

Abstract
Nascent polypeptides emerging into the lumen of the endoplasmic reticulum (ER) are N-glycosylated on asparagines in Asn-Xxx-Ser/Thr motifs. Processing of the core oligosaccharide eventually determines the fate of the associated polypeptide by regulating entry into and retention by the calnexin chaperone system, or extraction from the ER folding environment for disposal. Recent advances have shown that at least two N-glycans are necessary for protein access to the calnexin chaperone system and that polypeptide cycling in the system is a rather rare event, which, for folding-defective polypeptides, is activated only upon persistent misfolding. Additionally, dismantling of the polypeptide-bound N-glycan interrupts futile folding attempts, and elicits preparation of the misfolded chain for dislocation into the cytosol and degradation.
Introduction
Proteins destined for transport to organelles of the secretory and endocytic compartments, to the plasma membrane and to the extracellular space enter the endoplasmic reticulum (ER) lumen through a narrow protein-conducting channel, the Sec61 translocon complex. Nascent chains are covalently modified by the oligosaccharyltransferase (OST). This multimeric enzyme transfers preassembled core glycans composed of a glucose3-mannose9-N-acetyl-glucosamine2 (Glc3Man9GlcNAc2) structure (Figure 1a) from a lipid donor in the ER membrane to asparagines in Asn-Xxx-Ser/Thr motifs of growing nascent chains [1]. By increasing the overall hydrophilicity of the as yet unstructured nascent chains, N-glycans efficiently prevent aggregation, which would interfere with the polypeptide folding process. They rapidly become accessible to ER-resident sugar-processing enzymes and their subsequent modification determines the fate of the associated polypeptide chain. Sequential removal of the two outermost glucose residues enables association with ER lectin chaperones, calnexin (CNX) and calreticulin (CRT), whose involvement in polypeptide folding was established in the first half of the 1990s [2]. In some cases, protein folding might require cycling of the newly synthesized polypeptide in the lectin chaperone system, regulated by the repetitive removal and readdition of the innermost glucose residue. Slow removal of mannose residues eventually leads to the extraction of folding-defective and terminally misfolded polypeptides from the lectin chaperone system, and their preparation for disposal [3,4].
Figure 1.
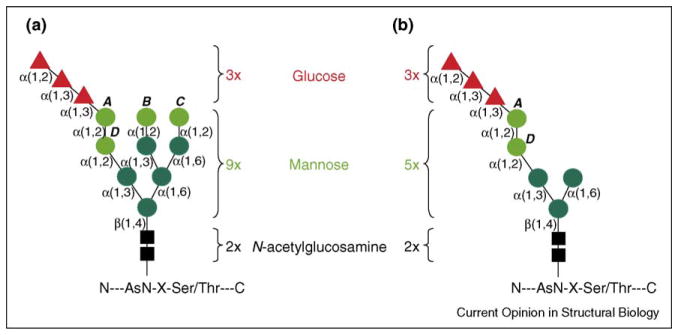
High-mannose precursor structures from (a) normal (e.g. HEK293) and (b) Dol-P-Man-deficient (B3F7) cell lines. The structure of the glycan that is initially transferred to the nascent polypeptide is indicated. N-acetylglucosamine (black boxes), core mannose residues (dark green circles), peripheral α1,2-mannose residues (light green circles) and glucose residues (red triangles) are shown with the linkages indicated. Individual α1,2-mannose residues are labeled A–D. The absence of Dol-P-Man in the B3F7 cell line results in the transfer of a precursor structure missing four of the nine mannose residues, but retaining α1,2-mannose residues A and D.
The focus of this review is the involvement of sugar-processing reactions in the two opposing aspects of ER quality control: entry and retention of nascent glycoproteins within the lectin chaperone system to aid protein folding versus conversion of glycan structures into the recognition signals necessary for misfolded glycoprotein disposal, leading to dislocation into the cytosol and proteasomal degradation.
The calnexin chaperone system
The maturation of newly synthesized glycoproteins expressed in the mammalian ER is assisted by a complex chaperone system composed of two glucose-removing enzymes (glucosidase I [GI] and glucosidase II [GII]), one folding sensor displaying reglucosylation activity (UDP-glucose:glycoprotein glucosyltransferase [UGT1]), two sugar-binding chaperones (CNX and CRT) and one dedicated oxidoreductase (ERp57) (Figure 2). Briefly, GI and GII sequentially remove the first and second glucose residues of the polypeptide-bound N-glycan (step 1 in Figure 2). The resulting monoglucosylated trimming intermediate can then interact with heterodimeric complexes of either the membrane-bound ER lectin CNX or the soluble homolog CRT, each associated with ERp57 (step 2, Figure 2). ERp57 catalyzes the rate-limiting step of polypeptide folding in the ER lumen, namely the formation of intramolecular and intermolecular disulfide bonds. The intermediates in the polypeptide folding process are eventually released from the CNX–ERp57 or CRT–ERp57 complexes, and the last glucose is removed by GII (step 3, Figure 2). The folding sensor UGT1 scans the structure of the polypeptide for exposed non-native determinants. If these are detected, UGT1 then adds a glucose residue to the terminal mannose in the same position on branch A of the original N-glycan precursor structure (Figures 1 and 2, step 4), thus returning the immature and misfolded polypeptide to the CNX/CRT chaperone system (Figure 2, step 2; reviewed in [3]).
Figure 2.

The role of glycan processing in regulating access to the lectin chaperone system or targeting for ERAD. The synthesis of glycoproteins is initiated by the co-translational extrusion of nascent polypeptides through the Sec61p pore. OST transfers preassembled core glycans from a dolichol-pyrophosphate intermediate (Dol-PP-OS) to peptide Asn-X-(Ser/Thr) sequons. Glycan trimming by glucose removal occurs immediately after transfer to the polypeptide through the action of GI and GII (step 1). The latter enzyme is composed of a catalytic α-subunit and a β-subunit with lectin activity. Folding intermediates with Glc1Man9GlcNAc2 (G1M9N2) structures (orange ribbon diagram with stick glycan structures) are ligands for interaction with the membrane-associated lectin CNX (not shown) or soluble CRT (green ribbon diagram; PDB code 1JHN), both of which are associated with the oxidoreductase ERp57 (step 2). Dissociation from the lectin is followed by further cleavage of glucose residues by GII (step 3). Correctly folded glycoproteins are packaged in anteriograde transport vesicles with the potential assistance of the mannose-binding lectin ERGIC53 (yellow ribbon diagram; PDB code 1GV9) or other homologous proteins. Proteins that have not completed the folding process are recognized by the folding sensor UGT1, which adds a glucose residue back to the glycan structure (step 4) and allows reassociation with CRT–ERp57 (step 2) and reintegration into the CNX cycle (first phase of ER retention). Terminally misfolded glycoproteins dissociate from the CNX cycle, and can be transferred to the BiP chaperone system (step 5, second phase of ER retention) or directly targeted for disposal (step 6). The signal for disposal is mediated by mannose trimming to Man8GlcNAc2 in yeast or Man5GlcNAc2 in mammalian cells through the action of class 1 α-mannosidases such as ERManI (blue ribbon diagram; PDB code 1X9D) and/or GolgiManIA/IB/IC and/or EDEM proteins (step 6). EDEM1–3 (blue ribbon diagram based on the structure of ERManI, a sequence homolog of the EDEM proteins) and a putative (yet to be identified) mammalian ortholog of the yeast mannose lectin Yos9p (pink ribbon diagram based on the cation-dependent mannose-6-phosphate receptor [PDB code 1M6P], a sequence homolog of Yos9p) may play a role in shuttling ERAD substrates to the site of dislocation. The precise mode of substrate dislocation into the cytosol remains unclear and may involve the putative translocon pores Sec61p and/or the Derlin homologs Der1–3p. The cytosolic ATPase p97 is involved in extraction of ERAD substrates from the ER before deglycosylation by a cytosolic PNGase, ubiquitination by a complex comprising AMFR, Y33K and HR23B, and degradation by the cytosolic proteasome [52].
Entry of nascent polypeptides into the calnexin chaperone system
Proteins synthesized in the ER lumen may associate with CNX even before termination of their synthesis [3]. With an elongation rate approaching 5 residues/s in mammalian cells, the synthesis of an average protein of about 500 residues takes less than 2 min. Thus, for co-translational association with CNX, the triglucosylated N-glycan added to nascent chains must be converted into the monoglucosylated trimming intermediate necessary for CNX association in a matter of a few seconds by the rapid and coordinated action of GI and GII. GI removes the outermost glucose residue immediately after N-glycan addition to the growing nascent chain. The removal of the ‘middle’ glucose by GII is regulated and requires trans-activation of GII by another N-glycan on the same polypeptide chain [5••]. Trans-activation is elicited when the regulatory β-subunit of GII, a sequence homolog of the mannose-6-phosphate receptor, binds to mannoses in the 6′-pentamannosyl branch of a core N-glycan. This would induce a change in the conformation of the catalytic α-subunit of GII and its correct positioning for removal of the middle glucose from another N-glycan unit [5••]. Consistent with this model, the presence of more than one N-glycan is required for the formation of a complex between CNX and nascent chains in canine microsomes and semi-permeabilized cells [6,7].
Cyclingf of folding-competent and folding-defective polypeptides in the calnexin chaperone system
One round of association with CNX/CRT may not be sufficient for effective folding. Substrate cycling in the lectin chaperone system might, to some extent, facilitate the folding process by transiently interrupting the physical constraints resulting from the anchoring of the substrate N-glycans to the lectin. In the case of influenza virus hemagglutinin, which is one of the best-studied models of chaperone-assisted glycoprotein folding in the mammalian ER, GII/UGT1-driven cycles of release from and reassociation with CNX are required for efficient folding (M Molinari et al., unpublished). However, this protein appears to be one of the very few proteins that obligatorily depend on assistance from members of the CNX chaperone system for efficient maturation [8–10]. For the bulk of glycoproteins expressed in mammalian cells, deletion of UGT1 does not affect maturation ([11••]; T Soldà and M Molinari, unpublished). Thus, as previously reported for polypeptides expressed in Schizosaccharomyces pombe [12], it is likely that most glycoproteins in mammalian cells acquire native structure in a single round of association with CNX. Deletion of UGT1 also does not affect the stringency of ER quality control. The CNX/CRT and BiP chaperone systems in fact act sequentially to prevent forward transport of non-native polypeptides (Figure 2, first and second phase of ER retention). In cells lacking UGT1, terminally misfolded glycoproteins eventually released from CNX/CRT do associate with BiP, and are prepared for dislocation into the cytosol and proteasome-mediated destruction. UGT1 deletion accelerates transfer of misfolded glycopolypeptides from the first to the second phase of ER retention, but it does so after a long time lag. This is consistent with the activation of substrate cycling in the CNX chaperone system only upon persistent misfolding [11••].
Protein folding dynamics: competition between conformational maturation and protein disposal
The kinetics of protein folding in the ER can be variable. Both ‘normal’ and ‘mutant’ proteins display a wide range of folding kinetics, even with the assistance of the ER chaperone machinery. Completion of protein folding and assembly of multimeric complexes is required in order to pass the ER quality control checkpoint that precedes transport through the secretory pathway [2]. Thus, proteins that are fully defective for folding as a result of mutation or truncation are retained and quantitatively targeted for ER-associated degradation (ERAD). This is in contrast to proteins with slow folding kinetics, which can be partially targeted for disposal. Contradictory data have been published on the actual fraction of wild-type proteins that are rapidly degraded in mammalian cells upon failure of their folding program, ranging from 20–30% [13] to a much lower amount [14]. Certainly, folding efficiency is strictly substrate dependent, and the percentage of nascent proteins that are folded as opposed to degraded is presumably controlled by competition between conformational maturation and recognition for ER disposal. Consistent with this model, overexpression of ER α-mannosidase I (ERManI, see next section), one of the regulators of ERAD kinetics, resulted in the accelerated disposal of both misfolded glycoproteins and a fraction of wild-type proteins that are normally targeted for secretion [15]. Many loss-of-function human genetic diseases can, therefore, be considered ‘protein misfolding disorders’, as the mutant proteins may not be fully defective, but their kinetics of conformational maturation are slowed such that normal recognition by the ERAD system results in disposal before effective folding is complete. Competition between maturation and disposal is illustrated by the use of ‘chemical and pharmacological chaperones’ to rescue loss-of-function disorders by nucleating accelerated conformational maturation in advance of ERAD targeting [16].
The interruption of futile folding attempts in the ER: protein degradation is timed by removal of mannose residues
The targeting of folding-defective polypeptides and orphan subunits of multimeric complexes for dislocation into the cytosol and proteasome-mediated destruction is crucial for maintenance of cell homeostasis [17]. The ERAD machinery is easily saturated [18] and a defective ability to adapt ERAD activity to the load of misfolded glycoproteins in the ER lumen results in the progressive loss of protein folding efficiency and secretion capacity [19]. Genetic evidence from the yeast Saccharomyces cerevisiae [20] has demonstrated the crucial role played by ERManI in the removal of a single mannose from N-glycan branch B as a ‘mannose timer’ for the initiation of the ERAD process. For mammalian cells, the role of mannose trimming in ERAD was first reported by Su et al. in 1993 [21]. They demonstrated that the α1,2-mannosidase inhibitor 1-deoxymannojirimycin (dMNJ) blocked the disposal of heterologously expressed yeast prepro-α-factor [21]. It was not until considerably later that numerous studies confirmed that selective α-mannosidase inhibition using dMNJ or kifunensine (Kif) could selectively delay ER disposal of a variety of misfolded mutant proteins (reviewed in [22]). These observations, combined with the known specificity of ERManI for the cleavage of mannose B [23], led to the extension of the yeast-specific mannose timer model to mammalian cells [22].
Extensive demannosylation as a signal for protein disposal from the mammalian ER
More recent findings highlight differences between the extent of mannose removal required for ERAD of folding-defective polypeptides in yeast and mammalian cells. In short, recent studies show that, during retention in the mammalian ER, N-glycans on folding-defective polypeptides are extensively processed to Man5–6 (step 6 in Figure 2; reviewed in [4]). Moreover, mannose removal remains obligatory to promote protein degradation in mannosyl phosphoryldolichol (Dol-P-Man)-deficient cell lines, which are characterized by the addition of aberrant oligosaccharides to nascent chains [24]. As a consequence of the peculiar structure of the N-linked glycans added to nascent chains in these cell lines (e.g. B3F7 [25] (Figure 1b) and MadIA214 [24]), the removal of mannose A was suggested to be the signal for disposal in mammalian cells. Importantly, mannose A is the only acceptor for UGT1-catalyzed protein reglucosylation, an activity that delays protein degradation by retaining misfolded polypeptides in the CNX/CRT chaperone system. The importance of the removal of mannose A for protein disposal in mammalian cells is contrasted with the targeting of proteins for ERAD in S. cerevisiae, which lacks both UGT1 homologs and mannosidase homologs capable of cleaving residue A.
Mannosidases and mannosidase-like proteins in the lumen of the ER
The observation that both yeast and mammals require α1,2-mannose trimming as a prerequisite for ERAD has highlighted the central roles of processing α1,2-mannosidases in key timing steps of misfolded glycoprotein disposal. The inhibitors dMNJ and Kif are both selective for the broad family of class 1 (GH47 [26]) α-mannosidases, which are conserved from yeast to mammals [23]. There are three subfamilies within the GH47 enzyme family, including ERManI, a collection of Golgi α-mannosidases (GolgiManIA, GolgiManIB and GolgiManIC in mammals, but absent in yeast) [23] and a collection of putative ER lectins termed EDEM proteins (EDEM1–3 in mammals and Htm1p/Mnl1p in yeast) [27••,28••]. The former two subfamilies of enzymes catalyze Manα1, 2-Man hydrolysis through a unique, Ca2+-dependent inverting glycosidase mechanism. The enzymes adopt an (αα)7 barrel structure, with the active site in the core of the barrel (Figure 3) [29••]. The domain structure enables the formation of an extensive matrix of interactions between the glycan substrate and the enzyme. This matrix, which extends across the cleft leading to the barrel core [29••,30,31,32••], confers the branch specificity of cleavage by the enzymes. Kinetic studies have shown that ERManI preferentially cleaves mannose B [23], but high enzyme concentrations or prolonged digestion can result in further cleavage of other α1,2-Man linkages [33]. In contrast, the collection of Golgi enzymes preferentially cleaves mannose A, C and D (Figure 1a), but cleaves mannose B with at least tenfold lower efficiency [23]. Thus, despite similarities of both sequence and structure, the ER and Golgi α-mannosidases unexpectedly engage their respective oligosaccharide substrates quite differently, and are essentially complementary in their recognition and cleavage of the four α1,2-Man linkages during glycan processing [23].
Figure 3.

Structural modeling of the EDEM proteins indicates conservation of residues involved in hydrolysis by ERManI. The structure of human ERManI (PDB code 1X9D, orange ribbon diagram) was used as a template for structural modeling of EDEM1 (GenBank accession number NP_055489, gray ribbon diagram), EDEM2 (GenBank accession number NP_060687, magenta ribbon diagram) and EDEM3 (GenBank accession number NP_079467, green ribbon diagram) using the Swiss-Model modeling server (swissmodel.expasy.org). Models of all three proteins were aligned with the ERManI structure using DeepView-Swiss-PdbViewer (www.expasy.org/spdbv/) and displayed as (a) an end-on view of the (αα)7 barrel or (b) a side view using MacPymol (pymol.sourceforge.net). The bound Ca2+ ion is shown as a blue sphere and the uncleaved thiodisaccharide pseudosubstrate in the core of the barrel as a white stick figure [29••]. Significant overlap of the four structures is revealed in the α-helical segments, whereas the loop regions are more divergent. For each of the EDEM proteins, certain loop regions were removed to facilitate structural modeling; the positions of these sequences are indicated by the red arrowheads. A subset of the residues involved in substrate binding and hydrolysis are indicated (c) in cartoon form [29••] and (d) in stick form. A structural equivalent of all six catalytic residues, with the exception of F659, was exactly conserved in sequence and position in all four proteins. The latter residue was conserved in sequence, but a distortion of the corresponding helix in the EDEM1 model resulted in repositioning of this sidechain (not shown). The function of each of the residues is indicated in red text [29••]. Residue numbering in (c,d) is for ERManI. Water molecules coordinating the Ca2+ ion are indicated by small red space-fill structures. Interactions with the Ca2+ ion are indicated by cyan dotted lines, as are interactions between the carbonyl oxygen and Oγ of Thr688 and the Ca2+ ion. Proposed acid, base and nucleophile trajectories are illustrated with magenta dotted lines, and hydrogen bonds are shown as green dotted lines.
The observation of cleavage intermediates smaller than Man8GlcNAc2 during ERAD in mammalian cells suggests several possibilities for glycan trimming before targeting for disposal. One possibility is local sequestration of ERAD intermediates in a compartment with relatively enriched ERManI activity; this could lead to more extensive trimming beyond Man8GlcNAc2. A second possibility for trimming to smaller glycan structures is the involvement of the Golgi subfamily of enzymes. Localization studies have detected GolgiManIA in the ER and ER-Golgi intermediate compartment (IC) in addition to the Golgi [34]. Evidence has also been presented that recycling of misfolded proteins between the ER, IC and Golgi is a prerequisite for ERAD [35–40]. A third possibility postulates α-mannosidase activity for the third subfamily of GH47 enzymes, the EDEM proteins. Initial studies on yeast EDEM (Htm1p/Mnl1p) [41,42] and two of the three mammalian orthologs (EDEM1 and EDEM2) [27••,28••,43–45] failed to reveal hydrolytic activity, despite the conservation of the catalytic residues employed by ERManI and the Golgi mannosidases (Figure 3). Recent data indicate that overexpression of EDEM1 (S Olivari and M Molinari, unpublished) or EDEM3 [46•] enhances mannose removal from the N-glycans of ERAD candidates, suggesting a potential role for this family of proteins in glycan trimming during ERAD.
Glycan recognition during ERAD
The hypothesis that mannose trimming plays a timing role in the ERAD process necessitates the decoding of the glycan signal by components of the disposal machinery. The main candidate lectins for this recognition role are the EDEM proteins, based on numerous criteria [27••,28••,41–45,46•]. Because of their extensive sequence homology to the processing mannosidases (Figure 3), the EDEMs are proposed to have a glycan-binding cleft capable of recognizing high-mannose intermediates [29••]. Both ERManI and the Golgi mannosidases have considerable affinity for their Man5GlcNAc2 enzymatic products [30,32••], and a direct role for ERManI in glycan recognition during ERAD in S. pombe was also proposed [47]. Although the mode of action of the EDEMs is still unclear, their roles during ERAD have been demonstrated by protein overexpression, which accelerates the disposal of misfolded ERAD substrates [27••,28••,43–45,46•], and by a reduction in expression through gene disruption [41] or RNAi knockdown [43], which prolongs the ER residence of disposal substrates. Direct interactions with misfolded glycoproteins have been demonstrated by co-immunoprecipitation [27••,28••,43–45,46•] and transfer of ERAD substrates from CNX to EDEMs has been shown [43,44]. Mammalian EDEMs are soluble proteins in the ER lumen [28••]. EDEM1 has been found in association with membrane-spanning Derlin-2 and -3, putative retrograde transport pores. Derlin-2 and -3 are associated with the cytosolic ATPase p97, which is also involved in dislocation of ERAD substrates [48•]. These data are consistent with a possible role for EDEMs in shuttling ERAD candidates from the ER folding environment to the site of dislocation into the cytosol for disposal. Despite the established roles of the EDEMs in the ERAD process and data showing that EDEM1 binds both the N-glycan and polypeptide moieties of misfolded proteins [43], the specificity of interactions between EDEMs and the glycans of ERAD substrates has not yet been described.
Recent studies have also implicated an additional lectin in ERAD-related recognition events in S. cerevisiae. Three research groups simultaneously published the characterization of the yeast protein Yos9p, a yeast ortholog of a gene amplified in human osteosarcomas and a sequence homolog of the mannose-6-phosphate receptor, and demonstrated its role in misfolded glycoprotein recognition during ERAD [49•–51•]. Similar to EDEMs, Yos9p is required for efficient misfolded glycoprotein disposal; direct interaction with ERAD substrates was indicated by co-immunoprecipitation. Association of Yos9p and misfolded glycoproteins required Man8GlcNAc2 or Man5-GlcNAc2 glycan structures, but not other processing intermediates. Selective recognition of specific glycosylation sites on ERAD substrates was indicated. The studies disagreed on the involvement of peptide determinants in the recognition of misfolded ERAD substrates by Yos9p, and on the sequential or parallel nature of the interactions between Yos9p and EDEMs with ERAD substrates. However, the studies clearly demonstrated that this novel lectin contributed to misfolded glycoprotein disposal.
Conclusions
The processing and recognition of N-glycan structures play critical roles in the two opposing functions of quality control in the ER. On one side, the processed glycans recruit the chaperone functions of lectin–oxidoreductase complexes to aid in the formation of fully folded and disulfide-bonded tertiary and quaternary structures. A bifunctional lectin–glucosidase complex plays a role in the creation of the glycan ligand for chaperone recognition. The other side of quality control is the recognition and disposal of terminally misfolded proteins that would otherwise accumulate and impair ER function. The kinetics of folding of a given protein in the ER determines its fate, with some proteins folding efficiently and others folding inefficiently even with chaperone assistance. In the latter case, competition between folding and disposal is mediated by a timing function in the lumen of the ER. A simple model of glycan-mediated timing of disposal is present in S. cerevisiae, whereby a single mannose cleavage step, presumably in the context of a misfolded polypeptide, results in recognition by the putative ER lectins EDEM (Htm1p) and Yos9p, and eventual targeting for dislocation into the cytosol. In mammalian cells, more extensive glycan processing has been detected on ERAD intermediates, calling into question the enzymes responsible for the trimming reactions and the lectins involved in their recognition. ERManI and EDEMs play rate-limiting roles in ERAD in animal cells, but the precise glycan structures recognized and the potential roles of mammalian Yos9 homologs remain to be resolved. Interactions between EDEMs and putative retrotranslocation pore components suggest a direct role in dislocation, but the mechanism of recognition and transfer to the pore also remains to be resolved.
Acknowledgments
KWM is supported by US Public Health Service grants GM047533 and RR005351. MM is supported by grants from the Max Cloetta Foundation, Foundation for Research on Neurodegenerative Diseases, Swiss National Center of Competence in Research on Neural Plasticity and Repair, Swiss National Science Foundation, Telethon, Silvio Leoni Foundation, Synapsis and Bangerter-Rhyner Foundation.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 2.Ellgaard L, Molinari M, Helenius A. Setting the standards: quality control in the secretory pathway. Science. 1999;286:1882–1888. doi: 10.1126/science.286.5446.1882. [DOI] [PubMed] [Google Scholar]
- 3.Hebert DN, Garman SC, Molinari M. The glycan code of the endoplasmic reticulum: asparagine-linked carbohydrates as protein maturation and quality-control tags. Trends Cell Biol. 2005;15:364–370. doi: 10.1016/j.tcb.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 4.Lederkremer GZ, Glickman MH. A window of opportunity: timing protein degradation by trimming of sugars and ubiquitins. Trends Biochem Sci. 2005;30:297–303. doi: 10.1016/j.tibs.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 5••.Deprez P, Gautschi M, Helenius A. More than one glycan is needed for ER glucosidase II to allow entry of glycoproteins into the calnexin/calreticulin cycle. Mol Cell. 2005;19:183–195. doi: 10.1016/j.molcel.2005.05.029. This paper provides interesting new insights into substrate access to the CNX chaperone system. Access requires the rapid and coordinated intervention of two distinct glucose-removing enzymes: GI, which acts immediately after N-glycan addition to nascent chains; and GII, for which association with one N-glycan promotes deglucosylation of a second glycan displayed by the same polypeptide chain (trans-activation) [DOI] [PubMed] [Google Scholar]
- 6.Rodan AR, Simons JF, Trombetta ES, Helenius A. N-linked oligosaccharides are necessary and sufficient for association of glycosylated forms of bovine RNase with calnexin and calreticulin. EMBO J. 1996;15:6921–6930. [PMC free article] [PubMed] [Google Scholar]
- 7.Wang N, Daniels R, Hebert DN. The cotranslational maturation of the type I membrane glycoprotein tyrosinase: the heat shock protein 70 system hands off to the lectin-based chaperone system. Mol Biol Cell. 2005;16:3740–3752. doi: 10.1091/mbc.E05-05-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Molinari M, Eriksson KK, Calanca V, Galli C, Cresswell P, Michalak M, Helenius A. Contrasting functions of calreticulin and calnexin in glycoprotein folding and ER quality control. Mol Cell. 2004;13:125–135. doi: 10.1016/s1097-2765(03)00494-5. [DOI] [PubMed] [Google Scholar]
- 9.Pieren M, Galli C, Denzel A, Molinari M. The use of calnexin and calreticulin by cellular and viral glycoproteins. J Biol Chem. 2005;280:28265–28271. doi: 10.1074/jbc.M501020200. [DOI] [PubMed] [Google Scholar]
- 10.Solda T, Garbi N, Hammerling GJ, Molinari M. Consequences of ERp57 deletion on oxidative folding of obligate and facultative clients of the calnexin cycle. J Biol Chem. 2006;281:6219–6226. doi: 10.1074/jbc.M513595200. [DOI] [PubMed] [Google Scholar]
- 11••.Molinari M, Galli C, Vanoni O, Arnold SM, Kaufman RJ. Persistent glycoprotein misfolding activates the glucosidase II/UGT1-driven calnexin cycle to delay aggregation and loss of folding competence. Mol Cell. 2005;20:503–512. doi: 10.1016/j.molcel.2005.09.027. This is the first study of protein folding and quality control in cells lacking the ER folding sensor UGT1. UGT1 deletion does not affect the tightness of ER quality control. It rather accelerates the transfer of terminally misfolded glycopolypeptides from a first (the CNX cycle) to a second (the BiP chaperone system) level of ER retention. [DOI] [PubMed] [Google Scholar]
- 12.Fanchiotti S, Fernandez F, D’Alessio C, Parodi AJ. The UDP-Glc:Glycoprotein glucosyltransferase is essential for Schizosaccharomyces pombe viability under conditions of extreme endoplasmic reticulum stress. J Cell Biol. 1998;143:625–635. doi: 10.1083/jcb.143.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–774. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- 14.Vabulas RM, Hartl FU. Protein synthesis upon acute nutrient restriction relies on proteasome function. Science. 2005;310:1960–1963. doi: 10.1126/science.1121925. [DOI] [PubMed] [Google Scholar]
- 15.Wu Y, Swulius MT, Moremen KW, Sifers RN. Elucidation of the molecular logic by which misfolded alpha 1-antitrypsin is preferentially selected for degradation. Proc Natl Acad Sci USA. 2003;100:8229–8234. doi: 10.1073/pnas.1430537100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernier V, Lagace M, Bichet DG, Bouvier M. Pharmacological chaperones: potential treatment for conformational diseases. Trends Endocrinol Metab. 2004;15:222–228. doi: 10.1016/j.tem.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 17.Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol. 2005;7:766–772. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- 18.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 19.Eriksson KK, Vago R, Calanca V, Galli C, Paganetti P, Molinari M. EDEM contributes to maintenance of protein folding efficiency and secretory capacity. J Biol Chem. 2004;279:44600–44605. doi: 10.1074/jbc.M407972200. [DOI] [PubMed] [Google Scholar]
- 20.Jakob CA, Burda P, Roth J, Aebi M. Degradation of misfolded endoplasmic reticulum glycoproteins in Saccharomyces cerevisiae is determined by a specific oligosaccharide structure. J Cell Biol. 1998;142:1223–1233. doi: 10.1083/jcb.142.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su K, Stoller T, Rocco J, Zemsky J, Green R. Pre-Golgi degradation of yeast prepro-alpha-factor expressed in a mammalian cell. Influence of cell type-specific oligosaccharide processing on intracellular fate. J Biol Chem. 1993;268:14301–14309. [PubMed] [Google Scholar]
- 22.Cabral CM, Liu Y, Sifers RN. Dissecting glycoprotein quality control in the secretory pathway. Trends Biochem Sci. 2001;26:619–624. doi: 10.1016/s0968-0004(01)01942-9. [DOI] [PubMed] [Google Scholar]
- 23.Moremen K. α-Mannosidases in asparagine-linked oligosaccharide processing and catabolism. In: Ernst B, Hart G, Sinay P, editors. Oligosaccharides in Chemistry and Biology: A Comprehensive Handbook. part 1. II. Wiley and Sons, Inc; 2000. pp. 81–117. [Google Scholar]
- 24.Ermonval M, Kitzmuller C, Mir AM, Cacan R, Ivessa NE. N-glycan structure of a short-lived variant of ribophorin I expressed in the MadIA214 glycosylation-defective cell line reveals the role of a mannosidase that is not ER mannosidase I in the process of glycoprotein degradation. Glycobiology. 2001;11:565–576. doi: 10.1093/glycob/11.7.565. [DOI] [PubMed] [Google Scholar]
- 25.Foulquier F, Harduin-Lepers A, Duvet S, Marchal I, Mir AM, Delannoy P, Chirat F, Cacan R. The unfolded protein response in a dolichyl phosphate mannose-deficient Chinese hamster ovary cell line points out the key role of a demannosylation step in the quality-control mechanism of N-glycoproteins. Biochem J. 2002;362:491–498. doi: 10.1042/0264-6021:3620491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henrissat B, Davies G. Structural and sequence-based classification of glycoside hydrolases. Curr Opin Struct Biol. 1997;7:637–644. doi: 10.1016/s0959-440x(97)80072-3. [DOI] [PubMed] [Google Scholar]
- 27••.Mast SW, Diekman K, Karaveg K, Davis A, Sifers RN, Moremen KW. Human EDEM2, a novel homolog of family 47 glycosidases, is involved in ER-associated degradation of glycoproteins. Glycobiology. 2005;15:421–436. doi: 10.1093/glycob/cwi014. This paper, together with those by Olivari et al. [28••] and Hirao et al. [46•], reports the identification and characterization of EDEM2 and EDEM3, two EDEM homologues involved in the regulation of terminally misfolded glycoprotein disposal. [DOI] [PubMed] [Google Scholar]
- 28••.Olivari S, Galli C, Alanen H, Ruddock L, Molinari M. A novel stress-induced EDEM variant regulating endoplasmic reticulum-associated glycoprotein degradation. J Biol Chem. 2005;280:2424–2428. doi: 10.1074/jbc.C400534200. This paper reports the identification and characterization of EDEM2 and EDEM3, and describes their induction upon ER stress. It was also found that EDEM, previously described as a membrane-bound protein, is actually a soluble protein in the ER lumen in most cell lines. [DOI] [PubMed] [Google Scholar]
- 29••.Karaveg K, Siriwardena A, Tempel W, Liu ZJ, Glushka J, Wang BC, Moremen KW. Mechanism of class 1 (glycosylhydrolase family 47) α-mannosidases involved in N-glycan processing and endoplasmic reticulum quality control. J Biol Chem. 2005;280:16197–16207. doi: 10.1074/jbc.M500119200. This paper examines the novel glycosidase mechanism of human ERManI, involving a unique glycone conformational distortion, an unusual general acid function and a role for an enzyme-bound Ca2+ ion in catalysis. The study provides a framework for understanding catalysis by the true hydrolases in this enzyme family and the structural basis of the compromised catalysis of the EDEM proteins. [DOI] [PubMed] [Google Scholar]
- 30.Tempel W, Karaveg K, Liu ZJ, Rose J, Wang BC, Moremen KW. Structure of mouse Golgi α-mannosidase IA reveals the molecular basis for substrate specificity among class 1 (family 47 glycosylhydrolase) α1,2-mannosidases. J Biol Chem. 2004;279:29774–29786. doi: 10.1074/jbc.M403065200. [DOI] [PubMed] [Google Scholar]
- 31.Vallee F, Karaveg K, Herscovics A, Moremen KW, Howell PL. Structural basis for catalysis and inhibition of N-glycan processing class I α1,2-mannosidases. J Biol Chem. 2000;275:41287–41298. doi: 10.1074/jbc.M006927200. [DOI] [PubMed] [Google Scholar]
- 32••.Karaveg K, Moremen KW. Energetics of substrate binding and catalysis by class 1 (glycosylhydrolase family 47) α-mannosidases involved in N-glycan processing and endoplasmic reticulum quality control. J Biol Chem. 2005;280:29837–29848. doi: 10.1074/jbc.M505130200. This study maps the interactions between human ERManI and high-mannose N-glycan substrates using surface plasmon resonance. The energetic contributions to glycan binding are demonstrated for this family of enzymes. [DOI] [PubMed] [Google Scholar]
- 33.Herscovics A, Romero PA, Tremblay LO. The specificity of the yeast and human class I ER α1,2-mannosidases involved in ER quality control is not as strict previously reported. Glycobiology. 2002;12:14G–15G. [PubMed] [Google Scholar]
- 34.Kamhi-Nesher S, Shenkman M, Tolchinsky S, Fromm SV, Ehrlich R, Lederkremer GZ. A novel quality control compartment derived from the endoplasmic reticulum. Mol Biol Cell. 2001;12:1711–1723. doi: 10.1091/mbc.12.6.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roth J, Brada D, Lackie PM, Schweden J, Bause E. Oligosaccharide trimming Man9-mannosidase is a resident ER protein and exhibits a more restricted and local distribution than glucosidase II. Eur J Cell Biol. 1990;53:131–141. [PubMed] [Google Scholar]
- 36.Caldwell SR, Hill KJ, Cooper AA. Degradation of endoplasmic reticulum (ER) quality control substrates requires transport between the ER and Golgi. J Biol Chem. 2001;276:23296–23303. doi: 10.1074/jbc.M102962200. [DOI] [PubMed] [Google Scholar]
- 37.Hammond C, Helenius A. Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J Cell Biol. 1994;126:41–52. doi: 10.1083/jcb.126.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haynes CM, Caldwell S, Cooper AA. An HRD/DER-independent ER quality control mechanism involves Rsp5p-dependent ubiquitination and ER-Golgi transport. J Cell Biol. 2002;158:91–101. doi: 10.1083/jcb.200201053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taxis C, Vogel F, Wolf DH. ER-golgi traffic is a prerequisite for efficient ER degradation. Mol Biol Cell. 2002;13:1806–1818. doi: 10.1091/mbc.01-08-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vashist S, Kim W, Belden WJ, Spear ED, Barlowe C, Ng DT. Distinct retrieval and retention mechanisms are required for the quality control of endoplasmic reticulum protein folding. J Cell Biol. 2001;155:355–368. doi: 10.1083/jcb.200106123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jakob CA, Bodmer D, Spirig U, Battig P, Marcil A, Dignard D, Bergeron JJ, Thomas DY, Aebi M. Htm1p, a mannosidase-like protein, is involved in glycoprotein degradation in yeast. EMBO Rep. 2001;2:423–430. doi: 10.1093/embo-reports/kve089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakatsukasa K, Nishikawa S, Hosokawa N, Nagata K, Endo T. Mnl1p, an α-mannosidase-like protein in yeast Saccharomyces cerevisiae, is required for endoplasmic reticulum-associated degradation of glycoproteins. J Biol Chem. 2001;276:8635–8638. doi: 10.1074/jbc.C100023200. [DOI] [PubMed] [Google Scholar]
- 43.Molinari M, Calanca V, Galli C, Lucca P, Paganetti P. Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science. 2003;299:1397–1400. doi: 10.1126/science.1079474. [DOI] [PubMed] [Google Scholar]
- 44.Oda Y, Hosokawa N, Wada I, Nagata K. EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science. 2003;299:1394–1397. doi: 10.1126/science.1079181. [DOI] [PubMed] [Google Scholar]
- 45.Hosokawa N, Tremblay LO, You Z, Herscovics A, Wada I, Nagata K. Enhancement of endoplasmic reticulum (ER) degradation of misfolded Null Hong Kong α1-antitrypsin by human ER mannosidase I. J Biol Chem. 2003;278:26287–26294. doi: 10.1074/jbc.M303395200. [DOI] [PubMed] [Google Scholar]
- 46•.Hirao K, Natsuka Y, Tamura T, Wada I, Morito D, Natsuka S, Romero P, Sleno B, Tremblay LO, Herscovics A, et al. EDEM3, a soluble EDEM homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J Biol Chem. 2006;281:9650–9658. doi: 10.1074/jbc.M512191200. This paper reports the identification and characterization of EDEM3 as an active α-mannosidase. [DOI] [PubMed] [Google Scholar]
- 47.Movsichoff F, Castro OA, Parodi AJ. Characterization of Schizosaccharomyces pombe ER α-mannosidase: a reevaluation of the role of the enzyme on ER-associated degradation. Mol Biol Cell. 2005;16:4714–4724. doi: 10.1091/mbc.E05-03-0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48•.Oda Y, Okada T, Yoshida H, Kaufman RJ, Nagata K, Mori K. Derlin-2 and Derlin-3 are regulated by the mammalian unfolded protein response and are required for ER-associated degradation. J Cell Biol. 2006;172:383–393. doi: 10.1083/jcb.200507057. Derlin homologs Derlin-2 and -3 were identified as unfolded protein response (UPR)-responsive gene products that are required for degradation of ERAD substrates in human cells. A direct interaction between both Derlin-2 and -3 and EDEM1 and p97 was detected, linking lumenal glycan binding and cytosolic extraction of ERAD substrates. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49•.Bhamidipati A, Denic V, Quan EM, Weissman JS. Exploration of the topological requirements of ERAD identifies Yos9p as a lectin sensor of misfolded glycoproteins in the ER lumen. Mol Cell. 2005;19:741–751. doi: 10.1016/j.molcel.2005.07.027. This paper, together with those of Kim et al. [50•] and Szathmary et al. [51•], reports the identification and characterization of Yos9p in S. cerevisiae, an ER lectin involved in high-mannose glycan recognition in ERAD. [DOI] [PubMed] [Google Scholar]
- 50•.Kim W, Spear ED, Ng DT. Yos9p detects and targets misfolded glycoproteins for ER-associated degradation. Mol Cell. 2005;19:753–764. doi: 10.1016/j.molcel.2005.08.010. This paper reports the identification and characterization of Yos9p in S. cerevisiae as a lectin involved in the recognition of high-mannose glycans in ERAD. The lectin recognizes lumenal but not membrane-associated ERAD substrates, and targets them for disposal. [DOI] [PubMed] [Google Scholar]
- 51•.Szathmary R, Bielmann R, Nita-Lazar M, Burda P, Jakob CA. Yos9 protein is essential for degradation of misfolded glycoproteins and may function as lectin in ERAD. Mol Cell. 2005;19:765–775. doi: 10.1016/j.molcel.2005.08.015. This paper reports the identification and characterization of Yos9p in S. cerevisiae as a lectin involved in the recognition of high-mannose glycans in ERAD. Interaction with ERAD substrates was dependent on the presence of Man8GlcNAc2 or Man5GlcNAc2 glycan structures. [DOI] [PubMed] [Google Scholar]
- 52.Li G, Zhao G, Zhou X, Schindelin H, Lennarz WJ. The AAA ATPase p97 links peptide N-glycanase to the endoplasmic reticulum-associated E3 ligase autocrine motility factor receptor. Proc Natl Acad Sci USA. 2006;103:8348–8353. doi: 10.1073/pnas.0602747103. [DOI] [PMC free article] [PubMed] [Google Scholar]