

Abstract
Patient: Female, 55
Final Diagnosis: Metastatic malignant pheochromocytoma
Symptoms: Chest pain • tachycardia • tachypnea
Medication: —
Clinical Procedure: —
Specialty: Oncology
Objective:
Rare disease
Background:
Malignant pheochromocytoma is defined as the occurrence of the tumor in an area that is normally devoid of chromaffin tissue, direct tumor invasion, and/or metastasis. Metastatic malignant pheochromocytoma is very rare. Malignant pheochromocytoma carries a poor prognosis with a 5-year survival rate of 44%. The majority of pheochromocytoma cases are sporadic, but a small portion (10%) can be associated with hereditary syndromes such as neurofibromatosis type 1 (NF1).
Case Report:
A 55-year-old Hispanic woman presented to our care with chest pain. Her past medical problems included hypertension, neurofibromatosis type 1 (NF1), and pheochromocytoma status after right laparoscopic adrenalectomy, which was converted to open procedure about 19 months ago. Initial vital signs were significant for tachycardia, tachypnea, and hypertension. Computed tomography (CT) angiography of the chest was performed to rule out a pulmonary embolism, but instead revealed multiple bilateral lung nodules measuring up to 8 mm, consistent with metastasis. CT of the abdomen and pelvis revealed a lytic lesion in the posterior aspect of the left pedicle and transverse process of L4, and the neck/greater trochanter of the left femur. MIBG scan revealed widespread metastatic disease. She received an outpatient chemotherapy regimen of cyclophosphamide, dacarbazine, and vincristine.
Conclusions:
Metaiodobenzylguanidine (MIBG) is an alkyl-guanidine derivative similar to noradrenaline, which accumulates in tissue derived from neural crest cells. Current medical literature suggests that therapeutic 131I MIBG has a response rate of 50–75%. Combination chemotherapy using cyclophosphamide, vincristine, and dacarbazine has been the most widely used regimen for malignant pheochromocytoma. This combination of chemotherapy has been shown to have a high response rate and symptomatic improvement. Numerous therapeutic regimens exist for metastatic malignant pheochromocytoma; however, no regimen has been shown to have a benefit significantly superior to the others.
MeSH Keywords: Neurofibromatosis 1; Pheochromocytoma; Drug Therapy, Combination
Background
Pheochromocytomas arise from chromaffin cells of the sympathetic nervous system (SNS). The incidence of pheochromocytoma is 0.001 in the general population (1). Pheochromocytomas most commonly arise from the adrenal medulla and 10% arise from an extra-adrenal site (paraganglioma). The incidence of malignant pheochromocytoma ranges from 2.5% to 13% [1]. Malignant pheochromocytoma is defined as the occurrence of the tumor in an area that is normally devoid of chromaffin tissue, direct tumor invasion, and/or metastasis. Metastasis frequently occurs in the lymph nodes, bone, liver, and lungs. Malignant pheochromocytoma carries a poor prognosis with a 5-year survival rate of 44% [2].
Three clinical predictors of metastasis have been established: the size of the primary tumor, its location, and germline mutations of the SDHB gene [3]. Pheochromocytomas that are larger than 5 cm are associated with an increased risk of metastasis and decreased survival [3]. Succinate dehydrogenase (SDHB) gene mutation is currently the most important contributor to hereditary malignant pheochromocytomas.
The classical clinical manifestations include palpitations, headaches, hypertension, sweating, lethargy, pallor, depression, and anxiety. The diagnosis is made by measuring metanephrines in the urine and plasma. No specific biochemical marker is available that can identify malignant pheochromocytoma. The most common causes of death in patients with metastatic pheochromocytoma are cardiac dysfunction, hypertensive crisis, and excessive tumor burden [3]. Metastatic pheochromocytomas have high morbidity and mortality rates due to excessive secretion of catecholamines, which predispose patients to cardiovascular and other endocrine complications. The current medical literature lacks reliable morphological or histological criteria; therefore the diagnosis of malignancy is based on the clinical behavior of the tumor [4].
Most cases pheochromocytoma are sporadic, but a small portion (10%) can be associated with hereditary syndromes, including von Hippel-Lindau (VHL), multiple endocrine neoplasia (MEN) type 2A or 2B, and neurofibromatosis (NF) type 1. Genetic testing is highly recommended, given the importance of hereditary factors in the development of pheochromocytoma. NF1 patients are more likely to develop tumors like gastrointestinal stromal tumor, paraganglioma, pheochromocytoma, and pancreatic neuroendocrine tumor [5].
Neurofibromatosis type 1 (NF1) is a genetic disorder characterized by the presence of multiple soft-tissue tumors of neural crest origin, café’-au-lait spots, and Lisch nodules [6]. NF-1 affects 1 in 3500 individuals worldwide. It has autosomal dominant inheritance and complete penetrance [7]. The NF-1 gene is a tumor suppressor gene located on chromosome 17q11.2 [7]. Pheochromocytoma occurs in 0.1–5.7% of patients with NF-1 [7]. Pheochromocytomas associated with NF-1 are commonly found in women in their forties. The majority of these NF-1 associated pheochromocytomas will be benign and 11% will be malignant [8]. Herein, we present a case of a patient with NF-1 and metastatic malignant pheochromocytoma treated with combination chemotherapy.
Case Report
A 55-year-old Hispanic woman presented to our care with chest pain of 6-h duration, described as being pressure-like, with severe intensity, and located at the substernal area with radiation to the left shoulder and back. Other complaints included nausea, vomiting, shortness of breath, and generalized weakness. She denied any palpitations, headache, sweating, or dizziness. Her past medical problems included hypertension, neurofibromatosis type 1 (NF1), and pheochromocytoma, status after right laparoscopic adrenalectomy that was converted to open procedure about 19 months ago after rupture of the capsule surrounding the tumor. Surgical pathology investigation reported a pheochromocytoma confined by a fibrous pseudocapsule without capsular invasion and extensive necrosis. After the surgery, she failed to follow-up with the endocrinologist as an outpatient.
Initial vital signs were significant for tachycardia (heart rate [HR]: 110), tachypnea (respiratory rate [RR)]: 22–26 breaths per minute), and hypertension (blood pressure [BP]: 224/116 mmHg). Pertinent physical examination findings included point tenderness on the left 4th rib, multiple neurofibromas, and café au lait spots. She also had a significant limited range of motion at the left hip joint. There were no other significant physical examination findings. Upon admission, she was started on a nicardipine drip for BP control and was changed to oral BP medications the following day. Hypertension was eventually controlled on the 5th hospital day on prazosin 2 mg 3 times a day and propranolol 20 mg 3 times a day. Initial laboratory workup (Table 1) on admission was only significant for leukocytosis, hemoconcentration, hyperglycemia, and a slightly elevated AST. The other laboratory findings were normal.
Table 1.
Initial laboratory work-up.
| White blood cell count | 17.14×103 uL (4.5–11.0×103/UL) |
| Hemoglobin | 15.8 g/dL (12.0–15.0 g/dL) |
| Hematocrit | 44.4% (36–47%) |
| Platelet count | 427×103/uL (150–450×103/UL |
| Sodium | 136 mmol/L (135–145 mmol/L) |
| Potassium | 4.1 mmol/L (3.5–5.1 mmol/L) |
| Chloride | 103 mmol/L (98–107 mmol/L) |
| CO2 | 19 mmol/L (21–32 mmol/L) |
| Serum glucose | 184 mg/dL (70–100 mg/dL) |
| BUN | 19 mg/dL (7–22 mg/dL) |
| Creatinine | 0.66 mg/dL (0.60–1.30 mg/dL) |
| Calcium | 10.0 mmol/L (8.5–10.1 mmol/L) |
| Albumin | 4.4 g/dL (3.4–5.0 g/dL) |
| Protein | 8.5 g/dL (8.5–10.1 mmol/L) |
| AST | 42 IU/L (15–37 IU/L) |
| ALT | 22 IU/L (12–78 IU/L) |
| Alk. phosphatase | 127 IU/L (50–136 IU/L) |
Computed tomography (CT) angiography of the chest was performed to rule out a pulmonary embolism, but instead revealed multiple bilateral lung nodules measuring up to 8 mm, consistent with metastasis. Also noted was a destructive lesion of the posterior aspect of the left 4th rib. Pelvic radiograph revealed a lytic expansile lesion with a focal cortical break in the left greater trochanter and left femoral neck. CT of the abdomen (Figure 1) and pelvis revealed a lytic lesion in the posterior aspect of the left pedicle and transverse process of L4, and the neck/greater trochanter of the left femur. Urine metanephrines was 711 mcg/g creat (21–153 mcg/g creat), normetanephrine was 12782 mcg/g creat (108–524 mcg/g creat). Urine 5-HIAA was 0.6 mg 24-h creatinine. Genetic analysis demonstrated a mutation of the succinate dehydrogenase (SDHB) gene. Metaiodobenzylguanidine (MIBG) scan (Figure 2) revealed widespread metastatic disease. On the 12th hospital day, she had an angiogram, which revealed embolization of the left circumflex artery, was immediately followed by a cephalomedullary fixation of the left femur with a trochanteric femoral nail. After many discussions with oncology specialists within our facility and MD Anderson, we eventually opted to proceed with outpatient chemotherapy. She had no complications during the postoperative period and was eventually discharged on the 18th hospital day.
Figure 1.
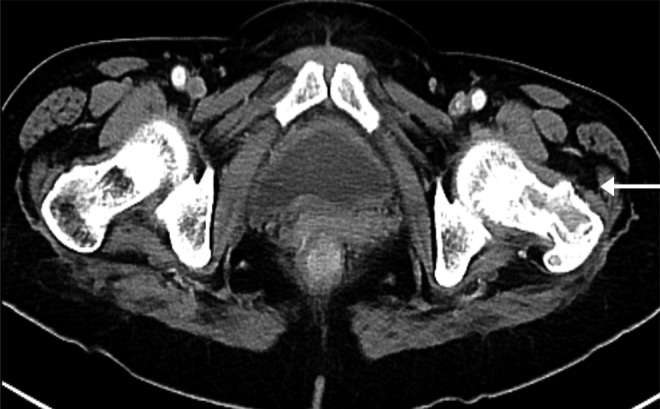
CT abdomen and pelvis: lytic lesion in the neck/greater trochanter of the left femur (white arrow).
Figure 2.

MIBG scan (before chemotherapy): widespread metastatic disease. F – front, B – back,
As an outpatient she received her first dose of chemotherapy (cyclophosphamide, dacarbazine, and vincristine) 1 month afterwards. Outpatient labs included plasma metanephrine 294 pg/mL (<57 pg/mL), normetanephrine 3512 pg/mL (<148 pg/mL), epinephrine 177 pg/mL, and norepinephrine 1657 pg/mL. The urine metanephrine was 2765 mcg/24 h (90–315 mcg/24 h) and normetanephrine was 9295 mcg/24 h (122–676 mcg/24 h). Follow-up MIBG scan (Figure 3) 3 months after admission (3 cycles of chemotherapy) showed interval progression of the metastatic disease with recurrence in the right adrenal surgical bed, a new lesion in the left shoulder, progression to the left posterior 4th rib, and thoracolumbar and pelvic metastasis. At the time of the MIBG scan she had received 3 cycles of chemotherapy with no significant improvement of metastatic disease.
Figure 3.
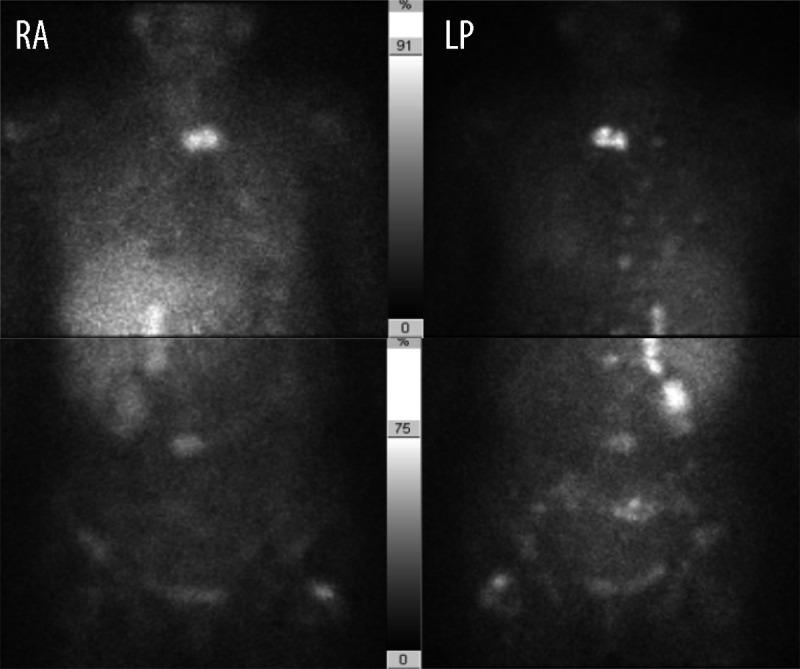
MIBG scan (post-chemotherapy): interval progression of the metastatic disease (L – left, R – eight, A – anterior, P – posterior).
Discussion
Magnetic resonance imaging (MRI) or computed tomography (CT) is recommended to evaluate metastasis to the thoracic, abdominal, and pelvic cavities. If metastasis cannot be detected through CT or MRI, then MIBG or 18F-fluorodeoxyglucose positron emission tomography (FDG-PET) scans could be utilized [9]. Bone metastases are very common and thus a full-body bone scan could help rule out skeletal metastases. Metaiodobenzylguanidine (MIBG) is an alkyl-guanidine derivative similar to noradrenaline, which accumulates in tissue derived from neural crest cells. MIBG scan with radio-labeled iodine (I), either 123I or 131I, can detect pheochromocytomas. MIBG scan aids in the diagnosis and follow-up of pheochromocytomas. It has a sensitivity of 77–100% and specificity of 95–100% [10]. MIBG is able to visualize functioning pheochromocytomas >0.2 grams.
Initially, the treatment of malignant pheochromocytoma should be focused on symptomatic control of the elevated catecholamine state through alpha and beta adrenergic blockade. The mainstay treatment of localized non-malignant pheochromocytoma is surgery. However, no effective treatment currently exists for metastatic malignant pheochromocytomas [11]. The initial therapeutic approach (if reasonable) for malignant pheochromocytoma is surgical excision (if in an appropriate location), or a debulking procedure. Surgical resection of the primary tumor or metastasis can decrease the hormonal effects of these tumors. No data currently exists that demonstrate improved survival or reduction of symptoms after surgical debulking procedure.
Radiotherapy is considered for control of inoperable tumors and palliation of painful osseous metastases [12]. Radiation has not shown much effect on tumor growth or hormone production, but chemotherapy has had some promising reported success [12]. Current medical literature suggests that therapeutic 131I MIBG has a response rate of 50–75% [13]. Radio-labeled 131I MIBG administered in much higher quantities and activity than for diagnostic imaging. MIBG scan will be positive in up to 60% of malignant pheochromocytomas [14].
Combination chemotherapy using cyclophosphamide, vincristine, and dacarbazine has been the most widely used regimen for malignant pheochromocytoma. This combination of chemotherapy has been shown to have a high response rate and symptomatic improvement [15]. It has been shown to be well tolerated and should be reserved for patients with metastasis that is not medically controlled and is unresectable. Patients treated with chemotherapy should have frequent follow-up visits that include reassessment of biochemical and imaging studies. Sunitinib is an oral receptor tyrosine kinase inhibitor with anti-angiogenic and anti-tumor activity that targets the PDGF, VEGF, c-KIT, and FLT3 receptors [16]. Sunitinib inhibits catecholamine synthesis and secretion in pheochromocytoma tumor cells by blocking VEGF receptor 2 via the PLC-γ pathway [16]. Sunitinib may prove useful in the treatment of malignant pheochromocytoma in the future [17].
Conclusions
It is important for the clinician to consider metastatic pheochromocytoma, especially in patients with a previous history of pheochromocytoma and a hereditary syndrome such as neurofibromatosis. Our patient had neurofibromatosis and tested positive for the SDHB gene mutation. Numerous therapeutic regimens exist for metastatic malignant pheochromocytoma; however, no regimen has been shown to have a benefit significantly superior to the others. Unfortunately, MIBG therapy was not the first-line therapy due to the limited financial resources of the patient. Future research investigations are highly recommended to establish better treatment options for this debilitating and fulminant disease. Our case demonstrates a rare disease of metastatic malignant pheochromocytoma associated with neurofibromatosis and its lack of response to chemotherapy.
Footnotes
Disclosures
All participating authors in this study declare no financial, professional, or personal conflicts.
No grant support was received for this case report.
All authors were involved in manuscript preparation and literature review.
References:
- 1.Yurt A, Arda MN, Vardar E. Metastatic pheochromocytoma of the thoracic spinal extradural space. Case report and review of the literature. Kobe J Med Sci. 2005;51(4):49–53. [PubMed] [Google Scholar]
- 2.Safford SD, Coleman RE, Gockerman JP, et al. Iodine-131 metaiodobenzylguanidine is an effective treatment for malignant pheochromocytotoma and paraganglioma. Surgery. 2003;134(6):956–63. doi: 10.1016/s0039-6060(03)00426-4. [DOI] [PubMed] [Google Scholar]
- 3.Jimenez C, Rohren E, Habra MA, et al. Current and future treatments for malignant pheochromocytoma and sympathetic paraganglioma. Cur Oncol Rep. 2013;15(4):356–71. doi: 10.1007/s11912-013-0320-x. [DOI] [PubMed] [Google Scholar]
- 4.Huang H, Abraham J, Hung E, et al. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: recommendation from a 22-year follow-up of 18 patients. Cancer. 2008;113(8):2020–28. doi: 10.1002/cncr.23812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hari Kumar KV, Shaikh A, Sandhu AS, Prusty P. Neurofibromatosis 1 with pheochromocytoma. Indian J Endocrinol Metab. 2011;15:S406–8. doi: 10.4103/2230-8210.86988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zinnamosca L, Petramala L, Cotesta D, et al. Neurofibromatosis type 1 (NF1) and pheochromocytoma: prevalence, clinical and cardiovascular aspects. Archi Dermatol Res. 2011;303(5):317–25. doi: 10.1007/s00403-010-1090-z. [DOI] [PubMed] [Google Scholar]
- 7.Zografos GN, Vasiliadis GK, Zagouri F, et al. Pheochromocytoma associated with neurofibromatosis type 1: concepts and current trends. World J Surg Oncol. 2010;8:14. doi: 10.1186/1477-7819-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lew JI, Jacome FJ, Solorzano CC. Neurofibromatosis-associated pheochromocytoma. J Am Coll Surg. 2006;202(3):550–51. doi: 10.1016/j.jamcollsurg.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 9.Szalat A, Fraenkel M, Doviner V, et al. Malignant pheochromocytoma: predictive factors of malignancy and clinical course in 16 patients at a single tertiary medical center. Endocrine. 2011;39(2):160–66. doi: 10.1007/s12020-010-9422-5. [DOI] [PubMed] [Google Scholar]
- 10.Wan WH, Tan KY, Ng C, et al. Metastatic malignant phaeochromocytoma: a rare entity that underlies a therapeutic quandary. Asian J Surg. 2006;29(4):294–302. doi: 10.1016/S1015-9584(09)60106-8. [DOI] [PubMed] [Google Scholar]
- 11.Zarnegar R, Kebebew E, Duh QY, Clark OH. Malignant Pheochromocytoma. Surg Oncol Clin N Am. 2006;15(3):555–71. doi: 10.1016/j.soc.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 12.Chrisoulidou A, Kaltsas G, Ilias I, Grossman AB. The diagnosis and management of malignant phaeochromocytoma and paraganglioma. Endocr Relat Cancer. 2007;14(3):569–85. doi: 10.1677/ERC-07-0074. [DOI] [PubMed] [Google Scholar]
- 13.de Wailly P, Oragano L, Rade F, et al. Malignant pheochromocytoma; new malignancy criteria. Langenbecks Arch Surg. 2012;397(2):239–46. doi: 10.1007/s00423-011-0850-3. [DOI] [PubMed] [Google Scholar]
- 14.Adjalle R, Plouin PF, Pacak K, Lehnert H. Treatment of malignant pheochromocytoma. Horm Metab Res. 2009;41(9):687–96. doi: 10.1055/s-0029-1231025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nomura K, Kimura H, Shimizu S, et al. Survivial of patients with metastatic malignant pheochromocytoma and efficacy of combined cyclophosphamide, vincristine, and dacarbazine chemotherapy. J Clin Endocrinol Metab. 2009;94(8):2850–56. doi: 10.1210/jc.2008-2697. [DOI] [PubMed] [Google Scholar]
- 16.Aita Y, Ishii KA, Saito Y, et al. Sunitinib inhibits catecholamine synthesis and secretion in pheochomocytoma tumor cells by blocking VEGF receptor 2 via PLC-γ-related Pathways. Am J Physiol Endocrinol Metab. 2012;303(8):E1006–14. doi: 10.1152/ajpendo.00156.2012. [DOI] [PubMed] [Google Scholar]
- 17.Gonias S, Goldsby R, Matthay KK, et al. Phase II study of high-dose [131I] metaiodobenzylguanidine therapy for patients with metastatic pheochromocytoma and paraganglioma. J Clin Oncol. 2009;27(25):4162–68. doi: 10.1200/JCO.2008.21.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]