

Abstract
Usher syndrome type 2 (USH2) is the predominant form of USH, a leading genetic cause of combined deafness and blindness. PDZD7, a paralog of two USH causative genes, USH1C and USH2D (WHRN), was recently reported to be implicated in USH2 and non-syndromic deafness. It encodes a protein with multiple PDZ domains. To understand the biological function of PDZD7 and the pathogenic mechanism caused by PDZD7 mutations, we generated and thoroughly characterized a Pdzd7 knockout mouse model. The Pdzd7 knockout mice exhibit congenital profound deafness, as assessed by auditory brainstem response, distortion product otoacoustic emission and cochlear microphonics tests, and normal vestibular function, as assessed by their behaviors. Lack of PDZD7 leads to the disorganization of stereocilia bundles and a reduction in mechanotransduction currents and sensitivity in cochlear outer hair cells. At the molecular level, PDZD7 determines the localization of the USH2 protein complex, composed of USH2A, GPR98 and WHRN, to ankle links in developing cochlear hair cells, likely through its direct interactions with these three proteins. The localization of PDZD7 to the ankle links of cochlear hair bundles also relies on USH2 proteins. In photoreceptors of Pdzd7 knockout mice, the three USH2 proteins largely remain unchanged at the periciliary membrane complex. The electroretinogram responses of both rod and cone photoreceptors are normal in knockout mice at 1 month of age. Therefore, although the organization of the USH2 complex appears different in photoreceptors, it is clear that PDZD7 plays an essential role in organizing the USH2 complex at ankle links in developing cochlear hair cells. GenBank accession numbers: KF041446, KF041447, KF041448, KF041449, KF041450, KF041451.
INTRODUCTION
Usher syndrome (USH) is the most common genetic disease characterized by hearing loss combined with retinitis pigmentosa (1–3). It occurs in ∼1 in 23 000 people worldwide. USH is categorized into three clinical types, according to its auditory and vestibular symptoms. Type 2 USH (USH2) is the most prevalent form, accounting for up to 60% of all USH cases (4). USH2 patients exhibit moderate congenital hearing loss, retinal degeneration, but normal vestibular function. To date, the molecular mechanisms underlying USH2 are not completely understood and there is no cure for this disease.
USH2A, GPR98 and WHRN are the three known USH2 causative genes, which are predicted to encode a cell adhesion protein, a G protein-coupled receptor and a scaffold protein, respectively (5–7). The N-terminal two PDZ domains of WHRN can bind to the PDZ-binding motif (PBM) at the C termini of USH2A and GPR98 in vitro. In photoreceptors, the three proteins are colocalized at the periciliary membrane complex between the outer and the inner segment (8). Loss of any one of these USH2 proteins causes mislocalization and reduced expression of the other two USH2 proteins (8). Delivery of WHRN into Whrn mutant photoreceptors can rescue the localization and expression defects of USH2A and GPR98 (9). These findings indicate that the three USH2 proteins are present in the same multiprotein complex, the USH2 complex, and that WHRN is involved in organizing this complex. In cochlear hair cells, the three USH2 proteins are colocalized at ankle links of the hair bundle (8,10–12). However, their subcellular localizations do not completely depend on each other, as they do in photoreceptors (8), indicating that the composition and formation of the USH2 complex may not occur exactly the same in cochlear hair cells as in photoreceptors.
Mutations in PDZD7 (PDZ domain-containing 7) were found to cause congenital non-syndromic hearing impairment, to exacerbate retinal symptoms in USH2 patients and to probably contribute to digenic USH (MIM, *612 971) (13,14). PDZD7 is a paralog of WHRN and harmonin (USH1C). It shares 55 and 35% similarity in protein sequence with WHRN and harmonin, respectively. PDZD7 was reported to interact in vitro with USH2A, GPR98, harmonin and SANS (the USH1G protein) (13,14). In zebrafish, PDZD7 is expressed beneath the kinocilia in hair cells and at the base of the outer segment in photoreceptors. Reduction in PDZD7 expression by morpholino injection results in bent hair bundles in hair cells and altered GPR98 expression in photoreceptors (14). In rats, PDZD7 was localized to ankle links of cochlear (CHCs) and vestibular (VHCs) hair cells, together with WHRN and GPR98 (15). Based on these findings, PDZD7 could be a new component of the USH2 complex. In this paper, we have generated a Pdzd7 knockout mouse. Using this mouse, we studied the expression, localization and function of PDZD7 in (CHCs) and VHCs and retinal photoreceptors. Our findings demonstrate that PDZD7 is an indispensible organizer of the USH2 complex in cochlear hair cells and that PDZD7 may play a little role in the organization of the USH2 complex in photoreceptors.
RESULTS
Pdzd7−/− mice do not express PDZD7
A Pdzd7 mutant mouse line was created using Pdzd7tm1a(EUCOMM)Wtsi embryonic stem cells. In this mutant mouse, a knockout-first allele (tm1a) of Pdzd7 was generated by inserting a gene trapping cassette between exons 1 and 2 and a loxP site between exons 5 and 6 of the gene (Fig. 1A and B). This targeted allele was designed to trap and truncate Pdzd7 transcripts after exon 1 using a robust engrailed-2 splice acceptor site and an efficient polyadenylation signal (16). Disruption of Pdzd7 transcription in the mutant mice was confirmed by reverse transcriptase–polymerase chain reaction (RT–PCR) using primers upstream and downstream of the inserted cassette (Fig. 1A and D and Supplementary Material, Table S1). Specifically, RT–PCR using primers positioned in exons that encode the individual three PDZ domains of Pdzd7 (l and m, n and o and j and k, Fig. 1A and Supplementary Material, Fig. S2) confirm the elimination of Pdzd7 transcripts with these domains. To examine protein expression of Pdzd7, we first generated antibodies against the N- or C-terminal region of PDZD7 (PDZD7_N or PDZD7_C) from rabbits and chickens (Supplementary Material, Fig. S1A). The specificity of these antibodies against PDZD7 but not its paralogs (harmonin or WHRN; Supplementary Material, Fig. S1B) and no cross-reaction among the N- and C-terminal antibodies (Supplementary Material, Fig. S1C) were verified in transfected HEK293 cells. Immunostaining using the rabbit PDZD7_N and chicken PDZD7_C antibodies detected signals at the cochlear hair bundle in the wild-type, but not mutant, mice (Fig. 1E). However, western blotting analysis using the PDZD7 antibodies could not detect PDZD7 expression in retinal or cochlear lysates during development or in adulthood, suggesting that the protein expression of PDZD7 had a very low level in these tissues. Therefore, we tried another approach to examine PDZD7 protein expression, immunoprecipitation using the rabbit PDZD7_C antibody followed by western blotting analysis using the chicken PDZD7_C antibody. Using this approach, a weak band was found at ∼120 kDa in the wild-type, but not mutant, retina and cochlea during development (Fig. 1F). The size of this band was the same as that of the recombinant full-length PDZD7 in HEK293 cells. These results demonstrated that the expression of PDZD7 was completely eliminated in Pdzd7 mutant mice. Therefore, the Pdzd7 mutant mice are Pdzd7 complete knockout mice (Pdzd7−/−). These Pdzd7−/− mice were vital and fertile. Their body weight at P21–P28 was similar to that of their wild-type littermates (9.40 ± 0.14 g for the wild-type and 9.51 ± 0.55 g for the knockout, mean ± SE, n = 5 each).
Figure 1.

Pdzd7−/− mice were generated. (A) A schematic diagram of the gene targeting Pdzd7tm1a(EUCOMM)Wtsi allele. Arabic numerals in rectangles denote the exon numbers. Arrows with italic lower case letters label the position of primers used in this study. En2 SA, mouse engrailed 2 gene splice acceptor; IRES, internal ribosomal entry segment; βgal, β-galactosidase gene; pA, simian virus 40 polyadenylation signal; βact, human β-actin promoter; neo, neomycin phosphotransferase gene. (B) Confirmation of insertion of the targeting cassette into the Pdzd7 gene by PCR using primer pairs of a/b and c/d. (C) Identification of the mutant allele by routine genotyping PCR using primers, f, h and g. (D) Disrupted transcription of the Pdzd7 gene shown by RT–PCR analysis in the retina using primers e, i, j, k, l, m, n and o. RT–PCR of rhodopsin was used here as a positive control. Note: primer pairs of l/m, n/o and j/k were designed to amplify PDZ1, PDZ2 and PDZ3 regions of PDZD7 cDNA, respectively. (E) Immunofluorescence demonstrates loss of PDZD7 expression in Pdzd7−/− cochlear hair cells at P4. Scale bars, 10 µm. (F) Western blotting (WB) following immunoprecipitation (IP) found loss of PDZD7 expression in the retina at P6 (middle) and in the cochlea at P9 (right) in Pdzd7−/− mice. The same procedure was performed using PDZD7-transfected HEK293 cells (left). The band just above 34 kDa on the P9 inner ear blot showed no difference between wild-type and Pdzd7−/− littermates in another experiment at P5 (data not shown). Thus, it is considered non-specific. Arrowheads point to the PDZD7 bands. Asterisks mark non-specific bands. IgG, non-immune rabbit immunoglobulin; rPDZD7_C, rabbit antibody against PDZD7_C ; cPDZD7_C, chicken antibody against PDZD7_C; rPDZD7_N, rabbit antibody against PDZD7_N. +/+, wild-type; −/−, Pdzd7 knockout. The DNA sequences of primers are shown in Supplementary material, Table S1.
Pdzd7−/− mice have normal vestibular function and congenital profound deafness
Pdzd7−/− mice exhibited no signs of head tossing/bobbing or circling. The trunk curl test showed that they were able to reach out to a presented horizontal surface as their wild-type littermates. They could also swim. These observations suggested that Pdzd7−/− mice had grossly normal vestibular function (17). To examine the auditory function of Pdzd7−/− mice, we performed the auditory brainstem response (ABR), distortion product otoacoustic emission (DPOAE) and cochlear microphonics (CM) tests. Compared with wild-type littermates, Pdzd7−/− mice were profoundly deaf at 3–4 weeks of age with severely elevated ABR and DPOAE thresholds across all frequencies tested (Fig. 2A–C). The increased DPOAE thresholds indicated abnormal outer hair cell (OHC) function. Pdzd7−/− mice showed reduced CMs at 3 weeks of age (Fig. 2D and E), suggestive of defective mechanotransduction. Three weeks of age is the time point when hair bundles and hair cells become functionally mature. Therefore, PDZD7 is involved in hair cell development and loss of its expression causes congenital profound deafness.
Figure 2.

Pdzd7−/− mice have congenital profound deafness. (A) Representative ABR responses from a pair of wild-type (WT) and Pdzd7−/− (KO) littermates at P21 in response to click stimuli. (B) ABR response thresholds at different test frequencies measured from wild-type and Pdzd7−/− littermates at P21–P28. (C) DPOAE response thresholds at different test frequencies measured from wild-type and Pdzd7−/− littermates at P21–P28. (D) Representative CMs from a pair of wild-type and Pdzd7−/− littermates at P21. (E) CM amplitudes at different click levels measured from wild-type and Pdzd7−/− littermates at P21–P28. Numbers in the parentheses are the numbers of mice tested. Error bars represent the SE.
PDZD7−/− mice have defective hair bundles in cochlear OHCs
Light microscopy showed that the gross morphology of the cochlea in Pdzd7−/− mice was indistinguishable from that in wild-type littermates at 1 month of age. The size and dimension of the cochlea and the arrangement of one-row inner hair cells (IHCs) and three-row OHCs appeared normal (data not shown). Staining of the kinocilium, using an antibody against acetylated α-tubulin, and staining of the stereociliary bundle, using phalloidin as a marker of actin filaments, revealed that planar cell polarity appeared close to normal in both IHCs and OHCs in Pdzd7−/− mice (Figs 3A and 4A). According to the above observations, hair cell proliferation and early differentiation may not be significantly affected in Pdzd7−/− mice. However, abnormal morphology of hair bundles was observed in the cochlear OHCs of Pdzd7−/− mice at postnatal day 4 (P4) and in adulthood (Figs 3 and 4 and Supplementary material, Tables S2 and S3). At P4, kinocilia were mislocalized in ∼6.6% of OHCs, and ∼30% of hair bundles had missing stereocilia (Fig. 3A and D and Supplementary Material, Table S2). These findings were further confirmed by scanning electron microscopy (SEM; Fig. 3B, Cc, Cd, Ce and D and Supplementary Material, Table S2). Moreover, SEM revealed details of hair bundle morphological defects at a high magnification. At P4, ∼78% of Pdzd7−/− OHCs had hair bundles bent at their base and tilting toward the shortest row of stereocilia (Fig. 3B, Cb, Cd, Cf, Cg and D and Supplementary Material, Table S2), while Pdzd7+/− littermates had normal stereocilia morphology indistinguishable from the wild-type (Fig. 3B and Ca and Supplementary Material, Table S2). The tilted stereocilia precluded us from further examining the stereocilia in the shorter rows. At the age of 1 month, less OHCs (∼29%) showed fallen stereocilia (Fig. 4E and Supplementary Material, Table S3), ∼5% of OHC bundles completely disappeared along the cochlea (Fig. 4B and E and Supplementary Material, Table S3) and ∼49% of OHC bundles had splayed and twisted stereocilia (Fig. 4Cb, Ce and E and Supplementary Material, Table S3). Further examination found stereocilia with abnormal length and thickness (Fig. 4Cc and Cf) and loss of stereocilia in the shortest row (Fig. 4Cc, Cd and Ce) in Pdzd7−/− OHC bundles. Therefore, knockout of Pdzd7 results in defects in OHC hair bundle morphogenesis, which subsequently leads to disorganization, abnormal dimensions and eventual loss of stereocilia in adult cochlear outer hair bundles. In IHCs, the morphological defects in Pdzd7−/− hair bundles appeared less obvious at P4 and at 1 month of age (Figs 3B and 4D), which may be because of inherent differences in the shape and organization of IHC bundles compared with OHC bundles.
Figure 3.

Pdzd7−/− mice have disorganized outer hair bundles in the cochlea during development. (A) Staining of phalloidin (green) and acetylated α-tubulin (red) demonstrated the close to normal orientation but disrupted morphology of hair bundles (phalloidin) in the Pdzd7−/− cochlea at P4. Arrows point to kinocilia (acetylated α-tubulin) in hair bundles. Some of them moved to the side instead of being in the middle of the V-shaped hair bundles in the knockout. (B) SEM shows the morphology of hair bundles in the cochlear mid turn of Pdzd7+/− (control) and Pdzd7−/− littermates at P4. (C) High magnification view of hair bundles at P4. Compared with the Pdzd7+/− outer hair bundle (a), Pdzd7−/− outer hair bundles showed stereocilia tilting at the base toward the cell center (b, d, f and g, different regions along the cochlea), missing stereocilia (d and e) and mislocalized kinocilia (c and d). The simultaneous view of tilting OHC bundles and standing IHC bundles in Cg indicated that the tilting phenotype was not an artifact due to the imaging angle. The tilting stereocilia completely blocked the view of the two rows of shorter stereocilia. In some Pdzd7−/−OHCs, undifferentiated microvilli remained on the apex of the cells (b, c, f and g). Arrows point to kinocilia. (D) Quantification of hair bundles with mislocalized kinocilia, partial hair bundles and tilted hair bundles from SEM and fluorescence staining analyses. Details are shown in Supplementary material, Table S2. Scale bars, 5 µm (A and B) and 1 µm (C).
Figure 4.

Pdzd7−/− mice show stereocilia degeneration in mature cochlear outer hair bundles. (A) The cochlear mid turns from Pdzd7+/+ and Pdzd7−/− littermates at 1 month of age were stained with phalloidin. Empty arrowheads point to the phalloidin-labeled hair bundles. (B–E) SEM analysis of hair bundles from Pdzd7+/+ and Pdzd7−/− littermates at 1 month of age. (B) Low magnification images of OHC bundles. The filled arrowhead points to a missing hair bundle. (C) Images of individual OHC bundles. Compared with the Pdzd7+/+ hair bundle (a), Pdzd7−/− hair bundles had several obvious defects: (i) the stereocilia were separated with each other (filled arrowheads in b and e); (ii) some neighboring stereocilia twisted against each other (empty arrowhead in b); (iii) the length of the stereocilia in the same row varied (filled arrowheads in c); (iv) the stereocilia in the shortest row were missing (empty arrowheads in c, d and e); (v) the stereocilia tended to lie down (filled arrowheads in e); (v) the stereocilia had different diameters in the same row (filled arrowheads in f). (D) Images of IHC bundles from Pdzd7+/+ and Pdzd7−/− littermates. (E) Quantification of defective OHC bundles from the SEM analysis. Details are shown in Supplementary material, Table S3. Scale bars, 20 µm (A), 5 µm (B) and 1 µm (C and D).
PDZD7−/− mice have defective MET in cochlear OHCs
Examination of mechanoelectrical transduction (MET) currents in cochlear OHCs of Pdzd7−/− mice at P4–P6 revealed a significant decrease in peak current amplitude, as well as a significant broadening of the 10–90% operating range relative to wild-type controls. MET currents were recorded from 16 OHCs from seven Pdzd7−/− mice and 8 OHCs from five wild-type mice. Representative families of transduction currents are shown in Figure 5 for wild-type (Figure 5A) and Pdzd7−/− (Figure 5B) OHCs. Loss of the Pdzd7 gene resulted in an average peak current reduction of 52% (Fig. 5C and D). We also found that the current reduction was accompanied by a reduction in sensitivity. Sensitivity was assayed from stimulus–response relationships (Fig. 5C), which were generated from peak transduction currents plotted as a function of stimulus amplitude and fitted with second-order Boltzmann equations. We measured sensitivity as the 10–90% operating range for wild-type and Pdzd7−/− OHCs and found an average increase of 0.22 µm (33%) in Pdzd7−/− OHCs (Fig. 5E), which indicated reduced sensitivity.
Figure 5.
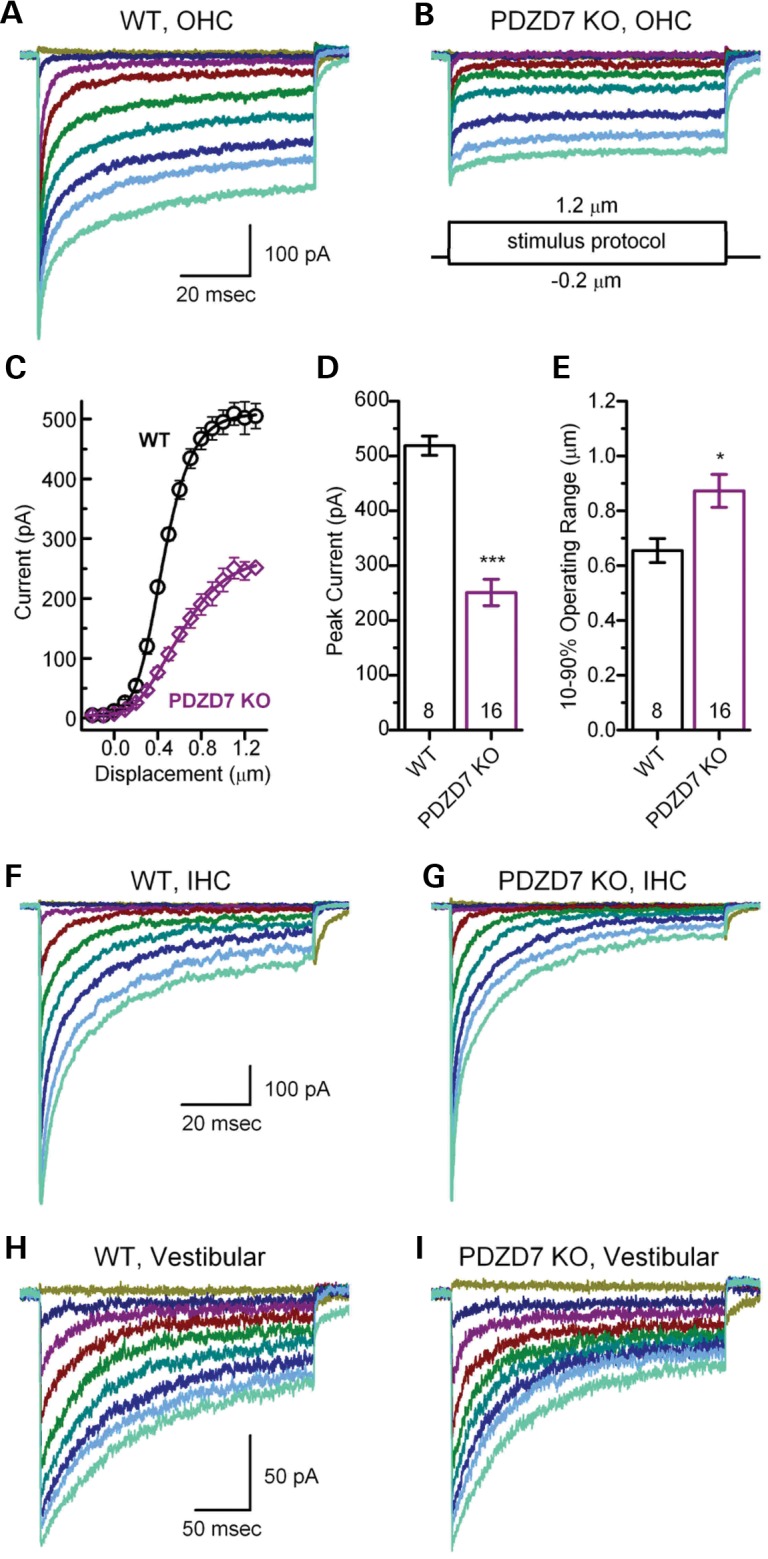
MET currents are abnormal in Pdzd7−/− cochlear OHCs. (A and B) Representative families of MET currents recorded from OHCs at P4 in response to mechanical hair bundle deflections that ranged from −0.2 to 1.2 µm in 0.05 µm increments. The scale bar and stimulus protocol, which shows the stimulus envelope, apply to both current families. Currents were recorded in voltage-clamp mode at −64 mV from the mid-apex. (C) Mean OHC stimulus response relations (n = 8 WT cells, 5 mice, circles; n = 16 KO cells, 7 mice, diamonds). Peak transduction currents were taken from current families and plotted as a function of stimulus amplitude. The data were shifted along the x-axis to align their mid-points, then averaged. The average data (±SE) were fitted with Boltzmann equations (lines). (D) Mean maximal MET currents (±SE) from OHCs. Genotype and number of hair cells are shown at the bottom. (E) Mean 10–90% operating range (±SE) was estimated from individual stimulus–response relations, like the one shown in (C). Genotype and number of OHCs are shown at the bottom. (F and G) Representative families of MET currents recorded from IHCs at P4 in response to mechanical hair bundle deflections. Currents were recorded in the voltage-clamp mode at −84 mV from the basal end. The scale bar applies to both panels. (H and I) Representative families of MET currents recorded from utricle type II hair cells at P4 in response to mechanical hair bundle deflections. Currents were recorded in the voltage-clamp mode at −64 mV. The scale bar applies to both panels.
We analyzed adaptation in Pdzd7−/− OHCs using current traces evoked by a half maximal stimulus, but found no significant differences relative to wild-type. Fast adaptation time constants based on double exponential fits were 0.7 ± 0.6 ms (n = 16 KO OHCs, 7 mice) and 0.8 ± 0.6 ms (n = 8 WT OHCs, 5 mice), respectively. Slow time constants for Pdzd7−/− OHCs (18 ± 13 ms) were not significantly different (P = 0.09) from wild-type controls (9 ± 7 ms).
Next, we examined cochlear IHCs from wild-type and Pdzd7−/− mice (Fig. 5F and G). We found no significant difference (P = 0.8) in the maximal current amplitudes between Pdzd7−/− (695 ± 102 pA, n = 14 IHCs, 2 mice) and wild-type (706 ± 126 pA, n = 16 IHCs, 4 mice) IHCs and there were no differences in sensitivity or adaptation between the two genotypes, consistent with the lack of morphological defects in Pdzd7−/− IHCs.
Lastly, we recorded from type II VHCs of the utricle, an otolith organ sensitive linear head movements and gravity. There was little difference in MET amplitude (Fig. 5H and I) between VHCs of wild-type (163 ± 17 pA, n = 10 VHCs, 9 mice) and Pdzd7−/− (182 ± 15 pA, n = 11 VHCs, 7 mice) animals, and we found no differences in adaptation or hair bundle sensitivity. These results were consistent with the lack of morphological defects and the lack of balance problems in Pdzd7−/− mice.
In summary, the Pdzd7−/− OHCs exhibited reduced MET currents and reduced sensitivity without a total loss of function, whereas IHCs and VHCs had normal MET. The data suggest that the structural defects in outer hair bundles are the primary deficits in the inner ears of Pdzd7−/− mice and that the reduction in MET current and decreased sensitivity are secondary consequences of the structural defects. Indeed, if the primary defect was in the mechanotransduction machinery itself, it might be expected to present in all hair cell types as is the case for Tmc1 and Tmc2 knockout mice (18). However, we found that both IHCs and VHCs of Pdzd7−/− mice retained structural integrity and normal mechanotransduction. An MET deficit that is restricted to OHCs is consistent with the reduced CMs and the elevated DPOAE and ABR thresholds we documented in Pdzd7−/− mice (Fig. 2).
PDZD7 expresses its full-length isoform at the protein level during development
Several splice variants of PDZD7 are expressed in both the inner ear and the retina (13,14). To thoroughly investigate the expression of PDZD7 at the mRNA and protein levels, we carried out RT–PCR and western blotting analyses. RT–PCR using primers located on the first and last (exon 16) exons of the Pdzd7 gene (Supplementary Material, Table S1) identified five Pdzd7 splice variants from 19 independent clones in the P30 wild-type retina (Supplementary Material, Fig. S2). All five Pdzd7 splice variants were predicted to be translated into proteins with stop codons after the second and before the third PDZ domain of PDZD7. Therefore, N-terminal but not full-length Pdzd7 splice variants were detected in the adult mouse retina. The same RT–PCR approach was used in the P5 and P24 cochleas. Both full-length and N-terminal Pdzd7 splice variants were found at these ages (Supplementary Material, Fig. S2). The prevalence of the full-length Pdzd7 variant was very low, 1 in 11 at P5 and 1 in 5 at P24. N-terminal Pdzd7 variants found at these two time points were all predicted to be translated into proteins with the N-terminal two PDZ domains similar to those in the retina.
Western blotting analysis was conducted in the retina and cochlea using all the PDZD7 antibodies we generated. Compared with the Pdzd7−/− tissues, which we considered a negative control, we could not find PDZD7-specific bands in the wild-type. However, as mentioned above, we were able to find a specific weak band of ∼120 kDa in the immunoprecipitates of the rabbit PDZD7_C antibody from the cochlea at P5–P9 and the retina at P6 (Fig. 1F). This weak band was not found in the adult retina or cochlea using the same approach. These results indicated that the full-length PDZD7 was probably expressed in the cochlea and retina at a low level during development and may be expressed at an even lower level in the adult cochlea. Furthermore, we could not find any PDZD7-specific bands in the immunoprecipitates of the rabbit PDZD7_N antibody from the retina or cochlea at various time points, even though this antibody could be successfully used for immunoprecipitation in cultured cells (Fig. 8B). Therefore, if N-terminal PDZD7 variants existed at the protein level, their expression might be very rare throughout the development and adulthood.
Figure 8.

PDZD7 and WHRN directly interact in HEK293 cells. (A) PDZD7 was coimmunoprecipitated with (green fluorescent protein) GFP-tagged WHRN from HEK293 cells double transfected with these two proteins. Immunoprecipitation (IP) was performed using a rabbit GFP antibody (rGFP). Cells double transfected with GFP and PDZD7 were used as a negative control. Left, the success of IP was confirmed by western blotting (WB) using the rGFP antibody. Right, the presence of PDZD7 in the immunoprecipitate was detected by western blotting using the chicken antibody against PDZD7_C (cPDZD7_C). (B) WHRN was coimmunoprecipitated with PDZD7 from HEK293 cells double transfected with these two proteins. IP was performed using the rabbit antibody against PDZD7_N. Cells transfected with only WHRN were used as a negative control. Left, the success of IP was confirmed by western blotting using the chicken antibody against PDZD7_C. Right, the presence of WHRN in the immunoprecipitate was detected by western blotting using a chicken antibody against the C-terminal region of WHRN (cWHRN_C). The filled arrowheads on the right of blots point to the targeted proteins. The empty arrowhead points to GFP bands. The asterisk labels the rabbit GFP antibody bands.
PDZD7 is located at ankle links in developing inner ear hair cells, but its localization cannot be determined in photoreceptors
Immunostaining with phalloidin demonstrated that PDZD7 was present at the base of hair bundles in cochlear IHCs and OHCs from P0 to P10 (Fig. 6A) and in VHCs at P8 (Fig. 6B). GPR98 and WHRN have been localized to ankle links at the base of hair bundles (10,11). To investigate whether PDZD7 was also localized at ankle links, we performed double immunostaining of PDZD7 with GPR98 and WHRN, respectively. PDZD7 signal was colocalized with GPR98 in both IHCs and OHCs (Fig. 6C). Pearson's coefficients were 0.88–0.91 for OHCs and 0.88–0.94 for IHCs. PDZD7 signal also overlapped with WHRN at the base but not the tip of hair bundles (Fig. 6D). Pearson's coefficients were 0.66–0.77 for OHCs and 0.78–0.88 for IHCs. The fractions of WHRN signal overlapping PDZD7 signal (M2) were 0.52–0.75, which were probably mainly from the base of hair bundles. Therefore, colocalization of PDZD7 with GPR98 and WHRN at the base of hair bundles indicates that PDZD7 is present at ankle links in hair cells. Ankle links exist in cochlear hair cells during hair bundle development (19). Consistent with this time line, the staining signal of PDZD7 disappeared at the base of hair bundles in adult hair cells (P23 in Fig. 6A) and its expression could not be detected in mature cochleas by the previously mentioned western blotting and immunoprecipitation studies. This localization of PDZD7 at ankle links in mouse inner ear hair cells is in agreement with a recent finding in rats (15). Additionally, double staining using PDZD7_N and PDZD7_C antibodies gave staining patterns similar to each other (Fig. 6E). Pearson's coefficients were 0.83–0.84 for chicken PDZD7_C and rabbit PDZD7_N combination and 0.78–0.87 for rabbit PDZD7_C and chicken PDZD7_N combination. This result indicates that full-length PDZD7 and N-terminal PDZD7, if present, have similar subcellular distributions.
Figure 6.

PDZD7 is localized at ankle links in mouse inner ear hair cells. (A) Double staining of PDZD7 (red) and phalloidin (green) in cochlear hair cells. PDZD7 was localized at the base of hair bundles (phalloidin) in both IHCs and OHCs during P0 and P10. The PDZD7 signal disappeared at P23. (B) Double staining of PDZD7 (red) and phalloidin (green) in VHCs at P8. A Pdzd7−/− mouse (KO) was used as a negative control. (C) Triple staining demonstrated that PDZD7 (red) was colocalized with GPR98 (blue) at ankle links of hair bundles (phalloidin, green) in cochlear IHCs and OHCs at P4. Note that the four cells at the low magnification (left) and one cell at the high magnification (right) are five different cells for IHCs and OHCs. Correlation analyses between PDZD7 and GPR98 were conducted using images at both low and high magnifications. Pearson's and Manders' coefficients are shown below images. (D) Double staining demonstrated that PDZD7 (green) was colocalized with WHRN (red) at ankle links but not at the tip (arrows) of stereocilia in cochlear IHCs and OHCs at P4. Single cells in the enlarged view (right) were chosen from the four cells in the low magnification view (left) for IHCs and OHCs. Correlation analyses between PDZD7 and WHRN were conducted using images at both low and high magnifications. Pearson's and Manders' coefficients are shown below images. (E) Costaining of PDZD7_N and PDZD7_C in P4 cochlear hair cells. Left, low magnification images of both IHCs and OHCs double labeled with two combinations of PDZD7_N and PDZD7_C antibodies. Right, images of individual IHCs triple labeled with PDZD7_N, PDZD7_C and phalloidin (green). These individual cells were not chosen from the low magnification images. The colors of PDZD7_N and PDZD7_C signals match those of their word labels. Correlation analyses between signals of PDZD7_N and PDZD7_C were conducted using images at both low and high magnifications. Pearson's and Manders' coefficients are shown below images. r, Pearson's coefficient; M1, Manders' coefficient, fraction of green or blue signals overlapping red signals; M2, Manders' coefficient, fraction of red signals overlapping green or blue signals; cPDZD7_C, chicken antibody against PDZD7_C; rPDZD7_N, rabbit antibody against PDZD7_N; rPDZD7_C, rabbit antibody against PDZD7_C; cPDZD7_N, chicken antibody against PDZD7_N; Pha, phalloidin; Scale bars, 5 µm (A, low magnification images in C, D and E), 10 µm (B) and 1 µm (single cell images in C, D and E).
In the adult retina, immunostaining using both rabbit and chicken PDZD7_N antibodies found a strong signal between the outer and inner segment next to but not exactly overlapping with the WHRN signal. This signal pattern was similar to that reported previously (14). However, we found the same signal pattern in Pdzd7−/− mice, indicating that this signal is not PDZD7-specific (data not shown). Immunostaining using rabbit and chicken PDZD7_C antibodies did not detect any obvious signals. These results support the notion that PDZD7 expression may be too low to be detected in the adult mouse retina. Currently, the localization of PDZD7 in the retina is unclear.
PDZD7 determines the subcellular localization of GPR98, WHRN and USH2A in cochlear hair cells but not in photoreceptors
We examined whether PDZD7 played a role in the normal localization of USH2 proteins and in maintaining the integrity of the USH2 complex. Immunostaining showed a clear signal distribution of GPR98, WHRN and USH2A at ankle links and additional WHRN at the tip of the hair bundle in wild-type cochlear IHCs and OHCs at P4 (Fig. 7A–C). However, in the Pdzd7 knockout of the same age, GPR98 moved up along the stereocilia and about half of GPR98 was present at the tip of stereocilia in both IHCs and OHCs (Fig. 7A, Supplementary Material, Fig. S3A and Table S4). The localization of WHRN at ankle links was largely eliminated in the cochlear IHCs and OHCs (Fig. 7B, Supplementary Material, Fig. S3B and Table S4). The changes in USH2A distribution were not exactly the same between IHCs and OHCs (Fig. 7C, Supplementary Material, Fig. S3C and Table S4). In 38% of the OHCs examined, USH2A signals, like GPR98, were moved to the tip of the stereocilia. The rest OHCs showed diffused USH2A signals at the base of the hair bundle. In IHCs, most USH2A signals disappeared, and only residual USH2A could be detected at ankle links. These mislocalizations of USH2 proteins in Pdzd7−/− hair bundles were believed not due to stereocilia degeneration, which was not severe at P4 as shown in the SEM study (Fig. 3). The distributions of MYO7A and SANS, the two proteins encoded by the USH1B and USH1G genes, were normal. MYO7A was located along the entire stereocilia in IHCs and at the apical region of the stereocilia in OHCs at P6 (Supplementary Material, Fig. S4A), while SANS was present at the apical region of the stereocilia in both IHCs and OHCs at P4 (Supplementary Material, Fig. S4B). MYO15A is a WHRN-interacting protein and localized at the tip of stereocilia (20). Knockout of Pdzd7 did not change the localization of MYO15A in hair cells (Supplementary Material, Fig. S4C). These findings demonstrated that PDZD7 was essential for the localization of USH2 proteins at ankle links in cochlear IHCs and OHCs. In addition, these results indicate that the role of PDZD7 in the localization of USH2 proteins may not be exactly the same in IHCs and OHCs. PDZD7 was previously reported to interact directly with GPR98 and USH2A (14). To investigate whether a direct interaction also existed between PDZD7 and WHRN, we transfected PDZD7 and WHRN into HEK293 cells. The reciprocal coimmunoprecipitation of these two proteins from the transfected cell lysate provided evidence to support their direct interaction (Fig. 8). Therefore, PDZD7 determines the subcellular localization of USH2 proteins probably through its direct interactions with these proteins.
Figure 7.

USH2 proteins are mislocalized in Pdzd7−/− cochlear hair cells. (A) GPR98 (red) was present at ankle links of hair bundles (phalloidin, green) in Pdzd7+/+ (WT) cochlear hair cells. In Pdzd7−/− mice (KO), only a small amount of GPR98 remained at ankle links. Most GPR98 moved upward toward the tip or along the stereocilia in IHC and OHC bundles. (B) WHRN (red) was present at the tip and ankle links of hair bundles (green, phalloidin) in Pdzd7+/+ cochlear hair cells. In Pdzd7−/− cochleas, the WHRN signals at the hair bundle tip stayed unchanged, while the signals at ankle links largely disappeared. (C) USH2A (red) was present at ankle links of the hair bundle (green, phalloidin) in Pdzd7+/+ cochlear hair cells. In about half of Pdzd7−/− OHCs, partial USH2A signal was mislocalized to the tip of the hair bundle, while the rest was still present at ankle links. In Pdzd7−/− IHCs, USH2A signals were significantly reduced at ankle links. Arrowheads point to the base of hair bundles. Scale bars, 1 µm.
Immunostaining signals of USH2A, WHRN and GPR98 in Pdzd7−/− photoreceptors were similar to those in wild-type photoreceptors. They were present at the periciliary membrane complex between the rootlet and the axonemal microtubules, labeled by antibodies against rootletin and acetylated α-tubulin, respectively (Fig. 9A–C). Therefore, PDZD7 plays a limited role in localizing USH2 proteins in photoreceptors. Additionally, both scotopic and photopic electroretinogram responses, which represent the vision function mainly from rod and cone photoreceptors, respectively, appeared normal in Pdzd7−/− mice when compared with their wild-type or heterozygous littermates at 1 month of age (data not shown). Because of the presence of an Rd8 mutation in the genetic background of our Pdzd7−/− mice, we could not examine the effect of Pdzd7 knockout on the vision function at older ages (21).
Figure 9.

USH2 proteins remain at the periciliary membrane complex in Pdzd7−/− photoreceptors. Compared with wild-type (Pdzd7+/+) photoreceptors, the distributions of WHRN (green, A), USH2A (green, B) and GPR98 (green, C), relative to the ciliary rootlet (rootletin, red, left panels) and the axonemal microtubules (acetylated α-tubulin, red, right panels), appeared normal in Pdzd7−/− photoreceptors. Similar results were obtained from three pairs of Pdzd7+/+ and Pdzd7−/− mice. Scale bars, 5 µm.
USH proteins play variable roles in the localization of PDZD7 in cochlear hair cells
Immunofluorescence was performed to study the localization of PDZD7 in a series of USH mutant mouse models (Fig. 10 and Supplementary Material, Fig. S5). Two Whrn mutant mouse lines were available in our laboratory. The whirler mouse (Whrnwi/wi) had a naturally occurring deletion between exons 6 and 9 of the Whrn gene (22). The Whrn targeted mutant mouse line (Whrnneo/neo) was generated by replacement of the 3′ part of exon 1 with a neomycin cassette (8). PDZD7 localization appeared close to normal in the majority of hair cells in both Whrn mutant mouse lines at P4 (Fig. 10A–C, Supplementary Material, Fig. S5A–C and Table S5). In a small proportion of hair cells in these two mutant mice, PDZD7 showed a slightly diffuse pattern at the base of OHC bundles and reduced signals in IHCs. In Gpr98 mutant mice at P4, PDZD7 signals were not restricted to the base of the hair bundle but diffused upward (Fig. 10D, Supplementary Material, Fig. S5D and Table S5). In IHCs, it was sometimes distributed along the entire stereocilia. In Ush2a knockout mice, PDZD7 distribution was similar to that found in Gpr98 mutant mice at P4 (Fig. 10E, Supplementary Material, Fig. S5E and Table S5). The PDZD7 signals eventually disappeared in OHCs at P7 (data not shown). In Sans2J2J mutant mice, which had a deletion in the Ush1g gene, the subcellular localization of PDZD7 was normal in both IHCs and OHCs (Fig. 10F, Supplementary Material, Fig. S5F and Table S5). Therefore, the localization of PDZD7 depends on GPR98 and USH2A, while slightly relying on WHRN. Localization of PDZD7 is independent on SANS.
Figure 10.

Localization of PDZD7 relies on USH2 proteins but not SANS in cochlear hair cells. Compared with wild-type mice (WT, A), distribution of PDZD7 appeared normal or slightly diffuse at the base of the hair bundle in IHCs and OHCs of both whirler (Whrnwi/wi, B) and Whrn targeted mutant (Whrnneo/neo, C) mice. In Gpr98−/− (D) and Ush2a−/− (E) mice, PDZD7 was mislocalized along the stereocilia in both IHC and OHC bundles. In Sans2J/2J mice (F), although the hair bundle was severely disrupted, the localization of PDZD7 at ankle links was normal. Mutant mice at P4 were shown in all the panels. Red, signals from the chicken antibody against PDZD7_C; Green, hair bundles labeled with phalloidin. Arrowheads point to the base of hair bundles. Scale bars, 1 µm.
On embryonic day 18 (E18), PDZD7 and USH2A were found to express in the wild-type cochlear IHCs and OHCs, while WHRN was not (Supplementary Material, Fig. S6). The signal patterns of PDZD7 and USH2A at this age were similar to that of GPR98 on E17 reported previously (10). In IHCs, they were at the base of the emerging hair bundle. In OHCs, they exhibited circular or semi-circular signals on the cellular apex. E17 is the time point before the emergence of ankle links in mouse cochlear hair cells. Therefore, PDZD7, USH2A and GPR98 but not WHRN may depend on each other and be crucial for the initial formation of the USH2 complex during the emergence of ankle links.
Digenic heterozygosity of PDZD7 with USH genes does not affect hearing function in young mice
Based on the previous report that PDZD7 contributed to digenic USH with USH genes (14), it was possible that digenic heterozygous mutant mice of Pdzd7 and USH genes had impaired hearing function. To test this possibility, we performed ABR tests in digenic heterozygous mice of Pdzd7 with Ush2a, Gpr98, Whrn or Sans, respectively. No elevated ABR thresholds were found in any of these digenic heterozygous mutant mice at P24 compared with their control single heterozygous littermates (Supplementary Material, Fig. S7). The involvement of Cdh23ahl allele in some of the digenic heterozygous colonies precluded us from testing ABRs at older ages. Therefore, these digenic heterozygous mutant mice did not exhibit congenital hearing impairment, which is typically found in human clinical cases of USH1 and USH2.
DISCUSSION
In mouse cochlear hair cells, we show that PDZD7 is localized at ankle links of the hair bundle during development, which confirms a recent study on PDZD7 in rats (15). Importantly, we discovered the dependence of USH2 proteins on PDZD7 for their normal localization in cochlear hair cells. In these cells, PDZD7 appears to be an organizer of the USH2 complex upstream of WHRN, another organizer of the USH2 complex. Therefore, disruption of PDZD7 expression affects the integrity of the USH2 complex in hair cells, which is believed to be the primary cause of disorganized hair bundles, abnormal MET and eventual profound deafness. The functional significance of PDZD7 as an organizer of the USH2 complex in photoreceptors appears to be less than in hair cells. Therefore, our findings shed light on the molecular basis underlying the pathogenesis of non-syndromic hearing impairment, retinal diseases and digenic USH2, caused or contributed to by PDZD7 mutations. Additionally, this study provides evidence supporting the notion that the formation and composition of the USH2 complex are not exactly the same in cochlear hair cells as in photoreceptors.
The localization of PDZD7 at ankle links in the inner ear hair cells is supported by the following lines of evidence. First, the spatiotemporal expression profile of PDZD7 in cochlear hair cells is similar to that of ankle links in mice (19). Second, PDZD7 is colocalized with GPR98 and WHRN at the base of hair bundles in cochlear hair cells, where GPR98 is an important component of ankle links (10,11) and interacts with WHRN (8,10,15). Third, PDZD7 interacts directly with WHRN as well as two other USH2 proteins, GPR98 and USH2A (14,15). Fourth, localization of the three USH2 proteins at ankle links depends on the presence of PDZD7, and localization of PDZD7 also relies on GPR98 and USH2A. Therefore, our study now firmly establishes that PDZD7 is a novel and essential component of the USH2 complex, which was previously known to be comprised of USH2A, GPR98 and WHRN (8,10). PDZD7 and WHRN are paralogs sharing multiple protein–protein interaction modules, such as PDZ domains, a proline-rich region and a harmonin_N module. Both their PDZ domains are able to bind to the PBMs of USH2A and GPR98 (8,12,23). Loss of WHRN causes mislocalization of USH2A in cochlear hair bundles (10). Thus, both PDZD7 and WHRN play an important role in the formation of the USH2 complex in vivo. However, the dependence of WHRN on PDZD7 to localize at ankle links (Fig. 7B) but not vice versa (Fig. 9B and C) indicates that PDZD7 plays a more upstream role than WHRN in this function. In addition, PDZD7 and SANS have been shown to interact directly (13) and are colocalized in cultured cells (15). However, we found that PDZD7 and SANS are not mutually dependent for their normal localization in hair bundles in the cochlea (Fig. 10 and Supplementary Material, Fig. S4B). This finding indicates that the interaction between PDZD7 and SANS may have little significance in vivo. Moreover, MYO7A does not rely on PDZD7 for its localization in hair bundles as well. Therefore, PDZD7 may not be important for the USH1 protein network in the cochlear hair bundle.
To date, several other proteins have been shown to interact with USH2 proteins and/or to be present at ankle links in hair cells. They include vezatin, espin and adenylyl cyclase 6 (10,24). Vezatin is an integral membrane protein associated with actin filaments and binds to the cytoplasmic region of USH2A (10,25). Espin interacts with WHRN, and this interaction has been shown to modulate the actin-binding/bundling activity of espin (24). Adenylyl cyclase 6 has been proposed to function downstream of GPR98 signaling, because loss of GPR98 affects the expression and localization of adenylyl cyclase 6 at ankle links (10). In view of these known and predicted functions of the proteins, the USH2 complex at ankle links could have a role related to actin bundle formation/dynamics in the stereocilia during hair bundle development. Actin bundles are present immediately beneath the plasma membrane of the stereocilia and provide crucial support to the stereocilia. Without the fully functional USH2 complex, the actin bundles could be less organized or less rigid to support the stereocilia, which could explain why many stereocilia are bent or fallen at the base of hair bundles in Pdzd7−/− mice, especially during development. Similar defects are also seen in the pdzd7a knockdown zebrafish and other USH2 mutant mice (10,11,14). Additionally, in wild-type mice, the stereocilia in OHC hair bundles seem to be more organized than in IHC hair bundles (Fig. 4C and D). In Ush2a, Gpr98, Whrn, Pdzd7 mutant mice (except the whirler mouse), hair bundle morphology and physiology are usually more severely affected in OHCs than in IHCs (8,10,11,26) (this study). This suggests that the USH2 complex plays a more important role in OHCs and it is probably involved in the organization of stereocilia in hair bundles. Furthermore, normal mechanotransduction found in Pdzd7−/− IHCs and VHCs supports and extends the conclusion obtained from previous studies on two different Gpr98 mutant mice (10,11) that ankle links and the USH2 complex do not directly participate in mechanotransduction. We believe that the abnormal mechanotransduction at P4 and the reduced CMs at P21–P30 found in Pdzd7−/− mice are secondary to the defective hair bundle organization. Finally, although USH2 proteins and PDZD7 are localized at ankle links in VHCs as well (11,15,27) (this study), it is unclear why vestibular function is unaffected in most USH2 mutant mice (all except the whirler mouse), Pdzd7−/− mice or USH2 patients.
PDZD7 expression varies in different tissues, with both N-terminal and full-length transcripts in the cochlea and probably only N-terminal transcripts in the mature retina. This expression difference may explain why the homozygous reciprocal translocation, 46,XY,t(10;11),t(10;11), causes hearing impairment but not retinal symptoms (13) and why the mutation, R56PfsX, accelerates and exacerbates the retinal symptoms in USH2A patients (14). The chromosomal translocation mutation creates a breakpoint in intron 10 of the PDZD7 gene, which should affect the full-length PDZD7 transcript but not the N-terminal PDZD7 transcripts (Supplementary Material, Fig. S2). In contrast, the mutation, R56PfsX, probably affects the expression of N-terminal PDZD7 in the retina and both the N-terminal and full-length PDZD7 in the cochlea (Supplementary Material, Fig. S2). At the protein level, only the full-length PDZD7 transcript was detectable during development in this study. However, we cannot rule out the existence of the N-terminal PDZD7 proteins. They may exist transiently or at an extremely low level, which may be a reason why we could not determine the localization of PDZD7 in photoreceptors. Interestingly, in the absence of PDZD7, the localization of the three USH2 proteins appears to be unperturbed in photoreceptors (Fig. 9). Therefore, unlike in the cochlear hair cells, PDZD7 may play a less important role in photoreceptors. Considering our previous findings on USH2 proteins in photoreceptors (8,9), we believe that WHRN but not PDZD7 is indispensible for the USH2 complex formation in photoreceptors.
In summary, this study demonstrates that PDZD7, a component of the USH2 complex at ankle links, plays an essential role in organization of this complex in developing cochlear hair cells, while PDZD7 is not directly involved in MET. Loss of PDZD7 expression leads to congenital hearing loss due to disorganized cochlear outer hair bundles. The function of PDZD7 in cochlear IHCs and VHCs may be subtle or redundant. In photoreceptors, the organization of the USH2 complex is probably different from that in cochlear hair cells. These findings provide valuable new insights into the molecular mechanism underlying deafness and blindness caused by mutations in PDZD7.
MATERIALS AND METHODS
Animals
All experiments involving animals were approved by the Institutional Animal Care and Use Committee at the University of Utah and in accordance with a protocol approved by the Institutional Animal Care and Use Committee of the Boston Children's Hospital (Protocol 11-04-1959).
Pdzd7tm1a(EUCOMM)Wtsi embryonic stem cells were purchased from EUCOMM and microinjected into blastocysts of an albino C57BL6 strain (Jax stock # 000058). The chimeras mice were backcrossed with the same strain of albino C57BL6 mice to generate Pdzd7 heterozygous mutant mice. This work was performed by the University of Utah Transgenic and Gene Targeting mouse core. The Pdzd7 homozygous mutant mice were obtained by mating the heterozygous mutant mice. To confirm the insertion of the large cassette into the Pdzd7 gene, PCRs were conducted using the genomic DNA isolated from the liver of the mutant mouse and primers located outside the 5′ and 3′ homology arms of the targeting vector (Fig. 1A and B and Supplementary Material, Table S1). The insertion of the third loxP between exons 5 and 6 was verified by sequencing the PCR product containing this loxP site. The routine genotyping of this mutant mouse was conducted by short range PCRs as shown in Figure 1C.
WHRN targeted mutant (Whrntm1Tili, referred to as Whrnneo in this paper), whirler (Whrnwi) and Ush2a knockout (Ush2atm1Tili, referred to as Ush2a−/− in this paper) mice were described previously (8,22,26). Gpr98 mutant mice (Mass1Frings, referred to as Gpr98−/− in this paper) were generated by crossing the BUB/BnJ mice (Jax stock #000653) with wild-type mice (mixed genetic background of C57BL6 and 129sv). The obtained Gpr98−/− mice were free of the Pderd1 mutation. Sans mutant mice (B6(Cg)-Ush1gjs−2J/J, referred to as Sans2J/2J in this paper) were purchased from the Jackson Laboratory (Jax stock #006111).
Total RNA isolation, RT–PCR and DNA constructs
Total RNA was isolated from the mouse retina or cochlea at various time points using the TRIzol reagent (Life Technologies, Grand Island, NY, USA). RT–PCR was conducted using the ThermoScript™ RT-PCR system (Life Technologies). Manufacturer's instructions were followed exactly in the above two experiments. PDZD7 full-length cDNA (2–1021 amino acids, NP_001182194) was generated by RT–PCR from the mouse cochlear total RNA and inserted into the pcDNA3.1(−) and pEGFP-C1 plasmids. PDZD7 cDNA fragments were amplified from the PDZD7 full-length cDNA by PCR. PDZD7_N (1–316 amino acids, NP_001182194) and PDZD7_C (680–1021 amino acids, NP_001182194) cDNA fragments were inserted into the pET28 vector. PDZD7_N (1–308 amino acids, NP_001182194) and PDZD7_C (835–1021 amino acids, NP_001182194) cDNA fragments were inserted into the pEGFP-C1 vector. Harmonin a isoform (1–584 amino acids, NP_076138) cDNA was generated by RT–PCR from P5 mouse inner ear total RNA and inserted in the pcDNA3.1(−) plasmid. USH2A (5053–5193 amino acids, NP_067383), GPR98 (1212–2211 amino acids, NP_473394), WHRN (375–800 amino acids, NP_082916), SANS (1–461 amino acids, NP_789817, aka USH1G), MYO7A (1–997 amino acids, NP_001243010) and MYO15A (2997–3511 amino acids, NP_034992) cDNA fragments were generated from the mouse retinal total RNA by RT–PCR and cloned into the pET28 vector. WHRN (721–907 amino acids, NP_082916) cDNA was also generated from the mouse retinal total RNA by RT–PCR, but cloned into the pGEX4t-1 vector. Green fluorescent protein (GFP) cDNA was taken from the enhanced green fluorescent protein (pEGFP)-C1 vector and inserted into the pET28 vector. All DNA constructs made in this study were confirmed by DNA sequencing. The WHRN full-length cDNA in thepcDNA3.1(−) and pEGFP-C2 plasmids were described previously (24).
Antibodies
Recombinant His-tagged PDZD7_N, PDZD7_C, GFP, USH2A, GPR98, WHRN, SANS, MYO7A and MYO15A proteins or protein fragments were expressed in BL21-CodonPlus (DE3)-arginine isoleucine, proline, leucine cells after isopropyl beta-D-1-thiogalactogyranoside induction (Agilent Technologies, Santa Clara, CA, USA) using their pET28 constructs. They were purified by chromatography using a Ni2+-charged His•Bind resin column (EMD Millipore, Billerica, MA, USA). The purified recombinant proteins were used to immunize rabbits, while the purified PDZD7 proteins were also used to immunize chickens. The rabbit PDZD7, USH2A, GPR98, SANS, MYO7A, MYO15A and GFP antibodies were purified by sequentially flowing through a column cross-linked with a His-tag to clear antibodies against the His-tag and a column cross-linked with their corresponding antigens. The rabbit WHRN antibody was affinity-purified against the glutathione S-transferase-tagged WHRN fragment. The chicken PDZD7 antibodies were affinity-purified from egg yolk extracts by Aves Labs, Inc. (Tigard, OR, USA). The specificity of PDZD7, USH2A, GPR98, WHRN, SANS and MYO7A antibodies was confirmed by comparing their immunofluorescence between the wild-type and their respective mutant cochlear hair cells. The specificity of the GFP antibody was verified by comparing the western blotting signals of HEK293 cells transfected with GFP and empty vectors. The polyclonal rabbit rootletin antibody and chicken whirlin antibody were described previously (8,28). Monoclonal mouse antibodies against acetylated α-tubulin and actin were purchased from Sigma-Aldrich (St Louis, MO, USA). Hoechst dye 33 342, Alexa fluorochrome-conjugated phalloidin and secondary antibodies were obtained from Life Technologies. Horseradish peroxidase (HRP)-conjugated secondary antibodies were obtained from the Jackson ImmunoResearch Laboratories, Inc. (AffiniPure, West Grove, PA, USA).
Cell culture
HEK293 cells were cultured in the Dulbecco's modified Eagle's medium medium supplemented with 5% fetal bovine serum and 1% penicillin-streptomycin (Life Technologies). Transient transfections were carried out using the TurboFect™ in vitro transfection reagent (Fermentas Life Sciences, Glen Burnie, MD) according to the manufacturer's instructions. Cells were collected at ∼24 h after transfection.
Immunoprecipitation and western blotting
Transfected HEK293 cells, mouse retinas or mouse cochleas were homogenized in lysis buffer [50 mm Tris–HCl, pH 8.0, 150 mm NaCl, 0.5% Triton X-100, 5 mm ethylenediaminetetraacetic acid (EDTA), 0.5 mm phenylmethylsulfonyl fluoride, 1× protease inhibitor and 1 mm dithiothreitol] and cleared by centrifugation at 21 000g for 10 min. The resulting lysate was pre-incubated with protein G sepharose (GE Healthcare, Waukesha, WI, USA) for 1 h, cleared by centrifugation at 21 000g for 10 min, incubated with a primary antibody for 3.5 h, cleared again by centrifugation at 21 000g for 10 min and incubated with protein G sepharose for another 1 h. After a brief spin-down at 2000 g, the pellet was washed with lysis buffer four times and boiled in the Laemmli sample buffer for 10 min. All the above procedures were performed at 4°C. For western blotting, two retinas from one mouse, pooled cochleas from several mice, or transfected HEK293 cells were homogenized in an appropriate amount of the RadioImmune Precipitation Assay buffer [10 mm Tris–HCl, pH 7.5, 150 mm NaCl, 1% Triton X-100, 1% Deoxy Cholate sodium salt, 0.1% sodium dodecyl sulphate (SDS)] and boiled with the Laemmli buffer for 10 min. The resulting mouse tissue samples, cell lysates or the above-generated immunoprecipitates were separated by SDS–polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride (PVDF) membrane. The PVDF membrane was sequentially subjected to blocking for 1 h, primary antibody incubation overnight at 4°C and HRP-conjugated secondary antibody incubation for 1 h. The protein signals were developed using the chemiluminescent substrate and detected using the AlphaInnotech AlphaView software in a FluorChem Q machine (Cell Biosciences, Inc., Santa Clara, CA, USA).
Immunofluorescence staining
Enucleated mouse eyes were frozen immediately in the Tissue-Tek® OCT compound on dry ice and sectioned at 10 µm. The retinal sections were then fixed in 4% formaldehyde/phosphate-buffered saline (PBS) for 10 min and permeabilized by 0.2% Triton X-100/PBS for 5 min. The mouse cochlea at various time points was dissected, fixed in 4% formaldehyde/PBS from 30 min to overnight depending on the antibodies and permeabilized by 0.5% Triton X-100/PBS for 15–20 min. Additional incubations of the cochlea in 50 mm NH4Cl for 15 min and in tween tris buffered saline (20 mm Tris, pH 7.5, 150 mm NaCl, 0.1% Tween 20) for 10 min were performed for the USH2A and WHRN antibodies. The retinal sections or whole-mount cochleas were then blocked in 5% goat serum/PBS for 1 h, incubated with primary antibodies in 5% goat serum/PBS at appropriate dilution ratios at 4°C overnight, washed several times with PBS and then incubated with the Alexa fluorochrome-conjugated secondary antibodies in 5% goat serum/PBS for 1 h. For the retinal staining, Hoechst dye 33 342 was added to the secondary antibody mixture. For the cochlear staining, Alexa fluorochrome-conjugated phalloidin was added to the secondary antibody mixture. For double and triple staining, primary antibodies from different species were used. Alexa Fluor® 488, 594 and 647 secondary antibodies and phalloidin were followed. The stained retinal sections or cochleas were viewed and photographed using a confocal laser scanning microscope with a PLAPON 60XO objective (NA: 1.42) (Model FV1000, Olympus, Tokyo, Japan). Most images were captured using a zoom factor of 3–6 by the XY acquisition mode. Only a small number of images were z-stacked images. Pearson's and Manders' coefficients were calculated through correlation analysis using Image J plugin, JACoP (29).
Scanning electron microscopy
After transcardial perfusion with 2% glutaraldehyde/PBS, mouse cochleas were dissected, fixed in 2% glutaraldehyde/PBS for 2 h and decalcified in 100 mm EDTA for several days. The transcardial perfusion and decalcification steps were skipped for P4 pups. The cochleas were further dissected, rinsed in PBS and post-fixed by alternative incubations in 0.5% osmium tetroxide, water and 0.3% thiocarbohydrizide. Each step was 2 min long. Then the cochlear tissues were dehydrated in graded ethanol series and critical-point-dried. Images were taken using a Hitachi S-4800 scanning electron microscope. To quantify morphological defects of hair bundles, regions along the cochlea were randomly imaged. Defective hair bundles were counted from SEM images. The following criteria were used to define the defects: (i) mislocalized kinocilia were kinocilia not at the middle point of the symmetrical V-shaped hair bundle (Fig. 3Cc and Cd); (ii) partial hair bundles were hair bundles with one arm of the V-shape partially or completely missing (Fig. 3Cd and Ce); (iii) tilted hair bundles were hair bundles whose two short rows of stereocilia were completely blocked by the longest row of stereocilia from a top and oblique view (Fig. 3Cb, Cd, Cf and Cg) and (iv) splayed hair bundles were hair bundles whose stereocilia were not connected to each other (Fig. 4Cb and Ce).
ABR, DPOAE, CM and electroretinogram tests
ABRs and DPOAEs were performed at 3–4 weeks of age. Mice were anesthetized with a combination drug of ketamine (100 mg/kg) and xylazine (10 mg/kg i.p.). ABRs/DPOAEs were done in a double-walled sound chamber (IAC, Bronx, NY, USA). The body temperature was maintained at ∼37°C via a heating lamp. Only the right ear from each animal was tested. A small incision was made at the tragus to allow better access to the ear canal. For ABRs, an electrostatic speaker (EC-1, Tucker-Davis Technology, Alachua, FL, USA) fitted with a 1.5 cm long polyethylene tube was placed abutting the ear canal. Recording electrodes were placed under the skin at the vertex and mastoid, with a remote ground in the rump area. ABR signals were amplified with a TDT RA4 pre-amplifier, filtered from 100 to 3000 Hz, averaged and digitized with a TDT RA16BA processor controlled by BioSigRP software. Acoustic stimuli were digitally generated and processed by a RX6 real-time processor and passed through a PA5 attenuator prior to delivery to the speaker amplifier at a rate 24–32 times/s. A series of responses to either clicks (0.1 ms) or tone pips (5 ms with 0.5 ms cos2 rise and fall) were collected with 5 or 10 dB intensity steps, over a 70–80 dB range.
To obtain CM, click polarities were not alternated and both rarefaction and condensation click ABR were acquired. ABR traces in response to tone pips were visually inspected after plotting the amplitude of each peak against stimulus intensity. Thresholds typically corresponded to a level one step below that at which the peak-to-peak response amplitude began to rise. The CMp-p was measured using a 1 ms window just before the downward deflection of the N1 peak on the rarefaction click ABR traces. The DPOAEs were measured using an ER-10B+ (Etymotic Research) microphone coupled with two EC1 speakers. Stimuli of two primary tones f1 and f2 (f2/f1 = 1.2) were presented with f2 = f1 – 10 dB. Primary tones were stepped from 30 to 80 dB sound pressure level (SBL) (for f1) in 10 dB increments and swept from 8 to 32 kHz in 1/2 octave steps. Stimuli were generated and attenuated digitally (200 kHz sampling). The ear canal sound pressure was preamplified and digitized. A fast Fourier transformation was computed, and the sound pressures at f1, f2 and 2f1 − f2 were extracted after spectral averaging from 50 serial waveform traces. The noise floor (average of 10 points in the fast fourier transformation on either side of 2f1 − f2) was also measured: it ranged between −25 and 0 dB SPL, depending on the test frequencies. All data were shown in mean ± SE. The procedure for electroretinogram tests was exactly the same as described previously (9).
Hair cell electrophysiology
Cochleas from Pdzd7−/− and wild-type (B6.129S6 or Swiss Webster) mice were harvested during the first postnatal week (P3–P6). The organs of Corti were gently isolated and tectorial membrane mechanically removed in minimum essential medium (Invitrogen) supplemented with 10 mm hydroxyethyl piperazineethanesulfonic acid (HEPES) and 0.05 mg/ml Ampicilin at pH 7.4. The tissue was then divided into quarter turns (base, mid base, mid apex and apex) and pinned beneath a pair of thin glass fibers adhered to a round glass coverslip. The hair cells were visualized from the apical surface using an upright Axioskop FS microscope (Carl Zeiss) equipped with an X63 water-immersion objective with differential interference contrast optics. Images were acquired with a C2400 CCD camera and Argus image processor (Hamamatsu).
Recordings were performed at a holding potential of −64 mV in standard artificial perilymph solution containing (in mm): 137 NaCl, 0.7 NaH2PO4, 5.8 KCl, 1.3 CaCl2, 0.9 MgCl2, 10 HEPES and 5.6-Glucose. Vitamins (1:50) and amino acids (1:100) were added from concentrates (Invitrogen) and the solution adjusted by NaOH to a final pH of 7.4 (310 mOsm/kg). Recording pipettes (3–5 mΩ) were pulled from R6 capillary glass (King Precision Glass) and filled with intracellular solution containing the following (in mm): 135 KCl, 5 HEPES, 5 ethylene glycol tetraacetic acid, 2.5 MgCl2, 2.5 K2-ATP, 0.1 CaCl2, adjusted with KOH to pH 7.4 (285 mOsm/kg). Currents were recorded at room temperature (22–24°C) using an Axopatch 200B amplifier (Molecular Devices), filtered at 10 kHz with a low-pass Bessel filter, digitized at ≥20 kHz with a 16-bit acquisition board (Digidata 1322A) and pClamp 8.2 software (Molecular Devices). Data were stored on disk for off-line analysis using OriginPro 7.5 (OriginLab) and the results are presented as the mean ± SD.
Mechanical stimulation
Mechanical stimuli were transmitted to the stereocilia bundle using a stiff glass probe with a fire polished tip ∼3–5 µm in diameter (30). The probe was mounted on a one-dimensional PICMA chip piezo actuator (Physik Instruments) driven by a 400 mA ENV400 amplifier (Piezosystem). Voltage steps were used to evoke bundle deflections with a stimulus filtered at 10 kHz by a low-pass 8-pole Bessel filter (Krohn-Hite) to eliminate residual probe resonance. Hair bundle deflections were monitored continuously via video microscopy during recording to ensure the probe and stereocilia bundle moved in unison. Images of the probe at static voltage steps were used to calibrate the stimulus displacement relative to the probe rest position to 2 µm in both the positive and the negative direction (spatial resolution of ∼4 nm). Video images (temporal resolution ∼30 ms) of the probe were recorded to confirm on-axis motion. The 10–90% rise time of the probe was ∼20 µs.
Statistical analysis
Student's t-tests were conducted in Microsoft Office Excel to compare values between two different groups. A P-value of <0.05 was considered to indicate a significant difference between groups.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the National Institutes of Health (EY020853 to J.Y., DC005439 to J.R.H., DC011720 to J.R.H. and EY014800 to core grant to the Department of Ophthalmology &Visual Sciences, University of Utah); Hearing Health Foundation to J.Z.; the National Organization for Hearing Research Foundation to J.Z.; Foundation Fighting Blindness to J.Y.; E. Matilda Ziegler Foundation for the Blind, Inc. to J.Y.; Research to Prevent Blindness to J.Y. and a startup package from the Moran Eye Center, University of Utah to J.Y.
Supplementary Material
ACKNOWLEDGEMENTS
We appreciate Dr Xiaohui Zhang, Ms Susan Tamowski and Ms Nancy Chandler for assisting us in assessing retinal fundus, generating the Pdzd7 mouse model and processing samples for scanning electron microscopy.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Boughman J.A., Vernon M., Shaver K.A. Usher syndrome: definition and estimate of prevalence from two high-risk populations. J. Chronic Dis. 1983;36:595–603. doi: 10.1016/0021-9681(83)90147-9. [DOI] [PubMed] [Google Scholar]
- 2.Hartong D.T., Berson E.L., Dryja T.P. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 3.Keats B.J., Corey D.P. The usher syndromes. Am. J. Med. Genet. 1999;89:158–166. [PubMed] [Google Scholar]
- 4.Eudy J.D., Weston M.D., Yao S., Hoover D.M., Rehm H.L., Ma-Edmonds M., Yan D., Ahmad I., Cheng J.J., Ayuso C., et al. Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science. 1998;280:1753–1757. doi: 10.1126/science.280.5370.1753. [DOI] [PubMed] [Google Scholar]
- 5.Reiners J., Nagel-Wolfrum K., Jurgens K., Marker T., Wolfrum U. Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp. Eye Res. 2006;83:97–119. doi: 10.1016/j.exer.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 6.Williams D.S. Usher syndrome: animal models, retinal function of Usher proteins, and prospects for gene therapy. Vision Res. 2008;48:433–441. doi: 10.1016/j.visres.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang J. In: Hearing Loss. Naz S., editor. Croatia: Intech Open Access; 2012. pp. 293–328. [Google Scholar]
- 8.Yang J., Liu X., Zhao Y., Adamian M., Pawlyk B., Sun X., McMillan D.R., Liberman M.C., Li T. Ablation of whirlin long isoform disrupts the USH2 protein complex and causes vision and hearing loss. PLoS Genet. 2010;6:e1000955. doi: 10.1371/journal.pgen.1000955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zou J., Luo L., Shen Z., Chiodo V.A., Ambati B.K., Hauswirth W.W., Yang J. Whirlin replacement restores the formation of the USH2 protein complex in whirlin knockout photoreceptors. Invest. Ophthalmol. Vis. Sci. 2011;52:2343–2351. doi: 10.1167/iovs.10-6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michalski N., Michel V., Bahloul A., Lefevre G., Barral J., Yagi H., Chardenoux S., Weil D., Martin P., Hardelin J.P., et al. Molecular characterization of the ankle-link complex in cochlear hair cells and its role in the hair bundle functioning. J. Neurosci. 2007;27:6478–6488. doi: 10.1523/JNEUROSCI.0342-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGee J., Goodyear R.J., McMillan D.R., Stauffer E.A., Holt J.R., Locke K.G., Birch D.G., Legan P.K., White P.C., Walsh E.J., et al. The very large G-protein-coupled receptor VLGR1: a component of the ankle link complex required for the normal development of auditory hair bundles. J. Neurosci. 2006;26:6543–6553. doi: 10.1523/JNEUROSCI.0693-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Wijk E., van der Zwaag B., Peters T., Zimmermann U., Te Brinke H., Kersten F.F., Marker T., Aller E., Hoefsloot L.H., Cremers C.W., et al. The DFNB31 gene product whirlin connects to the Usher protein network in the cochlea and retina by direct association with USH2A and VLGR1. Hum. Mol. Genet. 2006;15:751–765. doi: 10.1093/hmg/ddi490. [DOI] [PubMed] [Google Scholar]
- 13.Schneider E., Marker T., Daser A., Frey-Mahn G., Beyer V., Farcas R., Schneider-Ratzke B., Kohlschmidt N., Grossmann B., Bauss K., et al. Homozygous disruption of PDZD7 by reciprocal translocation in a consanguineous family: a new member of the Usher syndrome protein interactome causing congenital hearing impairment. Hum. Mol. Genet. 2009;18:655–666. doi: 10.1093/hmg/ddn395. [DOI] [PubMed] [Google Scholar]
- 14.Ebermann I., Phillips J.B., Liebau M.C., Koenekoop R.K., Schermer B., Lopez I., Schafer E., Roux A.F., Dafinger C., Bernd A., et al. PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J. Clin. Invest. 2010;120:1812–1823. doi: 10.1172/JCI39715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grati M., Shin J.B., Weston M.D., Green J., Bhat M.A., Gillespie P.G., Kachar B. Localization of PDZD7 to the stereocilia ankle-link associates this scaffolding protein with the Usher syndrome protein network. J. Neurosci. 2012;32:14288–14293. doi: 10.1523/JNEUROSCI.3071-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Testa G., Schaft J., van der Hoeven F., Glaser S., Anastassiadis K., Zhang Y., Hermann T., Stremmel W., Stewart A.F. A reliable lacZ expression reporter cassette for multipurpose, knockout-first alleles. Genesis. 2004;38:151–158. doi: 10.1002/gene.20012. [DOI] [PubMed] [Google Scholar]
- 17.Hardisty-Hughes R.E., Parker A., Brown S.D. A hearing and vestibular phenotyping pipeline to identify mouse mutants with hearing impairment. Nat. Protoc. 2010;5:177–190. doi: 10.1038/nprot.2009.204. [DOI] [PubMed] [Google Scholar]
- 18.Kawashima Y., Geleoc G.S., Kurima K., Labay V., Lelli A., Asai Y., Makishima T., Wu D.K., Della Santina C.C., Holt J.R., et al. Mechanotransduction in mouse inner ear hair cells requires transmembrane channel-like genes. J. Clin. Invest. 2011;121:4796–4809. doi: 10.1172/JCI60405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodyear R.J., Marcotti W., Kros C.J., Richardson G.P. Development and properties of stereociliary link types in hair cells of the mouse cochlea. J. Comp. Neurol. 2005;485:75–85. doi: 10.1002/cne.20513. [DOI] [PubMed] [Google Scholar]
- 20.Belyantseva I.A., Boger E.T., Naz S., Frolenkov G.I., Sellers J.R., Ahmed Z.M., Griffith A.J., Friedman T.B. Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat. Cell Biol. 2005;7:148–156. doi: 10.1038/ncb1219. [DOI] [PubMed] [Google Scholar]
- 21.Mattapallil M.J., Wawrousek E.F., Chan C.C., Zhao H., Roychoudhury J., Ferguson T.A., Caspi R.R. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest. Ophthalmol. Vis. Sci. 2012;53:2921–2927. doi: 10.1167/iovs.12-9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mburu P., Mustapha M., Varela A., Weil D., El-Amraoui A., Holme R.H., Rump A., Hardisty R.E., Blanchard S., Coimbra R.S., et al. Defects in whirlin, a PDZ domain molecule involved in stereocilia elongation, cause deafness in the whirler mouse and families with DFNB31. Nat. Genet. 2003;34:421–428. doi: 10.1038/ng1208. [DOI] [PubMed] [Google Scholar]
- 23.Adato A., Lefevre G., Delprat B., Michel V., Michalski N., Chardenoux S., Weil D., El-Amraoui A., Petit C. Usherin, the defective protein in Usher syndrome type IIA, is likely to be a component of interstereocilia ankle links in the inner ear sensory cells. Hum. Mol. Genet. 2005;14:3921–3932. doi: 10.1093/hmg/ddi416. [DOI] [PubMed] [Google Scholar]
- 24.Wang L., Zou J., Shen Z., Song E., Yang J. Whirlin interacts with espin and modulates its actin-regulatory function: an insight into the mechanism of Usher syndrome type II. Hum. Mol. Genet. 2012;21:692–710. doi: 10.1093/hmg/ddr503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bahloul A., Simmler M.C., Michel V., Leibovici M., Perfettini I., Roux I., Weil D., Nouaille S., Zuo J., Zadro C., et al. Vezatin, an integral membrane protein of adherens junctions, is required for the sound resilience of cochlear hair cells. EMBO Mol. Med. 2009;1:125–138. doi: 10.1002/emmm.200900015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu X., Bulgakov O.V., Darrow K.N., Pawlyk B., Adamian M., Liberman M.C., Li T. Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc. Natl Acad. Sci. USA. 2007;104:4413–4418. doi: 10.1073/pnas.0610950104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delprat B., Michel V., Goodyear R., Yamasaki Y., Michalski N., El-Amraoui A., Perfettini I., Legrain P., Richardson G., Hardelin J.P., et al. Myosin XVa and whirlin, two deafness gene products required for hair bundle growth, are located at the stereocilia tips and interact directly. Hum. Mol. Genet. 2005;14:401–410. doi: 10.1093/hmg/ddi036. [DOI] [PubMed] [Google Scholar]
- 28.Yang J., Liu X., Yue G., Adamian M., Bulgakov O., Li T. Rootletin, a novel coiled-coil protein, is a structural component of the ciliary rootlet. J. Cell Biol. 2002;159:431–440. doi: 10.1083/jcb.200207153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bolte S., Cordelieres F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 30.Stauffer E.A., Holt J.R. Sensory transduction and adaptation in inner and outer hair cells of the mouse auditory system. J. Neurophysiol. 2007;98:3360–3369. doi: 10.1152/jn.00914.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.