

Abstract
Dura mater graft-associated Creutzfeldt-Jakob disease (dCJD) can be divided into two subgroups that exhibit distinct clinical and neuropathological features, with the majority represented by a non-plaque-type of dCJD (np-dCJD) and the minority by a plaque-type of dCJD (p-dCJD). The two distinct phenotypes of dCJD had been considered to be unrelated to the genotype (methionine, M or valine, V) at polymorphic codon 129 of the PRNP gene or type (type 1 or type 2) of abnormal isoform of prion protein (PrPSc) in the brain, while these are major determinants of clinicopathological phenotypes of sporadic CJD (sCJD). The reason for the existence of two distinct subgroups in dCJD had remained elusive. Recent progress in research of the pathogenesis of dCJD has revealed that two distinct subgroups of dCJD are caused by infection with different PrPSc strains from sCJD, i.e., np-dCJD caused by infection with sCJD-MM1/MV1, and p-dCJD caused by infection with sCJD-VV2 or -MV2. These studies have also revealed previously unrecognized problems as follows: (i) the numbers of p-dCJD patients may increase in the future, (ii) the potential risks of secondary infection from dCJD, particularly from p-dCJD, may be considerable, and (iii) the effectiveness of the current PrPSc decontamination procedures against the PrPSc from p-dCJD is uncertain. To prevent secondary infection from p-dCJD, the establishment of effective decontamination procedures is an urgent issue. In this review, we summarize the past and future problems surrounding dCJD.
Keywords: Creutzfeldt-Jakob disease, Prion protein, Dura mater grafts, Humanized knock-in mouse
Introduction
Dura mater grafts used to repair the dural defects at neurosurgery can cause fatal disease years to decades later. The tragedy of dura mater graft-associated Creutzfeldt-Jakob disease (dCJD) was considered to be nearly over. However, recent progress in research of the pathogenesis of dCJD has revealed previously unrecognized problems. In this review, we summarize the past and future problems surrounding dCJD.
Creutzfeldt-Jakob disease (CJD) is a lethal transmissible neurodegenerative disease. The central event in the pathogenesis of CJD is a conformational change of the normal cellular isoform of prion protein (PrPC) into an abnormal infectious isoform of prion protein (PrPSc) [1]. The conformational change of PrPC can occur due to either one of three causes: spontaneous conversion in sporadic CJD (sCJD), mutations in the PRNP gene in genetic CJD, or infection with PrPSc in iatrogenic CJD and variant CJD.
One of the most frequent sources of iatrogenic PrPSc infection is dura mater grafts obtained from human cadavers undiagnosed as CJD. The sum of dCJD (228 cases) and growth hormone-associated CJD (226 cases) accounts for 97% of total iatrogenic CJD cases [2]. A single brand of dura mater graft, Lyodura®, was used for all the dCJD cases in whom the brand name was identified. Although the causative dura mater grafts were manufactured by a German company, 62% (142 cases) of total dCJD cases have been found in Japan [2,3]. Persistent efforts of a Japanese CJD surveillance team have clarified the outline of dCJD outbreaks. The onset of Japanese dCJD patients peaked in the late 1990s, and most of the patients had received the grafts during 1983–1987, while as many as 100,000 persons received the Lyodura® grafts during this period [4,5]. In the process of conducting this elaborate survey, a puzzling mystery about dCJD emerged.
A mystery about dCJD
There is growing evidence that dCJD can be divided into two subgroups that exhibit distinct clinical and neuropathological phenotypes, with the majority (68%) represented by a non-plaque-type of dCJD (np-dCJD) and the minority (32%) by a plaque-type of dCJD (p-dCJD) (Figure 1) [6-12]. The clinicopathological features of np-dCJD are identical to those of classical sCJD, whereas p-dCJD is characterized by (i) ataxic gait as an initial symptom, (ii) slow progression of neurological symptoms, (iii) absence or late occurrence of periodic sharp-wave complexes (PSWC) on electroencephalogram (EEG), and (iv) widespread PrPSc amyloid plaques in the brain [11-16]. There is no significant difference in gender composition, site of graft, age at grafting, and year of grafting between the two subgroups [11,12].
Figure 1.
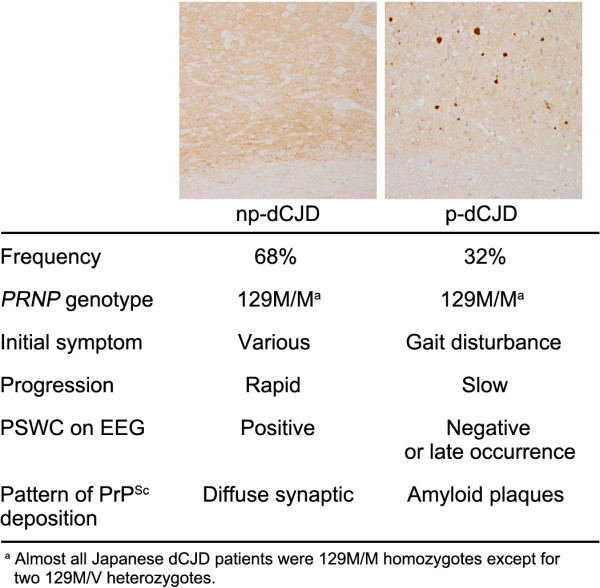
Clinicopathological features of the two subgroups of dCJD. The patients with dCJD can be divided into two subgroups, with the majority represented by a non-plaque-type of dCJD (np-dCJD) and the minority by a plaque-type of dCJD (p-dCJD) [11,12]. Neuropathological hallmark of p-dCJD is widespread PrPSc amyloid plaques, while np-dCJD shows diffuse synaptic-type PrPSc deposition.
sCJD also shows wide phenotypic heterogeneity, and its clinicopathological phenotypes are determined by the genotype at polymorphic codon 129 of the PRNP gene and type of PrPSc in the brain (Figure 2) [17,18]. The codon 129 of the PRNP gene shows methionine (M)/valine (V) polymorphism. Two types of PrPSc (type 1 and type 2) are distinguishable according to the size of the proteinase K-resistant core of unglycosylated PrPSc (21 and 19 kDa, respectively), reflecting differences in the proteinase K-cleavage site (at residues 82 and 97, respectively) [19]. On the other hand, the two distinct phenotypes of dCJD had been considered to be unrelated to their PRNP genotype or type of PrPSc in the brain [11]. In Japan, almost all dCJD patients had the same genotype, i.e., homozygous for methionine at codon 129 (129 M/M), except two heterozygotes [2], and the type of PrPSc in their brains had been reported as type 1 [6,10,11]. The reason for the existence of two distinct subgroups in dCJD had remained elusive.
Figure 2.

Two major determinants of the phenotypic heterogeneity of sCJD. (a) The PRNP genotype at polymorphic codon 129. (b) Type of PrPSc in the brain. Types 1 and 2 PrPSc are cleaved by proteinase K at different sites (at residues 82 and 97, respectively) [19]. (c) In western blot analysis, types 1 and 2 PrPSc are distinguishable by the size of the proteinase K-resistant core of the unglycosylated PrPSc (21 and 19 kDa, respectively) [17].
Solving the mystery
In 2003, an unusual p-dCJD case was reported [9]. This patient showed the accumulation of unusual PrPSc with intermediate electrophoretic mobility between types 1 and 2 PrPSc. Then, we reevaluated the biochemical properties of PrPSc in the two subgroups of dCJD and found that the size of PrPSc from p-dCJD was invariably smaller than that of type 1 PrPSc from np-dCJD (Figure 3) [20]. This intermediate-sized PrPSc was designated as type i PrPSc.
Figure 3.

Distinct biochemical properties of PrPSc in the two subgroups of dCJD. The intermediate-sized PrPSc in p-dCJD, designated as type i PrPSc, was smaller than type 1 PrPSc in np-dCJD [20].
To resolve the mystery of the existence of two distinct subgroups in dCJD, we hypothesized that they might be caused by infection with different PrPSc strains from distinct sCJD subgroups. According to the PRNP genotype and type of PrPSc in the brain, sCJD is classified into six subgroups (MM1, MV1, VV1, MM2, MV2, or VV2) [17]. MM1 and MV1, which are the predominant subgroups in sCJD, show the same clinicopathological features. Meanwhile, MM2 can be divided into three subgroups based on histopathological criteria (MM2T, thalamic form showing characteristic atrophy of thalamic and inferior olivary nuclei; MM2C, cortical form showing a predominant cortical pathology; or MM2T + C, mixed form) [17,21]. In addition, MV2 is also divided into three subgroups based on histopathological criteria (MV2K showing kuru type PrPSc amyloid plaques, MV2C showing a predominant cortical pathology, or MV2K + C showing mixed histopathology) [17,18]. The clinicopathological features of np-dCJD, such as short duration of illness, PSWC on EEG, or diffuse synaptic-type PrPSc deposition in the brain, are identical to those of sCJD-MM1/MV1. In contrast, the clinicopathological features of p-dCJD, such as ataxic gait as an initial symptom, slow progression of neurological symptoms, absence or late occurrence of PSWC on EEG, or formation of PrPSc plaques in the brain, are similar to those of sCJD-VV2, −MV2K, or -MV2K + C. These similarities raised the possibility that np-dCJD might be caused by infection with sCJD-MM1/MV1, whereas p-dCJD might be caused by infection with sCJD-VV2, −MV2K, or -MV2K + C.
To test this possibility, we examined the transmission properties of the dCJD and sCJD subgroups using humanized mice carrying human PrP with either the 129 M/M or V/V genotype [20,22,23]. In these transmission experiments, p-dCJD and sCJD-VV2, −MV2K, or -MV2K + C were identical in the transmissibility to the PrP-humanized mice (Table 1, Figure 4a) and in the neuropathological and biochemical features in the inoculated mice (Figure 4b, c). By contrast, np-dCJD showed the same transmission properties as sCJD-MM1. In particular, the 129 M/M mice inoculated with sCJD-VV2, −MV2K, or -MV2K + C material showed widespread PrPSc plaques and type i PrPSc accumulation similar to the p-dCJD patients, whereas the 129 M/M mice inoculated with sCJD-MM1 material showed diffuse synaptic-type PrPSc deposition and type 1 PrPSc accumulation similar to the np-dCJD patients. Thus, these animal models support the hypothesis that the origin of np-dCJD is sCJD-MM1/MV1 and that of p-dCJD is sCJD-VV2, −MV2K, or -MV2K + C. Indeed, the incidence rate of p-dCJD (32%) among total dCJD is close to the sum total of the incidence of sCJD-VV2 (15%), −MV2K (8%), and -MV2K + C (3%) [18].
Table 1.
Transmission of dCJD or sCJD to PrP-humanized mice
|
Inoculum (ID) |
Incubation period in days ± SEM (
n
/
n
0
)
a
|
||
|---|---|---|---|
| |
129 M/M |
129 V/V |
|
| |
Tg + Ki-Hu129 M/M
b
|
Ki-Hu129M/M
b
|
Ki-Hu129V/V
b
|
| (9.8×) c | (1×) | (1×) | |
| np-dCJD (GF) |
161 ± 5 (5/5) |
N.D. d |
N.D. |
| np-dCJD (TC) |
208 ± 2 (5/5) |
N.D. |
N.D. |
| p-dCJD (KR) |
420 ± 10 (5/5) |
685 ± 51 (5/5) |
259 ± 6 (6/6) |
| p-dCJD (KD) |
398 ± 10 (5/5) |
447 ± 51 (6/6) |
317 ± 8 (11/11) |
| sCJD-MM1 |
175 ± 4 (9/9) |
467 ± 24 (8/8) |
774 ± 32 (6/6) |
| sCJD-VV2 |
505 ± 14 (5/5) |
633 ± 49 (6/6) |
302 ± 9 (7/7) |
| sCJD-MV2K |
N.D. |
638 ± 57 (4/4) |
329 ± 3 (4/4) |
| sCJD-MV2K+C |
N.D. |
600 ± 22 (6/6) |
332 ± 15 (4/4) |
| 129 M/M mouse-passaged sCJD-VV2 | N.D. | 685 ± 17 (6/6) | 309 ± 3 (7/7) |
an, number of mice positive for PrP accumulation in the immunohistochemical analysis; n0, number of inoculated mice.
Ki-Hu129M/M, knock-in mice expressing human PrP with the 129 M/M genotype; Ki-Hu129V/V, knock-in mice expressing human PrP with the 129 V/V genotype; Tg + Ki-Hu129M/M, Ki-Hu129M/M crossed with transgenic mice overexpressing human PrP with the 129 M genotype.
dN.D., not done.
Figure 4.

Transmission of dCJD or sCJD to humanized mice carrying human PrP with either the 129 M/M or 129 V/V genotype. (a) p-dCJD and sCJD-VV2, −MV2K, or -MV2K + C were identical in the transmissibility to the PrP-humanized mice. (b) Immunohistochemical analysis of PrPSc in the mouse brain revealed that p-dCJD and sCJD-VV2, −MV2K, or -MV2K + C were indistinguishable with regard to the neuropathological phenotypes in the inoculated mice. G, gray matter; W, white matter. (c) Westernblot analysis of PrPSc in the mouse brain showed that p-dCJD and sCJD-VV2, −MV2K, or -MV2K + C were also indistinguishable with regard to the biochemical properties of PrPSc in the inoculated mice. 129 M/M, knock-in mice expressing human PrP with the 129 M/M genotype; 129 V/V, knock-in mice expressing human PrP with the 129 V/V genotype.
Molecular basis of the generation of two distinct subgroups in dCJD
At the molecular level, np-dCJD contains type 1 PrPSc with the codon 129 M genotype (denoted as M1 PrPSc), whereas p-dCJD contains type i PrPSc with the codon 129 M genotype (Mi PrPSc) (Table 2). Meanwhile, sCJD-MM1/MV1 contains M1 PrPSc, and sCJD-VV2 contains type 2 PrPSc with the codon 129 V genotype (V2 PrPSc). Recently, we found that sCJD-MV2K contains Mi PrPSc and V2 PrPSc, whereas sCJD-MV2K + C also contains type 2 PrPSc with the codon 129 M genotype and cortical pathology (M2C PrPSc) in addition to Mi PrPSc and V2 PrPSc (Table 2) [23]. M2 PrPSc can be divided into two subgroups based on histopathological phenotypes. M2C PrPSc causes a predominant cortical pathology in sCJD-MM2C, −MV2C, or -MV2K + C, whereas M2T PrPSc causes atrophy of thalamic and inferior olivery nuclei in sCJD-MM2T.
Table 2.
Molecular classification of dCJD and sCJD
| Classification a | Codon 129 genotype | PrP Sc type | Transmission type b | Original PrP Sc | Existing PrP Sc |
|---|---|---|---|---|---|
| np-dCJD |
M/M |
1 |
M1 |
M1 |
M1 |
| p-dCJD |
M/M |
i c |
V2 |
V2 d |
Mi |
| sCJD-MM1 |
M/M |
1 |
M1 |
M1 |
M1 |
| sCJD-VV2 |
V/V |
2 |
V2 |
V2 |
V2 |
| sCJD-MV2K |
M/V |
i + 2 |
V2 |
V2 |
Mi + V2 |
| sCJD-MV2K+C | M/V | i + 2 | V2 | M2Ce + V2 | M2C + Mi + V2 |
aAccording to Parchi, 1999 [17]; and Parchi, 2011 [18].
bAccording to Bishop, 2010 [24]; Kobayashi, 2007 [20]; and Kobayashi, 2013 [23].
cIntermediate type located between types 1 and 2 PrPSc.
dAlthough not only V2 PrPSc but also Mi PrPSc can cause p-dCJD if transmitted to the 129 M/M individuals, the primary origin of Mi PrPSc is V2 PrPSc (see text).
eM2 PrPSc can be divided into two subgroups based on histopathological phenotypes. M2C PrPSc causes a predominant cortical pathology, whereas M2T PrPSc causes atrophy of thalamic and inferior olivery nuclei.
The generation of M1 PrPSc in np-dCJD is simply due to the infection with M1 PrPSc from sCJD-MM1/MV1. On the other hand, the generation of Mi PrPSc in p-dCJD is rather complicated. Transmission of V2 PrPSc, i.e., sCJD-VV2, to the 129 M/M mice generated Mi PrPSc (Figure 4c) [20]. Similarly, transmission of Mi PrPSc, i.e., p-dCJD, to the 129 M/M mice also generated Mi PrPSc. Therefore, both V2 PrPSc and Mi PrPSc can generate Mi PrPSc if transmitted to individuals with the 129 M/M genotype. Indeed, transmission of sCJD-MV2K containing Mi PrPSc and V2 PrPSc to the 129 M/M mice also generated Mi PrPSc (Figure 4c). Meanwhile, sCJD-MV2K + C contains M2C PrPSc besides Mi PrPSc and V2 PrPSc (Table 2). However, M2C PrPSc lacks or has very low infectivity and does not affect the transmission properties of the coexisting PrPSc[23]. Therefore, the transmission of sCJD-MV2K + C to the 129 M/M mice can also result in the generation of Mi PrPSc (Figure 4c). Taken together, Mi PrPSc in p-dCJD is generated by infection with Mi PrPSc and/or V2 PrPSc from sCJD-VV2, −MV2K, or -MV2K + C. It is noteworthy that Mi PrPSc can be observed in the 129 M/M mice inoculated with V2 PrPSc but not in sCJD patients with the 129 M/M genotype, suggesting that Mi PrPSc in sCJD-MV2K or -MV2K + C is also generated by V2 PrPSc seed-dependent conversion but not by spontaneous conversion of the 129 M PrPC. Therefore, the primary origin of Mi PrPSc is V2 PrPSc. This can account for the similarities in transmission properties between Mi PrPSc and V2 PrPSc. Thus, M1 PrPSc in np-dCJD and Mi PrPSc in p-dCJD are completely different with regard to the neuropathological phenotypes, biochemical features, and transmission properties, reflecting their distinct PrPSc origins. In contrast to M1 PrPSc, which is the most common PrPSc observed in sCJD patients with the 129 M/M genotype, Mi PrPSc has never been observed in sCJD patients with the 129 M/M genotype. Therefore, the detection of Mi PrPSc can be sound evidence of iatrogenic infection in individuals with the 129 M/M genotype and would contribute to reliable surveillance of iatrogenic cases such as p-dCJD.
To verify experimentally that Mi PrPSc originates from V2 PrPSc and its transmission properties are identical to those of the parental V2 PrPSc, we performed a modeling study using PrP-humanized mice (Figure 5a) [25]. As described above, the 129 M/M mice inoculated with V2 PrPSc showed widespread PrPSc plaques and Mi PrPSc accumulation in the brain as an experimental model of p-dCJD. We then inoculated the Mi PrPSc from these mice into other PrP-humanized mice with either the 129 M/M or V/V genotype. This secondary passage revealed that the transmission properties of the Mi PrPSc, i.e., 129 M/M mouse-passaged sCJD-VV2, are identical to those of the parental V2 PrPSc. In particular, although the incompatibility of the codon 129 genotypes between host and inoculum usually results in a prolonged incubation period [20], the 129 V/V mice inoculated with the Mi PrPSc showed a shorter incubation period compared with the 129 M/M mice (Table 1). Moreover, the altered neuropathological phenotype and biochemical properties at the primary passage in the 129 M/M mice reverted to the original ones in the secondary passage in the 129 V/V mice (Figure 5b, c). Thus, this modeling study shows that (i) V2 PrPSc infection in a host with the incompatible codon 129 M/M genotype generates an unusual PrPSc with altered conformational properties, i.e., Mi PrPSc, (ii) the emerging Mi PrPSc retains the memory of the parental V2 PrPSc within its conformational properties, and (iii) the parental V2 PrPSc re-emerges and proliferates rapidly if the Mi PrPSc is transmitted to the original host with the codon 129 V/V genotype. This phenomenon, designated as traceback, can be a useful tool to identify the origin of PrPSc infection if atypical PrPSc emerges in the future [20,22,26].
Figure 5.

Modeling of p-dCJD and traceback phenomenon. (a) Schematic diagram of the modeling study. The 129 V/V mice were highly susceptible to the 129 M/M mouse-passaged sCJD-VV2, i.e., Mi PrPSc, despite their incompatible codon 129 genotypes. Moreover, the altered neuropathological and biochemical phenotypes in the primary passage in the 129 M/M mice reverted to the original ones in the secondary passage in the 129 V/V mice. This is because the origin of Mi PrPSc is V2 PrPSc. This phenomenon has been designated as traceback [20,26]. (b) Immunohistochemical analysis of PrPSc in the brains from the PrP-humanized mice infected with the 129 M/M mouse-passaged sCJD-VV2. G, gray matter; W, white matter. (c) Westernblot analysis of PrPSc in the brains from the PrP-humanized mice infected with sCJD-VV2 or the 129 M/M mouse-passaged sCJD-VV2. 129 M/M, knock-in mice expressing human PrP with the 129 M/M genotype; 129 V/V, knock-in mice expressing human PrP with the 129 V/V genotype.
Remaining problems
Our transmission studies resolved the complicated pathogenesis of dCJD. However, they have also revealed several issues surrounding dCJD that need to be addressed in the future.
First, the numbers of p-dCJD patients may increase in the future. The experimental p-dCJD model, i.e., the 129 M/M mice inoculated with Mi PrPSc and/or V2 PrPSc from sCJD-VV2, −MV2K, or -MV2K + C, showed a longer incubation period compared with the np-dCJD model, i.e., the 129 M/M mice inoculated with M1 PrPSc from sCJD-MM1 (Table 1). This raises the concern that additional p-dCJD patients, who are presenting still at the subclinical stage, may emerge after a longer incubation period in the future. Although the numbers of patients with newly developed dCJD have dropped off, continuous surveillance will be required to find remaining p-dCJD cases.
Second, the potential risks of secondary infection from dCJD, particularly from p-dCJD, may be considerable. As described above, the transmission studies raise a concern about the existence of subclinical p-dCJD patients. dCJD patients may undergo more than one neurosurgical operation due to their underlying diseases (the primary disease for which the neurosurgery was performed) [4]. In addition, p-dCJD patients may be more frequently autopsied because the clinical features of p-dCJD are atypical compared with those of classical sCJD [11]. These facts suggest that there may be considerable risk of secondary infection from p-dCJD patients. Individuals with the 129 V/V genotype may be more vulnerable to the infection with Mi PrPSc from p-dCJD, as suggested by the fact that the 129 V/V mice were highly susceptible to Mi PrPSc in the transmission study (Table 1). Additionally, 129 M/M individuals may be also affected after a prolonged incubation period, as suggested by the high attack rate (100%) of the 129 M/M mice inoculated with Mi PrPSc. Therefore, secondary infection from p-dCJD can occur regardless of the codon 129 genotype. Comprehensive analysis of the distribution of PrPSc in the peripheral tissues of p-dCJD patients will be also required to assess the potential risks of secondary infection.
Finally, the efficacy of the current PrPSc decontamination procedures against Mi PrPSc needs to be tested in the future. Mi PrPSc in p-dCJD and M1 PrPSc in np-dCJD differ in the sizes of the proteinase K-resistant core, suggesting their conformational differences. Moreover, their parental PrPSc strains are also different. Different PrPSc strains can show different thermostability [27,28] and different susceptibility to the decontamination procedures [29]. To prevent the spread of secondary infection from dCJD patients to medical workers or other patients, adequate decontamination and disinfection of the instruments used for neurosurgery or autopsy are essential. However, the current PrPSc decontamination procedures were developed using scrapie isolates and tested using CJD isolates other than p-dCJD [30-32]. Therefore, further studies using Mi PrPSc will be needed to assess the effectiveness of the current procedures. For this purpose, sensitive detection systems for Mi PrPSc are also prerequisite to evaluating quantitatively the reduction of infectivity after the decontamination procedures. Real-time quaking-induced conversion [33,34], protein misfolding cyclic amplification [35-39], or transgenic mice overexpressing human PrP with the 129 V genotype [20] might be useful to detect the reduced infectivity of Mi PrPSc at high sensitivity. Using such sensitive detection systems, effective decontamination procedures for Mi PrPSc can be established in the future.
Concluding remarks
Recent progress in the study of the pathogenesis of dCJD has revealed that the two distinct subgroups of dCJD are caused by infection with different PrPSc strains of sCJD, i.e., np-dCJD caused by M1 PrPSc from sCJD-MM1/MV1 and p-dCJD caused by Mi PrPSc and/or V2 PrPSc from sCJD-VV2, −MV2K, or -MV2K + C. Studies have also revealed previously unrecognized problems such as the considerable risks of secondary infection from dCJD, particularly from p-dCJD. To prevent secondary infection from p-dCJD, the effectiveness of the current decontamination procedures should be tested urgently using sensitive Mi PrPSc detection systems.
Competing interest
The authors declare that they have no competing interest.
Contributor Information
Atsushi Kobayashi, Email: kobayashi@med.tohoku.ac.jp.
Yuichi Matsuura, Email: zrxmatsu@affrc.go.jp.
Shirou Mohri, Email: shirou@med.tohoku.ac.jp.
Tetsuyuki Kitamoto, Email: kitamoto@med.tohoku.ac.jp.
Acknowledgements
We thank members of the Creutzfeldt-Jakob Disease Surveillance Committee in Japan, Creutzfeldt-Jakob disease specialists in the prefectures, and Creutzfeldt-Jakob disease patients and families for providing important clinical information. We thank Y. Ishikawa, H. Kudo, M. Yamamoto, and A. Yamazaki for their excellent technical assistance, and B. Bell for critical review of the manuscript. This study was supported by Grants-in-Aid from the Ministry of Health, Labor and Welfare of Japan (A.K., S.M., and T.K.), Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (A.K. and T.K.), and a grant for TSE research from the Ministry of Health, Labor and Welfare of Japan (H23-Shokuhin-Ippan-005) (T.K.).
References
- Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell. 1998;2:337–348. doi: 10.1016/S0092-8674(00)81163-0. [DOI] [PubMed] [Google Scholar]
- Brown P, Brandel JP, Sato T, Nakamura Y, MacKenzie J, Will RG, Ladogana A, Pocchiari M, Leschek EW, Schonberger LB. Iatrogenic Creutzfeldt-Jakob disease, final assessment. Emerg Infect Dis. 2012;2:901–907. doi: 10.3201/eid1806.120116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaguchi T, Sakai K, Noguchi-Shinohara M, Nozaki I, Takumi I, Sanjo N, Sadakane A, Nakamura Y, Kitamoto T, Saito N, Mizusawa H, Yamada M. Insight into the frequent occurrence of dura mater graft-associated Creutzfeldt-Jakob disease in Japan. J Neurol Neurosurg Psychiatry. 2013;2:1171–1175. doi: 10.1136/jnnp-2012-304850. [DOI] [PubMed] [Google Scholar]
- Nakamura Y Uehara R Watanabe M Sadakane A Yamada M Mizusawa H Maddox R Sejvar MPH J Belay E Schonberger L Update: Creutzfeldt-Jakob disease associated with cadaveric dura mater grafts - Japan, 1978–2008 MMWR Morb Mortal Wkly Rep 200821152–1154.18946463 [Google Scholar]
- Nozaki I, Hamaguchi T, Sanjo N, Noguchi-Shinohara M, Sakai K, Nakamura Y, Sato T, Kitamoto T, Mizusawa H, Moriwaka F, Shiga Y, Kuroiwa Y, Nishizawa M, Kuzuhara S, Inuzuka T, Takeda M, Kuroda S, Abe K, Murai H, Murayama S, Tateishi J, Takumi I, Shirabe S, Harada M, Sadakane A, Yamada M. Prospective 10-year surveillance of human prion diseases in Japan. Brain. 2010;2:3043–3057. doi: 10.1093/brain/awq216. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Hoshi K, Muramoto T, Homma M, Ironside JW, Kuzuhara S, Sato T, Yamamoto T, Kitamoto T. Creutzfeldt-Jakob disease with florid-type plaques after cadaveric dura mater grafting. Arch Neurol. 1999;2:357–362. doi: 10.1001/archneur.56.3.357. [DOI] [PubMed] [Google Scholar]
- Hoshi K, Yoshino H, Urata J, Nakamura Y, Yanagawa H, Sato T. Creutzfeldt-Jakob disease associated with cadaveric dura mater grafts in Japan. Neurology. 2000;2:718–721. doi: 10.1212/WNL.55.5.718. [DOI] [PubMed] [Google Scholar]
- Mochizuki Y, Mizutani T, Tajiri N, Oinuma T, Nemoto N, Kakimi S, Kitamoto T. Creutzfeldt-Jakob disease with florid plaques after cadaveric dura mater graft. Neuropathology. 2003;2:136–140. doi: 10.1046/j.1440-1789.2003.00489.x. [DOI] [PubMed] [Google Scholar]
- Kretzschmar HA, Sethi S, Földvári Z, Windl O, Querner V, Zerr I, Poser S. Iatrogenic Creutzfeldt-Jakob disease with florid plaques. Brain Pathol. 2003;2:245–249. doi: 10.1111/j.1750-3639.2003.tb00025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh K, Muramoto T, Tanaka T, Kitamoto N, Ironside JW, Nagashima K, Yamada M, Sato T, Mohri S, Kitamoto T. Association of an 11–12kDa protease-resistant prion protein fragment with subtypes of dura graft-associated Creutzfeldt-Jakob disease and other prion diseases. J Gen Virol. 2003;2:2885–2893. doi: 10.1099/vir.0.19236-0. [DOI] [PubMed] [Google Scholar]
- Noguchi-Shinohara M, Hamaguchi T, Kitamoto T, Sato T, Nakamura Y, Mizusawa H, Yamada M. Clinical features and diagnosis of dura mater graft associated Creutzfeldt-Jakob disease. Neurology. 2007;2:360–367. doi: 10.1212/01.wnl.0000266624.63387.4a. [DOI] [PubMed] [Google Scholar]
- Yamada M, Noguchi-Shinohara M, Hamaguchi T, Nozaki I, Kitamoto T, Sato T, Nakamura Y, Mizusawa H. Dura mater graft-associated Creutzfeldt-Jakob disease in Japan: clinicopathological and molecular characterization of the two distinct subtypes. Neuropathology. 2009;2:609–618. doi: 10.1111/j.1440-1789.2008.00987.x. [DOI] [PubMed] [Google Scholar]
- Lane KL, Brown P, Howell DN, Chain BJ, Hulette CM, Burger PC, DeArmond SJ. Creutzfeldt-Jakob disease in a pregnant woman with an implanted dura mater graft. Neurosurgery. 1994;2:737–740. doi: 10.1227/00006123-199404000-00026. [DOI] [PubMed] [Google Scholar]
- Kopp N, Streichenberger N, Deslys JP, Laplanche JL, Chazot G. Creutzfeldt-Jakob disease in a 52-year-old woman with florid plaques. Lancet. 1996;2:1239–1240. doi: 10.1016/s0140-6736(05)65510-9. [DOI] [PubMed] [Google Scholar]
- Takashima S, Tateishi J, Taguchi Y, Inoue H. Creutzfeldt-Jakob disease with florid plaques after cadaveric dural graft in a Japanese woman. Lancet. 1997;2:865–866. doi: 10.1016/S0140-6736(05)62035-1. [DOI] [PubMed] [Google Scholar]
- Kimura K, Nonaka A, Tashiro H, Yaginuma M, Shimokawa R, Okeda R, Yamada M. Atypical form of dura graft associated Creutzfeldt-Jakob disease: report of a postmortem case with review of the literature. J Neurol Neurosurg Psychiatry. 2001;2:696–699. doi: 10.1136/jnnp.70.5.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichemberger N, Julien J, Vital C, Ghetti B, Gambetti P, Kretzschmar H. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;2:224–233. doi: 10.1002/1531-8249(199908)46:2<224::AID-ANA12>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Parchi P, Strammiello R, Giese A, Kretzschmar H. Phenotypic variability of sporadic human prion disease and its molecular basis: past, present, and future. Acta Neuropathol. 2011;2:91–112. doi: 10.1007/s00401-010-0779-6. [DOI] [PubMed] [Google Scholar]
- Parchi P, Zou W, Wang W, Brown P, Capellari S, Ghetti B, Kopp N, Schulz-Schaeffer WJ, Kretzschmar HA, Head MW, Ironside JW, Gambetti P, Chen SG. Genetic influence on the structural variations of the abnormal prion protein. Proc Natl Acad Sci USA. 2000;2:10168–10172. doi: 10.1073/pnas.97.18.10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A, Asano M, Mohri S, Kitamoto T. Cross-sequence transmission of sporadic Creutzfeldt-Jakob disease creates a new prion strain. J Biol Chem. 2007;2:30022–30028. doi: 10.1074/jbc.M704597200. [DOI] [PubMed] [Google Scholar]
- Parchi P, Strammiello R, Notari S, Giese A, Langeveld JP, Ladogana A, Zerr I, Roncaroli F, Cras P, Ghetti B, Pocchiari M, Kretzschmar H, Capellari S. Incidence and spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: an updated classification. Acta Neuropathol. 2009;2:659–671. doi: 10.1007/s00401-009-0585-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A, Asano M, Mohri S, Kitamoto T. A traceback phenomenon can reveal the origin of prion infection. Neuropathology. 2009;2:619–624. doi: 10.1111/j.1440-1789.2008.00973.x. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Iwasaki Y, Otsuka H, Yamada M, Yoshida M, Matsuura Y, Mohri S, Kitamoto T. Deciphering the pathogenesis of sporadic Creutzfeldt-Jakob disease with codon 129M/V and type 2 abnormal prion protein. Acta Neuropathol Commun. 2013;2:74. doi: 10.1186/2051-5960-1-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop MT, Will RG, Manson JC. Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc Natl Acad Sci USA. 2010;2:12005–12010. doi: 10.1073/pnas.1004688107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A, Sakuma N, Matsuura Y, Mohri S, Aguzzi A, Kitamoto T. Experimental verification of a traceback phenomenon in prion infection. J Virol. 2010;2:3230–3238. doi: 10.1128/JVI.02387-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano M, Mohri S, Ironside JW, Ito M, Tamaoki N, Kitamoto T. vCJD prion acquires altered virulence through trans-species infection. Biochem Biophys Res Commun. 2006;2:293–299. doi: 10.1016/j.bbrc.2006.01.149. [DOI] [PubMed] [Google Scholar]
- Taylor DM, Fraser H, McConnell I, Brown DA, Brown KL, Lamza KA, Smith GR. Decontamination studies with the agents of bovine spongiform encephalopathy and scrapie. Arch Virol. 1994;2:313–326. doi: 10.1007/BF01310794. [DOI] [PubMed] [Google Scholar]
- Rutala WA, Weber DJ. Creutzfeldt-Jakob disease: recommendations for disinfection and sterilization. Clin Infect Dis. 2001;2:1348–1356. doi: 10.1086/319997. [DOI] [PubMed] [Google Scholar]
- Rogez-Kreuz C, Yousfi R, Soufflet C, Quadrio I, Yan ZX, Huyot V, Aubenque C, Destrez P, Roth K, Roberts C, Favero M, Clayette P. Inactivation of animal and human prions by hydrogen peroxide gas plasma sterilization. Infect Control Hosp Epidemiol. 2009;2:769–777. doi: 10.1086/598342. [DOI] [PubMed] [Google Scholar]
- Brown P, Gibbs CJ Jr, Amyx HL, Kingsbury DT, Rohwer RG, Sulima MP, Gajdusek DC. Chemical disinfection of Creutzfeldt-Jakob disease virus. N Engl J Med. 1982;2:1279–1282. doi: 10.1056/NEJM198205273062107. [DOI] [PubMed] [Google Scholar]
- Taguchi F, Tamai Y, Uchida K, Kitajima R, Kojima H, Kawaguchi T, Ohtani Y, Miura S. Proposal for a procedure for complete inactivation of the Creutzfeldt-Jakob disease agent. Arch Virol. 1991;2:297–301. doi: 10.1007/BF01310679. [DOI] [PubMed] [Google Scholar]
- Tateishi J, Tashima T, Kitamoto T. Practical methods for chemical inactivation of Creutzfeldt-Jakob disease pathogen. Microbiol Immunol. 1991;2:163–166. doi: 10.1111/j.1348-0421.1991.tb01544.x. [DOI] [PubMed] [Google Scholar]
- Wilham JM, Orrú CD, Bessen RA, Atarashi R, Sano K, Race B, Meade-White KD, Taubner LM, Timmes A, Caughey B. Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog. 2010;2:e1001217. doi: 10.1371/journal.ppat.1001217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi R, Satoh K, Sano K, Fuse T, Yamaguchi N, Ishibashi D, Matsubara T, Nakagaki T, Yamanaka H, Shirabe S, Yamada M, Mizusawa H, Kitamoto T, Klug G, McGlade A, Collins SJ, Nishida N. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat Med. 2011;2:175–178. doi: 10.1038/nm.2294. [DOI] [PubMed] [Google Scholar]
- Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;2:810–813. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- Suyama K, Yoshioka M, Akagawa M, Murayama Y, Horii H, Takata M, Yokoyama T, Mohri S. Assessment of prion inactivation by fenton reaction using protein misfolding cyclic amplification and bioassay. Biosci Biotechnol Biochem. 2007;2:2069–2071. doi: 10.1271/bbb.70085. [DOI] [PubMed] [Google Scholar]
- Yoshioka M, Murayama Y, Miwa T, Miura K, Takata M, Yokoyama T, Nishizawa K, Mohri S. Assessment of prion inactivation by combined use of Bacillus-derived protease and SDS. Biosci Biotechnol Biochem. 2007;2:2565–2568. doi: 10.1271/bbb.70257. [DOI] [PubMed] [Google Scholar]
- Beekes M, Lemmer K, Thomzig A, Joncic M, Tintelnot K, Mielke M. Fast, broad-range disinfection of bacteria, fungi, viruses and prions. J Gen Virol. 2010;2:580–589. doi: 10.1099/vir.0.016337-0. [DOI] [PubMed] [Google Scholar]
- Takeuchi A, Komiya M, Kitamoto T, Morita M. Deduction of the evaluation limit and termination timing of multi-round protein misfolding cyclic amplification from a titration curve. Microbiol Immunol. 2011;2:502–509. doi: 10.1111/j.1348-0421.2011.00340.x. [DOI] [PubMed] [Google Scholar]