to the primitive orthorhombic space group P212121, with unit-cell parameters a = 37.7, b = 74.7, c = 75.1 Å, and diffracted to 1.55 Å resolution. There was one molecule in the asymmetric unit. The preliminary structure was determined by the molecular-replacement method.
1. Introduction
The synthetic pathway of peptidoglycan, which is the major component of the bacterial cell wall, has been extensively investigated as a target for the design of antibacterial agents including β-lactam antibiotics. The peptidoglycan layer is responsible for protecting bacteria from osmotic pressure and maintaining cell morphology (Vollmer et al., 2008 ▶; Popham & Young, 2003 ▶). The peptidoglycan layer is a mesh-like structure of alternating N-acetylglucosamine and N-acetylmuramic acid moieties that are cross-linked by peptapeptide chains (Lovering et al., 2012 ▶). The assembly of peptidoglycan chains is carried out by penicillin-binding proteins (PBPs) through transglycosylation and transpeptidation reactions (Sauvage et al., 2008 ▶). PBPs are classified into two groups: high-molecular-weight (HMW) and low-molecular-weight (LMW) PBPs (Sauvage et al., 2008 ▶). The HMW PBPs are further divided into class A and class B PBPs. The class A PBPs are bifunctional enzymes which have transglycosylase and transpeptidase activities, whereas the class B PBPs exhibit only transpeptidase activity. The LMW PBPs generally act as a dd-carboxypeptidase and are involved in the control of cell morphology and cell division (Ghosh et al., 2008 ▶; Nelson & Young, 2001 ▶).
Listeria monocytogenes is a Gram-positive pathogenic bacterium that causes a serious food-borne infectious disease called listeriosis with a fatality rate of 25–30% (Southwick & Purich, 1996 ▶). For the treatment of listeriosis, ampicillin is used either alone or in combination with other medications (Hof et al., 1997 ▶). Although the penicillin-related β-lactams are used as a gold standard for the treatment of listeriosis, the molecular basis of the action of the antibiotics is largely unknown.
In a previous study, five PBPs in L. monocytogenes were found using radiolabelled β-lactams: two HMW class A enzymes (PBP1 and PBP4), two HMW class B enzymes (PBP2 and PBP3) and one LMW enzyme (PBP5) (Vicente et al., 1990 ▶). Recently, an additional four PBPs including PBPD2 were identified by both in silico analysis and binding assays using the fluorescent antibiotic BOCILLIN FL (Korsak et al., 2010 ▶). LmPBPD2 encoded by lmo2812 from L. monocytogenes was found to be a LMW PBP exhibiting dd-carboxypeptidase activity (Korsak et al., 2010 ▶). The overall fold of LmPBPD2 may be most similar to the structure of the Escherichia coli LMW PBP6 (PDB entry 3it9; Chen et al., 2009 ▶), which shows the highest sequence identity to LmPBPD2 (32%) among solved PBP structures. However, the structural characteristics of LMW PBPs in L. monocytogenes have not been reported.
Here, we report the cloning, expression, purification, crystallization and preliminary X-ray crystallographic analysis of LmPBPD2 (Lmo2812). The structural information on LmPBPD2 will contribute to the design of better inhibitory agents against PBPs, therefore achieving more effective treatment for infections by bacteria, including L. monocytogenes.
2. Materials and methods
2.1. Cloning, expression and purification of PBPD2 (Lmo2812)
The gene encoding LmPBPD2 (UniProt accession No. Q8Y3M3) was amplified by the polymerase chain reaction (PCR) with Pfu DNA polymerase from genomic DNA of L. monocytogenes EGD. The primers were 5′-CCGAAGGATCCTCTACGGAGCAACCAAACTTATAC-3′ (forward) and 5′-CCGAACTCGAGCTAATCTTCTTTAAACCCAACCTCC-3′ (reverse), containing BamHI and XhoI restriction sites, respectively (bold). The PCR product was subcloned into the BamHI- and XhoI-digested pProExHTb vector (Invitrogen, USA) to generate an expression plasmid encoding residues 21–272 of LmPBPD2 with an N-terminal His6 tag. The lmo2812 gene insertion was confirmed by DNA sequencing. The resulting plasmid was transformed into the expression host E. coli B834(DE3) cells (Novagen, USA) harbouring the pRIL plasmid obtained from BL21(DE3)RIL cells (Stratagene, USA). The transformed cells were then grown at 310 K in Luria–Bertani medium containing 100 µg ml−1 ampicillin and 34 µg ml−1 chloramphenicol until the OD600 reached 0.5–0.6. Expression of recombinant protein was induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside and the cells were incubated for 18 h at 295 K. The cells were harvested by centrifugation at 5000g at 277 K and were resuspended in ice-cold buffer A (30 mM Tris–HCl pH 8.0, 0.3 M NaCl). The resuspended cells were lysed by sonication and the lysate was centrifuged at 15 000g for 30 min.
The supernatant was loaded onto a 5 ml HisTrap FF column (GE Healthcare, Sweden) equilibrated with buffer A. After washing with buffer A supplemented with 20 mM imidazole, the bound protein was eluted with a solution consisting of 30 mM Tris–HCl pH 8.0, 50 mM NaCl, 200 mM imidazole. To remove the hexahistidine tag, the eluted fraction was digested overnight with TEV protease (Invitrogen, USA) at a ratio of 1:100(w:w) at 277 K. The target protein was then applied onto a 5 ml HiTrap Q column (GE Healthcare, Sweden) and eluted with a linear gradient of 50–1000 mM NaCl in a buffer consisting of 30 mM Tris–HCl pH 8.0. The eluted protein was further purified by size-exclusion chromatography on a Superdex 200 column (GE Healthcare, Sweden) equilibrated with 10 mM Tris–HCl pH 8.0, 100 mM NaCl. The fractions containing LmPBPD2 were pooled and concentrated to 25 mg ml−1. Aliquots were flash-frozen in liquid nitrogen and stored at 193 K for subsequent use in crystallization.
2.2. Crystallization
Truncated LmPBPD2 (residues 21–272) with an additional five amino acids (GHMGS) at the N-terminus was crystallized at 295 K using the sitting-drop vapour-diffusion method in 96-well plates (Hampton Research, USA). Initial crystallization trials were carried out by the sparse-matrix method (Jancarik et al., 1991 ▶) using commercial kits from Hampton Research (Crystal Screen HT, SaltRX HT, Index HT and PEG/Ion HT) and Emerald Bio (Wizard I, Wizard II, Cryo I and Cryo II). Drops consisting of 0.4 µl protein solution and 0.4 µl reservoir solution were equilibrated against 70 µl reservoir solution. After 7 d, crystals appeared in condition No. 8 of PEG/Ion HT (20% PEG 3350, 0.2 M KCl). The LmPBPD2 crystals grew to approximate dimensions of 30 × 15 × 200 µm.
2.3. X-ray data collection
For X-ray data collection, a single crystal was transferred into reservoir solution supplemented with 20% glycerol and immediately flash-cooled in a nitrogen-gas stream at 100 K. Native X-ray diffraction data were collected using a Pilatus 2M detector on beamline BL-1A at the Photon Factory (PF), Japan. A data set consisting of 180 images of 1° oscillation at a 178 mm crystal-to-detector distance and an exposure time of 0.5 s per image was collected to 1.55 Å resolution. The data were indexed, integrated and scaled using the programs iMosflm and SCALA from the CCP4 package (Battye et al., 2011 ▶; Evans, 2006 ▶). The data-collection statistics are shown in Table 1 ▶.
Table 1. Diffraction statistics.
Values in parentheses are for the highest resolution shell. A 0σ cutoff filter was applied during the scaling process.
| X-ray source | Beamline BL-1A, PF |
| Wavelength (Å) | 1.100 |
| Resolution (Å) | 37.68–1.55 (1.63–1.55) |
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 37.7, b = 74.7, c = 75.1 |
| Completeness (%) | 96.9 (80.6) |
| R merge † (%) | 9.1 (39.4) |
| Multiplicity | 5.3 (3.6) |
| Average I/σ(I) | 10.8 (3.3) |
R
merge = 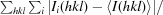
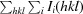 , where Ii(hkl) and 〈I(hkl)〉 are the observed intensity and the mean intensity of related reflections, respectively.
, where Ii(hkl) and 〈I(hkl)〉 are the observed intensity and the mean intensity of related reflections, respectively.
3. Results and discussion
The N-terminal 20 residues of LmPBPD2, which contain a putative transmembrane helix, were not included in the expression construct to facilitate its purification and crystallization. LmPBPD2 was successfully overexpressed in E. coli B834(DE3)RIL cells and purified to 98% purity using a nickel-affinity column and anion-exchange chromatography followed by gel-filtration chromatography (Fig. 1 ▶). Each litre of E. coli culture yielded ∼22 mg of LmPBPD2. The crystals were obtained using the sitting-drop vapour-diffusion method at 295 K with reservoir solution composed of 0.2 M KCl, 20%(w/v) PEG 3350. The LmPBPD2 crystals reached dimensions of about 30 × 15 × 200 µm within 7 d (Fig. 2 ▶). The crystal diffracted to a resolution of 1.55 Å using synchrotron radiation. The observation of split spots in the diffraction patterns suggests that the mounted crystal might be twinned (Fig. 3 ▶). Fortunately, the diffraction spots from the two crystalline domains did not overlap, and one crystalline domain mainly contributed to the diffraction. Thereby, the data were indexed and integrated as if the crystal were single by excluding one of the non-overlapping reflections. A twinning analysis for the processed data using phenix.xtriage estimated the twin fraction to be 0.021, which was a very small value that could be ignored (Adams et al., 2010 ▶). Therefore, the data were not detwinned for further processing. The crystal belonged to space group P212121, with unit-cell parameters a = 37.7, b = 74.7, c = 75.1 Å. The diffraction data set had a resolution range of 37.68–1.55 Å with a completeness of 96.9% and an R merge of 9.1% (Table 1 ▶). Assuming the presence of one LmPBPD2 molecule per asymmetric unit, the Matthews coefficient V M (Matthews, 1968 ▶) was 1.81 Å3 Da−1 with a solvent content of 31.9%, indicating that the crystal had a tightly packed lattice with a low solvent content (Trillo-Muyo et al., 2013 ▶). Molecular replacement was carried out using Phaser (McCoy, 2007 ▶) with the structure of PBP6 from E. coli (PDB entry 3it9; Chen et al., 2009 ▶), which is the most closely related structure to Lmo2812, sharing 32% amino-acid sequence identity, as a search model. An initial search model yielded a solution with a translation-function Z-score of 10.8 and a rotation-function Z-score of 7.4. Further structural refinement is now in progress.
Figure 1.
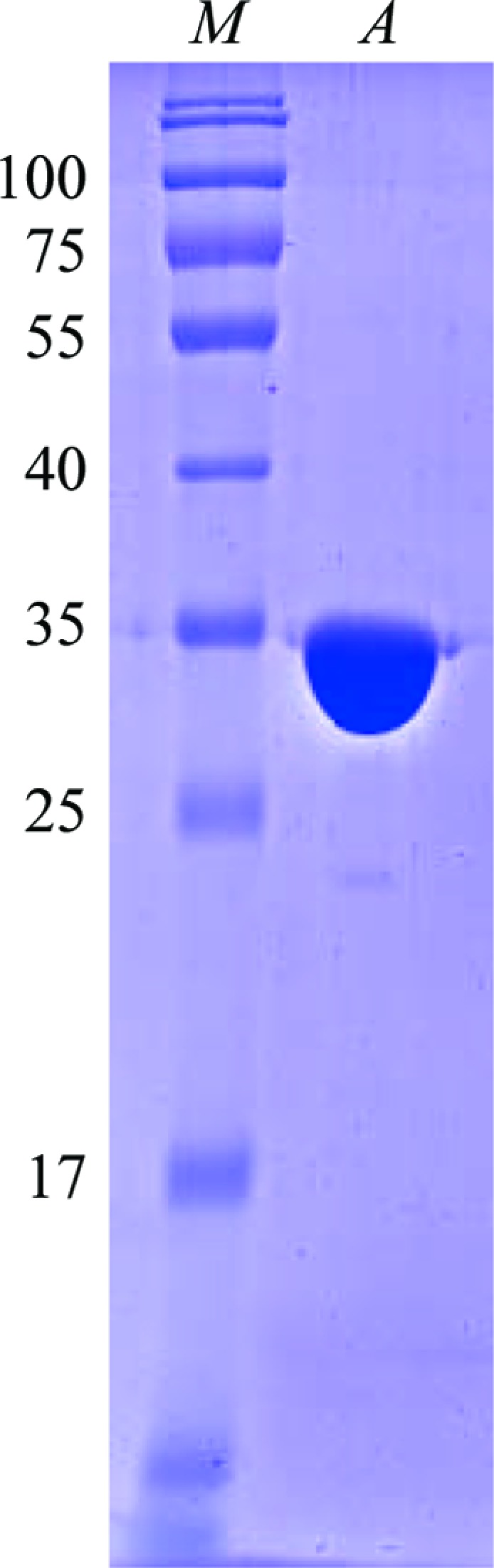
SDS–PAGE of purified LmPBPD2. Lane M, molecular-weight markers (labelled in kDa); lane A, purified LmPBPD2.
Figure 2.

Crystals of LmPBPD2. The crystals reached approximate dimensions of 30 × 15 × 200 µm within 7 d.
Figure 3.

An X-ray diffraction image from an LmPBPD2 crystal. The crystal was exposed for 0.5 s over a 1° oscillation range. In the magnified inset, diffraction spots extending to 1.55 Å resolution can be observed.
Acknowledgments
This research was part of the project titled ‘Marine Extreme Genome Research Center Program’ funded by the Ministry of Oceans and Fisheries, Korea.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Battye, T. G. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. W. (2011). Acta Cryst. D67, 271–281. [DOI] [PMC free article] [PubMed]
- Chen, Y., Zhang, W., Shi, Q., Hesek, D., Lee, M., Mobashery, S. & Shoichet, B. K. (2009). J. Am. Chem. Soc. 131, 14345–14354. [DOI] [PMC free article] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Ghosh, A. S., Chowdhury, C. & Nelson, D. E. (2008). Trends Microbiol. 16, 309–317. [DOI] [PubMed]
- Hof, H., Nichterlein, T. & Kretschmar, M. (1997). Clin. Microbiol. Rev. 10, 345–357. [DOI] [PMC free article] [PubMed]
- Jancarik, J., Scott, W. G., Milligan, D. L., Koshland, D. E. Jr & Kim, S.-H. (1991). J. Mol. Biol. 221, 31–34. [DOI] [PubMed]
- Korsak, D., Markiewicz, Z., Gutkind, G. O. & Ayala, J. A. (2010). BMC Microbiol. 10, 239. [DOI] [PMC free article] [PubMed]
- Lovering, A. L., Safadi, S. S. & Strynadka, N. C. (2012). Annu. Rev. Biochem. 81, 451–478. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J. (2007). Acta Cryst. D63, 32–41. [DOI] [PMC free article] [PubMed]
- Nelson, D. E. & Young, K. D. (2001). J. Bacteriol. 183, 3055–3064. [DOI] [PMC free article] [PubMed]
- Popham, D. L. & Young, K. D. (2003). Curr. Opin. Microbiol. 6, 594–599. [DOI] [PubMed]
- Sauvage, E., Kerff, F., Terrak, M., Ayala, J. A. & Charlier, P. (2008). FEMS Microbiol. Rev. 32, 234–258. [DOI] [PubMed]
- Southwick, F. S. & Purich, D. L. (1996). N. Engl. J. Med. 334, 770–776. [DOI] [PubMed]
- Trillo-Muyo, S., Jasilionis, A., Domagalski, M. J., Chruszcz, M., Minor, W., Kuisiene, N., Arolas, J. L., Solà, M. & Gomis-Rüth, F. X. (2013). Acta Cryst. D69, 464–470. [DOI] [PMC free article] [PubMed]
- Vicente, M. F., Pérez-Dáz, J. C., Baquero, F., Angel de Pedro, M. & Berenguer, J. (1990). Antimicrob. Agents Chemother. 34, 539–542. [DOI] [PMC free article] [PubMed]
- Vollmer, W., Blanot, D. & de Pedro, M. A. (2008). FEMS Microbiol. Rev. 32, 149–167. [DOI] [PubMed]