

Abstract
Prader–Willi syndrome (PWS) and Angelman syndrome (AS) are two neurodevelopmental disorders most often caused by deletions of the same region of paternally inherited and maternally inherited human chromosome 15q, respectively. AS is a single gene disorder, caused by the loss of function of the ubiquitin ligase E3A (UBE3A) gene, while PWS is still considered a contiguous gene disorder. Rare individuals with PWS who carry atypical microdeletions on chromosome 15q have narrowed the critical region for this disorder to a 108 kb region that includes the SNORD116 snoRNA cluster and the Imprinted in Prader–Willi (IPW) non-coding RNA. Here we report the derivation of induced pluripotent stem cells (iPSCs) from a PWS patient with an atypical microdeletion that spans the PWS critical region. We show that these iPSCs express brain-specific portions of the transcripts driven by the PWS imprinting center, including the UBE3A antisense transcript (UBE3A-ATS). Furthermore, UBE3A expression is imprinted in most of these iPSCs. These data suggest that UBE3A imprinting in neurons only requires UBE3A-ATS expression, and no other neuron-specific factors. These data also suggest that a boundary element lying within the PWS critical region prevents UBE3A-ATS expression in non-neural tissues.
INTRODUCTION
Angelman syndrome (AS) and Prader–Willi syndrome (PWS) are neurodevelopmental disorders that are most commonly caused by large deletions of human chromosome 15q11–q13 (1). Individuals with AS suffer from cognitive impairment, absent speech, seizures, ataxic gait and a happy demeanor, while individuals with PWS initially present with hypotonia and failure-to-thrive (2). PWS individuals then go through a phase of normal growth before progressing to a period of above-normal weight gain, and finally, a period of intense food seeking behavior and hyperphagia (3). PWS leads to morbid obesity without diet, exercise and often growth hormone intervention. Individuals with PWS also suffer mild cognitive impairment, short stature and behavioral issues including obsessive-compulsive disorder (4). While the two disorders are very different from one another, they can result from exactly the same deletion, but the deletion differs in its parent-of-origin. AS is caused by deletions of maternal chromosome 15q11–q13 and PWS is caused by the same deletion that occurs on the paternally inherited allele (1).
The parent-of-origin specific effects of chromosome 15q11–q13 deletions occur due to the regulation of the locus by genomic imprinting. A map of this region is shown in Figure 1. The imprinting control region is a differentially methylated region encompassing the promoter and first exon of SNURF-SNRPN (5,6). It is methylated on the paternal allele and unmethylated on the maternal allele. Several genes are expressed exclusively from the paternally inherited allele, including MKRN3 (7), MAGEL2 (8), NDN (9), NPAP1 (10), SNURF-SNRPN (11,12), SNORD107 (13), SNORD64 (14), SNORD108 (13), SNORD109A (13), SNORD116 (14), IPW (15), SNORD115 (14), SNORD109B (13) and UBE3A-ATS (16). Several paternally expressed genes, including the SNORD genes, IPW and UBE3A-ATS are non-coding RNAs (ncRNAs) driven from the SNURF-SNRPN promoter (13). In humans, SNORD107, SNORD64, SNORD108, SNORD109A, SNORD116 and IPW are expressed broadly across several tissues, but SNORD115, SNORD109B and UBE3A-ATS are expressed exclusively in neurons, concomitant with the use of upstream exons of the SNURF-SNRPN gene (17). Only one imprinted gene is expressed exclusively from the maternally inherited allele: UBE3A (18). This gene is expressed from both parental alleles in most tissues, but is expressed exclusively from the maternally inherited allele in neurons. The tissue-specific imprinted expression of UBE3A occurs to due to the tissue-specific expression of UBE3A-ATS from the paternal allele (16,17,19). Mutations of UBE3A are sufficient to cause AS (20).
Figure 1.
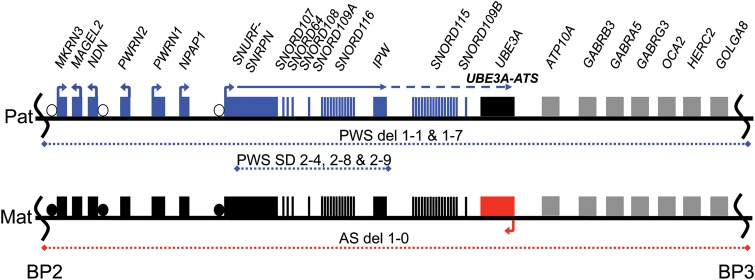
Map of chromosome 15q11–q13 region. Map of chromosome 15q11–q13 between common break points 2 and 3 (BP2 and BP3). Blue dotted lines represent the regions deleted on the paternal allele and red dotted line represents the regions deleted on the maternal allele for the indicated cell lines. Blue and red boxes denote genes expressed exclusively from paternal and maternal alleles, respectively. Gray boxes denote genes expressed biallelically. Differential methylated regions (DMRs) are shown using circles where open and closed circles represent unmethylated and methylated alleles, respectively. Arrows indicate the direction of transcription. A solid blue line represents paternal SNURF/SNRPN transcripts expressed in most cell types, whereas a dashed blue line indicates neuron-specific transcripts, including UBE3A-ATS.
Recently, several individuals with PWS caused by rare, atypical smaller deletions have been reported in the literature (21–23). Together these individuals have delineated a smallest region of deletion overlap that underlies the PWS phenotype. This smallest region of deletion overlap includes the SNORD116 cluster along with other singleton snoRNAs and the ncRNA, IPW. Interestingly, lymphoblastoid lines from two of these individuals were shown to express either SNORD115 or SNORD109B, transcripts that are usually restricted to neurons (21,22). We sought to determine whether non-neural cells from one of these individuals expressed the neuron-specific UBE3A-ATS and showed imprinted UBE3A expression. Here we report the derivation and characterization of induced pluripotent stem cells from an individual with a small atypical deletion spanning the SNORD116 cluster and IPW ncRNAs. We demonstrate that induced pluripotent stem cells (iPSCs) from this individual express UBE3A-ATS and show imprinted UBE3A expression, phenomena that are usually restricted to neurons. We show that this occurs without the expression of the neuron-specific upstream exons of SNURF-SNRPN, and results in a shortened SNURF-SNRPN/UBE3A-ATS ncRNA that is resistant to silencing by the topoisomerase inhibitor, Topotecan. These data suggest that the expression of UBE3A-ATS is sufficient for imprinted UBE3A expression and that a boundary element lying within the smallest region of deletion overlap for the PWS phenotype prevents the expression of UBE3A-ATS in non-neural tissues.
RESULTS
Derivation of iPSCs from PWS small deletion patient
We obtained PWS patient fibroblasts with a 187 kb microdeletion of paternal 15q11–q13 (21), and reprogrammed them into iPSCs using the human polycistronic STEMCCA lentiviral vector encoding OCT4, KLF4, SOX2 and CMYC (24). The boundaries of this deletion are diagrammed in Figure 1. Reprogrammed colonies were initially identified morphologically and were subsequently validated using immunocytochemistry to verify the expression of the pluripotency markers NANOG, SSEA-4, TRA1-60 and TRA1-81 by immunocytochemistry (Fig. 2A). PWS SD iPSCs had normal karyotypes of 46 XY (Supplementary Material, Fig. S1), because the microdeletion cannot be detected by G-banded karyotyping. We confirmed the deletion by quantitative RT–PCR for gene expression analysis in PWS SD iPSCs for genes within the microdeletion, as described in de Smith et al. (21). Expression of SNORD116 and IPW was undetectable in the PWS SD iPSCs, compared with the normal iPSC line NML 1-0, while genes located outside of the deleted region (NDN, ATP10A and GABRB3) were expressed in PWS SD iPSCs (Fig. 2B). IPSC lines derived from a PWS patient harboring a large deletion of paternal 15q11–q13 (PWS del 1-7), as well as from an AS patient with a large deletion of maternal 15q11–q13 (AS del 1-0), were used as controls (17). The boundaries of these deletions are also shown in Figure 1. PWS del 1-7 iPSCs did not express the paternally expressed genes NDN, SNORD116 and IPW as expected, but expressed ATP10A and GABRB3, while AS del 1-0 iPSCs expressed SNORD116 and IPW, as well as NDN, ATP10A and GABRB3 (Fig. 2B). To determine whether the differential methylation that marks the PWS-IC was maintained during reprogramming, the methylation imprint of the PWS-IC was assessed in PWS SD fibroblasts and iPSCs by methylation-sensitive restriction endonuclease quantitative PCR. Only an unmethylated paternal allele was observed in AS del 1-0 iPSCs, while only a methylated maternal allele was observed in PWS del 1-7 iPSCs, as previously described (17). Both a methylated maternal allele and an unmethylated paternal allele were present in all of the PWS SD fibroblasts and iPSC lines (Fig. 2C), with percent methylation ranging from 42 to 50% in most iPSC lines. The PWS SD 2-1 iPSC line appeared to have aberrant methylation of the PWS-IC, and was excluded from further study. The remaining PWS SD iPSC lines had methylation levels approximately equal to the PWS SD fibroblast line. To confirm that the PWS SD iPSC lines were capable of multi-lineage differentiation, the cells were differentiated into embryoid bodies (EBs). After 16 days of spontaneous differentiation, the EBs were collected and analyzed by qRT–PCR for lineage markers for all three germ layers, using a custom-designed TaqMan® scorecard that incorporated lineage markers described in Bock et al. (25). AS del 1-0, PWS del 1-7 and NML 1-0 iPSCs were used as positive controls in these assays. Early lineage markers representing each of the three embryonic germ layers were expressed in the EBs derived from each of the PWS SD iPSCs (Supplementary Material, Fig. S2), demonstrating that they are capable of multi-lineage differentiation.
Figure 2.

Characterization of PWS SD iPSC lines. (A) Immunocytochemistry for the pluripotency markers NANOG, SSEA-4, TRA1-80 and TRA1-81. Cell nuclei were counterstained with DAPI. Scale bar = 100 μm. (B) Expression of genes within 15q11–q13 region iPSCs to confirm deletion in PWS SD iPSCs. GAPDH was used as en endogenous control, and data were normalized to NML 1-0 iPSCs. (C) Methylation analysis of PWS-IC within SNPRN, using a methylation-sensitive restriction endonuclease quantitative PCR assay, confirming the maintenance of genomic imprinting at the PWS-IC following reprogramming in PWS SD iPSCs. PWS del 1-7 and AS del 1-0 iPSCs were used as controls. Percent methylation was reported plus or minus the SD of three replicates.
Paternal UBE3A is imprinted in non-neuronal cells
Two recent studies reported the expression of either SNORD115 or SNORD109B, transcripts that are normally restricted to neurons, in PWS SD patient lymphocytes (21,22). To determine whether the neuron-specific gene was also expressed in PWS SD iPSCs, we performed qRT–PCR for SNORD115. In agreement with the expression data from lymphocytes, robust expression of SNORD115 was observed in PWS SD iPSCs (Fig. 3A). We then performed conventional RT–PCR for UBE3A-ATS to determine whether this transcript extended into the UBE3A gene. Strong expression of UBE3A-ATS was observed using primers anchored in exons exclusive to UBE3A-ATS (Fig. 3B). Since UBE3A-ATS was expressed, we hypothesized that UBE3A expression would be imprinted; the expression of paternal UBE3A-ATS in neurons should repress-paternal UBE3A expression (16,17,19). We first tested this hypothesis using qRT–PCR to determine UBE3A mRNA levels in PWS SD iPSCs. Normal iPSCs have twice as much UBE3A mRNA, when compared with AS and PWS iPSCs with large deletions of chromosome 15q11–q13 (Fig. 3C). This most likely occurs because UBE3A expression is bi-allelic in iPSCs. Normal iPSCs have two copies of UBE3A, while AS and PWS iPSCs harbor a large deletion of chromosome 15q11–q13 with only a single copy of UBE3A. Although PWS SD iPSCs have two intact copies of UBE3A, its relative expression is half of the normal iPSCs and similar to both AS and PWS iPSCs, where there is only one copy of UBE3A (Fig. 3C). This suggests that UBE3A may be imprinted in the PWS SD iPSCs.
Figure 3.

Imprinting of paternal UBE3A in non-neuronal cells. (A) Gene expression analysis of SNORD115 snoRNAs in iPSCs by qRT–PCR. GAPDH was used as an endogenous control, and data were normalized to NML 1-0 10 week neurons. (B) Analysis of UBE3A-ATS expression in PWS SD iPSCs. RT-19 primers were used to analyze the expression of the UBE3A-ATS (13), and GAPDH was used as a control. NML 1-0 10 week neurons were used as a positive control, since the UBE3A-ATS is exclusively expressed in neurons. (C) Expression analysis of UBE3A in iPSCs. GAPDH was used as an endogenous control, and data were normalized to NML 1-0 iPSCs. PWS del 1-7 and AS del 1-0 iPSCs were used as controls. (D) Allele-specific RT–PCR showed equal expression of UBE3A in NML 1-0 iPSCs. In PWS SD iPSCs, the paternal UBE3A is repressed while UBE3A-ATS is expressed (S = sense, ATS = antisense and GM = genomic DNA). (E) RNA FISH using a riboprobe that specifically detects UBE3A sense transcripts (green) and a BAC probe that detects SNORD115 transcripts (red). UBE3A is actively transcribed from both alleles, and SNORD115 transcripts are not detected in NML 1-0 iPSCs. Scale bars = 5 μm. In PWS SD iPSCs, 83% of SNORD115-positive nuclei showed monoallelic expression of UBE3A and 17% of that showed biallelic expression of UBE3A, as indicated by the asterisk.
In order to determine whether one copy of UBE3A is repressed, we performed allele-specific RT–PCR analysis of UBE3A expression. We first screened most of the UBE3A cDNA to identify allele-specific polymorphisms. Upon finding no polymorphisms in the cDNA, we then scanned the introns in the hopes that a polymorphism in the pre-mRNA could be used to determine allele-specific expression. We identified a polymorphism in intron 9 of UBE3A where one allele has 5 copies of a 26 nucleotide repeat and the other allele has 4 copies of this repeat in PWS SD iPSCs. The normal iPSC line is also polymorphic at this site, with different numbers of repeats. We performed semi-quantitative strand-specific RT–PCR using primers flanking this polymorphism to determine the allele-specific expression of UBE3A sense pre-mRNA transcripts. As expected, the UBE3A sense is transcribed equally from both alleles in the normal iPSC line. In PWS SD iPSCs, however, most of the UBE3A sense transcript comes from only a single allele, presumably due to the aberrant expression of UBE3A-ATS from the paternal allele in these iPSCs (Fig. 3D).
To further confirm that the paternally inherited allele of UBE3A is silenced and the maternally inherited allele is expressed in PWS SD iPSCs, we performed RNA FISH using a riboprobe that specifically detects UBE3A sense transcripts and a BAC probe that detects SNORD115 transcripts. In normal iPSCs, UBE3A is actively transcribed from both alleles, and SNORD115 transcripts are not detected, as expected (Fig. 3E, left panel). In PWS SD iPSCs, most of the cells have a single expressed copy of UBE3A that does not co-localize with the single expressed copy of SNORD115, demonstrating that paternal UBE3A is repressed, presumably by the expression of UBE3A-ATS (Fig. 3E, middle and right panels). Notably, a small portion of cells (17% ± 2%) still express UBE3A from both parental alleles even though they produce SNORD115, indicated by the asterisk (Fig. 3E, right panel).
SNRPN upstream exon usage is not affected in PWS SD iPSCs and neurons
In brain, expression of UBE3A-ATS and imprinted UBE3A expression occur concomitantly with the use of several upstream exons of SNURF-SNRPN, diagrammed in Figure 4A. The use of these upstream exons is thought to shift the expression from the protein coding exons of SNURF-SNRPN to the non-coding downstream exons (26). In humans, at least one upstream SNURF-SNRPN exon is used in many non-neural tissues. Coincidently, downstream ncRNAs between and SNURF-SNRPN and IPW (inclusive of IPW) are expressed in many tissues, including iPSCs. To determine whether the aberrant expression of UBE3A-ATS correlated with the use of the neuron-specific upstream exons of SNURF-SNRPN, we differentiated the PWS SD iPSCs into neurons to compare upstream exon usage between neural and non-neural tissue.
Figure 4.

SNRPN upstream exon usage in PWS SD iPSCs neurons. (A) Map showing the organization of upstream SNURF-SNRPN exons (34). Upstream exons U1B, U1Bʹ, U1A and U2 are largely neuron specific, as indicated by the bracket. Exon U4 is expressed at low levels in a variety of tissues, including iPSCs. It is unclear whether U3 and U5 are brain specific. The location of the PWS-IC is indicated by a half-filled circle. (B) Immunocytochemistry for the neural marker MAP2 in PWS SD 2-8 10 week neurons. Cell nuclei were counterstained with DAPI. Scale bar = 100 μm. (C) Expression analysis of upstream SNRPN exons. GAPDH was used as an endogenous control, and data were normalized to NML 1-0 10 week neurons, since these exons are predominately expressed in neurons.
In order to choose PWS SD iPSC lines that are capable of efficient differentiation into neurons, we examined the expression levels of genes critical for specification into the neural lineage in d16 EBs (Supplementary Material, Fig. S3A), using the scorecard described above. In addition to the lineage markers used to verify multi-lineage differentiation potential, this card also had probe-primer sets for an abbreviated list of widely used neural lineage marker genes (Supplementary Material, Table S1) (25). Probe-primer sets to the pluripotency genes NANOG and ZFP42/REX1 were also included in the scorecard to determine whether pluripotency genes were appropriately turned off during differentiation (Supplementary Material, Fig. S3B). We used AS del 1-0 as a calibrator sample for these assays, since this AS iPSC line differentiates robustly into neurons using conventional neural differentiation assays (17). The PWS del 1-1 iPSC line was used as a negative control for these assays, since it does not robustly differentiate into neurons. Differentiation propensity of the PWS SD iPSC lines into neurons was determined based on two criteria: (i) robust expression of neural lineage markers and (ii) silencing of the pluripotency genes. PWS SD 2-8 and PWS SD 2-9 iPSC lines were found to have the highest propensities to differentiate into neurons due to the robust expression of neural lineage markers and silencing of the pluripotency genes. Despite the robust expression of the neural lineage markers in the PWS SD 2-4 iPSC line, this line may have limited differentiation potential in conventional neural differentiation assays due to the inappropriate expression of the pluripotency genes in d16 EBs. PWS SD 2-8 and PWS 2-9 iPSC lines were chosen for further analysis.
Both PWS SD 2-8 and PWS SD 2-9 iPSC lines were differentiated into neurons, as previously described (17). Following 10 weeks of differentiation, MAP2-positive neurons were identified by immunocytochemistry. A representative image is shown for 10 week PWS SD 2-8 iPSC-derived neurons (Fig. 4B). The expression of the neuron-specific upstream U1A, U2 and U4 exons was compared between PWS SD iPSCs and PWS SD iPSC-derived neurons by quantitative RT–PCR (Fig. 4C). The U1A and U2 exons were almost exclusively expressed in the neuron samples and were nearly undetectable in all six iPSC lines. Exon U4, which showed robust expression in NML 1-0, PWS SD 2-8 and PWS SD 2-9 iPSC-derived neurons, was expressed at low levels in NML 1-0 and AS del 1-0 iPSCs, was barely detectable in PWS SD iPSCs, and was undetectable in PWS del 1-7 iPSCs. Thus, imprinted expression of UBE3A in PWS SD iPSCs was not caused by the aberrant expression of the neuron-specific upstream SNRPN exons. Moreover, these results suggest that expression of UBE3A-ATS is sufficient for imprinted expression of UBE3A and that additional neuron-specific events were not required for this imprinting in non-neural tissues.
The shortened UBE3A-ATS in PWS SD neurons is less amenable to silencing by topotecan
Topoisomerase inhibitors have been shown to repress Ube3a-ats in mouse (27). This effect is length dependent and is shared between mouse and human across the genome, with longer genes showing greater repression by topoisomerase inhibitors (28). Since the UBE3A-ATS transcript is reduced by 187 kb in PWS SD iPSCs, we sought to determine whether a topoisomerase inhibitor was less effective on the shortened UBE3A-ATS transcript. We treated 10-week-old AS and PWS SD iPSC-derived neurons with increasing doses of Topotecan, a campothecin-type topoisomerase inhibitor that was previously shown to be effective at reducing mouse Ube3a-ats. We then performed qRT–PCR using a probe-primer set that recognizes the SNURF-SNRPN ncRNA in the region of SNORD115. This SNORD115 host transcript can be used as a proxy for UBE3A-ATS, and thus avoids possible confusion with the sense UBE3A transcript. Topotecan was highly effective in reducing the SNURF-SNRPN ncRNA (which includes UBE3A-ATS) in AS iPSC-derived neurons, showing 90% reduction of the transcript at a 10 μM dose (Fig. 5). However, topotecan was less effective in repressing the shortened SNURF-SNRPN ncRNA in PWS SD neurons, only showing a maximum 46% reduction compared with the 90% reduction seen in AS neurons (Fig. 5).
Figure 5.

Topotecan is less effective in repressing the UBE3A-ATS in PWS SD neurons. Relative expression of SNORD115 by qRT–PCR in 10 week neurons derived from PWS SD and AS iPSC lines treated with various concentrations of Topotecan. GAPDH was used as an endogenous control. **P-value ≤ 0.05.
DISCUSSION
Here we show that iPSCs derived from patients with PWS due to a small deletion on paternal chromosome 15q11–q13 express UBE3A-ATS and have imprinted expression of UBE3A. UBE3A-ATS expression is typically restricted to neurons, and consequently so is UBE3A imprinting. Work over the past decade has steadily focused on UBE3A-ATS as the mediator of UBE3A imprinted expression (16,17,19,29). Thus, understanding the neuron-specific regulation of UBE3A-ATS is critical for understanding the regulation of UBE3A imprinted expression.
UBE3A-ATS is within the neuron-specific portion of the SNURF-SNRPN ncRNA, which also includes SNORD115 and SNORD109B (13). In non-neuronal tissues and cell types, transcription of the SNURF-SNRPN locus usually stops at IPW, and thus SNORD115, SNORD109B and UBE3A-ATS are not expressed (17). In contrast to normal iPSCs where the neuron-specific portion of SNURF-SNRPN is not expressed, PWS SD iPSCs showed robust expression of SNORD115 and UBE3A-ATS (Fig. 3A and B). In normal iPSCs, the polyadenylation sites that lie at the 3′ end of IPW are likely transcriptional stops for the SNURF-SNRPN transcript. These sites, as well as additional as-of-yet unidentified regulatory elements, have been removed in the PWS SD patient-derived iPSCs, placing the SNURF-SNRPN promoter and exon 1 in direct association with the SNORD115 portion of the transcript. The identification of the regulatory elements that stop SNURF-SNRPN transcription will be an important clue in understanding the neuron-specific regulation of UBE3A-ATS and imprinted UBE3A expression. Alternatively, the SNURF-SNRPN transcript is terminated at IPW because that is the maximum length that can be transcribed during the iPSC cell cycle.
The aberrant expression of UBE3A-ATS transcript in PWS SD iPSCs leads to imprinted UBE3A expression (Fig. 3). We demonstrated this in three ways. First, we determined UBE3A expression levels. UBE3A mRNA levels in PWS SD iPSCs are consistent with having only a single expressed allele of the gene, like PWS and AS iPSCs harboring large deletions of paternal and maternal chromosome 15q11–q13, respectively. Normal iPSCs have UBE3A mRNA levels consistent with having two expressed copies of the gene (Fig. 3C). Secondly, we determined whether UBE3A is expressed from one or two alleles in PWS SD iPSCs using strand-specific RT–PCR. UBE3A is primarily expressed from a single allele in these cells and UBE3A-ATS is expressed exclusively from the opposite allele (Fig. 3D). Finally, we used RNA FISH to confirm that UBE3A was coming from a single allele that did not produce the SNORD115 transcript (Fig. 3E). These data suggest that UBE3A-ATS expression is sufficient for the imprinted expression of UBE3A, and that additional neuron-specific factors are not necessary for the repression of paternal UBE3A.
Interestingly, imprinting of UBE3A is not complete in every cell, as evidenced by both the RT–PCR and RNA FISH data. In fact, the RNA FISH data demonstrated that ∼20% of the cells showed juxtaposed UBE3A and SNORD115 expression in the same cell (Fig. 3E). While we do not know the exact reason for this, we speculate that in the dividing PWS SD iPSCs, replication may cause the premature termination of the SNURF-SNRPN/SNORD115 transcript prior to overlapping UBE3A, thus allowing UBE3A transcription in the sense direction. Indeed, paternal Ube3a has been shown to retain active histone modifications, even in the brain (30). The PWS SD iPSCs described here show that imprinted UBE3A expression can be maintained by the UBE3A-ATS, even in the presence of a transcriptionally competent paternal UBE3A promoter. It is therefore not necessary that paternal UBE3A undergoes repressive chromatin changes, even in neurons where it is normally imprinted.
There was a slight discrepancy between the FISH and RT–PCR data. The FISH data showed that 17%+/−2% of cells showed detectable expression from the paternal allele of UBE3A (Fig. 3E, lower panel), while the RT–PCR data showed that 27% of the total UBE3A RNA came from the paternal allele (Fig. 3D). There are at least three possible explanations for this: (i) the FISH assay failed to detect UBE3A in some cells where it was expressed from the paternal allele at a level lower than the detection limit; (ii) the active paternal allele is expressing more UBE3A than the maternal allele in some or all of the cells, so that ∼17% of the cells are producing ∼27% of the RNA or (iii) the semi-quantitative RT–PCR experiment may suffer from amplification bias or measurement bias (i.e. the genomic DNA in PWS SD 2-8 was measured as 54% paternal and 46% maternal).
Topoisomerase inhibitors were shown to reactivate the paternal allele of Ube3a in mouse neurons by repressing Snurf-Snrpn/Ube3a-ats (27). This occurs because topoisomerases are required for efficient transcription of long genes in both human and mouse neurons (28). The 187 kb deletion of the SNORD116 cluster enabled us to examine the effect of a topoisomerase inhibitor, topotecan, on a shortened SNURF-SNRPN/UBE3A-ATS transcript. We found that topotecan was not as effective on the shortened UBE3A-ATS transcript as it was on the full-length transcript, further supporting the observation that topoisomerases are important for the transcription of UBE3A-ATS and other long genes (Fig. 5).
Altogether, these data demonstrate that the SNURF-SNRPN transcript is regulated by a boundary element in non-neurons, and that in its absence, the expression of UBE3A-ATS is sufficient to cause UBE3A imprinted expression in the absence of additional neural factors. We do not know what comprises this boundary element, but we speculate that either the polyadenylation sites in the IPW gene act as transcriptional terminators, that CTCF protein-binding sites downstream of IPW separate active versus inactive chromatin territories or a combination of those two possibilities work to perform the boundary function. These findings provide important clues as to the regulation of the SNURF-SNRPN ncRNA in humans. Furthermore, they demonstrate that iPSCs derived from rare individuals with atypical deletions can teach us important lessons about the mechanisms of gene regulation.
MATERIALS AND METHODS
Cell lines and culture conditions
AS del 1-0, PWS del 1-1, PWS del 1-7 and NML 1-0 iPSC lines used in this study were cultured as described (17). Moreover, PWS small deletion (PWS SD) iPSC lines were derived from patient fibroblasts (21) using the human polycistronic STEMCCA lentiviral vector encoding OCT4, KLF4, SOX2, CMYC and cultured following previously published protocols (24). The iPSC lines were cultured in the hESC medium, i.e. Dulbecco's modified Eagle's medium/F-12 containing 20% KnockOut serum replacer, 0.1 mm non-essential amino acids, 1 mm l-glutamine (all from Life Technologies, Grand Island, NY), 0.1 mm β-mercaptoethanol (Sigma-Aldrich, St. Louis, MO, USA), supplemented with 4 ng/ml bFGF (Millipore, Billerica, MA, USA).
Immunocytochemistry
Immunocytochemistry was performed as previously described (31), using rabbit anti-NANOG (Stemgent, Cambridge, MA, USA), mouse anti-SSEA-4 (Developmental Studies Hybridoma Bank, Iowa City, IA, USA), mouse anti-TRA1-60 or mouse anti-TRA1-81 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or mouse anti-MAP2 (Millipore) antibodies. Cell nuclei were counterstained with DAPI.
qRT–PCR
RNA was isolated from cells using RNA-Bee (Tel Test, Inc., Friendswood, TX, USA) and cDNA was synthesized using High Capacity cDNA Reverse Transcription Kit (Life Technologies). Gene expression was analyzed using TaqMan® Gene Expression Assays, and the GAPDH Endogenous Control TaqMan® Assay was used as an endogenous control. Experiments were performed in triplicate. The data were analyzed using RQ2.1 software (Applied Biosystems), normalized to either NML 1-0 iPSCs or NML 1-0 10 week neurons. The Sybr qRT–PCR primers used to examine the expression of SNRPN upstream U2 and U4 exons, relative to the GAPDH endogenous control, are listed in Supplementary Material, Table S2.
Methylation-sensitive restriction endonuclease quantitative PCR assay
These assays were performed as described (17). The Methyl Profiler enzyme kit and SNRPN primer set (Qiagen) were used according to the manufacturer's instructions to assay methylation at the PWS-IC. Percent methylation was reported plus or minus the SD of three replicates. No intermediate methylation was observed in any of the fibroblast or iPSC lines with this assay. The SNRPN primer set assays seven different CpG sites within the PWS-IC.
RT–PCR for UBE3A-ATS
To examine whether the UBE3A-ATS was expressed in the PWS SD iPSC lines, conventional PCR primers were used. RT-19 primers described in Runte et al. (13) were used to analyze the expression of the UBE3A-ATS, and GAPDH was used as a loading control. NML 1-0 10 week neurons were used as a positive control, since the UBE3A-ATS is exclusively expressed in neurons.
Allele-specific RT–PCR
RNA was extracted from iPSCs using RNA-Bee (Tel Test, Inc.), followed by DNase treatment using TURBO DNA-free Kit (Life Technologies AM1907). DNase-treated RNA of 1 μg of was reverse transcribed using Superscript III (Life Technologies) at 53°C with strand-specific primers with tag sequences (GGAAACAGCTATGACCATAACAATTTTCCCATTCAGAT for sense, and CAGTCGGGCGTCATCATTTTCGTTATTGTTCCTTAGAA for anti-sense; tag sequences are shown in bold). PCR was performed with annealing temperature at 60°C for 27 cycles using Advantage 2 Polymerase Mix (Clonetech, Mountain View, CA, USA) with primer sequences as the following: CATGAGCTTAGACTTCACCTTTCA and GGGACCTCATGATGGCAATA. PCR products were run on precast Novex TBE 6% polyacrylamide gel (Life Technologies) and post-stained with SYBR Gold (Life Technologies). Images were taken using LAS-3000 and the bands were quantified using Multigauge V3.0 software (Fujifilm, Tokyo, Japan).
RNA FISH
iPSCs were grown on MEF-covered 12 mm coverslips for 4–6 days. Prior to 4% paraformaldehyde/PBS fixation for 10 min, coverslips were treated with CSK buffer for 30 s, 1% Triton-X100/CSK buffer for 10 min, and CSK buffer for 30 s. Coverslips were then dehydrated with 70, 85, 95 and 100% EtOH for 2 min each and air dried before hybridizing with probes. SNORD115 probes were made by labeling BAC RP11-37A4 using ENZO Nick Translation DNA Labeling System (Enzo, Farmingdale, NY, USA) with Alexa Fluor 594 dUTPs (Life Technologies) according to the manufacturer's instructions. UBE3A sense riboprobes were made from C7-3 cDNA (32) cloned in pBluescript SK+ and in vitro transcribed using the MAXIscript T7/T3 Kit (Life Technologies) with Alexa Fluor 488 UTPs (Life Technologies) according to the manufacturer's instructions. Transcribed riboprobes of 1 μg were hydrolyzed in carbonate buffer (60 mm Na2CO3 and 40 mm NaHCO3) at 60°C for 30 min and neutralized in 1/20 volume of 10% acetic acid prior to purifying labeled riboprobes by Micro Bio-Spin P-30 Tris column (Bio-Rad, Hercules, CA, USA). One hundred nanagrams of labeled SNORD115 was added to the UBE3A purified riboprobes along with 5 μg of human Cot-1 DNA, 5 μg of salmon sperm DNA and 2.5 volumes of 100% EtOH. Probes were precipitated at −20°C for 30 min and centrifuged at 12 000 rpm for 30 min. Probe pellets were air dried in the dark for 15 min and reconstituted in 100% formamide before heat denaturing at 85°C for 10 min. Two times hybridization buffer (4× SSC, 20% dextran sulfate, 2 mg/ml BSA) was added to the probes and applied equally to coverslips for overnight hybridization at 37°C in a dark humid chamber containing 2× SSC/50% formamide. Coverslips were washed three times with 2× SSC/50% formamide, three times with 2× SSC and three times with 1× SSC for 7 min each at 39°C prior to mounting with DAPI (Vectashield, Burlingame, CA, USA). Images were taken using Zeiss 780 confocal microscrope.
Neural differentiation
Ten-week neurons were differentiated from iPSCs using the monolayer protocol as described (33). Briefly, one-day post-split iPSCs were growing in N2B27 medium supplemented with 500 ng/ml Noggin (R&D Systems, Minneapolis, MN, USA) for 10 days. These colonies were then spilt onto poly-l-lysine and laminin-coated plates using StemPro EZ Passage tool (Life Technologies) and kept in N2B27 medium for 7 days. Once rosettes were forming in these colonies, the cells were trypsinized and replated onto poly-l-lysine and laminin-coated plates at higher density in N2B27 medium supplemented with ROCK inhibitor overnight. The cells were kept in N2B27 medium for another 6 days and split onto poly-l-lysine and laminin-coated plates with 1:4 to 1:6 ratio in neural differentiation media for 2 weeks. The cells were then split with 1:6 ratio for another 5 weeks in neural differentiation media before topoisomerase inhibitor treatment.
Topoisomerase inhibitor treatment
Topoisomerase inhibitor, topotecan (Molcan Corporation, Toronto, Ontario, Canada), was dissolved in DMSO (Sigma-Aldrich) to make a 100 mm stock. A series of dilutions was made to achieve the final concentrations of 100, 10, 1 μm, 100 and 10 nm containing 0.1% DMSO for dose–response curve experiments. The cells were treated with various concentrations of topoisomerase inhibitor for continuous 6 days prior to collecting RNA for experiments.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by grants from the National Institutes of Health (R01HD068730 to S.J.C.); Foundation for Prader–Willi Syndrome (to M.L.); Connecticut Stem Cell Research Grant (11SCA01 to K.M.-T and 12SCBUCHC to S.J.C.) and the Raymond and Beverly Sackler Foundation.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Dr. Susan Holder for collecting the skin biopsy from the patient, and members of the laboratory for their helpful discussions and critical reading of the manuscript. The contents in this work are solely the responsibility of the authors and do not necessarily represent the official views of the state of Connecticut.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Cassidy S.B., Dykens E., Williams C.A. Prader–Willi and Angelman syndromes: sister imprinted disorders. Am. J. Med. Genet. 2000;97:136–146. doi: 10.1002/1096-8628(200022)97:2<136::aid-ajmg5>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 2.Williams C.A., Beaudet A.L., Clayton-Smith J., Knoll J.H., Kyllerman M., Laan L.A., Magenis R.E., Moncla A., Schinzel A.A., Summers J.A., et al. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am. J. Med. Genet. A. 2006;140:413–418. doi: 10.1002/ajmg.a.31074. [DOI] [PubMed] [Google Scholar]
- 3.Miller J.L., Lynn C.H., Driscoll D.C., Goldstone A.P., Gold J.A., Kimonis V., Dykens E., Butler M.G., Shuster J.J., Driscoll D.J. Nutritional phases in Prader–Willi syndrome. Am. J. Med. Genet. Part A. 2011;155A:1040–1049. doi: 10.1002/ajmg.a.33951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cassidy S.B., Driscoll D.J. Prader–Willi syndrome. Eur. J. Hum. Genet. 2009;17:3–13. doi: 10.1038/ejhg.2008.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reis A., Dittrich B., Greger V., Buiting K., Lalande M., Gillessen-Kaesbach G., Anvret M., Horsthemke B. Imprinting mutations suggested by abnormal DNA methylation patterns in familial Angelman and Prader–Willi syndromes. Am. J. Hum. Genet. 1994;54:741–747. [PMC free article] [PubMed] [Google Scholar]
- 6.Saitoh S., Buiting K., Rogan P.K., Buxton J.L., Driscoll D.J., Arnemann J., Konig R., Malcolm S., Horsthemke B., Nicholls R.D. Minimal definition of the imprinting center and fixation of chromosome 15q11-q13 epigenotype by imprinting mutations. Proc. Natl. Acad. Sci. USA. 1996;93:7811–7815. doi: 10.1073/pnas.93.15.7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jong M.T., Carey A.H., Caldwell K.A., Lau M.H., Handel M.A., Driscoll D.J., Stewart C.L., Rinchik E.M., Nicholls R.D. Imprinting of a RING zinc-finger encoding gene in the mouse chromosome region homologous to the Prader–Willi syndrome genetic region. Hum. Mol. Genet. 1999;8:795–803. doi: 10.1093/hmg/8.5.795. [DOI] [PubMed] [Google Scholar]
- 8.Boccaccio I., Glatt-Deeley H., Watrin F., Roeckel N., Lalande M., Muscatelli F. The human MAGEL2 gene and its mouse homologue are paternally expressed and mapped to the Prader–Willi region. Hum. Mol. Genet. 1999;8:2497–2505. doi: 10.1093/hmg/8.13.2497. [DOI] [PubMed] [Google Scholar]
- 9.Jay P., Rougeulle C., Massacrier A., Moncla A., Mattei M.G., Malzac P., Roeckel N., Taviaux S., Lefranc J.L., Cau P., et al. The human necdin gene, NDN, is maternally imprinted and located in the Prader–Willi syndrome chromosomal region. Nat. Genet. 1997;17:357–361. doi: 10.1038/ng1197-357. [DOI] [PubMed] [Google Scholar]
- 10.Buiting K., Nazlican H., Galetzka D., Wawrzik M., Gross S., Horsthemke B. C15orf2 and a novel noncoding transcript from the Prader–Willi/Angelman syndrome region show monoallelic expression in fetal brain. Genomics. 2007;89:588–595. doi: 10.1016/j.ygeno.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 11.Cattanach B.M., Barr J.A., Evans E.P., Burtenshaw M., Beechey C.V., Leff S.E., Brannan C.I., Copeland N.G., Jenkins N.A., Jones J. A candidate mouse model for Prader–Willi syndrome which shows an absence of Snrpn expression. Nat. Genet. 1992;2:270–274. doi: 10.1038/ng1292-270. [DOI] [PubMed] [Google Scholar]
- 12.Glenn C.C., Porter K.A., Jong M.T., Nicholls R.D., Driscoll D.J. Functional imprinting and epigenetic modification of the human SNRPN gene. Hum. Mol. Genet. 1993;2:2001–2005. doi: 10.1093/hmg/2.12.2001. [DOI] [PubMed] [Google Scholar]
- 13.Runte M., Huttenhofer A., Gross S., Kiefmann M., Horsthemke B., Buiting K. The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum. Mol. Genet. 2001;10:2687–2700. doi: 10.1093/hmg/10.23.2687. [DOI] [PubMed] [Google Scholar]
- 14.Cavaille J., Buiting K., Kiefmann M., Lalande M., Brannan C.I., Horsthemke B., Bachellerie J.P., Brosius J., Huttenhofer A. Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proc. Natl. Acad. Sci. USA. 2000;97:14311–14316. doi: 10.1073/pnas.250426397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wevrick R., Kerns J.A., Francke U. Identification of a Novel Paternally Expressed Gene in the Prader–Willi-Syndrome Region. Hum. Mol. Genet. 1994;3:1877–1882. doi: 10.1093/hmg/3.10.1877. [DOI] [PubMed] [Google Scholar]
- 16.Rougeulle C., Cardoso C., Fontes M., Colleaux L., Lalande M. An imprinted antisense RNA overlaps UBE3A and a second maternally expressed transcript. Nat. Genet. 1998;19:15–16. doi: 10.1038/ng0598-15. [DOI] [PubMed] [Google Scholar]
- 17.Chamberlain S.J., Chen P.F., Ng K.Y., Bourgois-Rocha F., Lemtiri-Chlieh F., Levine E.S., Lalande M. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader–Willi syndromes. Proc. Natl. Acad. Sci. USA. 2010;107:17668–17673. doi: 10.1073/pnas.1004487107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rougeulle C., Glatt H., Lalande M. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat. Genet. 1997;17:14–15. doi: 10.1038/ng0997-14. [DOI] [PubMed] [Google Scholar]
- 19.Meng L., Person R.E., Beaudet A.L. Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum. Mol. Genet. 2012;21:3001–3012. doi: 10.1093/hmg/dds130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kishino T., Lalande M., Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 1997;15:70–73. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- 21.de Smith A.J., Purmann C., Walters R.G., Ellis R.J., Holder S.E., Van Haelst M.M., Brady A.F., Fairbrother U.L., Dattani M., Keogh J.M., et al. A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum. Mol. Genet. 2009;18:3257–3265. doi: 10.1093/hmg/ddp263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sahoo T., del Gaudio D., German J.R., Shinawi M., Peters S.U., Person R.E., Garnica A., Cheung S.W., Beaudet A.L. Prader–Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat. Genet. 2008;40:719–721. doi: 10.1038/ng.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duker A.L., Ballif B.C., Bawle E.V., Person R.E., Mahadevan S., Alliman S., Thompson R., Traylor R., Bejjani B.A., Shaffer L.G., et al. Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader–Willi syndrome. Eur. J. Hum. Genet. 2010;18:1196–1201. doi: 10.1038/ejhg.2010.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Somers A., Jean J.C., Sommer C.A., Omari A., Ford C.C., Mills J.A., Ying L., Sommer A.G., Jean J.M., Smith B.W., et al. Generation of transgene-free lung disease-specific human induced pluripotent stem cells using a single excisable lentiviral stem cell cassette. Stem Cells. 2010;28:1728–1740. doi: 10.1002/stem.495. doi:1710.1002/stem.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bock C., Kiskinis E., Verstappen G., Gu H., Boulting G., Smith Z.D., Ziller M., Croft G.F., Amoroso M.W., Oakley D.H., et al. Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell. 2011;144:439–452. doi: 10.1016/j.cell.2010.12.032. doi:410.1016/j.cell.2010.1012.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Landers M., Bancescu D.L., Le Meur E., Rougeulle C., Glatt-Deeley H., Brannan C., Muscatelli F., Lalande M. Regulation of the large (approximately 1000 kb) imprinted murine Ube3a antisense transcript by alternative exons upstream of Snurf/Snrpn. Nuc. Acids Res. 2004;32:3480–3492. doi: 10.1093/nar/gkh670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang H.S., Allen J.A., Mabb A.M., King I.F., Miriyala J., Taylor-Blake B., Sciaky N., Dutton J.W., Jr, Lee H.M., Chen X., et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature. 2012;481:185–189. doi: 10.1038/nature10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.King I.F., Yandava C.N., Mabb A.M., Hsiao J.S., Huang H.S., Pearson B.L., Calabrese J.M., Starmer J., Parker J.S., Magnuson T., et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature. 2013;501:58–62. doi: 10.1038/nature12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chamberlain S.J., Brannan C.I. The Prader–Willi syndrome imprinting center activates the paternally expressed murine Ube3a antisense transcript but represses paternal Ube3a. Genomics. 2001;73:316–322. doi: 10.1006/geno.2001.6543. [DOI] [PubMed] [Google Scholar]
- 30.Rougeulle C., Navarro P., Avner P. Promoter-restricted H3 Lys 4 di-methylation is an epigenetic mark for monoallelic expression. Hum. Mol. Genet. 2003;12:3343–3348. doi: 10.1093/hmg/ddg351. [DOI] [PubMed] [Google Scholar]
- 31.Zeng H., Guo M., Martins-Taylor K., Wang X., Zhang Z., Park J.W., Zhan S., Kronenberg M.S., Lichtler A., Liu H.X., et al. Specification of region-specific neurons including forebrain glutamatergic neurons from human induced pluripotent stem cells. PLoS One. 2010;5:e11853. doi: 10.1371/journal.pone.0011853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kishino T., Wagstaff J. Genomic organization of the UBE3A/E6-AP gene and related pseudogenes. Genomics. 1998;47:101–107. doi: 10.1006/geno.1997.5093. [DOI] [PubMed] [Google Scholar]
- 33.Germain N.D., Banda E.C., Becker S., Naegele J.R., Grabel L.B. Derivation and Isolation of NKX2.1-Positive Basal Forebrain Progenitors from Human Embryonic Stem Cells. Stem Cells Dev. 2013;22:1477–1489. doi: 10.1089/scd.2012.0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Farber C., Dittrich B., Buiting K., Horsthemke B. The chromosome 15 imprinting centre (IC) region has undergone multiple duplication events and contains an upstream exon of SNRPN that is deleted in all Angelman syndrome patients with an IC microdeletion. Hum. Mol. Genet. 1999;8:337–343. doi: 10.1093/hmg/8.2.337. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.