

Abstract
DGKs (diacylglycerol kinases) catalyse the conversion of diacylglycerol into PA (phosphatidic acid), a positive modulator of mTOR (mammalian target of rapamycin). We have found that chenodeoxycholic acid and the synthetic FXR (farnesoid X receptor) ligand GW4064 induce the mRNA and protein expression of DGKθ in the HepG2 cell line and in primary human hepatocytes. Reporter gene studies using 1.5 kB of the DGKθ promoter fused to the luciferase gene revealed that bile acids increase DGKθ transcriptional activity. Mutation of putative FXR-binding sites attenuated the ability of GW4046 to increase DGKθ luciferase activity. Consistent with this finding, ChIP (chromatin immunoprecipitation) assays demonstrated that bile acid signalling increased the recruitment of FXR to the DGKθ promoter. Furthermore, GW4064 evoked a time-dependent increase in the cellular concentration of PA. We also found that GW4064 and PA promote the phosphorylation of mTOR, Akt and FoxO1 (forkhead box O1), and that silencing DGKθ expression significantly abrogated the ability of GW4046 to promote the phosphorylation of these PA-regulated targets. DGKθ was also required for bile-acid-dependent decreased glucose production. Taken together, our results establish DGKθ as a key mediator of bile-acid-stimulated modulation of mTORC2 (mTOR complex 2), the Akt pathway and glucose homoeostasis.
Keywords: bile acid, diacylglycerol kinase θ (DGKθ), farnesoid X receptor (FXR), hepatocyte, phosphatidic acid
INTRODUCTION
Bile acids are amphipathic molecules that regulate the elimination of cholesterol. In humans there are two major bile acid biosynthetic pathways, neutral and acidic, that are initiated by cholesterol 7α-hydroxylase (CYP7A1) and mitochondrial sterol 27-hydroxylase (CYP27A1) respectively. The neutral pathway produces primary bile acids [CA (cholic acid) and CDCA (chenodeoxycholic acid)] and secondary bile acids {DCA (deoxycholic acid) and lithocholic acid [1]}, whereas CDCA is the only product in the acidic pathway. Under normal physiological conditions, the neutral pathway is the primary bile acid biosynthetic pathway [2]. Besides their well-established roles in cholesterol metabolism, bile acids are emerging as key signalling mediators that evoke changes in various physiological processes by serving as ligands for TGR5, a G-protein-coupled receptor [3] and the nuclear receptor FXR (farnesoid X receptor; NR1H4) [4]. As the endogenous ligand for FXR, CDCA is a more potent ligand for FXR than DCA and LCA (lithocholic acid) [5]. However, FXR is also activated by synthetic agonists, such as GW4064 [6] and 6α-ECDCA (6α-ethyl-chenodeoxycholic acid) [7].
FXR is expressed in liver, intestine, pancreas, kidney and adrenal glands [6,8,9] and regulates the expression of target genes by binding to their promoters in a ligand-dependent manner. A pivotal function of FXR is to induce the expression of SHP (small heterodimer partner), a nuclear receptor that suppresses CYP7A1 [10]. FXR also regulates cholesterol production and lipid metabolism [11–14], a function that was demonstrated in FXR-null mice, where hepatic cholesterol, triacylglycerol and high-density lipoprotein expression are increased [14,15]. Furthermore, FXR plays an important role in the regulation of carbohydrate metabolism [17]. FXR suppresses PEPCK (phosphoenolpyruvate carboxykinase) and G6Pase (glucose 6-phosphatase), thereby regulating gluconeogenesis and glucose homoeostasis [18].
DGKs (diacylglycerol kinases) phosphorylate DAG (diacylglycerol) to produce PA (phosphatidic acid) [19–21]. There are ten mammalian isoforms of DGK, which are organized into five categories on the basis of the presence of specific domains identified in their primary sequence. DGKθ is the sole member of group V and harbours three cysteine-rich domains [22,23], compared with two for other DGK isoforms. DGKθ has been implicated in various physiological processes [24–28]. Translocation of DGKθ from the cytosol to the plasma membrane facilitates interaction with the EGFR (epidermal growth factor receptor) and inhibits the ability of protein kinase C to terminate EGFR signalling [29]. The synergistic interaction of DGKθ with acidic phospholipids and polybasic proteins such as Tau and histone H1 promote phospholipid-dependent activation of the enzyme [30]. Finally, adenosine A2a receptor-initiated repression of DGKθ by the small GTPase RhoA confers tolerance to ischaemia/reperfusion injury in hepatocytes [31].
The functional importance of PA as a second messenger that mediates various intracellular signalling pathway by regulation of protein kinases and phosphatases is well established [32]. PA has been shown to positively [33,34] and negatively [35] regulate mTOR (mammalian target of rapamycin) signalling. Moreover, a role for DGK activity in the regulation of mTOR-dependent pathways has been reported [36,37]; however, the precise mechanism or specific DGK isoform that regulates PA-dependent mTOR signalling was unclear. In the present study, we show that DGKθ gene expression (encoded by DGKQ) and activity are in response to bile acid stimulation. The DGKθ-produced PA increases the phosphorylation of mTOR, Akt and FoxO1 (forkhead box O1), thus suppressing gluconeogenic gene expression. In summary, we show that DGKθ plays an important role in hepatic bile acid-regulated glucose production.
EXPERIMENTAL
Materials
GW4064 and CDCA were obtained from Sigma–Aldrich), and PA (egg PA; catalogue number 840101) was from Avanti Polar Lipids.
Cell culture
HepG2 cells were obtained from the A.T.C.C. (Manassas, VA, U.S.A.) and cultured in Eagle’s minimum essential medium (Mediatech) supplemented with 10 % FBS (Mediatech) and antibiotics (1× penicillin-streptomycin/amphotericin B; Lonza). DGKθ-knockdown cells were generated as described previously [38]. The following lots of primary human hepatocyte cultures were purchased from XenoTech: lot 1147 (35-year-old male), lot 1152 (69-year-old female), lot 1155 (43-year-old female) and lot 1157 (59-year-old female). Primary human hepatocytes were plated in William’s E medium (Invitrogen) containing FBS, ITS (insulin-transfemin-selenium) (Mediatech), dexamethasone (0.1 μM) and antibiotics (1× penicillin-streptomycin/amphotericin B; Lonza).
Transient transfection and reporter gene analysis
HepG2 cells were subcultured on to 24-well plates and transfected with 20 ng of pGL3-DGKθ [38], 0.5 ng of pRL-CMV (Promega) and 25 ng of pcDNA3.1-FXR (provided by Dr David D. Moore, Baylor College of Medicine, Houston, TX, U.S.A.) using Genejuice® (Novagen). At 24 h after transfection, the cells were treated with 1 nM GW4064 for 24 h and the transcriptional activity of the DGKθ reporter gene was measured using a dual luciferase assay kit (Promega). Firefly (pGL3-DGKθ) luciferase activity were normalized to Renilla luciferase activity (pRL-CMV) and expressed as fold change over the mean of the untreated control group.
RNA isolation and real-time RT-PCR
Cells were subcultured on to 12-well plates and 24 h later were treated with 100 μM CDCA, 1 nM GW4064 or 10 μM PA for 24 h. Total RNA was extracted using Iso-RNA Lysis Regent (5 Prime) and amplified using a One-Step SYBR Green RT-PCR Kit (Thermo Fisher Scientific) and the following primers: DGKθ forward, 5′-CGTTCTCCGTACTGCTGTC-3′ and reverse, 5′-G-TCTGCCGTGTCGTTCTC-3′; DGKα forward, 5′-GCGAGGA-GGCTGGTGAGTC-3′ and reverse, 5′-TGGAAGATGGGAGG-CAGGATG-3′; DGKδ forward, 5′-GCACAGAACCTACAGA-ACC-3′ and reverse, 5′-GCGACCACCTCCAGAATC-3′, DGKζ forward, 5′-AGCAAGAAGAAGAAGAGG-3′ and reverse, 5′-GGATTGAGATACCAGAGG-3′; and β-actin forward, 5′-ACG-GCTCCGGCATGTGCAAG-3′ and reverse, 5′-TGACGATGCC-GTGCTGCATG-3′. DGKθ expression was normalized to β-actin content and calculated using the cycle threshold (ΔΔCT) method.
Western blotting
Cells were subcultured on to six-well plates and treated with 100 μM CDCA or 1 nM GW4064 for 48 h and harvested into RIPA buffer [50 mM Tris/HCl, pH 7.4, 1 % Nonidet P40, 0.25 % sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 150 nM aprotinin, 1 mM leupeptin, 1 mM E-64 and 500 mM 4-(2-aminoethyl)benzenesulfonylfluoride]. Cells were then lysed by sonication (one 5 s burst) followed by incubation on ice for 30 min. Lysates were centrifuged at 13 800 g for 10 min at 4 °C and the supernatant was collected for analysis by SDS/PAGE. Protein concentrations were determined using the BCA Protein Assay (Pierce). Aliquots of each sample (25 μg of protein) were run on SDS/PAGE (8 % gels) and transferred on to PVDF membranes (Millipore). Blots were probed with primary antibodies overnight. Anti-DGKθ antibody (1:1000 dilution, HPA026797) was obtained from Sigma–Aldrich. Anti-Akt antibody (1:1000 dilution, sc-8312), anti-phospho-Akt (1:1000 dilution, sc-7985) and anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (1:5000 dilution, sc-25778) antibodies were obtained from Santa Cruz Biotechnology. Anti-(total mTOR) (1:1500 dilution, 2983P), anti-phospho-mTOR(Ser2448) (1:1000 dilution, 2971S), anti-(total FoxO1) (1:1500 dilution, 2880S) and anti-phospho-FoxO1(Thr24) (1:1000 dilution, 9464P) antibodies were obtained from Cell Signaling Technology. Anti-rabbit secondary antibody was obtained from GE Healthcare (ECF Western Blotting Reagent). Blots were scanned on a VersaDoc 4000 (Bio-Rad Laboratories) and densitometric analysis was carried out using Quantity One software (Bio-Rad Laboratories).
ChIP (chromatin immunoprecipitation)
ChIP assays were performed as described previously [39]. Briefly, HepG2 cells were subcultured on to 15-cm-diameter dishes and treated with 1 nM GW4064 for 60 min. Cells were treated with 1 % formaldehyde for 10 min at room temperature and then incubated with 0.125 M glycine for 5 min. After washing twice with PBS, cells were harvested into RIPA buffer. The purified chromatin solutions were immunoprecipitated using 5 μg of anti-(acetyl-histone H3) (17-615, Millipore), anti-FXR (Santa Cruz Biotechnology) or anti-IgG (Millipore) antibodies and Protein A/G PLUS (Santa Cruz Biotechnology). Real-time PCR was carried out to amplify region − 1000 to − 700 bp of the DGKθ promoter [38] using the following primer sets: forward 5′-CAGAGTCCACAGCCCCCAGCCCCTTTCAGG-3′ and reverse 5′-CTGCCTCGTGCGCGCCACGGGTCTTGTTCA-3′. Output (immunoprecipitated promoter region) was normalized to input DNA. PCR products were run on 2 % agarose gels.
PA assay
HepG2 cells were grown on six-well plates and then treated with 1 nM GW4064 for 24 h to 72 h and total or nuclear (Nuclei Pure Kit; Sigma) lipids were harvested. PA content was quantified using a Total PA kit (Cayman Chemical) and a SpectraMax M5 MultiMode Microplate Reader (Molecular Devices) at an excitation wavelength of 530–540 nM and an emission wavelength of 585–595 nm. Data were quantified using SoftMax Pro Software (Molecular Devices).
DAG assay
The DAG assay was performed as described previously [38]. HepG2 cells or human hepatocytes were cultured on to six-well plates and treated with 1 nM GW4064 for 72 h. The amount of DAG in each sample was determined using a Human DAG ELISA kit (MyBioSource).
Glucose measurement assay
Wild-type and DGKθ knockdown HepG2 cells were seeded on to a 96-well plate with the normal growth medium at 37 °C in 5 % CO2. The next day, the medium was removed and changed to 100 μl of glucose-free medium and treated with GW4064 or PA for 48 h. A volume of 50 μl of medium was collected from each well and glucose output in HepG2 cells was determined by using the Amplex Red glucose kit (Invitrogen) [40]. A standard calibration curve of glucose (0.9–18 ng/ml) was prepared using the stock solution of glucose (72 μg/ml) in a volume of 50 μl. Standards and samples were both incubated with Amplex Red working solution in the dark for 30 min. The fluorescence was measured at a SpectraMax M5 Multi-Mode Microplate Reader (Molecular Devices).
Statistical analysis
One-way ANOVA and Tukey-Kramer multiple comparisons were performed using GraphPad Prism version 5.00 (GraphPad Software). The significant difference value was set as P < 0.05.
RESULTS
Bile acids induce DGKθ gene expression
As discussed above, bile acid signalling has been linked to PA metabolism [41]. Therefore we treated HepG2 cells with 100 μM CDCA or 1 nM GW4064 and assessed the effect on DGKθ expression. As shown in Figure 1(A), CDCA and GW4064 increased the mRNA expression of DGKθ 2.5- and 4-fold respectively. Consistent with an induction in the expression of mRNA, the protein levels of DGKθ were also increased in response to bile acid signalling, with GW4064 resulting in a 1.7-fold increase in the protein expression of DGKθ (Figure 1B). Significantly, the stimulatory effect of bile acids was also observed in primary human hepatocytes, where GW4064 evoked a 3.9-and 2.1-fold increase in DGKθ mRNA (Figure 1C) and protein expression (Figure 1D) respectively. In contrast, other DGK isoforms that have been shown to be expressed in hepatocytes [31] were not induced by GW4064 treatment.
Figure 1. Bile acids increase DGKθ mRNA and protein expression.

(A) HepG2 cells were cultured on to 12-well plates and treated for 24 h with 100 μM CDCA or 1 nM GW4064. Total RNA was isolated for analysis of DGKθ and β-actin mRNA expression by qRT-PCR (quantitative reverse transcription–PCR). Data are shown as fold change in DGKθ mRNA expression and normalized to the mRNA expression of β-actin. The data are the means ± S.E.M. for three separate experiments, each performed in triplicate. *P < 0.05, statistically different from untreated control group. (B) Representative blots from analysis of lysates isolated from HepG2 cells that were treated for 48 h with 100 μM CDCA or 1 nM GW4064. The graph depicts densitometric analysis of DGKθ (DGKq) protein expression normalized to GAPDH. The data are the means ± S.E.M. for three separate experiments, each carried out in triplicate. *P < 0.05, a statistically significant difference compared with untreated controls. (C) RNA was isolated from human hepatocytes that were treated for 24 h with 1 nM GW4064 and subjected to qRT-PCR. Data are shown as fold change in DGKα (DGKa), DGKσ (DGKd), DGKζ (DGKz) and DGKθ mRNA expression, normalized to the mRNA expression of β-actin, and represent the means ± S.E.M. of four donors with cells plated in triplicate. (D) Representative immunoblot of DGKθ protein expression in human hepatocytes that were treated for 72 h with 1 nM GW4064. The data are the means ± S.E.M. of four donors, with each experiment carried out in triplicate. *P < 0.05, a statistically significant difference compared with untreated controls.
FXR stimulates DGKθ expression
To examine further the mechanism by which bile acids induce DGKθ expression, HepG2 cells were transfected with a reporter construct that contained 1.5 kB of the DGKθ promoter fused to the luciferase gene [38]. As shown in Figure 2(A), GW4064 increased the transcriptional activity of the reporter gene by 3.9-fold. Overexpression of FXR increased luciferase activity by 3.7-fold, and GW4064 further increased FXR-dependent reporter gene activity by 9.2-fold (Figure 2A). In silico analysis of the promoter revealed the presence of two putative FXR response elements at − 910/− 904 (M1) and − 817/− 809 (M2) upstream of the translation start site. Although mutation of the M1 site had no significant effect on the ability of GW4064 or FXR to increase DGKθ reporter gene activity, mutation of M2 abrogated GW4064-or FXR-dependent luciferase activity (Figure 2B). However, mutation of the M1 and M2 sites decreased the ability of GW4064 to activate FXR-dependent reporter gene activity by 42 % and 60 % respectively. Reporter gene studies were consistent with ChIP assays where we found that GW4064 increased acetylation of histone H3 at a region of the DGKθ promoter that encompasses the FXR-binding site, which was concomitant with a 5-fold increase in the recruitment of FXR (Figure 3).
Figure 2. FXR increases DGKθ reporter gene activity.
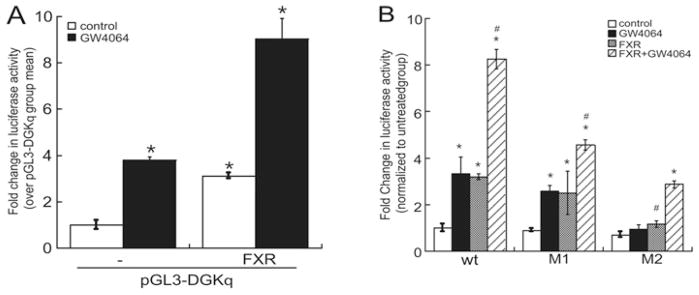
(A) HepG2 cells were transiently transfected with pGL3-DGKθ (pGL3-DGKq), pRL-CMV and pCMX-hFXR, and then treated with 1 nM GW4064 for 16 h. Luciferase activity in lysates isolated from control and GW4064-treated cells was measured by luminometry. The results were normalized to the luciferase activity of Renilla gene and expressed as the fold change in pGL3-DGKθ reporter gene activity over the untreated control group mean and represent the means ± S.E.M. for three separate experiments, each performed in triplicate. *P < 0.05, a statistically significant difference from the untreated control group. (B) HepG2 cells were transiently transfected with wild-type (wt) or mutant (M1 or M2) pGL3-DGKθ, pRL-CMV and pCMX-hFXR expression plasmids. * and # indicate a statistically significant difference (P < 0.05) from untreated control group and untreated FXR group respectively.
Figure 3. GW4064 stimulates FXR recruitment to the DGKθ promoter.

HepG2 cells were incubated with 1 nM GW4064 for 60 min, and then cross-linked with 1 % formaldehyde. The sheared chromatin was harvested and immunoprecipitated with antibodies against FXR or acetyl-histone H3 and recruitment to the DGKθ promoter (− 1000/− 700) assessed by qPCR. DNA purified was quantified by real-time PCR and normalized to the ΔΔCT values of input DNA. The results are expressed as fold change over untreated control. *P < 0.05, a statistically significant difference from untreated control group.
Bile acids induce PA concentration in HepG2 cells
Next we assessed the effect of FXR activation on the cellular concentration of PA and found that stimulation of HepG2 cells with 1 nM GW4064 increased the production of PA in a time-dependent manner, with a 3.7-fold increase observed after 72 h of stimulation (Figure 4A). The treatment with the DGK inhibitor R59949 reduced basal PA production and attenuated GW4064-stimulated PA biosynthesis, suggesting a prominent role for DGK activity in conferring increased PA production in response to bile acid synthesis. Since R59949 inhibits all DGK isoforms, we generated a cell line where the expression of DGKθ was stably silenced using shRNAs targeted against the lipid kinase (Figure 4B) and determined the relative contribution of DGKθ to GW4064-stimulated PA production. As shown in Figure 4(C), suppressing DGKθ expression resulted in a 55 % reduction in PA production, suggesting that DGKθ plays a predominant role in the response to GW4064 and that other isoforms contribute to the response. However, analysis of the mRNA expression of all other DGKθ isoforms in expressed in HepG2 cells and in human primary hepatocytes revealed that DGKθ was the sole isoform that exhibited an induction in mRNA expression in response to GW4064 (Figure 1C). Thus it is possible that bile acid signalling may regulate other DGK isoforms via non-transcriptional mechanisms. Concomitant with an increase in cellular PA, DAG content was reduced by 40 %with the GW4064 treatment, and the decrease was less in DGKθ-knockdown cells (Figure 4D). Consistent with the effect of GW4064 in HepG2 cells, the FXR agonist increased PA levels by 2.1-fold (Figure 4E) while decreasing DAG levels by 34 % (Figure 4F) in human hepatocytes.
Figure 4. Bile acids induce PA production.

(A) HepG2 cells were treated with 1 nM GW4064 and R59949 from 24 h to 72 h and PA levels were quantified by ELISA. (B) Lysates from wild-type (wt) and DGKθ-knockdown (DGKqkd) cells were harvested and analysed using SDS/PAGE and Western blotting using anti-DGKθ (top) and anti-GAPDH (bottom) antibodies. DGKθ mRNA expression was quantified in wild-type and DGKθ-knockdown HepG2 cells by qRT-PCR (quantitative reverse transcription–PCR). Data are shown as fold change in DGKθ mRNA expression and normalized to β-actin mRNA expression. (C) Wild-type and DGKθ-knockdown cells were treated with 1 nM GW4064 and the cellular amount of PA was measured and normalized to the protein concentration. (D) HepG2 cells and DGKθ-knockdown HepG2 cells were treated with 1 nM GW4064 for 72 h. Cellular DAG levels were measured by ELISA kit. The graph shows the means ± S.E.M. for three separate experiments, each performed in triplicate. *P < 0.05 indicate a statistically significant difference from the untreated wild-type control group. Human hepatocytes from donor 4 were cultured on to six-well plates and incubated with 1 nM GW4064 for 72 h. Cellular PA (E) and DAG (F) concentration were quantified by PA assay and DAG assay respectively. *P < 0.05, a statistically significant difference from the untreated control group.
Effect of silencing of DGKθ on GW4064-stimulated target gene activity and expression
PA is a well-established second messenger that regulates multiple proteins, including phosphatidylinositol 4-phosphate 5-kinase [42], Raf kinase [43], PKCε (protein kinase Cε) and PKCζ [44,45], sphingosine kinase [46] and the tyrosine phosphatase SHP-1 [47]. In addition, PA activates mTOR-dependent downstream signalling [33] and DGKζ-produced PA mediates mTOR signalling in HEK (human embryonic kidney)-293 cells [36]. On the basis of these previously published data and our findings from the present study demonstrating that bile acids induce DGKθ expression, we sought to investigate the functional significance of DGKθ-produced PA as it relates to mTOR signalling. As shown in Figure 5(A), both GW4064 and PA increase the cellular content of Ser2448-phosphorylated mTOR [49] in wild-type cells; however, GW4064 is unable to stimulate mTOR phosphorylation in DGKθ-knockdown cells. Consistent with these data, GW4064 activates phosphorylation of the mTOR target Akt at Thr473 [50,51], which plays a central role in inhibition of FoxO1 and leads to constitutive suppression of glucose production [52] in wild-type HepG2 cells, but not in cells where DGKθ expression is silenced. In contrast, we were unable to detect an increase in the phosphorylation of the mTORC1 (mTOR complex 1) downstream target p70 S6 kinase in HepG2 cells or primary human hepatocytes (results not shown), suggesting selective activation of the mTORC2 signalling pathway.
Figure 5. DGKθ is required for bile-acid-stimulated regulation of the mTOR pathway.

(A) Wild-type (WT) and DGKθ-knockdown (DGKqkd) HepG2 cells were treated with 1 nM GW4064 or 10 μM DLPA for 48 h respectively. The levels of phosphorylated (p) and total mTOR, Akt and FoxO1 were evaluated by Western blotting using the antibodies described in the Experimental section. (B) RNA was isolated from wild-type and DGKθ-knockdown HepG2 cells that were treated with 1 nM GW4064 or 10 μM DLPA for 24 h. The mRNA expression of PEPCK and G6Pase was normalized to β-actin. Data are graphed as fold change over wild-type control and represent the means ± S.E.M. for three independent experiments, each performed in triplicate. *P < 0.05, a statistically significant difference from untreated group. (C) HepG2 cells (wild-type and DGKθ-knockdown) were cultured in glucose-free medium and endogenous glucose production was measured after treatment with 1 nM GW4064 or 10 μM PA for 48 h. Data are graphed as the mean of duplicate measurements and represent three independent experiments, each performed in at least triplicate. *P < 0.05, a statistically significant difference from the wild-type untreated control group. (D) Primary human hepatocytes were incubated with 1 nM GW4064 or 10 μM PA for 48 h and proteins were isolated using SDS/PAGE and Western blot analysis. Blots were incubated with antibodies against phosphorylated (p) and total mTOR, Akt and FoxO1. (E) Primary human hepatocytes were treated with 1 nM GW4064 or 10 μM PA for 24 h and RNA was isolated for real-time RT (reverse transcription)–PCR. G6Pase and PEPCK mRNA expression was normalized to β-actin and data are shown as means ± S.E.M. for four donors, with each independent experiment carried out in triplicate. *P < 0.05, a statistically significant difference compared with untreated controls.
Bile acids have been reported to down-regulate gluconeogenic genes, including PEPCK and G6Pase [53]. Thus we assessed the role of DGKθ-catalysed PA production in regulating gluconeogenic gene expression. As shown in Figure 5(B), both GW4064 and PA potently suppressed the mRNA expression of PEPCK and G6Pase in wild-type HepG2 cells. GW4064 suppressed G6Pase expression by 62 % in wild-type cells and by 29 % in DGKθ-knockdown cells. Unexpectedly, silencing DGKθ reduced the mRNA expression of G6Pase by 46 %. Although GW4064 reduced PEPCK mRNA expression by 68 % in wild-type HepG2 cells, silencing DGKθ resulted in only a 43 % decrease in PEPCK mRNA expression, suggesting a role for DGKθ-dependent PA production in regulating hepatic gluconeogenic gene expression. These expression data were supported by glucose measurements, which indicated that GW4064 and PA suppressed glucose production in wild-type cells by 35 % and 65 % respectively (Figure 5C). In contrast, GW406 had no significant effect on glucose concentrations in the DGKθ-knockdown cell line, whereas PA reduced glucose production by 47 %. Importantly, GW4064 and PA increased the phosphorylation of mTOR, Akt and FoxO1 (Figure 5D) and suppressed the mRNA expression of G6Pase and PEPCK (Figure 5E) in primary human hepatocytes.
DISCUSSION
Bile acids are important signalling molecules that regulate hepatic cholesterol, triacylglycerol and glucose homoeostasis [2,14,54]. Previous studies have demonstrated that bile acids and insulin act co-operatively to regulate glucose storage in primary rodent hepatocytes [54,55]. In particular, a clinical study has shown that the activation of FXR is beneficial for the treatment of hypertriacylglycerolaemia [54]. DGKs play a pivotal role in balancing the cellular concentrations of two lipid second messengers, PA and DAG [21,33,56–58]. Since the first DGK was discovered [59], ten DGKs isoforms have been identified. Aberrant DGK function has been associated with several pathophysiological states, an expected consequence of the regulation of these enzymes in multiple tissues. Hepatic DAG accumulation can activate the predominant PKC isoform (PKCε) in liver, which has been strongly associated with hepatic insulin resistance [60,61], a major risk factor of Type 2 diabetes [62]. Mounting evidence indicates that bile acids regulate glucose homoeostasis. Recent research has demonstrated PEPCK and G6Pase are repressed by taurocholate in primary rat hepatocytes [53]. Moreover, the Akt pathway has been shown to have a central role in bile-acid-regulated glucose production [53,55].
DGKθ is modulated by PKCε [64], protein–protein interaction with RhoA [65] and phospholipid activation [66]. Moreover, recent studies have shown that the interaction of DGKθ with polybasic proteins such as histone H1 and Tau positively regulate the enzyme [30]. Our previous studies established a role for DGKθ in adrenocortical glucocorticoid production, where the kinase binds to the nuclear receptor SF-1 (steroidogenic factor-1) [67]. We have also recently shown that the expression of DGKθ in human adrenocortical cells is transcriptionally regulated by the cAMP signalling pathway, which induces the expression of DGKθ via a mechanism that requires SREBP1 (sterol-regulatory-element-binding protein 1) and SF-1 [38]. However, less is known about the factors that regulate the expression of DGKθ in liver or about the functional significance of the lipid kinase in hepatic cellular processes. Work by Baldanzi et al. [31] showed that DAG accumulation during adenosine-dependent hepatocyte preconditioning is regulated by DGKθ. In the present study we show that CDCA and GW4064 positively regulate the mRNA and protein expression of DGKθ (Figure 1) and identify a novel regulatory pathway whereby the lipid kinase contributes to the regulation of glucose homoeostasis (Figure 6). Significantly, we did not observe a similar effect on other DGK isoforms in primary human hepatocytes or HepG2 cells. It is worth noting that published studies have demonstrated that DGKθ is most abundant DGK isoform expressed in human hepatocytes and was identified as the major isoform that mediates the response to signalling pathways that result in DAG accumulation [31].
Figure 6. Model for the role of DGKθ in bile-acid-mediated regulation of glucose production.

Bile acids induce DGKθ (DGKq) gene expression resulting in increased cellular PA. PA activates mTOR signalling leading to increased phosphorylation of Akt and FoxO1, and ultimately decreased glucose production owing to a suppression of gluconeogenic gene expression.
Our luciferase reporter assays revealed that FXR positively regulated the transcriptional activity of the DGKθ reporter gene (Figure 2), a finding that was supported by ChIP studies, which found that GW4064 stimulated the recruitment of FXR to the endogenous DGKθ promoter (Figure 3). FXR regulates multiple genes involved in bile acid metabolism, including SHP [53]. Moreover, the links between bile acid signalling and lipid metabolism are well established [11,70,71], and bile acid receptors are emerging as attractive targets for the treatment of varied pathophysiological disease states, including cardiovascular disease and dyslipidaemia [72,73].
We found that the cellular PA concentration increases in response to GW4064 treatment and that DGKθ plays a predominant role in bile acid-stimulated PA production in HepG2 cells (Figure 4). As discussed earlier, PA increases the phosphorylation of Akt at Ser473 [34]. PLD1 (phospholipase D1) and PLD2 have been shown to be required for PA-dependent mTOR activation in skeletal muscle [75]. Moreover, the increase in PLD activity induced by amino acids and glucose confers elevated mTOR activation in multiple cancer cell lines, including MDA-MB-231 breast cancer cells and T24 bladder cancer cells [76]. The present study identifies that DGKθ is another source for PA production and mTORC2 activation in hepatocytes. A role for DGK activity in mTOR activation has been previously demonstrated in HEK-293 cells where overexpression of DGKθ identified a role for the isoform in mTOR signalling [36]. As described above, hepatic mTOR plays a critical role in regulation of the Akt–FoxO1 axis [77,78]. The physiological function of hepatic Akt is essential for maintaining glucose homoeostasis through increasing the phosphorylation of FoxO1, which abolished the transcriptional regulation effect of FoxO1 on gluconeogenesis genes [52]. The effect of silencing DGKθ on mTORC2 signalling and gluconeogenic gene expression (Figure 5) provides support for the role that this lipid kinase plays in regulating hepatic glucose homoeostasis. However, the inability of suppressing DGKθ to completely abrogate the repressive effects of GW4064 on gluconeogenic gene expression suggest that other DGK isoforms contribute to the PA-dependent hepatic glucose metabolism. Additionally, the effect of silencing DGKθ on the basal expression of G6Pase (Figure 5B) suggests that there are multiple mechanisms by which DGKθ may regulate glucose metabolism. In conclusion, we identify a novel role for DGKθ in hepatic bile acid signalling and glucose homoeostasis.
Acknowledgments
FUNDING
This work is supported by the National Institutes of Health [grant number DK084178 (to M.B.S.)].
Abbreviations used
- CDCA
chenodeoxycholic acid
- ChIP
chromatin immunoprecipitation
- DAG
diacylglycerol
- DCA
deoxycholic acid
- DGK
diacylglycerol kinase
- EGFR
epidermal growth factor receptor
- FoxO1
forkhead box O1
- FXR
farnesoid X receptor
- G6Pase
glucose 6-phosphatase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HEK
human embryonic kidney
- mTOR
mammalian target of rapamycin
- mTORC
mTOR complex
- PA
phosphatidic acid
- PEPCK
phosphoenolpyruvate carboxykinase
- PKC
protein kinase C
- PLD
phospholipase D
- SHP
small heterodimer partner
- SF-1
steroidogenic factor-1
Footnotes
AUTHOR CONTRIBUTION
Kai Cai and Marion Sewer participated in research design. Kai Cai conducted the experiments. Kai Cai and Marion Sewer performed data analysis and wrote the paper.
References
- 1.Mathis C, Poussin C, Weisensee D, Gebel S, Belcastro V, Xiang Y, Sewer A, Hengstermann A, Ansari S, Wagner S, et al. Smoker bronchial epithelium: a multi-dimensional in vivo/in vitro comparison. Toxicol Lett. 2012;211:S194–S194. [Google Scholar]
- 2.Fiorucci S, Mencarelli A, Palladino G, Cipriani S. Bile-acid-activated receptors: targeting TGR5 and farnesoid-X-receptor in lipid and glucose disorders. Trends Pharmacol Sci. 2009;30:570–580. doi: 10.1016/j.tips.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 3.Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 4.Huber RM, Murphy K, Miao B, Link JR, Cunningham MR, Rupar MJ, Gunyuzlu PL, Haws TF, Kassam A, Powell F, et al. Generation of multiple farnesoid-X-receptor isoforms through the use of alternative promoters. Gene. 2002;290:35–43. doi: 10.1016/s0378-1119(02)00557-7. [DOI] [PubMed] [Google Scholar]
- 5.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 6.Maloney PR, Parks DJ, Haffner CD, Fivush AM, Chandra G, Plunket KD, Creech KL, Moore LB, Wilson JG, Lewis MC, et al. Identification of a chemical tool for the orphan nuclear receptor FXR. J Med Chem. 2000;43:2971–2974. doi: 10.1021/jm0002127. [DOI] [PubMed] [Google Scholar]
- 7.Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, Morelli A, Parks DJ, Willson TM. 6α-Ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem. 2002;45:3569–3572. doi: 10.1021/jm025529g. [DOI] [PubMed] [Google Scholar]
- 8.Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 9.Downes M, Verdecia MA, Roecker AJ, Hughes R, Hogenesch JB, Kast-Woelbern HR, Bowman ME, Ferrer JL, Anisfeld AM, Edwards PA, et al. A chemical, genetic, and structural analysis of the nuclear bile acid receptor FXR. Mol Cell. 2003;11:1079–1092. doi: 10.1016/s1097-2765(03)00104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 11.Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147–191. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- 12.Pellicciari R, Costantino G, Fiorucci S. Farnesoid X receptor: from structure to potential clinical applications. J Med Chem. 2005;48:5383–5403. doi: 10.1021/jm0582221. [DOI] [PubMed] [Google Scholar]
- 13.Fiorucci S, Rizzo G, Donini A, Distrutti E, Santucci L. Targeting farnesoid X receptor for liver and metabolic disorders. Trends Mol Med. 2007;13:298–309. doi: 10.1016/j.molmed.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, Auwerx J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113:1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lambert G, Amar MJ, Guo G, Brewer HB, Jr, Gonzalez FJ, Sinal CJ. The farnesoid X-receptor is an essential regulator of cholesterol homeostasis. J Biol Chem. 2003;278:2563–2570. doi: 10.1074/jbc.M209525200. [DOI] [PubMed] [Google Scholar]
- 16.Reference deleted
- 17.Stayrook KR, Bramlett KS, Savkur RS, Ficorilli J, Cook T, Christe ME, Michael LF, Burris TP. Regulation of carbohydrate metabolism by the farnesoid X receptor. Endocrinology. 2005;146:984–991. doi: 10.1210/en.2004-0965. [DOI] [PubMed] [Google Scholar]
- 18.Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest. 2006;116:1102–1109. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cai J, Abramovici H, Gee SH, Topham MK. Diacylglycerol kinases as sources of phosphatidic acid. Biochim Biophys Acta. 2009;1791:942–948. doi: 10.1016/j.bbalip.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merida I, Avila-Flores A, Merino E. Diacylglycerol kinases: at the hub of cell signalling. Biochem J. 2008;410:631–631. doi: 10.1042/BJ20071040. [DOI] [PubMed] [Google Scholar]
- 21.Loewen CJ, Gaspar ML, Jesch SA, Delon C, Ktistakis NT, Henry SA, Levine TP. Phospholipid metabolism regulated by a transcription factor sensing phosphatidic acid. Science. 2004;304:1644–1647. doi: 10.1126/science.1096083. [DOI] [PubMed] [Google Scholar]
- 22.Los AP, van Baal J, de Widt J, Divecha N, van Blitterswijk WJ. Structure-activity relationship of diacylglycerol kinase θ. Biochim Biophys Acta. 2004;1636:169–174. doi: 10.1016/j.bbalip.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 23.Sakane F, Imai S, Kai M, Yasuda S, Kanoh H. Diacylglycerol kinases: why so many of them? Biochim Biophys Acta. 2007;1771:793–806. doi: 10.1016/j.bbalip.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 24.Raben DM, Tu-Sekine B. Nuclear diacylglycerol kinases: regulation and roles. Front Biosci. 2008;13:590–597. doi: 10.2741/2704. [DOI] [PubMed] [Google Scholar]
- 25.Shulga YV, Topham MK, Epand RM. Regulation and functions of diacylglycerol kinases. Chem Rev. 2011;111:6186–6208. doi: 10.1021/cr1004106. [DOI] [PubMed] [Google Scholar]
- 26.Topham MK, Epand RM. Mammalian diacylglycerol kinases: molecular interactions and biological functions of selected isoforms. Biochim Biophys Acta. 2009;1790:416–424. doi: 10.1016/j.bbagen.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tu-Sekine B, Raben DM. Regulation of DGK-θ. J Cell Physiol. 2009;220:548–552. doi: 10.1002/jcp.21813. [DOI] [PubMed] [Google Scholar]
- 28.van Blitterswijk WJ, Houssa B. Properties and functions of diacylglycerol kinases. Cell Signalling. 2000;12:595–605. doi: 10.1016/s0898-6568(00)00113-3. [DOI] [PubMed] [Google Scholar]
- 29.van Baal J, de Widt J, Divecha N, van Blitterswijk WJ. Diacylglycerol kinase θ counteracts protein kinase C-mediated inactivation of the EGF receptor. Int J Biochem Cell Biol. 2012;44:1791–1799. doi: 10.1016/j.biocel.2012.06.021. [DOI] [PubMed] [Google Scholar]
- 30.Tu-Sekine B, Raben DM. Dual regulation of diacylglycerol kinase (DGK)-θ: polybasic proteins promote activation by phospholipids and increase substrate affinity. J Biol Chem. 2012;287:41619–41627. doi: 10.1074/jbc.M112.404855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baldanzi G, Alchera E, Imarisio C, Gaggianesi M, Dal Ponte C, Nitti M, Domenicotti C, van Blitterswijk WJ, Albano E, Graziani A, Carini R. Negative regulation of diacylglycerol kinase θ mediates adenosine-dependent hepatocyte preconditioning. Cell Death Differ. 2010;17:1059–1068. doi: 10.1038/cdd.2009.210. [DOI] [PubMed] [Google Scholar]
- 32.English D, Cui Y, Siddiqui RA. Messenger functions of phosphatidic acid. Chem Phys Lipids. 1996;80:117–132. doi: 10.1016/0009-3084(96)02549-2. [DOI] [PubMed] [Google Scholar]
- 33.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 34.Toschi A, Lee E, Xu L, Garcia A, Gadir N, Foster DA. Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol Cell Biol. 2009;29:1411–1420. doi: 10.1128/MCB.00782-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang C, Wendel AA, Keogh MR, Harris TE, Chen J, Coleman RA. Glycerolipid signals alter mTOR complex 2 (mTORC2) to diminish insulin signaling. Proc Natl Acad Sci USA. 2012;109:1667–1672. doi: 10.1073/pnas.1110730109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Avila-Flores A, Santos T, Rincon E, Merida I. Modulation of the mammalian target of rapamycin pathway by diacylglycerol kinase-produced phosphatidic acid. J Biol Chem. 2005;280:10091–10099. doi: 10.1074/jbc.M412296200. [DOI] [PubMed] [Google Scholar]
- 37.Gorentla BK, Wan CK, Zhong XP. Negative regulation of mTOR activation by diacylglycerol kinases. Blood. 2011;117:4022–4031. doi: 10.1182/blood-2010-08-300731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cai K, Sewer MB. cAMP-stimulated transcription of diacylglycerol kinase theta (DGKθ) requires steroidogenic factor-1 (SF-1) and sterol responsive element binding protein 1 (SREBP1) J Lipid Res. 2013;54:2121–2132. doi: 10.1194/jlr.M035634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dammer EB, Leon A, Sewer MB. Coregulator exchange and sphingosine-sensitive cooperativity of steroidogenic factor-1, general control nonderepressed 5, p54, and p160 coactivators regulate cyclic adenosine 3 ′,5′-monophosphate-dependent cytochrome P450c17 transcription rate. Mol Endocrinol. 2007;21:415–438. doi: 10.1210/me.2006-0361. [DOI] [PubMed] [Google Scholar]
- 40.de Raemy-Schenk AM, Trouble S, Gaillard P, Page P, Gotteland JP, Scheer A, Lang P, Yeow K. A cellular assay for measuring the modulation of glucose production in H4IIE cells. Assay Drug Dev Technol. 2006;4:525–533. doi: 10.1089/adt.2006.4.525. [DOI] [PubMed] [Google Scholar]
- 41.Angelin B, Bjorkhem I, Einarsson K. Influence of bile acids on the soluble phosphatidic acid phosphates in rate liver. Biochem Biophys Res Commun. 1981;100:606–612. doi: 10.1016/s0006-291x(81)80219-7. [DOI] [PubMed] [Google Scholar]
- 42.Luo B, Prescott SM, Topham MK. Diacylglycerol kinase θ regulates phosphatidylinositol 4-phosphate 5-kinase Iα by a novel mechanism. Cell Signalling. 2004;16:891–897. doi: 10.1016/j.cellsig.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 43.Ghosh S, Strum JC, Sciorra VA, Daniel L, Bell RM. Raf-1 kinase possesses distinct binding domains for phosphatidylserine and phosphatidic acid. Phosphatidic acid regulates the translocation of Raf-1 in 12-O-tetradecanoylphorbol-13-acetate-stimulated Madin-Darby canine kidney cells. J Biol Chem. 1996;271:8472–8480. doi: 10.1074/jbc.271.14.8472. [DOI] [PubMed] [Google Scholar]
- 44.Lopez-Andreo MJ, Gomez-Fernandez JC, Corbalan-Garcia S. The simultaneous production of phosphatidic acid and diacylglycerol is essential for the translocation of protein kinase C ε to the plasma membrane in RBL-2H3 cells. Mol Biol Cell. 2003;14:4885–4895. doi: 10.1091/mbc.E03-05-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Limatola C, Schaap D, Moolenaar WH, van Blitterswijk WJ. Phosphatidic acid activation of protein kinase C-ζ overexpressed in COS cells: comparison with other protein kinase C isotypes and other acidic lipids. Biochem J. 1994;304:1001–1008. doi: 10.1042/bj3041001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delon C, Manifava M, Wood E, Thompson D, Krugmann S, Pyne S, Ktistakis NT. Sphingosine kinase 1 is an intracellular effector of phosphatidic acid. J Biol Chem. 2004;279:44763–44774. doi: 10.1074/jbc.M405771200. [DOI] [PubMed] [Google Scholar]
- 47.Frank C, Keilhack H, Opitz F, Zschornig O, Bohmer FD. Binding of phosphatidic acid to the protein-tyrosine phosphatase SHP-1 as a basis for activity modulation. Biochemistry. 1999;38:11993–12002. doi: 10.1021/bi982586w. [DOI] [PubMed] [Google Scholar]
- 48.Reference deleted
- 49.Navé BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999;344:427–431. [PMC free article] [PubMed] [Google Scholar]
- 50.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 51.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 52.Lu MJ, Wan M, Leavens KF, Chu QW, Monks BR, Fernandez S, Ahima RS, Ueki K, Kahn CR, Birnbaum MJ. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med. 2012;18:388–395. doi: 10.1038/nm.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cao RS, Cronk ZX, Zha WB, Sun LX, Wang X, Fang YW, Studer E, Zhou HP, Pandak WM, Dent P, et al. Bile acids regulate hepatic gluconeogenic genes and farnesoid X receptor via Gαi-protein-coupled receptors and the AKT pathway. J Lipid Res. 2010;51:2234–2244. doi: 10.1194/jlr.M004929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duran-Sandoval D, Cariou B, Percevault F, Hennuyer N, Grefhorst A, van Dijk TH, Gonzalez FJ, Fruchart JC, Kuipers F, Staels B. The farnesoid X receptor modulates hepatic carbohydrate metabolism during the fasting-refeeding transition. J Biol Chem. 2005;280:29971–29979. doi: 10.1074/jbc.M501931200. [DOI] [PubMed] [Google Scholar]
- 55.Han SL, Studer E, Gupta S, Fang YW, Qiao LA, Li WQ, Grant S, Hylemon PB, Dent P. Bile acids enhance the activity of the insulin receptor and glycogen synthase in primary rodent hepatocytes. Hepatology. 2004;39:456–463. doi: 10.1002/hep.20043. [DOI] [PubMed] [Google Scholar]
- 56.Brose N, Betz A, Wegmeyer H. Divergent and convergent signaling by the diacylglycerol second messenger pathway in mammals. Curr Opin Neurobiol. 2004;14:328–340. doi: 10.1016/j.conb.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 57.Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer. 2007;7:281–294. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- 58.Baillie GS, Huston E, Scotland G, Hodgkin M, Gall I, Peden AH, MacKenzie C, Houslay ES, Currie R, Pettitt TR, et al. TAPAS-1, a novel microdomain within the unique N-terminal region of the PDE4A1 cAMP-specific phosphodiesterase that allows rapid, Ca2+-triggered membrane association with selectivity for interaction with phosphatidic acid. J Biol Chem. 2002;277:28298–28309. doi: 10.1074/jbc.M108353200. [DOI] [PubMed] [Google Scholar]
- 59.Hokin LE, Hokin MR. Effects of acetylcholine on phosphate turnover in phospholipides of brain cortex in vitro. Biochim Biophys Acta. 1955;16:229–237. doi: 10.1016/0006-3002(55)90208-0. [DOI] [PubMed] [Google Scholar]
- 60.Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, Shulman GI. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 61.Cantley JL, Yoshimura T, Camporez JP, Zhang D, Jornayvaz FR, Kumashiro N, Guebre-Egziabher F, Jurczak MJ, Kahn M, Guigni BA, et al. CGI-58 knockdown sequesters diacylglycerols in lipid droplets/ER-preventing diacylglycerol-mediated hepatic insulin resistance. Proc Natl Acad Sci USA. 2013;110:1869–1874. doi: 10.1073/pnas.1219456110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106:171–176. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reference deleted
- 64.van Baal J, de Widt J, Divecha N, van Blitterswijk WJ. Translocation of diacylglycerol kinase theta from cytosol to plasma membrane in response to activation of G protein-coupled receptors and protein kinase C. J Biol Chem. 2005;280:9870–9878. doi: 10.1074/jbc.M409301200. [DOI] [PubMed] [Google Scholar]
- 65.McMullan R, Hiley E, Morrison P, Nurrish SJ. Rho is a presynaptic activator of neurotransmitter release at pre-existing synapses in C. elegans. Genes Dev. 2006;20:65–76. doi: 10.1101/gad.359706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tu-Sekine B, Ostroski M, Raben DM. Modulation of diacylglycerol kinase θ activity by α-thrombin and phospholipids. Biochemistry. 2007;46:924–932. doi: 10.1021/bi061170c. [DOI] [PubMed] [Google Scholar]
- 67.Li D, Urs AN, Allegood J, Leon A, Merrill AH, Jr, Sewer MB. Cyclic AMP-stimulated interaction between steroidogenic factor 1 and diacylglycerol kinase θ facilitates induction of CYP17. Mol Cell Biol. 2007;27:6669–6685. doi: 10.1128/MCB.00355-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reference deleted
- 69.Reference deleted
- 70.Li T, Chiang JY. Bile acid signaling in liver metabolism and diseases. J Lipids. 2012;2012:754067. doi: 10.1155/2012/754067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nguyen A, Bouscarel B. Bile acids and signal transduction: role in glucose homeostasis. Cell Signaling. 2008;20:2180–2197. doi: 10.1016/j.cellsig.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 72.Pols TW, Noriega LG, Nomura M, Auwerx J, Schoonjans K. The bile acid membrane receptor TGR5 as an emerging target in metabolism and inflammation. J Hepatol. 2011;54:1263–1272. doi: 10.1016/j.jhep.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Porez G, Prawitt J, Gross B, Staels B. Bile acids as targets for the treatment of dyslipidemia and cardiovascular disease. J Lipid Res. 2012;53:1723–1737. doi: 10.1194/jlr.R024794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reference deleted
- 75.Hornberger TA, Chu WK, Mak YW, Hsiung JW, Huang SA, Chien S. The role of phospholipase D and phosphatidic acid in the mechanical activation of mTOR signaling in skeletal muscle. Proc Natl Acad Sci USA. 2006;103:4741–4716. doi: 10.1073/pnas.0600678103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu L, Salloum D, Medlin PS, Saqcena M, Yellen P, Perrella B, Foster DA. Phospholipase D mediates nutrient input to mammalian target of rapamycin complex 1 (mTORC1) J Biol Chem. 2011;286:25477–25486. doi: 10.1074/jbc.M111.249631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Leavens KF, Easton RM, Shulman GI, Previs SF, Birnbaum MJ. Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metab. 2009;10:405–418. doi: 10.1016/j.cmet.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nakae J, Kitamura T, Silver DL, Accili D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J Clin Invest. 2001;108:1359–1367. doi: 10.1172/JCI12876. [DOI] [PMC free article] [PubMed] [Google Scholar]