

Abstract
Background
Renovascular hypertension (RVHT) increases cardiovascular morbidity and mortality. Renal revascularization with percutaneous transluminal renal angioplasty and stenting (PTRS) may reverse RVHT but may not fully regress cardiac remodeling and damage, possibly due to persistent myocardial insults. Bendavia is a mitochondrial targeted peptide that reduces ischemic cardiomyopathy by improving mitochondrial function. However, its potential for attenuating residual myocardial damage after reversal of RVHT has not been explored. We hypothesized that treatment with Bendavia as an adjunct to PTRS would improve cardiac function and oxygenation, and decrease myocardial injury in swine RVHT.
Methods and results
After 6 weeks of RVHT (unilateral renal artery stenosis) or control, pigs underwent PTRS (or sham), with adjunct continuous infusion of Bendavia (0.05 mg/kg intravenously, 30 min before to 3.5 h after PTRS) or vehicle (n =7 each). Four weeks later, systolic and diastolic function were assessed by multidetector computed tomography, myocardial oxygenation by blood oxygen level-dependent MRI, and myocardial morphology, apoptosis, mitochondrial biogenesis, and fibrosis evaluated ex vivo. PTRS restored blood pressure in both groups, yet E/A ratio remained decreased. Myocardial oxygenation and mitochondrial biogenesis improved, and myocardial inflammation, oxidative stress, and fibrosis normalized in association with improvement in diastolic function in RVHT +PTRS +Bendavia animals.
Conclusion
Adjunct Bendavia during PTRS in swine RVHT improved diastolic function and oxygenation and reversed myocardial tissue damage. This approach may allow a novel strategy for preservation of cardiac function and structure in RVHT.
Keywords: mitochondria, myocardium, renal hypertension, revascularization
INTRODUCTION
Secondary hypertension due to narrowing of the lumen of the renal artery is mainly consequent to atherosclerosis, has an incidence of about 0.4%, and its prevalence increases with age [1]. Hypoperfusion of the renal parenchyma often triggers renovascular hypertension (RVHT) by activating the renin–angiotensin–aldosterone system. This in turn leads to abnormalities in cardiac structure and function, as well as increased prevalence of peripheral artery disease [2].
In patients with RVHT, restoration of blood pressure levels using percutaneous renal angioplasty and stenting (PTRS) may fail to prevent cardiac remodeling and damage, likely due to persistent myocardial insults. Clinical studies have shown that despite significant reductions in blood pressure and cardiac remodeling, coronary perfusion [3] and diastolic function [4] remain unchanged after revascularization. Furthermore, renal artery revascularization in patients with RVHT improved heart failure control and reduced heart failure hospitalization, but not mortality [5]. Therefore, novel adjunct therapies are needed to attenuate myocardial injury and improve cardiovascular outcomes after renal revascularization.
We have previously shown in swine RVHT that renal revascularization triggers the release from the stenotic kidney of inflammatory cytokines such as monocyte chemoattractant protein (MCP)-1 [6], a key mediator of myocardial inflammation, fibrosis, vascular remodeling, and microvascular rarefaction induced by hypertension [7]. We have also previously shown that selective improvement in renal function reduced renal and systemic oxidative stress and inflammation, preserving myocardial microvascular function and architecture, which underscores the importance of cardiorenal cross-talk mediated by renal injury signals [8]. Therefore, strategies tailored to protect the kidney during reperfusion may potentially have a substantial impact in ameliorating myocardial damage.
Bendavia is a novel cell-permeable positively charged tetrapeptide with a dimethyltyrosine moiety that localizes to the inner mitochondrial membrane, and inhibits apoptosis and oxidative stress in several experimental models of ischemia reperfusion injury [9,10]. This drug inhibits reactive oxygen species (ROS) production and consequently prevents formation of the mitochondrial permeability transition pore (mPTP) [9,11], an important trigger for apoptosis. Recent studies have shown that treatment with Bendavia prevents cardiac hypertrophy, fibrosis, and left ventricular diastolic dysfunction in experimental cardiomyopathy [12–14], but its potential for attenuating myocardial damage after reversal of RVHT remains to be elucidated.
We have recently shown in swine RVHT that a continuous systemic infusion of Bendavia at the time of PTRS ameliorates subsequent renal parenchymal inflammation, oxidative stress, apoptosis, and microvascular damage, improving renal functional outcomes 4 weeks after vascular intervention [6]. However, whether these noteworthy effects on the stenotic kidneys are associated with significant improvement in cardiac function after revascularization remains unknown. Therefore, we hypothesized that treatment with Bendavia as an adjunct to PTRS would improve cardiac function and decrease myocardial inflammation, oxidative stress, and fibrosis in swine RVHT.
METHODS
Twenty-eight domestic female pigs were studied after 16 weeks of observation (Fig. 1). At baseline, animals were randomized in two groups: normal (n =7, fed normal pig diet) and RVHT (n =21, fed a high-cholesterol diet). Six weeks later, we induced unilateral renal artery stenosis (RAS) in RVHT pigs by placing an irritant coil in the main renal artery under fluoroscopy, which leads to a gradual narrowing of the renal artery, as previously described [15]. Normal animals underwent a sham procedure. We have previously shown in this model that blood pressure increases after coil implantation reaching hypertensive levels in 1–2 weeks [16,17]. The high-cholesterol diet served to emulate atherosclerosis.
FIGURE 1.
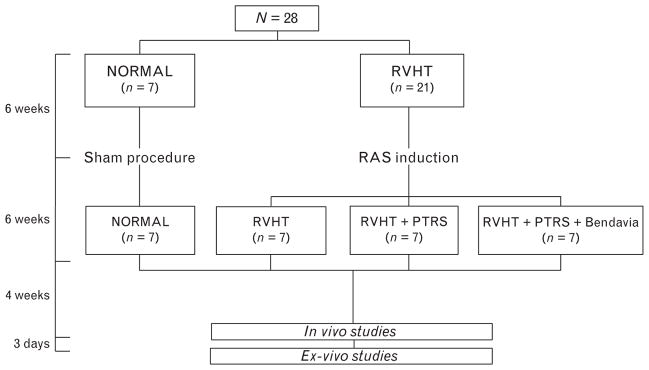
Schematic of the experimental protocol. PTRS, percutaneous transluminal renal angioplasty and stenting; RAS, renal artery stenosis; RVHT, renovascular hypertension.
After 6 weeks of RVHT or sham, animals were anesthetized with 0.5 g of intramuscular ketamine and xylazine, maintained with intravenous ketamine (0.2 mg/kg per min) and xylazine (0.03 mg/kg per min), and the degree of stenosis was angiographically determined. Subsequently, seven normal and seven RVHT pigs underwent a sham procedure, whereas the other 14 were treated with PTRS (Fig. 1) with or without a continuous intravenous infusion of Bendavia (0.050 mg/kg; Stealth Peptides Inc., Newton Centre, Massachusetts, USA) or physiological saline from 30 min before to 3.5 h after PTRS or sham [6].
Four weeks later, myocardial oxygenation was evaluated by blood oxygen level-dependent MRI (BOLD-MRI), whereas single-kidney glomerular filtration rate (GFR) and cardiac function were assessed by multidetector computed tomography (MDCT). Inferior vena cava (IVC) samples were collected for cholesterol panels, plasma renin activity (PRA), and isoprostane levels. Rate–pressure product (RPP) was calculated by heart rate ×SBP ×10−2 as an index of myocardial oxygen consumption [18].
Three days after completion of in-vivo studies, animals were euthanized with sodium pentobarbital (100 mg/kg). The heart was removed, dissected, and samples from the left ventricle (LV) preserved at −80°C for ex-vivo studies.
In-vivo studies
Percutaneous transluminal renal angioplasty and stenting
Renal revascularization was performed by expanding a 7F balloon catheter in the proximal middle section of the renal artery under fluoroscopy, as previously described [16,17]. The expansion of a tantalum stent to full balloon diameter restores renal artery patency. Then, the balloon was deflated and removed, leaving the stent embedded in the vascular wall.
Myocardial oxygenation
BOLD-MRI was performed at 3 Tesla (Signa Echo Speed; GE Medical Systems, Milwaukee, Wisconsin, USA). Anesthesia was maintained with inhaled isoflurane (1–2%) throughout the course of the scanning. Myocardial oxygenation was measured using 4–5 BOLD-MRI slices, prescribed along the heart short axis. Images were acquired during suspended respiration using Fast Gradient Echo sequence with repetition time/echo time/number of echoes/matrix size/ field of view/slice thickness/flip angle equal to 6.8 ms/ 1.6–4.8 ms/8/128 ×128/35/0.5 cm/30°. For data analysis, regions of interest were manually traced in the left ventricular wall myocardium on the 7-ms echo time image (Fig. 1a). Myocardial R2* values were estimated in each voxel by fitting the magnetic resonance (MR) signal intensity vs. echo times to a single exponential function and calculating the MR intensity decay rate, as previously shown [18].
Cardiac function
Cardiac function and structure were assessed in vivo using MDCT under intravenous ketamine (0.2 mg/kg per min) and xylazine (0.03 mg/kg per min) anesthesia. Following a bolus injection of nonionic, low osmolar contrast medium (Isovue-370; 0.33 ml/kg) into the right atrium, a 50-s flow study was performed in selected left ventricular sections to assess myocardial perfusion. In addition, the entire LV was scanned 20 times throughout the cardiac cycle to assess systolic and diastolic function. Left ventricular muscle mass (LVMM), cardiac output, early (E) and late (A) ventricular filling rates, and E/A ratio were calculated [19,20]. The images were analyzed with the Analyze software package (Biomedical Imaging Resource; Mayo Clinic, Rochester, Minnesota, USA).
Renal function
Stenotic-kidney GFR was measured using MDCT, as previously described [16,21]. In brief, 160 consecutive scans were performed following a central venous injection of iopamidol (0.5 ml/kg per 2 s). MDCT images were reconstructed and displayed with Analyze, and regions of interest (selected from cross-sectional images from the aorta and renal cortex) were traced to generate tissue attenuation curves. GFR was assessed from the cortical curve using the slope of the proximal tubular curve [21].
Ex-vivo studies
Apoptosis and mitochondrial biogenesis
Apoptosis was assessed in myocardial sections stained with double fluorescent terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and connexin-43 staining (Abcam, Cambridge, Massachusetts, USA; catalog#ab79010), as well as single fluorescent caspase-3 (1 : 200; Santa Cruz Biotechnology, Inc., Dallas, Texas, USA) staining. Mitochondrial biogenesis was evaluated by expression of peroxisome proliferator-activated receptor gamma coactivator (PGC)-1α (1 : 1000; Abcam), guanine adenine-binding protein (GABP, 1 : 1000; Abcam), and peroxisome proliferator-activated receptor (PPAR)-α (1 : 1000; Abcam), as described [6].
Microvascular architecture and remodeling
The heart was perfused with an intravascular contrast agent through a branch of the left circumflex coronary artery. Myocardial samples (2 ×1 ×1 cm) were prepared, scanned, and analyzed with Analyze in 7 slices obtained at equal intervals in the subepicardium or subendocardium to calculate the spatial density of myocardial microvessels (diameters 20–500 μm), as previously described [7,22].
Microvascular wall thickening was assessed in randomly selected intramyocardial vessels stained with antihuman α-smooth muscle actin (SMA) antibody (×40 magnification; DakoCytomation, Glostrup, Denmark). Media wall thickness to lumen diameter ratio was calculated, as described [20].
Oxidative stress and inflammation
Systemic oxidative stress was assessed by IVC levels of 8-epi-isoprostane (EIA kit) [23]. Myocardial oxidative stress was assessed by in-situ production of superoxide anion with dihydroethidium (DHE) and by expression of the NADPH-oxidase subunits p47 and p67 (1 : 200; both Santa Cruz), and by nitrotyrosine (1 : 200; Cayman Chemical Company, Ann Arbor, Michigan, USA) [23,24].
Inflammation was evaluated in myocardial sections by standard immunostaining for MCP-1 (1 : 7500; MyBioSource Inc., San Diego, California, USA) and double staining for pro-inflammatory CD68+/iNOS+ (M1) (Abnova Inc., Walnut, California, USA; catalog#: ab15323, 1 : 100) and reparative CD68+/Arinase-1 (M2) (sc-20150 cat#: HPA004114, 1 : 100) macrophages. In addition, myocardial expression of tumor necrosis factor (TNF)-α (Santa Cruz, 1 : 200), interleukin (IL)-6 (1 : 500; Abcam), and IL-10 (1 : 200; Santa Cruz) was quantified by western blot.
Myocardial fibrosis and collagen deposition
Myocardial fibrosis was evaluated in 5-μm sections of the LV stained with trichrome and Sirius red, and semiautomatically quantified using AxioVision 4.8.2.0 (Carl Zeiss SMT, Oberkochen, Germany), in 10–15 random fields, and expressed as percentage of staining of total surface area. In addition, gene expression of collagen I and III was assessed by real-time quantitative RT-PCR analysis, as previously described [25,26]. Fold change of collagen I and collagen III relative to housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was calculated using 2−ΔΔCT method. To evaluate the number of myofibroblasts, LV cross-sections were stained with αSMA, and the percentage of immunostaining area (excluding vessels) calculated, as previously described [27]. Finally, myocardial expression of transforming growth factor (TGF)-β, tissue inhibitor of metalloproteinase (TIMP)-1 (1 : 200; both Santa Cruz), and matrix metalloproteinase (MMP)-2 (Abcam, 1 : 1000) were measured by western blot.
Statistical analysis
Results were expressed as mean ±standard deviation. Parametric [analysis of variance (ANOVA) and unpaired Student’s t-test] and nonparametric (Wilcoxon and Kruskal–Wallis) tests were used as appropriate. P values less than 0.05 were considered statistically significant. Data were analyzed using JMP 0.9 (SAS Institute Inc., Cary, North Carolina, USA).
RESULTS
Six weeks after induction of RVHT, similar and significant degree of RAS was observed in animals with RVHT, RVHT +PTRS, and RVHT +PTRS +Bendavia, and their mean arterial pressure (MAP) was similarly elevated compared with normal (all P <0.05 vs. normal; data not shown).
Some of the systemic characteristics and renal function measured in these animals at 10 weeks were previously published [6]. Four weeks after PTRS, renal artery patency was restored and MAP normalized in all revascularized pigs (Table 1). Total cholesterol and low-density lipoprotein levels were similarly higher in all animals with RVHT compared with normal. However, stenotic-kidney GFR was similarly lower in RVHT and RVHT +PTRS PTRS compared with normal, but improved in RVHT + PTRS +Bendavia pigs (Table 1, P <0.01 vs. RVH, P <0.003 vs. RVHT +PTRS, P >0.27 vs. normal). RPP was elevated in RVHT, not different from normal levels in either RVHT +PTRS or RVHT +PTRS +Bendavia pigs, but further reduced in RVHT +PTRS +Bendavia compared with RVHT +PTRS (P =0.013). PRA levels were similar among the groups (data not shown).
TABLE 1.
Systemic characteristics of normal, hypertensive, hypertensive + revascularization, and hypertensive + revascularization + Bendavia pigs (n=7 each) 4 weeks after percutaneous transluminal renal angioplasty or sham
| Normal | Hypertensive | Hypertensive + revascularization | Hypertensive + revascularization +Bendavia | |
|---|---|---|---|---|
| Number of animals | 7 | 7 | 7 | 7 |
| Body weight (kg) | 45.5 ±2.3 | 43.8 ±1.9 | 42.6 ±1.5 | 47.7 ±2.4 |
| Degree of stenosis (%) | 0 | 87.5 ±7.5*,‡ | 0 | 0 |
| Mean arterial pressure (mmHg) | 87.2 ±4.6 | 150.9 ±7.6* | 90.5 ±6.8 | 91.5 ±8.5 |
| Total cholesterol (mg/dl) | 92.2 ±17.9 | 498.9 ±61.3* | 504.3 ±190.2* | 469.8 ±97.0* |
| Low-density lipoprotein (mg/dl) | 47.8 ±9.5 | 314.8 ±78.4* | 253.7 ±86.4* | 247.8 ± 142.4* |
| 8-Epi-isoprostane (pg/ml) | 83.2 ±6.7 | 134.9 ±9.8*,‡ | 142.2 ±14.0*,‡ | 120.2 ±15.8 |
| Stenotic-kidney GFR (ml/min) | 82.8 ±10.4 | 50.0 ±4.2* | 45.4 ±2.8*,‡ | 69.4 ±4.8 |
| Heart rate (bpm) | 71.7 ±9.5 | 78.4 ±9.1 | 78.1 ±6.4 | 74.1 ±9.1 |
| Rate–pressure product (mmHg × bpm) | 177.1 ±29.5 | 356.6 ±53.3*,‡ | 196.0 ±17.7‡ | 159.2 ±25.6 |
| Stroke volume | 44.0 ±7.4 | 41.6 ±7.6 | 42.4 ±3.0 | 44.2 ±6.3 |
| Cardiac output (l/min) | 3.1 ±0.6 | 3.5 ±1.1 | 3.2 ±0.2 | 3.4 ±0.7 |
| E/A ratio | 1.3 ±0.2 | 0.7 ±0.2*,‡ | 0.9 ±0.2*,‡ | 1.2 ±0.3 |
| LVMM (g/kg body weight) | 0.8 ±0.1 | 1.3 ±0.2*,‡ | 0.9 ±0.1 | 0.9 ±0.2 |
| Myocardial perfusion (ml/min g−1) | 0.8 ±0.3 | 0.7 ±0.1 | 0.8 ±0.1 | 0.8 ±0.1 |
E/A, early (E) and atrial (A) ventricular filling; GFR, glomerular filtration rate; LVMM, left ventricular muscle mass.
P ≤0.05 vs. normal;
P <0.05 vs. RVHT +PTRS +Bendavia.
Cardiac function and oxygenation
Heart rate, stroke volume, and cardiac output were similar among the groups (Table 1, P >0.05, ANOVA). LVMM was higher in RVHT compared with normal, but restored to normal levels in both PTRS-treated groups (Table 1). However, E/A ratio that was similarly lower in RVHT and RVHT +PTRS compared with normal did not differ from normal levels in PTRS +Bendavia-treated animals (P =0.03 vs. RVHT, P =0.05 vs. RVHT +PTRS, P =0.37 vs. normal). CT-derived myocardial perfusion was not different among the groups (Table 1). R2* values were equally elevated in RVHT and RVHT +PTRS compared with normal, indicating decreased myocardial oxygenation, but were normalized in animals treated with PTRS +Bendavia (Fig. 2a).
FIGURE 2.

(a) Representative left ventricular cross-sectional images of blood oxygen level-dependent (BOLD) MRI, showing hypoxic myocardium (red) in renovascular hypertension (RVHT) and quantification of R2* signal in the study groups. (b) Double immunofluorescence staining with terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL, green) and connexin-43 (red), and single caspase-3 (red) staining (top), and their quantification (bottom). (c) Myocardial expression of peroxisome proliferator-activated receptor gamma coactivator (PGC)-1α, guanine adenine-binding protein (GABP), and peroxisome proliferator-activated receptor (PPAR)-α. *P ≤0.05 vs. normal; ‡P <0.05 vs. RVHT +PTRS +Bendavia. PTRS, percutaneous transluminal renal angioplasty and stenting.
Apoptosis and mitochondrial biogenesis
The number of TUNEL (green) and caspase-3 (red) apoptotic cells was higher in RVHT and RVHT +PTRS compared with normal, but treatment with PTRS + Bendavia normalized TUNEL and decreased caspase-3 staining (Fig. 2b). Furthermore, cells with TUNEL-positive nuclei were also stained with connexin-43, confirming its cardiomyocyte origin. Myocardial expression of PGC-1α was upregulated only in RVHT animals treated with Bendavia (Fig. 2c). Expression of GABP was down-regulated in RVHT and RVHT +PTRS pigs, yet normalized in RVHT +PTRS +Bendavia animals (P =0.04 vs. RVHT, P =0.002 vs. RVHT +PTRS). Expression of PPAR-α showed a similar pattern and improved in RVHT +PTRS +Bendavia animals (Fig. 2c, P <0.04 vs. RVHT, P <0.01 vs. RVHT + PTRS), although it remained below normal.
Microvascular architecture
Microvascular subepicardial density was similar among the groups (Fig. 3a–b). Subendocardial density of medium size (200–300 μm in diameter) and large (>300 μm) microvessels was also similar among the groups. However, sub-endocardial density of small microvessels (20–200 μm) was lower in RVHT and RVHT +PTRS, but normalized in animals treated with Bendavia (Fig. 3a–c). Microvascular wall was thickened in RVHT and RVHT +PTRS vs. normal, and improved in Bendavia-treated pigs (Fig. 3d), although it remained higher than normal.
FIGURE 3.

(a) Representative three-dimensional computed tomography (CT) image of the left ventricle (left) demonstrating schematically sampling for myocardial micro-CT analysis (white box) and subsequent three-dimensional micro-CT reconstruction of myocardial microvascular architecture (right). Quantification of spatial density of microvessels in the subepicardium (b) and subendocardium (c). (d) Myocardial vessel wall-to-lumen ratio and its quantification in α-smooth muscle actin stained slides (40×). (e) In-situ production of superoxide anion with dihydroethidium (DHE). (f) Myocardial expression of the NADPH-oxidase subunits p47 and p67, and nitrotyrosine. *P ≤0.05 vs. normal; ‡P <0.05 vs. RVHT +PTRS +Bendavia. PTRS, percutaneous transluminal renal angioplasty and stenting; RVHT, renovascular hypertension.
Oxidative stress
Systemic isoprostane levels were similarly elevated in RVHT and RVHT +PTRS compared with normal, but did not differ from normal levels in RVHT +PTRS +Bendavia (Table 1). Likewise, in-situ production of superoxide anion in the myocardium was augmented in RVHT and RVHT +PTRS, but was restored to normal levels in animals treated with PTRS +Bendavia (Fig. 3e). Myocardial expression of p47 was upregulated in RVHT, and similarly downregulated in RVHT +PTRS and RVHT +PTRS +Bendavia groups, whereas expressions of p67 and nitrotyrosine remained upregulated in RVHT +PTRS, but normalized in Bendavia-treated pigs (Fig. 3f).
Inflammation
MCP-1 immunoreactivity was upregulated in all RVHT animals compared with normal, but ameliorated only in pigs treated with Bendavia (Fig. 4a). The number of M1 macrophages was elevated in all RVHT animals compared with normal, but reduced in RVHT +PTRS +Bendavia compared with RVHT and RVHT +PTRS. In contrast, the number of reparative M2 macrophages was higher than normal only in Bendavia-treated pigs (Fig. 4b, P = 0.01 vs. normal, P =0.008 vs. RVHT, and P =0.009 vs. RVHT +PTRS). Consequently, the M1/M2 ratio, indicating propensity for inflammatory activity, was higher in RVHT and RVHT +PTRS compared with normal, but normalized in RVHT +PTRS +Bendavia animals (Fig. 4b). Moreover, myocardial expression of TNF-α and IL-6 was similarly upregulated in RVHT and RVHT +PTRS, yet normalized in Bendavia-treated pigs (Fig. 4c). Furthermore, myocardial expression of IL-10 was downregulated in RVHT and RVHT +PTRS compared with normal, but normalized in RVHT +PTRS +Bendavia pigs (Fig. 4c).
FIGURE 4.

(a) Representative monocyte chemoattractant protein (MCP)-1 immunostaining (red, white arrow) and its quantification. (b) Double immunostaining for CD68 and iNOS (M1 macrophages, yellow) and CD68 and Arginase-1 (M2 macrophages, yellow) and their ratio. (c) Myocardial expression of tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-10. *P ≤0.05 vs. normal; ‡P <0.05 vs. RVHT +PTRS +Bendavia. PTRS, percutaneous transluminal renal angioplasty and stenting; RVHT, renovascular hypertension.
Fibrosis and collagen deposition
Myocardial collagen deposition (Sirius red) and the number of myofibroblasts were increased in RVHT and RVHT +PTRS compared with normal, but restored to normal levels in RVHT +PTRS +Bendavia pigs (Fig. 5a and 5c). Consequently, myocardial fibrosis (trichrome) was greater in RVHT, unchanged by PTRS, and improved in RVHT +PTRS +Bendavia animals (Fig. 5b). Myocardial expression of TGF-β and TIMP-1 was upregulated in RVHT and RVHT +PTRS, but TIMP-1 was downregulated to levels not different from normal in PTRS +Bendavia-treated pigs, while TGF-β remained unaltered. In addition, collagen I and III mRNA expression was similar among the groups (P =0.63 and P =0.91, ANOVA, data not shown). Contrarily, myocardial expression of MMP-2 was upregulated in RVHT and RVHT +PTRS, but normalized in RVHT +PTRS +Bendavia pigs (Fig. 5d).
FIGURE 5.

(a) Representative staining and quantification of Sirius red and trichrome (b, blue; both ×40). (c) Representative α-smooth muscle actin (α-SMA) staining and its quantification. (d) Myocardial expression of transforming growth factor (TGF)-β, tissue inhibitor of metalloproteinase (TIMP)-1, and matrix metalloproteinase (MMP)-2. *P ≤0.05 vs. normal; ‡P <0.05 vs. RVHT +PTRS +Bendavia. PTRS, percutaneous transluminal renal angioplasty and stenting; RVHT, renovascular hypertension.
DISCUSSION
This study shows that treatment with PTRS alone normalized blood pressure without substantial improvement of cardiac diastolic function, possibly due to ongoing myocardial inflammation or kidney dysfunction (decreased stenotic-kidney GFR). Intravenous infusion of Bendavia at the time of PTRS in swine RVHT normalized GFR, restored cardiac function and oxygenation, and attenuated myocardial remodeling, apoptosis, oxidative stress, inflammation, and fibrosis 4 weeks later. The beneficial effect of Bendavia in the myocardium may be partly attributed to its ability to improve renal function and attenuate PTRS-induced renal and myocardial injury pathways. The current study supports a role for Bendavia as an adjuvant therapy for cardiac and renal protection during PTRS.
RVHT induces cardiovascular structural changes and impairs cardiac function, leading to increased morbidity and mortality rates. In RVHT patients, left ventricular hypertrophy, congestive heart failure [28], and ischemic heart disease [29] may aggravate cardiovascular morbidity and mortality, as well as progression to renal insufficiency. Therefore, adequate therapeutic interventions aimed to decrease the incidence of cardiac complications in patients with RVHT are needed.
The use of renal artery angioplasty remains controversial among clinicians caring for patients with RVHT. Indeed, clinical studies have shown that cardiac remodeling and damage may persist after treatment with endovascular revascularization, despite significant reductions in blood pressure and left ventricular mass. Heart failure remains an important predictor of mortality after PTRS [5]. In 19 patients with RVHT, although SBP and left ventricular hypertrophy improved after PTRS, myocardial perfusion and coronary flow reserve remained unchanged [3]. Similarly, decreases in blood pressure and left ventricular mass may be accompanied by persistent diastolic dysfunction [4], warranting additional therapies to attenuate tissue injury and cardiac dysfunction in RVHT patients beyond a decrease in blood pressure.
We have previously shown in a swine model of nonatherosclerotic RAS that revascularization successfully decreases blood pressure, improves coronary microvascular architecture, and reverses myocardial hypertrophy and diastolic dysfunction [30]. However, simulation of the clinical scenario by superimposition of atherosclerosis aggravates myocardial microvascular dysfunction and oxidative stress in experimental RVHT [31]. In turn, increased myocardial injury secondary to atherosclerosis may lead to irreversible damage and compromise recovery upon reversal of RVHT.
In this study, we treated animals with atherosclerosis and RVHT with systemic infusion of Bendavia during PTRS, and evaluated cardiac structure and function 4 weeks later. Previously, this compound has demonstrated the unique ability to reduce myocardial infarct sizes when administered prior to the onset of cardiac reperfusion in animal models [32,33]. Bendavia also prevented cardiac hypertrophy, fibrosis, and left ventricular diastolic dysfunction in a mouse model of angiotensin II-induced cardiomyopathy [12]. Importantly, we have recently demonstrated its ability to improve renal structure and function after PTRS in swine RVHT [6]. The current study extends our previous observations, demonstrating a unique potential for Bendavia also in attenuating myocardial injury and improving diastolic dysfunction after revascularization.
The mechanisms by which Bendavia delivery resulted in improved cardiac function and structure 4 weeks after PTRS may be speculated. In swine RVHT, abrupt restoration of renal artery patency triggers release of the inflammatory cytokine MCP-1 [6], an important mediator of cardiac injury. Treatment with the MCP inhibitor bindarit improves left ventricular hypertrophy and diastolic function, and inhibits myocardial inflammation and microvascular wall thickening, underscoring a central role of this cytokine in regulating cardiac microvascular function and structure in experimental RVHT [7]. Furthermore, selective preservation of renal function even without PTRS reduces renal and systemic oxidative stress and inflammation, blunting cardiac dysfunction in experimental RVHT [8]. Taken together, these observations suggest that adjunctive renoprotective and cardioprotective strategies have the potential to prevent myocardial remodeling and dysfunction in swine RVHT.
Cellular loss is an important mechanism implicated in myocardial injury. Previous studies have demonstrated cardiac apoptosis in patients with essential hypertension [34] and in rats with RVHT [35]. Pertinently, the cardioprotective effect of Bendavia seems to be partly mediated by its antiapoptotic properties, disclosed by reduced number of TUNEL/connexin-43 (cardiomyocytes) and caspase-3 apoptotic cells. Bendavia ameliorated apoptosis subsequent to PTRS, possibly by preventing formation of the mPTP and the consequent release of cytochrome c to the cytoplasm [11].
Moreover, the data suggest that this drug can promote mitochondrial biogenesis, evidenced by upregulation of PGC-1α (a master regulator of this pathway) and its receptor GABP. This effect is likely secondary to improved mitochondrial respiration by Bendavia [36]. PGC-1-α plays an important role in upregulating expression of myocardial mitochondrial antioxidants such as superoxide dismutase-2 and thioredoxin-2 protecting hearts against myocardial oxidative stress, hypertrophy, and dysfunction [37]. Similarly, myocardial expression of PPAR-α was blunted in RVHT, but improved in Bendavia-treated pigs. Our findings are underscored by a recent study, showing augmented antioxidant defenses and cardiac function following PPAR-α stimulation in a rat model of myocardial ischemia [38]. In line with these observations, systemic oxidative stress was normalized in RVHT +PTRS +Bendavia pigs, indicated by lowered circulating isoprostane levels. Furthermore, myocardial expression of the oxidative markers p67 and nitrotyrosine was downregulated in Bendavia-treated animals, confirming its strong antioxidant capacity.
We have previously shown in swine RVHT that increased oxidative stress and inflammation modulates myocardial microvascular architecture [39]. In line with this notion, we found a selective decrease in the number of small microvessels in the subendocardium in RVHT pigs that PTRS alone failed to restore. This selective loss might be functionally significant, as patients with microvascular disease may show preferential subendocardial myocardial hypoperfusion [40]. However, Bendavia normalized sub-endocardial spatial density, possibly through decreased myocardial oxidative stress. Importantly, myocardial microvascular remodeling, assessed by media-to-lumen ratio, increased in RVHT and RVHT +PTRS pigs, but was improved in Bendavia-treated animals, underscoring the potential of this drug for improving not only the number, but also the quality of the microvessels.
Inflammation also represents an important determinant of microvascular damage in RVHT, and previous studies from our group have shown increased myocardial expression of MCP-1 in RVHT pigs [20,39]. In agreement, MCP-1 immunoreactivity was higher in both RVHT and RVHT +PTRS compared with normal, but attenuated (though not normalized) only in animals treated with Bendavia. Interestingly, our study shows that RVHT leads to an increase in the ratio of M1 (pro-inflammatory)/M2 (reparative) myocardial macrophages, indicating a predominant inflammatory phenotype. Interestingly, this ratio was reversed by Bendavia, accompanied by downregulation of protein expression of the proinflammatory cytokines TNF-α and IL-6. Furthermore, Bendavia restored the expression of IL-10, which might have contributed to the increase in the number of M2 macrophages. Importantly, preservation of microvascular structure and attenuation of injurious pathways such as inflammation and oxidative stress might have resulted in improved myocardial oxygenation, reflected by normalized myocardial R2* values. Amelioration of myocardial hypoxia in the RVHT + PTRS +Bendavia group may have been partly modulated by changes in oxygen consumption, as RPP decreased further in the Bendavia-treated RVHT group. Furthermore, ROS, which remain abundant in RVHT, can decrease the efficiency of oxygen utilization independent of blood pressure [41], and might have thereby affected the levels of R2*. In addition to oxidative stress, microvascular rarefaction and interstitial fibrosis also persisted in RVHT after PTRS and might have contributed to inefficient nutrient and oxygen supply despite unchanged myocardial perfusion.
RVHT induces myocyte hypertrophy, interstitial fibrosis, and consequent cardiac hypertrophy (increased LVMM) and impaired diastolic function (decreased E/A ratio) [7,30]. We found that myocardial fibrosis and collagen deposition were accentuated in RVHT and RVH +PTRS, but reduced in RVHT +PTRS +Bendavia. Although Bendavia did not affect the expression of TGF-β protein or collagen genes, it normalized protein expression of MMP-2, a key regulator of cardiac extracellular matrix turnover [42]. In agreement, expression TIMP-1, a major inhibitor of MMPs [43], was downregulated in PTRS + Bendavia-treated pigs, suggesting that Bendavia can modulate extracellular matrix remodeling by increasing its degradation. Nevertheless, we cannot rule out the possibility of posttranslational regulation of collagen I and III in Bendavia-treated pigs.
Diastolic function was impaired in RVHT compared with normal and did not improve after PTRS alone, unlike our previous observations in a nonatherosclerotic RVHT model [30], underscoring the contribution of atherosclerosis to oxidative and inflammatory injury. Notably, diastolic function was normalized in Bendavia-treated pigs, suggesting that the combination of PTRS and Bendavia confers cardioprotection by not only normalizing blood pressure but also reducing fibrosis and thereby improving cardiac compliance.
Limitations
Our study is limited by the use of young pigs, duration of the disease, and lack of comorbidities compared with clinical RVHT. Nevertheless, myocardial injury and cardiac dysfunction in swine RVHT are similar to humans, enhancing the clinical relevance of our findings. In addition, we did not collect blood or tissue samples in the first few days after angioplasty, limiting our ability to elucidate the mechanisms by which Bendavia attenuates reperfusion injury. Nonetheless, in a previous study, we detected an increase only in MCP-1 levels immediately after revascularization [6]. Additional studies are needed to evaluate different regimens and validate the clinical efficacy of Bendavia therapy in RVHT.
Conclusion
Randomized clinical trials have demonstrated little benefit in RVHT patients treated with renal artery revascularization compared with those treated with medical therapy alone in terms of cardiovascular morbidity and mortality, warranting development of new therapies to prevent tissue injury and preserve cardiac function. Bendavia shows a unique therapeutic potential for reducing cardiac apoptosis, oxidative stress, inflammation, and fibrosis following PTRS in chronic experimental RVHT. Furthermore, a single intravenous infusion of Bendavia during PTRS normalized diastolic function 4 weeks after revascularization. Therefore, novel cardioprotective properties of Bendavia enhance cardiac recovery after reversal of experimental RVHT, and may provide a novel therapeutic option.
Acknowledgments
This study was supported by a grant from Stealth Peptides Inc., and partly supported by NIH grant HL77131.
Abbreviations
- BOLD-MRI
blood oxygen level-dependent MRI
- DHE
dihydroethidium
- GABP
guanine adenine-binding protein
- GFR
glomerular filtration rate
- IVC
inferior vena cava
- LV
left ventricle
- LVMM
left ventricular muscle mass
- MCP
monocyte chemoattractant protein
- MDCT
multidetector computed tomography
- mPTP
mitochondrial permeability transition pore
- PGC
peroxisome proliferator-activated receptor gamma coactivator
- PPAR
peroxisome proliferator-activated receptor
- PRA
plasma renin activity
- PTRS
percutaneous transluminal renal angioplasty and stenting
- RVHT
renovascular hypertension
- SMA
smooth muscle actin
- TGF
transforming growth factor
- TIMP
tissue inhibitor of metalloproteinase
- TNF
tumor necrosis factor
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
Footnotes
Conflicts of interest
This study was supported by a grant from Stealth Peptides Inc. (Newton Centre, Massachusetts, USA).
This study has not been published and is not being considered for publication elsewhere in whole or in part in any language except as an abstract.
References
- 1.Kalra PA, Guo H, Kausz AT, Gilbertson DT, Liu J, Chen SC, et al. Atherosclerotic renovascular disease in United States patients aged 67 years or older: risk factors, revascularization, and prognosis. Kidney Int. 2005;68:293–301. doi: 10.1111/j.1523-1755.2005.00406.x. [DOI] [PubMed] [Google Scholar]
- 2.Green D, Kalra PA. The heart in atherosclerotic renovascular disease. Front Biosci (Elite Ed) 2012;4:856–864. doi: 10.2741/E424. [DOI] [PubMed] [Google Scholar]
- 3.Koivuviita N, Tertti R, Luotolahti M, Raitakari O, Vahlberg T, Nuutila P, et al. The effect of revascularization of atherosclerotic renal artery stenosis on coronary flow reserve and peripheral endothelial function. Nephron Clin Pract. 2011;118:c241–c248. doi: 10.1159/000321643. [DOI] [PubMed] [Google Scholar]
- 4.Corriere MA, Hoyle JR, Craven TE, D’Agostino RB, Jr, Edwards MS, Moore PS, Hansen KJ. Changes in left ventricular structure and function following renal artery revascularization. Ann Vasc Surg. 2010;24:80–84. doi: 10.1016/j.avsg.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 5.Kane GC, Xu N, Mistrik E, Roubicek T, Stanson AW, Garovic VD. Renal artery revascularization improves heart failure control in patients with atherosclerotic renal artery stenosis. Nephrol Dial Transplant. 2010;25:813–820. doi: 10.1093/ndt/gfp393. [DOI] [PubMed] [Google Scholar]
- 6.Eirin A, Li Z, Zhang X, Krier JD, Woollard JR, Zhu XY, et al. A mitochondrial permeability transition pore inhibitor improves renal outcomes after revascularization in experimental atherosclerotic renal artery stenosis. Hypertension. 2012;60:1242–1249. doi: 10.1161/HYPERTENSIONAHA.112.199919. [DOI] [PubMed] [Google Scholar]
- 7.Lin J, Zhu X, Chade AR, Jordan KL, Lavi R, Daghini E, et al. Monocyte chemoattractant proteins mediate myocardial microvascular dysfunction in swine renovascular hypertension. Arterioscler Thromb Vasc Biol. 2009;29:1810–1816. doi: 10.1161/ATVBAHA.109.190546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Urbieta-Caceres VH, Zhu XY, Jordan KL, Tang H, Textor K, Lerman A, Lerman LO. Selective improvement in renal function preserved remote myocardial microvascular integrity and architecture in experimental renovascular disease. Atherosclerosis. 2012;221:350–358. doi: 10.1016/j.atherosclerosis.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng FY, et al. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol. 2011;22:1041–1052. doi: 10.1681/ASN.2010080808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szeto HH. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid Redox Signal. 2008;10:601–619. doi: 10.1089/ars.2007.1892. [DOI] [PubMed] [Google Scholar]
- 11.Baines CP. The molecular composition of the mitochondrial permeability transition pore. J Mol Cell Cardiol. 2009;46:850–857. doi: 10.1016/j.yjmcc.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dai DF, Chen T, Szeto H, Nieves-Cintron M, Kutyavin V, Santana LF, Rabinovitch PS. Mitochondrial targeted antioxidant peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol. 2011;58:73–82. doi: 10.1016/j.jacc.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kloner RA, Hale SL, Dai W, Gorman RC, Shuto T, Koomalsingh KJ, et al. Reduction of ischemia/reperfusion injury with bendavia, a mitochondria-targeting cytoprotective peptide. J Am Heart Assoc. 2012;1:e001644. doi: 10.1161/JAHA.112.001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heusch G, Boengler K, Schulz R. Inhibition of mitochondrial permeability transition pore opening: the Holy Grail of cardioprotection. Basic Res Cardiol. 2010;105:151–154. doi: 10.1007/s00395-009-0080-9. [DOI] [PubMed] [Google Scholar]
- 15.Lerman LO, Schwartz RS, Grande JP, Sheedy PF, Romero JC. Noninvasive evaluation of a novel swine model of renal artery stenosis. J Am Soc Nephrol. 1999;10:1455–1465. doi: 10.1681/ASN.V1071455. [DOI] [PubMed] [Google Scholar]
- 16.Eirin A, Ebrahimi B, Zhang X, Zhu XY, Tang H, Crane JA, et al. Changes in glomerular filtration rate after renal revascularization correlate with microvascular hemodynamics and inflammation in swine renal artery stenosis. Circ Cardiovasc Interv. 2012;5:720–728. doi: 10.1161/CIRCINTERVENTIONS.112.972596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eirin A, Zhu XY, Urbieta-Caceres VH, Grande JP, Lerman A, Textor SC, Lerman LO. Persistent kidney dysfunction in swine renal artery stenosis correlates with outer cortical microvascular remodeling. Am J Physiol Renal Physiol. 2011;300:F1394–F1401. doi: 10.1152/ajprenal.00697.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li ZL, Woollard JR, Ebrahimi B, Crane JA, Jordan KL, Lerman A, et al. Transition from obesity to metabolic syndrome is associated with altered myocardial autophagy and apoptosis. Arterioscler Thromb Vasc Biol. 2012;32:1132–1141. doi: 10.1161/ATVBAHA.111.244061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodriguez-Porcel M, Herrman J, Chade AR, Krier JD, Breen JF, Lerman A, Lerman LO. Long-term antioxidant intervention improves myocardial microvascular function in experimental hypertension. Hypertension. 2004;43:493–498. doi: 10.1161/01.HYP.0000111834.03000.e4. [DOI] [PubMed] [Google Scholar]
- 20.Zhu XY, Daghini E, Chade AR, Napoli C, Ritman EL, Lerman A, Lerman LO. Simvastatin prevents coronary microvascular remodeling in renovascular hypertensive pigs. J Am Soc Nephrol. 2007;18:1209–1217. doi: 10.1681/ASN.2006090976. [DOI] [PubMed] [Google Scholar]
- 21.Daghini E, Primak AN, Chade AR, Krier JD, Zhu XY, Ritman EL, et al. Assessment of renal hemodynamics and function in pigs with 64-section multidetector CT: comparison with electron-beam CT. Radiology. 2007;243:405–412. doi: 10.1148/radiol.2432060655. [DOI] [PubMed] [Google Scholar]
- 22.Zhu XY, Daghini E, Chade AR, Versari D, Krier JD, Textor KB, et al. Myocardial microvascular function during acute coronary artery stenosis: effect of hypertension and hypercholesterolaemia. Cardiovasc Res. 2009;83:371–380. doi: 10.1093/cvr/cvp140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eirin A, Zhu XY, Krier JD, Tang H, Jordan KL, Grande JP, et al. Adipose tissue-derived mesenchymal stem cells improve revascularization outcomes to restore renal function in swine atherosclerotic renal artery stenosis. Stem Cells. 2012;30:1030–1041. doi: 10.1002/stem.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ebrahimi B, Li Z, Eirin A, Zhu XY, Textor SC, Lerman LO. Addition of endothelial progenitor cells to renal revascularization restores medullary tubular oxygen consumption in swine renal artery stenosis. Am J Physiol Renal Physiol. 2012;302:F1478–F1485. doi: 10.1152/ajprenal.00563.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu XY, Rodriguez-Porcel M, Bentley MD, Chade AR, Sica V, Napoli C, et al. Antioxidant intervention attenuates myocardial neovascularization in hypercholesterolemia. Circulation. 2004;109:2109–2115. doi: 10.1161/01.CIR.0000125742.65841.8B. [DOI] [PubMed] [Google Scholar]
- 26.Chade AR, Mushin OP, Zhu X, Rodriguez-Porcel M, Grande JP, Textor SC, et al. Pathways of renal fibrosis and modulation of matrix turnover in experimental hypercholesterolemia. Hypertension. 2005;46:772–779. doi: 10.1161/01.HYP.0000184250.37607.da. [DOI] [PubMed] [Google Scholar]
- 27.Chade AR, Zhu XY, Grande JP, Krier JD, Lerman A, Lerman LO. Simvastatin abates development of renal fibrosis in experimental renovascular disease. J Hypertens. 2008;26:1651–1660. doi: 10.1097/HJH.0b013e328302833a. [DOI] [PubMed] [Google Scholar]
- 28.Wright JR, Shurrab AE, Cooper A, Kalra PR, Foley RN, Kalra PA. Progression of cardiac dysfunction in patients with atherosclerotic renovascular disease. QJM. 2009;102:695–704. doi: 10.1093/qjmed/hcp105. [DOI] [PubMed] [Google Scholar]
- 29.Harding MB, Smith LR, Himmelstein SI, Harrison K, Phillips HR, Schwab SJ, et al. Renal artery stenosis: prevalence and associated risk factors in patients undergoing routine cardiac catheterization. J Am Soc Nephrol. 1992;2:1608–1616. doi: 10.1681/ASN.V2111608. [DOI] [PubMed] [Google Scholar]
- 30.Urbieta-Caceres VH, Zhu XY, Gibson ME, Favreau FD, Jordan K, Lerman A, Lerman LO. Reversal of experimental renovascular hypertension restores coronary microvascular function and architecture. Am J Hypertens. 2011;24:458–465. doi: 10.1038/ajh.2010.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodriguez-Porcel M, Lerman A, Herrmann J, Schwartz RS, Sawamura T, Condorelli M, et al. Hypertension exacerbates the effect of hypercholesterolemia on the myocardial microvasculature. Cardiovasc Res. 2003;58:213–221. doi: 10.1016/s0008-6363(03)00250-5. [DOI] [PubMed] [Google Scholar]
- 32.Orrenius S. Reactive oxygen species in mitochondria-mediated cell death. Drug Metab Rev. 2007;39:443–455. doi: 10.1080/03602530701468516. [DOI] [PubMed] [Google Scholar]
- 33.Cho S, Szeto HH, Kim E, Kim H, Tolhurst AT, Pinto JT. A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36. J Biol Chem. 2007;282:4634–4642. doi: 10.1074/jbc.M609388200. [DOI] [PubMed] [Google Scholar]
- 34.Gonzalez A, Ravassa S, Loperena I, Lopez B, Beaumont J, Querejeta R, et al. Association of depressed cardiac gp130-mediated antiapoptotic pathways with stimulated cardiomyocyte apoptosis in hypertensive patients with heart failure. J Hypertens. 2007;25:2148–2157. doi: 10.1097/HJH.0b013e32828626e2. [DOI] [PubMed] [Google Scholar]
- 35.Buzello M, Boehm C, Orth S, Fischer B, Ehmke H, Ritz E, et al. Myocyte loss in early left ventricular hypertrophy of experimental renovascular hypertension. Virchows Arch. 2003;442:364–371. doi: 10.1007/s00428-003-0775-5. [DOI] [PubMed] [Google Scholar]
- 36.Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009;119:573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu Z, Xu X, Hu X, Fassett J, Zhu G, Tao Y, et al. PGC-1 alpha regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxid Redox Signal. 2010;13:1011–1022. doi: 10.1089/ars.2009.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ibarra-Lara L, Hong E, Soria-Castro E, Torres-Narvaez JC, Perez-Severiano F, Del Valle-Mondragon L, et al. Clofibrate PPARalpha activation reduces oxidative stress and improves ultrastructure and ventricular hemodynamics in no-flow myocardial ischemia. J Cardiovasc Pharmacol. 2012;60:323–334. doi: 10.1097/FJC.0b013e31826216ed. [DOI] [PubMed] [Google Scholar]
- 39.Zhu XY, Daghini E, Chade AR, Rodriguez-Porcel M, Napoli C, Lerman A, Lerman LO. Role of oxidative stress in remodeling of the myocardial microcirculation in hypertension. Arterioscler Thromb Vasc Biol. 2006;26:1746–1752. doi: 10.1161/01.ATV.0000227469.40826.01. [DOI] [PubMed] [Google Scholar]
- 40.Panting JR, Gatehouse PD, Yang GZ, Grothues F, Firmin DN, Collins P, Pennell DJ. Abnormal subendocardial perfusion in cardiac syndrome X detected by cardiovascular magnetic resonance imaging. N Engl J Med. 2002;346:1948–1953. doi: 10.1056/NEJMoa012369. [DOI] [PubMed] [Google Scholar]
- 41.Welch WJ, Mendonca M, Aslam S, Wilcox CS. Roles of oxidative stress and AT1 receptors in renal hemodynamics and oxygenation in the postclipped 2K,1C kidney. Hypertension. 2003;41:692–696. doi: 10.1161/01.HYP.0000052945.84627.8F. [DOI] [PubMed] [Google Scholar]
- 42.Tousoulis D, Kampoli AM, Papageorgiou N, Antoniades C, Siasos G, Latsios G, et al. Matrix metallopropteinases in heart failure. Curr Top Med Chem. 2012;12:1181–1191. doi: 10.2174/1568026611208011181. [DOI] [PubMed] [Google Scholar]
- 43.Gomez DE, Alonso DF, Yoshiji H, Thorgeirsson UP. Tissue inhibitors of metalloproteinases: structure, regulation and biological functions. Eur J Cell Biol. 1997;74:111–122. [PubMed] [Google Scholar]