

Abstract
RNase-L is a mediator of type 1 interferon-induced antiviral activity that has diverse and critical cellular roles, including the regulation of cell proliferation, differentiation, senescence and apoptosis, tumorigenesis, and the control of the innate immune response. Although RNase-L was originally shown to mediate the endonucleolytic cleavage of both viral and ribosomal RNAs in response to infection, more recent evidence indicates that RNase-L also functions in the regulation of cellular mRNAs as an important mechanism by which it exerts its diverse biological functions. Despite this growing body of work, many questions remain regarding the roles of mRNAs as RNase-L substrates. This review will survey known and putative mRNA substrates of RNase-L, propose mechanisms by which it may selectively cleave these transcripts, and postulate future clinical applications.
Introduction
In the decades since its discovery, the role of RNase-L has expanded far beyond its initial characterization as part of the interferon (IFN)-regulated antiviral response. Seminal works have identified additional roles for RNase-L, including proapoptotic, antiproliferative, anti-bacterial, and both tumor suppressive and oncogenic activities (Hassel and others 1993; Castelli and others 1997; Zhou and others 1997; Carpten and others 2002; Li and others 2008; Chakrabarti and others 2011; Ezelle and Hassel 2012; Long and others 2013). Disruption of the RNASEL gene in mice and knockdown of its expression in cells validated these roles for RNase-L; however, the mechanisms by which it mediates these biologic activities have remained elusive. As RNase-L is thought to exert its effects in cells through its endonucleolytic cleavage of RNA substrates, identification of the RNA targets of RNase-L is essential to dissect its mechanisms of action. With the exception of ribosomal RNA (rRNA) cleavage, which is typically associated with widespread RNase-L activation in apoptotic cells, little is known about the identities of cellular RNase-L substrates. The absence of a definitive profile of RNase-L target RNAs has prevented the development of an activity assay based on substrate cleavage, which is critical to determine the full spectrum of physiologic and pathologic conditions in which it functions, and to exploit RNase-L as a potential biomarker and therapeutic target. Despite the technical challenges associated with isolating RNase-L-substrate intermediates, several transcripts that exhibit RNase-L-dependent regulation have been reported. In this review, we introduce the biochemical and biologic activities of RNase-L, evaluate the candidate RNase-L substrates identified to date, and examine potential mechanisms of substrate targeting. Additionally, we discuss how these mechanistic insights can be used to develop RNase-L-directed therapeutic strategies.
RNase-L Activation Through the 2-5A Pathway
RNase-L is the terminal component of the 2-5A system, an RNA cleavage pathway that mediates the IFN-α/β-induced antiviral activity. IFN induces the expression of a family of 2′,5′-oligoadenylate synthetases (OAS) that are activated by double-stranded RNA (dsRNA) to polymerize ATP into 2-5A [px5′A(2′p5′A)n; x=1–2; n≥2] (Hovanessian 2007; Kristiansen and others 2011). 2-5A binds latent RNase-L monomers to induce its dimerization and activation (Baglioni and others 1978; Clemens and Williams 1978; Zhou and others 1993; Dong and Silverman 1995) (Fig. 1A). Mutational and structural analysis revealed that 2-5A binding occurs in the ankyrin repeat domains 2–4 that are located in the N-terminal portion of RNase-L (Hassel and others 1993; Dong and others 1997; Nakanishi and others 2004; Tanaka and others 2004). This interaction leads to a conformational change that exposes the pseudokinase domain within the C-terminal kinase-extension nuclease domain to permit oligomerization of RNase-L monomers and expose the ribonuclease domain for enzymatic activity (Dong and Silverman 1997) (Fig. 1B). Previous studies indicated that activation led solely to RNase-L dimerization; however, a recent study suggested that RNase-L oligomerization can also occur through a complex of 4 interacting 2-5A-bound RNase-L monomers with enhanced activity (Han and others 2012). Activated RNase-L cleaves single-stranded microbial RNAs, ribosomal RNAs, and mRNAs to mediate its various biological activities (Floyd-Smith and others 1981; Silverman and others 1983). 2-5A pathway activity is attenuated by a 2′-phosphodiesterase (2′PDE) and cellular phosphatases that can inactivate 2-5A (Kubota and others 2004). In addition, RNase-L is inhibited by the RNase-L inhibitor (RLI), and the expression of RNase-L is maintained at low basal levels through transcriptional and post-transcriptional mechanisms in most cell types (Bisbal and others 1995; Li and others 2000; Zhou and others 2005). This stringent regulation limits the duration of expression and activity to protect cells from deleterious effects of widespread RNase-L activity and indiscriminate RNA cleavage. Studies to date are consistent with a model in which RNase-L functions in a post-transcriptional mechanism to rapidly reprogram cellular gene expression in response to diverse antiproliferative, immunologic, and microbial stimuli. RNase-L is thought to mediate these distinct biologic activities through the regulation of specific transcript profiles in different physiologic settings. Accordingly, identification of RNase-L substrates and its means of target recognition will provide important insights into the mechanisms underlying RNase-L biologic activities.
FIG. 1.

(A) Activation of RNase-L-mediated cleavage of substrates by the 2-5A pathway. 2′PDE, 2′-phosphodiesterase; dsRNA, double-stranded RNA; IFN, interferon; p'tase, phosphatase; RLI, RNase-L inhibitor; ssRNA, single-stranded RNA. (B) Map of the domains of RNase-L including 2-5A binding. KEN, kinase-extension nuclease.
RNase-L-Dependent Regulation of Gene Expression
Microarray analyses by our group and others have identified distinct RNase-L-regulated transcripts in different biologic settings (Malathi and others 2005; Li and others 2008; Andersen and others 2009; Domingo-Gil and others 2010). These findings provide evidence that RNase-L has the capacity to selectively target specific cellular RNAs and suggest that the context-specific profile of RNase-L targets is responsible for its diversity of biologic activities. RNase-L-dependent upregulation and downregulation of cellular transcripts have been reported and are thought to reflect both direct and indirect effects of RNase-L on gene expression. Direct cleavage by RNase-L can result in a downregulation of a particular mRNA substrate and its encoded protein, positively or negatively impacting gene expression downstream of the cleaved target. For example, if an RNase-L substrate encodes a regulator of gene expression (eg, transcription factor), its RNase-L-mediated downregulation may lead to indirect secondary effects because of changes in the expression of the transcription factor-regulated genes. Indeed, the RNase-L-dependent induction of transcriptional signaling through c-Jun N-terminal kinase and extracellular signal-regulated kinase was reported in prostate cancer cells (Malathi and others 2005). Endonucleolytic cleavage of cellular RNAs is typically followed by the rapid degradation of the cleavage products; thus, RNA cleavage by endonucleases has historically been associated with an increased turnover of the substrate RNA and downregulation of its encoded product. In contrast to this paradigm, a subset of RNase-L-generated cleavage products of hepatitis C virus (HCV) RNA and unidentified cellular transcripts are stable, likely because of the presence of extensive secondary structure (Han and others 2004). These cleavage products can activate cytosolic retinoic acid-inducible gene 1 (RIG-I)-like RNA sensors to induce signaling and transcription of IFN-β, thereby providing a novel mechanism by which RNase-L indirectly modulates cellular gene expression and amplifies IFN-induced activities (Malathi and others 2007). As evidenced by these examples, the selective cleavage of primary RNase-L substrates can influence gene expression at multiple levels to exert profound effects on cellular gene regulatory networks and mediate its biologic functions. Therefore, it is essential to determine the mechanisms by which RNase-L targets the cleavage of specific cellular RNAs to identify strategies that manipulate its activity for therapeutic applications. As a first step toward dissecting the mechanisms of RNase-L-substrate targeting, investigators have focused on identifying direct targets of RNase-L cleavage. Criteria to evaluate putative RNase-L substrates include an inverse relationship between RNase-L expression or activity and steady-state RNA concentration and half-life. A physical association between RNase-L and a candidate RNA substrate as analyzed by ribonucleoprotein immunoprecipitation (RNP-IP) provides further evidence that the RNA is an authentic RNase-L substrate. Recently developed technologies that combine RNA–protein interaction-based screening with deep sequencing [eg, photoactivatable-ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP)] provide a potential strategy to directly identify candidate substrates that interact with RNase-L and gain information on the sites of RNase-L–RNA interactions. Thus far, technical barriers have hampered its immediate application to the identification of RNase-L substrates (Kishore and others 2011). Specifically, RNase-L is a low-abundance protein and antibodies that efficiently immunoprecipitate endogenous RNase-L are not available; therefore, screening is limited to cell lines stably transfected with epitope-tagged RNase-L. Furthermore, to efficiently isolate cellular RNAs that associate with activated RNase-L, cleavage of the bound RNA must be inhibited. This presents a challenge in the case of RNase-L as 2-5A binding, RNase-L oligomerization/enzyme activation, and RNA cleavage involve overlapping residues and domains (Dong and others 1997). For example, catalytically-deficient mutants of RNase-L fail to dimerize (K392R; Dong and Silverman 1999) or form active heterodimers with the native RNase-L (R667A; Dong and others 2001). Accordingly, these mutants may not mimic the substrate interaction properties of the native enzyme and their use does not represent a practical approach to stabilize enzyme-substrate interactions. Efforts to identify RNase-L-associated RNAs have employed conditions in which RNase-L activity is reduced to inhibit substrate cleavage (LeRoy and others 2005); in addition, crosslinking has been used to trap RNase-L catalytic intermediates (Li and others 2007). Using these strategies, specific cellular RNase-L substrates have been reported; below we evaluate the evidence of their substrate status and describe their proposed roles in the biologic activities of RNase-L (Table 1).
Table 1.
Criteria and Evidence for Substrate Status of Reported RNase-L Targets
| mRNA target | Steady-state expression | mRNA stability | Physical association | References | |
|---|---|---|---|---|---|
| mRNA stability and translation | HuR | Decreased with RNase-L overexpression | Stabilized in RNase-L−/− cells | RNP-IP | Al-Ahmadi and others (2009), Al-Haj and others (2012) |
| TTP | Increased in RNase-L−/− cells and decreased with RNase-L overexpression | Destabilized in RNase-L−/− cells and stabilized in RNase-L overexpression | RNP-IP | Al-Haj and others (2012) | |
| Ribosomal proteins | Increased in RNase-L−/− cells | Destabilized during 2-5A–RNase-L activation | RNP-IP | Andersen and others (2009) | |
| Differentiation(myogenesis) | MyoD | Decreased with RNase-L overexpression | Stabilized indirectly by RNase-L activation through RLI overexpression | RNP-IP | Bisbal and others (2000), Andersen and others (2009), Salehzada and others (2009) |
| Myogenin | Decreased indirectly by RNase-L activation through RLI overexpression | — | — | Bisbal and others (2000) | |
| Stat3, Hdac5 | Decreased with RNase-L overexpression | Destabilized with RNase-L overexpression | — | Salehzada and others (2009) | |
| Differentiation (adipogenesis) | Chop-10 | Decreased in RNase-L+/+ cells during differentiation | Destabilized in RNase-L+/+ cells during differentiation | RNP-IP | Fabre and others (2012) |
| Aebp1 | Decreased with RNase-L overexpression | Destabilized with RNase-L overexpression | — | Salehzada and others (2009) | |
| Interferon-stimulated genes | ISG43 | Decreased with RNase-L overexpression | Stabilized in RNase-L−/− cells | — | Li and others (2000) |
| ISG15 | — | Stabilized in RNase-L−/− cells | — | Li and others (2000) | |
| PKR | Increased in RNase-L−/− cells | Stabilized in RNase-L−/− cells | — | Khabar and others (2003) | |
Aebp1, AE-binding protein-1; Chop-10, C/EBP homologous protein-10; HuR, Hu-antigen R; ISG, interferon-stimulated gene; MyoD, myogenic differentiation 1; PKR, RNA-dependent protein kinase; RLI, RNase-L inhibitor; RNP-IP, ribonucleoprotein immunoprecipitation; TTP, tristetraprolin.
mRNA stability and translation
Hu-antigen R
Hu-antigen R (HuR) preferentially interacts with A-U-rich elements (AREs) that are often located in the 3′-untranslated regions (3′UTR) of a wide array of labile mRNAs, including those encoding growth factors, oncogenes, and cytokines. As an ARE-binding protein (AREBP), HuR prevents destabilization and promotes translation of a subset of mRNAs by occluding the binding sites of negative regulators such as miRNAs and other RNA-binding proteins (RNABPs) (Brennan and Steitz 2001; Mazan-Mamczarz and others 2003; Galban and others 2008; Trojanowicz and others 2011). The cytoplasmic translocation, post-translational regulation, and function of HuR are modulated by diverse stimuli such as insulin, glucose deprivation, UV light, and hypoxia (Wang and others 2000; Galban and others 2008; Chu and others 2012; Paukku and others 2012). Overexpression of RNase-L decreases the expression levels of HuR mRNA and protein, resulting in a corresponding inhibition of cell proliferation. This effect was dependent upon both cell cycle phases and the cytoplasmic localization of RNase-L (Al-Ahmadi and others 2009). In this study, Al Ahmadi and others (2009) also observed enhanced stability of HuR mRNA in cells lacking RNase-L by actinomycin-D (act-D) time-course assays in HeLa cells and mouse embryonic fibroblasts (MEFs). Further studies verified the interaction of HuR mRNA with RNase-L by RNP-IP assay in HEK293-T cells (Al-Haj and others 2012). RNase-L-dependent negative regulation, enhanced mRNA turnover, and interaction with HuR mRNA provide evidence that HuR is an RNase-L substrate. Additional cell types and conditions beyond cell cycle regulation remain to be investigated. Interestingly, RNase-L mRNA is stabilized by HuR, enhancing RNase-L-mediated antiviral activity (Li and others 2007). This reciprocal regulation between RNase-L and HuR suggests that a regulatory network exists that may function to regulate common biological activities such as cell proliferation. For example, the RNase-L targeting of HuR, an RNABP that enhances the expression of cell cycle- and proliferation-associated proteins, results in the downregulation of these targets to mediate the antiproliferative activity. As an HuR target, RNase-L mRNA will be destabilized, resulting in its own downregulation and subsequent derepression of HuR expression, completing this regulatory loop.
Tristetraprolin
HuR stabilizes mRNA targets, whereas tristetraprolin (TTP) recognizes a distinct, but overlapping, population of ARE mRNA targets to direct their decay. TTP expression is rapidly and transiently induced by a myriad of stimuli, including phorbol esters, insulin, serum, lipopolysaccharide (LPS), growth factors, and hypoxia (Varnum and others 1989; DuBois and others 1990; Lai and others 1990; Nakajima and Wall 1991; Yin and Yang 1993; Fujihara and others 2003; Ogawa and others 2003; Suzuki and others 2003; Ross and others 2012). A vast population of transcripts is targeted by TTP for degradation, including tumor-necrosis factor-α, cyclooxgenase-2, vascular endothelial growth factor, granulocyte macrophage colony-stimulating factor, proto-oncogene serine/threonie protein kinase-1, and various interleukins (Lai and others 1999; Carballo and others 2000; Sawaoka and others 2003; Suswam and others 2008; Mahat and others 2012; Ross and others 2012; Brooks and Blackshear 2013). A recent study by Al-Haj and others (2012) reported that the expression of RNase-L decreased TTP mRNA levels in both MEFs and HeLa cells. TTP mRNA was detected in an RNP complex containing RNase-L, indicating an interaction. Counterintuitively, the authors also found that TTP mRNA was stabilized when RNase-L was present in both cell models. Subsequent analysis, however, demonstrated that this phenomenon resulted from elevated levels of TTP autoregulation; the absence of RNase-L allowed for increased levels of TTP protein and degradation of its own mRNA. Experiments in which the 3′UTR of the TTP mRNA was removed to prevent TTP autoregulation verified that RNase-L enhanced TTP mRNA decay. As the expression and stability of TTP mRNA is transiently induced and rapidly attenuated, further investigation is required to assess the role of RNase-L in regulating TTP in basal and induced conditions. TTP mediates tumor-suppressive activities, including enhanced apoptosis and decreased proliferation (Brennan and others 2009; Ross and others 2012). Given that the expression of RNase-L correlated to the same phenotypes, RNase-L-mediated regulation of TTP is predicted to be transient and condition specific.
Ribosomal proteins
Ribosomal proteins assist in ribosome structure assembly through interactions with rRNA. These proteins mediate critical functions in translation regulation and were identified as potential RNase-L substrates in a study examining its role in senescence (Andersen and others 2009). A microarray analysis determined an RNase-L-dependent negative regulation of several transcripts encoding ribosomal proteins (RP mRNAs), including RPLP0, RPL3, RPL7, RPL41, and RPS12 mRNAs. The negative regulation following RNase-L activation, enhanced mRNA half-lives in RNase-L−/− MEFs, and the presence of these transcripts in an immunoprecipitable complex with RNase-L provided evidence that these RP mRNAs are RNase-L substrates. In addition, isoleucyl-tRNA synthetase mRNA was negatively regulated by RNase-L and stabilized in the absence of RNase-L. RNase-L-mediated degradation of RP mRNAs may assist in the suppression of translation that accompanies proliferative arrest in senescent cells. The impact of RNase-L-dependent regulation of RP mRNAs on translation in other cellular conditions remains to be examined. The combination of decreased expression of several RPs and the RNase-L-mediated cleavage of rRNA would lead to a potent loss of ribosome function and biogenesis, which would have drastic effects on cell survival. Interestingly, rRNA cleavage was not observed following 2-5A-induced senescence and downregulation of RP mRNAs, suggesting that targeting RP mRNAs may result in a limited inhibition of protein synthesis, whereas rRNA cleavage leads to ribosome dissociation and apoptosis (Andersen and others 2009).
Differentiation
Myogenic differentiation-1 (MyoD) and myogenin
As a mediator of antiproliferative activities, numerous reports have identified roles for RNase-L and 2-5A pathway components in cellular differentiation. Accordingly, several groups have explored differentiation models to screen for RNase-L substrates. During the differentiation of C2C12 mouse myoblasts, the expression of RNase-L and RLI was sequentially induced (Bisbal and others 2000). These observations led Bisbal and others (2000) to manipulate RNase-L activity via ectopic expression of RLI to assess its role in myogenesis. Overexpression of RLI decreased RNase-L activity, which corresponded with an increase in the expression of the muscle regulators, myogenic differentiation 1 (MyoD) and myogenin. The knockdown of RLI by siRNA increased RNase-L activity and led to a decrease in expression and stability of MyoD mRNA. A separate study, using RNP-IP assays to detect mRNAs associated with RNase-L, identified MyoD mRNA in a complex with RNase-L protein (Andersen and others 2009). Experiments to directly knockdown RNase-L expression would complement the RLI-targeted approach and provide additional evidence of its role in myogenesis. Consistent with the cleavage of MyoD, a master transcriptional regulator, RNase-L profoundly impacts myoblast differentiation, likely through indirectly regulating downstream genes (Salehzada and others 2009). Further investigation into RNase-L-dependent destabilization of other transcriptional regulators will provide a better understanding of the extent to each mechanism accounts for RNase-L-dependent changes in gene expression.
C/EBP homologous protein-10
C/EBP homologous protein (Chop)-10, a member of the CCAAT/enhancer-binding protein family, is involved in terminal adipocyte differentiation and expression of many adipocyte-specific genes (Tang and Lane 2000). Steady-state Chop-10 mRNA was unchanged during the differentiation of RNase-L−/− MEFs into adipocytes. This contrasted with RNase-L+/+ MEFs, where Chop-10 mRNA was markedly decreased by day 6 of differentiation. RNase-L mediated a decrease in Chop-10 mRNA stability, which was required to direct terminal adipocyte differentiation, lipid storage, and insulin sensitivity. Chop-10 mRNA was detected in an RNP complex with RNase-L, indicating interaction and providing further evidence that Chop-10 mRNA is an RNase-L substrate (Fabre and others 2012). MyoD and Chop-10 both promote differentiation, and their regulation by RNase-L provides a mechanism by which it regulates this process. Indeed, a study by Salehzada and others (2009) used Serial Analysis of Gene Expression (SAGE) data analysis to identify genes downregulated by RNase-L in the myogenic C2C12 cells and found targets common to both myoblast and adipocyte differentiation. Inhibition of myogenesis and increased adipogenesis in C2C12 cells overexpressing RNase-L could be a result of RNase-L-dependent regulation of the myogenic genes, MyoD, signal transducer and activator of transcription-3 (Stat3), and histone deacetylase-5 (Hdac5), as well as the adipogenic genes, Chop-10 and adipocyte-enhancing binding protein-1 (Aebp1). The physical association between these transcripts and RNase-L remains to be determined.
Interferon-stimulated genes
ISG43 and ISG15
IFN-stimulated genes (ISGs) are transiently induced by IFN and encode proteins that drive the IFN response in cells. This process is tightly regulated in order to prevent the deleterious effects of their prolonged expression (Castelli and others 1997; Sadler and Williams 2008). Li and others (2000) previously reported that RNase-L increased the mRNA turnover of ISG43 and ISG15 mRNAs. The negative regulation of ISGs by RNase-L was discovered using an RNase-L-deficient murine neuroblastoma cell line, N1E-115. Restoration of RNase-L expression in this cell line followed by differential display–PCR analysis found a negatively regulated mRNA that was cloned and determined to be a 43-kDa ubiquitin-specific protease (UBP), UBP43, also referred to as ISG43 (Liu and others 1999; Li and others 2000). RNase-L-dependent destabilization of both ISG43 and ISG15 mRNAs was observed using act-D time-course assays in RNase-L+/+ and−/− MEFs. Additional experiments to verify that these ISGs can be recovered in an RNP complex with RNase-L need to be completed to validate these as true RNase-L substrates. RNase-L regulation of these ISGs may function to attenuate the IFN response, and loss of RNase-L expression or function could produce an exaggerated and dangerous response to IFN.
Double-stranded RNA-dependent protein kinase
Also induced by IFN, RNA-dependent protein kinase (PKR) mediates antiviral and antiproliferative responses. PKR acts as a sensor of dsRNA and functions in numerous roles by integrating the signaling response to different stimuli, including virus, bacteria, growth factors, Toll-like receptor activation, and cell stress (Stark and others 1998; Williams 2001). The outcomes of PKR induction are a complex regulation of both transcription and translation to drive antiviral and antiproliferative activities (Garcia and others 2006). The similarities in the activation and the roles for RNase-L and PKR are interesting because RNase-L targets PKR mRNA for cleavage (Khabar and others 2003). Khabar and others (2003) examined PKR as an RNase-L substrate using RNase-L−/− MEFs to show RNase-L-dependent negative regulation and enhanced mRNA turnover of PKR mRNA. To validate PKR mRNA as a true RNase-L substrate, experiments showing interactions such as RNP-IP assays are necessary. The ultimate consequence of RNase-L-mediated cleavage of PKR mRNA may be to limit PKR expression to prevent chronic inflammation.
Potential Mechanisms of RNase-L Substrate Targeting
Although the RNase-L substrates discussed above encode mediators of diverse cellular activities, common features of their regulation may provide clues into the mechanisms by which they are targeted for cleavage by RNase-L (Table 1). For example, most of the substrates are transiently expressed in an inducible manner, suggesting that polysome association and active translation may be one feature of RNase-L targeting; indeed, polysome-associated RNase-L has been reported (Salehzada and others 1991). The presence of cis-acting RNA elements that may function in RNase-L targeting has not been examined for any of the reported substrates above; however, the majority of substrates contain AREs (TTP, HuR, Chop-10, and MyoD), suggesting that RNase-L may associate with AREBPs as a possible targeting mechanism (Ubeda and others 1999; Figueroa and others 2003; Brooks and others 2004; Al-Ahmadi and others 2009). In the following sections, we discuss three models that incorporate contextual features of RNase-L-dependent regulation into potential mechanisms of substrate targeting. These models are not mutually exclusive and aspects of each one may function alone, or in combination, in cells. This framework may provide a means to view data from future studies and generate models that more accurately depict the mechanism(s) of RNase-L-substrate targeting.
Localized activation model
The production of the RNase-L activator 2-5A by OAS requires dsRNA and suggests a mechanism by which RNase-L targets RNAs containing secondary structure that is sufficient to activate OAS. Specifically, the localized production of 2–5A is proposed to result in a limited activation of RNase-L and the selective cleavage of proximal RNAs (Fig. 2A; Nilsen and Baglioni 1979). This localized activation and targeting model is consistent with observations that modulation of RNase-L expression or activity does not result in global changes to the cellular mRNA profile. Experiments using synthetic single-stranded RNAs that contained double-stranded regions showed that they were preferentially cleaved after IFN induction, suggesting that an IFN-induced protein locally activated RNase-L-directed cleavage; however, this model requires further testing with endogenous substrates (Nilsen and Baglioni 1979). As mentioned previously, several RNase-L substrates contain AREs that have been observed to form double-stranded regions with hairpin loops (Putland and others 2002; Fialcowitz and others 2005; Paschoud and others 2006). Consistent with this targeting model, the cellular levels of 2-5A seem to reflect the selective capacity of RNase-L. At low, potentially localized concentrations of 2-5A, RNase-L reduced viral RNA without degradation of cellular mRNAs or ribosomal cleavage, whereas higher 2-5A levels produced widespread degradation of cellular RNAs (Li and others 1998). 2-5A is a rapidly inactivated, short-lived molecule, which suggests that proximity may be an efficient mechanism for RNase-L targeting. RNA components of the viral life cycle are thought to provide the activating dsRNA in infected cells, whereas double-stranded regions within endogenous, cellular RNAs may activate OAS in uninfected cells. In response to HCV infection, RNase-L cleaves HCV RNA into 200–500 base fragments at UA and UU dinucleotide sequences within single-stranded (ss) regions of predicted stem-loop structures (Malathi and others 2010). Consistent with the idea of RNase-L activation in close proximity to sites of 2-5A production, the stem-loop structure of HIV can activate OAS to produce 2-5A (Maitra and others 1994). RNase-L is also known to cleave encephalomyocarditis virus (EMCV) RNA, and OAS has been detected in a physical complex with ECMV-replicative intermediate RNA, providing additional support of the localized activation model (Gribaudo and others 1991). The only known cellular activators of OAS to date are the mRNAs that encode the Raf kinase inhibitor protein and poly(rC)-binding protein 2; however, it is not known if RNase-L cleaves these transcripts (Molinaro and others 2006). The localized activation of RNase-L remains a viable model for RNase-L-substrate targeting, and the identification of cellular substrates now permits experiments to directly test the potential of these transcripts to form double-stranded (ds) structures capable of activating OAS. Importantly, the capacity of mRNAs to form ds structures is likely to be dynamically regulated by RNABPs, potentially adding another mechanism to regulate OAS/RNase-L activities.
FIG. 2.
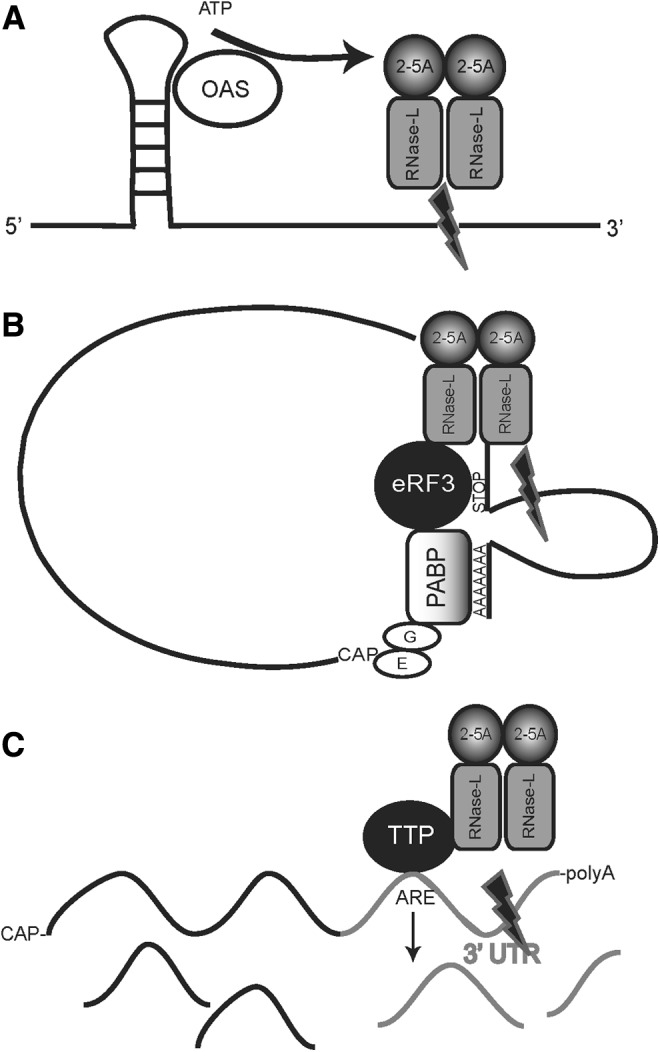
Models of RNase-L substrate targeting. (A) Localized activation of OAS and RNase-L results in the cleavage of contiguous or proximal RNAs. OAS, 2′,5′-oligoadenylate synthetase. (B) RNase-L-mediated cleavage of polysome-bound RNAs via association with eRF3. PABP, poly-A-binding protein. (C) RNase-L-mediated cleavage is targeted through its interaction with RNABPs (TTP is shown as the RNABP in this figure). 3′UTR, 3′-untranslated region; RNABP, RNA-binding protein; TTP, tristetraprolin.
Ribosome-associated targeting
The localized activation model emphasizes the role of substrate-associated elements in target selection and provides a means for OAS activation and production of 2-5A; however, the subcellular sites of RNase-L activation and substrate cleavage are not addressed. Indeed, while RNase-L is typically diffusely distributed in the cytosol, it has also been detected in specific cellular compartments, including mitochondria, nucleus, cytoskeleton, and ribosomes (Wreschner and others 1981a; Silverman and others 1983; Salehzada and others 1991; Tnani and others 1998; Le Roy and others 2001; Chase and others 2003). Of these sites, several lines of evidence suggest that polysome-associated RNase-L may be involved in substrate targeting. The identification of discrete, RNase-L-generated rRNA cleavage products and the observation that RNase-L activation could cleave polysome-bound RNAs to dissociate polysomes suggested that rRNA is an important target of RNase-L and provided a means to monitor its activity (Nilsen and others 1981, 1982). However, rRNA cleavage is typically detected only in the presence of high levels of 2-5A that are associated with virus infection and apoptosis. Furthermore, RNase-L-mediated antiproliferative and antiviral activities can occur in the absence of detectable rRNA cleavage. Together, these observations suggest that viral and cellular mRNAs may be the primary targets of RNase-L, with rRNA cleavage limited to conditions of robust virus infection and maximal production of 2-5A. Indeed, degradation of rRNA is an inefficient mechanism to inhibit translation because of the high energetic cost to re-synthesize new ribosomes. Accordingly, cells sequester vacant ribosomes rather than destroying them to reduce protein synthesis in response to stress (Henras and others 2008; Pisarev and others 2010; Pisareva and others 2011; Nurenberg and Tampe 2013). It may be that under circumstances of extreme stress, the higher levels of activated RNase-L drive the cleavage of rRNA for a final stress response before or during apoptosis.
An alternative to the targeting of ribosome-associated rRNA is the RNase-L-mediated cleavage of polysome-bound transcripts. This strategy of targeting polysome-bound transcripts has been demonstrated for the Polysomal Ribonuclease-1 endonuclease (PMR-1; Yang and Schoenberg 2004). Consistent with this idea, many of the RNase-L substrates identified to date are transiently expressed and translated in an inducible manner, suggesting that ribosome association may be one component of RNase-L substrate selectivity. However, RNase-L does not cleave all actively translating RNAs, suggesting that other factors are involved. In this regard, RLI and eRF3 are ribosome-associated proteins that interact with RNase-L and function in translation regulation. RLI was isolated in an RNA complex with RNase-L and determined to be an inhibitor of RNase-L activity (Bisbal and others 1995). RLI has been implicated in diverse functions beyond RNase-L inhibition, including mRNA metabolism, mRNA processing, translation, and ribosome recycling (Dong and others 2004; Kispal and others 2005; Nurenberg and Tampe 2013). eRF3 and eRF1 mediate translation termination, and the interaction of eRF3 with poly-A-binding protein (PABP) promotes ribosome recycling. Le Roy and others (2005) demonstrated that RNase-L competes with PABP for eRF3 binding and that overexpression or activation of RNase-L functionally inhibited translation termination. Importantly, eRF3 preferentially interacted with 2-5A-activated RNase-L, suggesting that the interaction may serve to direct RNase-L enzymatic activity. Le Roy and others (2005) propose that the inhibition of RNase-L activation by ribosome-associated RLI may delay mRNA cleavage until translation is complete. For example, release of RLI as the ribosome is separated from mRNA could assist in mRNA degradation while it is still in a ribosome-associated eRF3 complex (Pisareva and others 2011). Mapping and mutagenesis of RNase-L–eRF3 interaction domains may provide reagents to rigorously test this model.
Related to the idea of ribosome association as a mechanism of RNase-L targeting is the observation that actively translating RNAs adopt a closed-loop conformation in which the 3′UTR forms an exposed loop that may be accessible to regulatory factors, including RNABPs and endoribonucleases (Sonenberg and Dever 2003). The binding of activated RNase-L to eRF3 could potentially displace PABP, extend the exposed loop, and provide a section of mRNA to cleave, thereby disrupting the closed-loop structure, diminishing translation efficiency and enhancing mRNA decay (Uchida and others 2002). The prominence of an exposed-loop structure formed during active translation is dependent on the length of the 3′UTR and possible secondary structure, and represents a potential site of cleavage by RNase-L (Fig. 2B). This mechanism may provide accessibility to mRNA targets but not specificity as eRF3 and RLI function in the termination of all actively translating ribosomes, so the mechanism that drives the specificity of RNase-L targeting is not clear. Perhaps, in line with the localized activation model, specific mRNAs containing dsRNA regions provide this selectivity. Another possibility is the interaction of RNase-L with other proteins that could target specific mRNAs. Indeed, during active translation, the looped out 3′UTR of certain mRNAs would contain target sites for various RNABPs and regulatory RNAs that could assist or occlude RNase-L-directed RNA cleavage. Testing the model by examining polysome association of target and nontarget transcripts in the presence and absence of RNase-L activation is necessary.
RNABP interaction model
A well-characterized mechanism for cellular mRNA decay is through RNABPs, particularly AREBPs, that function to recruit or occlude RNA decay machinery to promote or inhibit mRNA turnover (Chen and Shyu 1995; Ross 1995; Guhaniyogi and Brewer 2001). In vitro mapping studies and analysis of viral RNAs have demonstrated that RNase-L preferentially cleaves ssRNA after UU and UA dinucleotides. This overlap in the composition of RNase-L target sequences and AREBP binding sites suggested that AREBPs may associate with RNase-L to target the cleavage of specific transcripts that contain these elements (Floyd-Smith and others 1981; Wreschner and others 1981a, 1981b) (Fig. 2C). Consistent with this prediction, we determined that the AREBP, TTP, associates with RNase-L in cells (Fig. 3). Furthermore, a subset of TTP targets were identified as candidate RNase-L substrates, eg, IL-8, HMGA2, and TTP mRNAs, supporting a model in which TTP, as well as potentially other RNABPs, may direct RNase-L cleavage (Brooks and others 2004; Malathi and others 2005; Suswam and others 2008; Al-Haj and others 2012; Kim and others 2012). In support of a model of RNABP-targeted endonucleolytic cleavage, an interesting and recently characterized nuclease, Regnase-1, contains both endoribonucleolytic activity and a domain comprised of a CCCH-type zinc finger similar to the 2 zinc fingers that mediate binding of TTP to its ARE mRNA targets (Lee and others 2013a; Uehata and Akira 2013). It is unknown whether Regnase-1 requires additional factors to recognize substrate mRNAs; however, its domain structure suggests that Regnase-1 evolved to simultaneously target and cleave mRNAs, comparable to a 2-step mechanism of TTP interaction with RNase-L to target the cleavage of specific substrates. A thorough analysis of mRNAs that are regulated by both RNase-L and TTP will provide further insights into potential mechanisms involved. Experiments using recombinant components are required to directly assess the impact of TTP on RNase-L binding and cleavage of RNAs. An interaction between RNase-L and RNABPs such as TTP and eRF3 suggests a mechanism by which RNABPs recruit RNase-L to cleave specific mRNAs; however, only a subset of TTP targets exhibit RNase-L-dependent regulation and eRF3 is associated with all actively translating ribosomes, indicating that additional factors are required to direct RNase-L target recognition. In this regard, components of RNase-L-targeting models that invoke localized activation, ribosome association, and RNABPs may function together to mediate enhanced selectivity. For example, the 3′UTR structure of several ARE-containing transcripts can contain stem-loop structures with double-stranded regions that could potentially activate RNase-L through the localized activation mechanism (Putland and others 2002; Fialcowitz and others 2005; Paschoud and others 2006). Furthermore, ribosome-associated OAS has been reported, potentially linking this targeting mechanism to the ribosome (Hovanessian and others 1987).
FIG. 3.
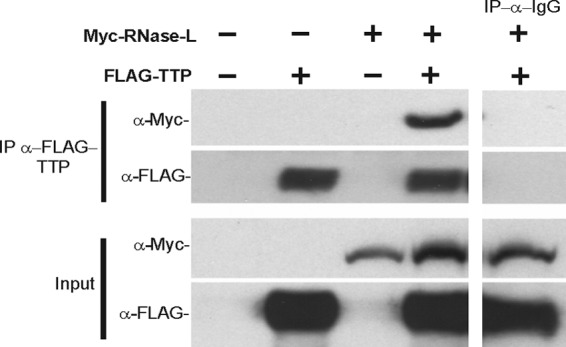
Co-immunoprecipitation (IP) of Myc-tagged RNase-L and FLAG-tagged TTP in 293-T cells using FLAG and IgG control antibodies.
An overlap of TTP-directed RNase-L targeting and the ribosomal association model is also plausible; for example, TTP interacts with PABP, a protein that assists in the closed-loop structure of translation (Worthington and others 2005). TTP could bind to an ARE in the exposed loop of the actively translating mRNA, thereby displacing the eRF3–PABP interaction and potentially allowing RNase-L-directed cleavage of the mRNA. This scenario would disrupt the closed-loop structure while rapidly degrading the mRNA for immediate effects on the translation of select proteins. AREBPs, including TTP, have also been reported to modulate translation and may thus interact with RNase-L to modulate cleavage of ribosome-associated transcripts (Anderson and Kedersha 2002; Mazan-Mamczarz and others 2003; Qi and others 2012; Tiedje and others 2012). It will be of great interest to determine if AREBPs form a complex with RNase-L in the presence of eRF3 and how interactions of RNase-L with these proteins may affect substrate binding. Relatedly, eRF3 preferentially interacts with 2-5A-bound RNase-L (Le Roy and others 2005), yet it is unknown if RNase-L activation impacts its interaction with TTP. Alternatively, interaction of RNABPs with RNase-L may function to both activate RNase-L in a 2-5A-independent manner and mediate substrate specificity. Thus, RNase-L-interacting RNABPs may modulate its activity in the absence of 2-5A whereas canonical 2-5A-mediated activation limited to specific conditions in which OAS is activated by endogenous or pathogen-derived sources. This intriguing scenario requires further investigation. A second potential link between AREBP-directed and ribosome-associated RNase-L targeting strategies is suggested by the observation that RNase-L activation, as well as the expression of specific AREBPs and target RNAs, is induced in response to an overlapping set of stimuli (eg, IFN and LPS). Thus, the association of RNase-L and AREBPs with ribosomes, the coordinate induction of their expression and the targeting of actively translating mRNAs suggests that ribosomes may be a key site of RNase-L cleavage. Consistent with a requirement for the stringent regulation and transient expression profile of induced target transcripts, TTP may direct RNase-L targeting at specific times when immediate decay is necessary, as endoribonucleolytic decay is thought to be a more rapid process compared with de-adenylation-dependent decay that typically involves 3′ deadenylation, 5′ decapping, and exonucleolytic cleavage (Tomecki and Dziembowski 2010). Indeed, the inverse regulation of TTP expression and activation to temporally restrict its activity following LPS stimulation have been well documented. Furthermore, an integrated ribosome-targeted mechanism may clarify how mRNAs are selectively cleaved at lower concentrations of activated RNase-L, while elevated levels of 2-5A may flood the system and lead to the cleavage of proximal rRNA during cellular apoptosis.
Finally, although rRNA cleavage and ribosome dissociation appear to be energetically unfavorable strategies to inhibit translation that may be reserved for apoptotic cells, it is formally possible that the RNABP targeting mechanism may recruit RNase-L to actively translating targets, but that the rRNA, rather than the translating mRNA, is the primary RNase-L target. In this scenario, following rRNA cleavage the mRNA is released from the ribosome and exposed to default cellular decay mechanisms. In this manner, RNase-L would indirectly impact the stability of mRNAs through cleavage of rRNA in their associated ribosomes. Given that RNase-L may target only a subset of translating mRNAs, the rRNA cleavage products resulting from this mechanism may be below the limits of detection for established rRNA cleavage assays. Rigorous testing of this and other targeting models using biochemical approaches with recombinant components and defined substrates is required.
Future Directions and Clinical Implications
Studies to date have demonstrated that RNase-L can exert potent effects on the cellular gene expression program to mediate diverse biologic activities. New technologies (eg, PAR-CLIP) and developing reagents (eg, a robust panel of RNase-L antibodies) provide the means to generate a more comprehensive profile of RNase-L substrates (Kishore and others 2011). An important question in the application of such novel screening strategies to identify RNase-L substrates is the choice of experimental system. Thus far, a determination of the physiologic and pathologic settings in which RNase-L is thought to mediate critical cellular functions has relied on studies that correlated phenotypic changes (eg, antiviral activity, differentiation, apoptosis) with ectopic overexpression or knockdown of RNase-L in cultured cells, or RNase-L deficiency in knockout mice. However, given its established roles in response to diverse stimuli (eg, innate immune and antiproliferative signals), it is likely that RNase-L mediates important functions in a broader spectrum of physiologic and pathologic settings that have yet to be tested. For example, RNase-L is thought to contribute to IFN induction in the contexts of viral and bacterial infection; therefore, RNase-L may also function in the numerous biological processes and disease states attributed to type 1 IFNs (Li and others 2000; Bisbal and others 2007). Determining the impact of RNase-L in type 1 IFN-related pathologies, such as inflammatory and autoimmune diseases and intestinal homeostasis, could delineate novel physiologic and pathologic settings for RNase-L (Gonzalez-Navajas and others 2012). Similarly, in light of its established role in the production of the RNase-L activator 2-5A, conditions in which OAS is dysregulated may implicate a function for RNase-L as its downstream effector. For example, OAS is a potential biomarker in systemic lupus erythemasus, because of its increased expression and the differential expression of specific OAS isoforms (Feng and others 2006; Ye and others 2007). Conversely, the expression of OAS is decreased in prostate cancer. A single-nucleotide polymorphism (SNP) in OAS, already associated with increased infections, is correlated with increased incidence of prostate cancer (Mandal and others 2011; Chaudhary 2012). The downstream loss of RNase-L activity because of dysregulation of OAS may contribute to these pathologies. Indeed, mutations linking RNase-L to prostate cancer have been reported. Specifically, the RNASEL gene maps to the hereditary prostate cancer 1 (HPC1) allele, and the germ-line mutation R462Q, which reduces RNase-L activity, occurs in 13% of prostate cancers (Casey and others 2002; Xiang and others 2003). In addition, SNPs in and downstream from the RNASEL gene correlate with increased risk of head and neck, cervical, and breast cancer, further supporting the necessity to define the roles of RNase-L activation in these tissues. While a role for xenotropic murine leukemia virus-related virus in chronic fatigue syndrome (CFS) has been disproven (Simmons and others 2011), a truncated version of RNase-L has been associated with this disease (Shetzline and Suhadolnik 2001). In this regard, a knock-in strategy to express the truncated form in mice may help to dissect its contribution to the etiology and pathogenesis of CFS. Beyond mutations in the RNase-L gene, the report of its regulation by microRNA-29 revealed an interesting oncogenic role in chronic myelogenous leukemia (Lee and others 2013b). Finally, our lab has identified a novel role for RNase-L in response to gastrointestinal injury in a model of colitis using RNase-L-deficient mice (Long and others 2013). These recent findings suggest that RNase-L, like other regulators of cell proliferation, may function in oncogenic or tumor suppressive activities in distinct contexts; this may reflect different substrate profiles in these settings, further highlighting the need to define its targeting mechanism.
A full understanding of the scope of RNase-L biologic functions is required to assess its potential as a biomarker or therapeutic target; however, the lack of a means to directly detect activated RNase-L in cells has hampered our ability to gain a more complete appreciation of RNase-L functions. Indeed, analysis of RNase-L substrates as an indirect indication of activity may provide only a context-specific readout, and detection of rRNA cleavage is typically limited to conditions of widespread RNase-L activation. Furthermore, reagents to reliably detect 2-5A at the subnanomolar levels at which it is present in cells are not available. A potential approach to measure active RNase-L was suggested by mutational and structural analyses that identified conformational changes associated with RNase-L activation (Dong and Silverman 1997; Tanaka and others 2004; Han and others 2012). These 2-5A-induced alterations are likely to generate novel epitopes that can be used to produce antibodies that selectively detect the activated enzyme. Although the large-scale production of recombinant RNase-L has been successfully accomplished only in a few laboratories, the production of 2-5A-bound RNase-L antigen is feasible and likely to yield antisera specific for this form of the protein (Rusch and others 2001; Pandey and Rath 2004). Such an activation-state-specific reagent would be invaluable for studies of RNase-L activation kinetics and investigations into the subcellular location and associated protein/RNA components of the active enzyme. In addition, information on RNase-L activation in patient samples will provide evidence of its potential roles in response to antimicrobial and cancer chemotherapeutic agents. In view of the potential great utility of an activation-state-specific antibody, the development of strategies for the sensitive detection of RNase-L activation in vivo is an area of active investigation.
A more comprehensive understanding of the relationship between RNase-L biologic functions and substrate profile will be provided by the future studies described above; however, mechanistic insights into RNase-L substrate targeting and cleavage require a rigorous dissection of an RNase-L mRNA-ribonucleoprotein (mRNP) complex. Although RNase-L can cleave specific sites in purified viral RNAs in the absence of accessory factors, it is likely that RNABPs and potentially regulatory RNAs influence the cleavage of cellular RNAs by RNase-L in cells. Accordingly, proteomic analysis of proteins that immunoprecipitate with active RNase-L in cells represents a first approach to identify components of an RNase-L mRNP complex; the antisera to activated RNase-L discussed above would be valuable in this strategy. Subsequent validation of a direct interaction between RNase-L and putative binding partners in vitro and mapping of the binding sites will permit mutagenesis to disrupt the interaction and test its functional significance for target cleavage in cells. In a similar structure–function approach, deletion analysis of the RNA substrate can be used to identify RNA elements that are involved in RNase-L recognition and cleavage. The development of a cell-free system that recapitulates the selective RNase-L-dependent regulation of target RNAs would augment transfection studies to test the roles of specific components of an RNase-L mRNP complex. This classical in vitro approach has been successfully used to dissect the mechanisms of other endonucleases (e.g. PMR-1, Yang and Schoenberg 2004) and is necessary to fully elucidate the requirements for RNase-L-directed cleavage.
To develop RNase-L-directed clinical applications, it is essential to know the physiologic and pathologic conditions in which RNase-L is active and to identify strategies to modulate its activity. As discussed above, novel reagents to detect active RNase-L will provide a more complete view of its activity profile. Beyond 2-5A activation, post-translational modifications may modulate RNase-L activity. The only published modification of RNase-L is the hydroxylation at asparagine-233 by factor inhibiting hypoxia-inducible factor, which indicates a potential role in response to oxygen signaling (Cockman and others 2009). The RNase-L paralog, IRE1, undergoes autophosphorylation to induce a conformational change and unmask the ribonuclease domain (Zhou and others 2006; Ali and others 2011). Neither kinase activity nor phosphorylation has been reported for RNase-L; however, treatment of cells with the phosphatase inhibitor okadaic acid inhibited 2-5A binding and protein kinase-C activation by TPA-reduced degradation (Sukumar 1991). Interestingly, a separate report demonstrated that TPA induced proteasome-dependent regulation of the RNase-L protein but failed to detect an ubiquitin-modified form (Chase and others 2003). These observations suggested that phosphorylation of RNase-L may function to inhibit activity through protein stability by the ubiquitin–proteasome pathway as has been observed for other proteins, but this model has not yet been directly demonstrated. Alternatively, RNase-L-interacting partners such as TTP, androgen receptor, and RLI can be phosphorylated to modulate their activities. This modification could provide a potential mechanism by which phosphorylation indirectly alters the activity of RNase-L, but this remains to be validated. The pseudokinase domain of RNase-L has emerged as a critical platform for modulation of RNase-L activity by 2-5A-induced dimerization and pharmacologic agents (eg, sunitinib) and may mediate the interaction of RNase-L with RNABPs to target substrate cleavage (Fig. 2) (Jha and others 2011). Accordingly, studies to more precisely map interactions in this region and determine how manipulation of these interactions impacts substrate binding and cleavage are required. Such studies will provide data to evaluate strategies that target the pseudokinase domain as a means to modulate RNase-L activity and function. In light of our model in which the interaction of RNase-L with RNABPs targets its cleavage to specific RNAs, thereby determining the substrate profile and context-specific biologic activities, it is intriguing to speculate that small molecule manipulation of RNABP–RNase-L interactions could alter substrate cleavage and direct RNase-L activity for therapeutic applications. A mechanistic understanding of RNase-L-substrate targeting will permit rigorous testing of this model and potentially reveal novel clinical applications.
Acknowledgments
Research described in this review was funded in part by NIH Grant AI077556 (B.A.H.), a VA Merit Award (B.A.H.), and the NIH T32 Cancer Biology Training Grant (S.E.B.-L.). The project described was supported by Award No. T32CA154274 from the National Cancer Institute. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Author Disclosure Statement
B.A.H. is an inventor on patents relating to RNase-L licensed to Alios BioPharma. No competing financial interests exist for all other authors.
References
- Al-Ahmadi W, Al-Haj L, Al-Mohanna FA, Silverman RH, Khabar KSA. 2009. RNase L downmodulation of the RNA-binding protein, HuR, and cellular growth. Oncogene 28(15):1782–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Haj L, Blackshear PJ, Khabar KSA. 2012. Regulation of p21/CIP1/WAF-1 mediated cell-cycle arrest by RNase L and tristetraprolin, and involvement of AU-rich elements. Nucleic Acids Res 40(16):7739–7752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali MMU, Bagratuni T, Davenport EL, Nowak PR, Silva-Santisteban MC, Hardcastle A, McAndrews C, Rowlands MG, Morgan GJ, Aherne W, Collins I, Davies FE, Pearl LH. 2011. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. Embo J 30(5):894–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen JB, Mazan-Mamczarz K, Zhan M, Gorospe M, Hassel BA. 2009. Ribosomal protein mRNAs are primary targets of regulation in RNase-L-induced senescence. RNA Biol 6(3):305–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, Kedersha N. 2002. Visibly stressed: the role of eIF2, TIA-1, and stress granules in protein translation. Cell Stress & Chaperones 7(2):213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baglioni C, Minks MA, Maroney PA. 1978. Interferon action may be mediated by activation of a nuclease by pppA2′p5′A2′p5′A. Nature 273(22):3. [DOI] [PubMed] [Google Scholar]
- Bisbal C, Martinand C, Silhol M, Lebleu B, Salehzada T. 1995. Cloning and characterization of a Rnase-L inhibitor—a new Component of the interferon-regulated 2-5a pathway. J Biol Chem 270(22):13308–13317 [DOI] [PubMed] [Google Scholar]
- Bisbal C, Silhol M, Laubenthal K, Kaluza T, Carnac G, Milligan L, Le Roy F, Salehzada T. 2000. The 2′-5′ oligoadenylate/RNase L/RNase L inhibitor pathway regulates both MyoD mRNA stability and muscle cell differentiation. Mol Cell Biol 20(14):4959–4969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisbal C, Silverman RH. 2007. Diverse functions of RNase L and implications in pathology. Biochimie 89(6–7):789–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CM, Steitz JA. 2001. HuR and mRNA stability. Cell Mol Life Sci 58(2):266–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan SE, Kuwano Y, Alkharouf N, Blackshear PJ, Gorospe M, Wilson GM. 2009. The mRNA-destabilizing protein tristetraprolin is suppressed in many cancers, altering tumorigenic phenotypes and patient prognosis. Cancer Res 69(12):5168–5176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks SA, Blackshear PJ. 2013. Tristetraprolin (TTP): interactions with mRNA and proteins, and current thoughts on mechanisms of action. Biochim Biophys Acta-Gene Regul Mech 1829(6–7):666–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks SA, Connolly JE, Rigby WF. 2004. The role of mRNA turnover in the regulation of tristetraprolin expression: evidence for an extracellular signal-regulated kinase-specific, AU-rich element-dependent, autoregulatory pathway. J Immunol 172(12):7263–7271 [DOI] [PubMed] [Google Scholar]
- Carballo E, Lai WS, Blackshear PJ. 2000. Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony-stimulating factor messenger RNA deadenylation and stability. Blood 95(6):1891–1899 [PubMed] [Google Scholar]
- Carpten J, Nupponen N, Isaacs S, Sood R, Robbins C, Xu J, Faruque M, Moses T, Ewing C, Gillanders E, Hu P, Bujnovszky P, Makalowska I, Baffoe-Bonnie A, Faith D, Smith J, Stephan D, Wiley K, Brownstein M, Gildea D, Kelly B, Jenkins R, Hostetter G, Matikainen M, Schleutker J, Klinger K, Connors T, Xiang Y, Wang Z, De Marzo A, Papadopoulos N, Kallioniemi OP, Burk R, Meyers D, Gronberg H, Meltzer P, Silverman R, Bailey-Wilson J, Walsh P, Isaacs W, Trent J. 2002. Germline mutations in the ribonuclease L gene in families showing linkage with HPC1. Nat Genet 30(2):181–184 [DOI] [PubMed] [Google Scholar]
- Casey G, Neville PJ, Plummer SJ, Xiang Y, Krumroy LM, Klein EA, Catalona WJ, Nupponen N, Carpten JD, Trent JM, Silverman RH, Witte JS. 2002. RNASEL Arg462Gln variant is implicated in up to 13% of prostate cancer cases. Nat Genet 32(4):582–583 [DOI] [PubMed] [Google Scholar]
- Castelli JC, Hassel BA, Wood KA, Li XL, Amemiya K, Dalakas MC, Torrence PF, Youle RJ. 1997. A study of the interferon antiviral mechanism: Apoptosis activation by the 2-5A system. J Exp Med 186(6):967–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti A, Jha BK, Silverman RH. New insights into the role of RNase L in innate immunity. J Interferon Cytokine Res 31(1):49–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase BI, Zhou Y, Xiang Y, Silverman RH, Zhou A. 2003. Proteasome-mediated degradation of RNase L in response to phorbol-12-myristate-13-acetate (PMA) treatment of mouse L929 cells. J Interferon Cytokine Res 23(10):565–573 [DOI] [PubMed] [Google Scholar]
- Chaudhary SKMaJ 2012. Abstract C57: The expression of 2′5′-oligoadenylate synthetase in prostate cancer and its effect on prostate cancer cell cycle. Cancer Res 72 [Google Scholar]
- Chen CY, Shyu AB. 1995. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci 20(11):465–470 [DOI] [PubMed] [Google Scholar]
- Chu PC, Chuang HC, Kulp SK, Chen CS. 2012. The mRNA-stabilizing factor HuR protein is targeted by beta-TrCP protein for degradation in response to glycolysis inhibition. J Biol Chem 287(52):43639–43650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens MJ, Williams BR. 1978. Inhibition of cell-free protein synthesis by pppA2′p5′A2′p5′A: a novel oligonucleotide synthesized by interferon-treated L cell extracts. Cell 13(3):565–572 [DOI] [PubMed] [Google Scholar]
- Cockman ME, Webb JD, Ratcliffe PJ. 2009. FIH-dependent asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Hypoxia and Consequences from Molecule to Malady 1177:9–18 [DOI] [PubMed] [Google Scholar]
- Domingo-Gil E, Gonzalez JM, Esteban M. 2010. Identification of cellular genes induced in human cells after activation of the OAS/RNaseL pathway by vaccinia virus recombinants expressing these antiviral enzymes. J Interferon Cytokine Res 30(3):171–188 [DOI] [PubMed] [Google Scholar]
- Dong B, Niwa M, Walter P, Silverman RH. 2001. Basis for regulated RNA cleavage by functional analysis of RNase L and Ire1p. RNA 7(3):361–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong B, Silverman RH. 1995. 2-5A-dependent RNase molecules dimerize during activation by 2-5A. J Biol Chem 270(8):4133–4137 [DOI] [PubMed] [Google Scholar]
- Dong B, Silverman RH. 1997. A bipartite model of 2-5A-dependent RNase L. J Biol Chem 272(35):22236–22242 [DOI] [PubMed] [Google Scholar]
- Dong B, Silverman RH. 1999. Alternative function of a protein kinase homology domain in 2′,5′-oligoadenylate-dependent RNase L. Nucleic Acids Res 27(2):439–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Lai R, Nielsen K, Fekete CA, Qiu H, Hinnebusch AG. 2004. The essential ATP-binding cassette protein RLI1 functions in translation by promoting preinitiation complex assembly. J Biol Chem 279(40):42157–42168 [DOI] [PubMed] [Google Scholar]
- DuBois RN, McLane MW, Ryder K, Lau LF, Nathans D. 1990. A growth factor-inducible nuclear protein with a novel cysteine/histidine repetitive sequence. J Biol Chem 265(31):19185–19191 [PubMed] [Google Scholar]
- Ezelle HJ, Hassel BA. 2012. Pathologic effects of RNase-L dysregulation in immunity and proliferative control. Front Biosci (Schol Ed) 4:767–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre O, Salehzada T, Lambert K, Boo Seok Y, Zhou A, Mercier J, Bisbal C. 2012. RNase L controls terminal adipocyte differentiation, lipids storage and insulin sensitivity via CHOP10 mRNA regulation. Cell Death Differ 19(9):1470–1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng XB, Wu H, Grossman JM, Hanvivadhanakul P, FitzGerald JD, Park GS, Dong X, Chen WL, Kim MH, Weng HH, Furst DE, Gorn A, McMahon M, Taylor M, Brahn E, Hahn BH, Tsao BP. 2006. Association of increased interferon-inducible gene expression with disease activity and lupus nephritis in patients with systemic lupus erythematosus. Arthritis Rheum 54(9):2951–2962 [DOI] [PubMed] [Google Scholar]
- Fialcowitz EJ, Brewer BY, Keenan BP, Wilson GM. 2005. A hairpin-like structure within an AU-rich mRNA-destabilizing element regulates trans-factor binding selectivity and mRNA decay kinetics. J Biol Chem 280(23):22406–22417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa A, Cuadrado A, Fan J, Atasoy U, Muscat GE, Munoz-Canoves P, Gorospe M, Munoz A. 2003. Role of HuR in skeletal myogenesis through coordinate regulation of muscle differentiation genes. Mol Cell Biol 23(14):4991–5004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd-Smith G, Slattery E, Lengyel P. 1981. Interferon action: RNA cleavage pattern of a (2′-5′)oligoadenylate–dependent endonuclease. Science 212(4498):1030–1032 [DOI] [PubMed] [Google Scholar]
- Fujihara M, Muroi M, Tanamoto K, Suzuki T, Azuma H, Ikeda H. 2003. Molecular mechanisms of macrophage activation and deactivation by lipopolysaccharide: roles of the receptor complex. Pharmacol Ther 100(2):171–194 [DOI] [PubMed] [Google Scholar]
- Galban S, Kuwano Y, Pullmann R, Martindale JL, Kim HH, Lal A, Abdelmohsen K, Yang XL, Dang YJ, Liu JO, Lewis SM, Holcik M, Gorospe M. 2008. RNA-Binding proteins HuR and PTB promote the translation of hypoxia-inducible factor 1 alpha. Mol Cell Biol 28(1):93–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M. 2006. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev 70(4):1032–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Navajas JM, Lee J, David M, Raz E. 2012. Immunomodulatory functions of type I interferons. Nat Rev Immunol 12(2):125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribaudo G, Lembo D, Cavallo G, Landolfo S, Lengyel P. 1991. Interferon action - binding of viral-Rna to the 40-kilodalton 2′-5′-oligoadenylate synthetase in interferon-treated hela-cells infected with encephalomyocarditis virus. J Virol 65(4):1748–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guhaniyogi J, Brewer G. 2001. Regulation of mRNA stability in mammalian cells. Gene 265(1–2):11–23 [DOI] [PubMed] [Google Scholar]
- Han YC, Whitney G, Donovan J, Korennykh A. 2012. Innate immune messenger 2-5A tethers human RNase L into active high-order complexes. Cell Rep 2(4):902–913 [DOI] [PubMed] [Google Scholar]
- Hassel BA, Zhou A, Sotomayor C, Maran A, Silverman RH. 1993. A dominant negative mutant of 2-5A-dependent RNase suppresses antiproliferative and antiviral effects of interferon. EMBO J 12(8):3297–3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henras AK, Soudet J, Gerus M, Lebaron S, Caizergues-Ferrer M, Mougin A, Henry Y. 2008. The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell Mol Life Sci 65(15):2334–2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovanessian AG. 2007. On the discovery of interferon-inducible, double-stranded RNA activated enzymes: the 2′-5′oligoadenylate synthetases and the protein kinase PKR. Cytokine Growth Factor Rev 18(5–6):351–361 [DOI] [PubMed] [Google Scholar]
- Hovanessian AG, Laurent AG, Chebath J, Galabru J, Robert N, Svab J. 1987. Identification of 69-Kd and 100-Kd Forms of 2-5a Synthetase in Interferon-Treated Human-Cells by Specific Monoclonal-Antibodies. EMBO J 6(5):1273–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha BK, Polyakova I, Kessler P, Dong B, Dickerman B, Sen GC, Silverman RH. 2011. Inhibition of RNase L and RNA-dependent protein kinase (PKR) by sunitinib impairs antiviral innate immunity. J Biol Chem 286(30):26319–26326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khabar KS, Siddiqui YM, al-Zoghaibi F, al-Haj L, Dhalla M, Zhou A, Dong B, Whitmore M, Paranjape J, Al-Ahdal MN, Al-Mohanna F, Williams BR, Silverman RH. 2003. RNase L mediates transient control of the interferon response through modulation of the double-stranded RNA-dependent protein kinase PKR. J Biol Chem 278(22):20124–20132 [DOI] [PubMed] [Google Scholar]
- Kim CW, Vo MT, Kim HK, Lee HH, Yoon NA, Lee BJ, Min YJ, Joo WD, Cha HJ, Park JW, Cho WJ. 2012. Ectopic over-expression of tristetraprolin in human cancer cells promotes biogenesis of let-7 by down-regulation of Lin28. Nucleic Acids Res 40(9):3856–3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishore S, Jaskiewicz L, Burger L, Hausser J, Khorshid M, Zavolan M. 2011. A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat Methods 8(7):559–564 [DOI] [PubMed] [Google Scholar]
- Kispal G, Sipos K, Lange H, Fekete Z, Bedekovics T, Janaky T, Bassler J, Netz DJA, Balk J, Rotte C, Lill R. 2005. Biogenesis of cytosolic ribosomes requires the essential iron-sulphur protein Rli1p and mitochondria. EMBO J 24(3):589–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansen H, Gad HH, Eskildsen-Larsen S, Despres P, Hartmann R. 2011. The oligoadenylate synthetase family: an ancient protein family with multiple antiviral activities. J Interferon Cytokine Res 31(1):41–47 [DOI] [PubMed] [Google Scholar]
- Kubota K, Nakahara K, Ohtsuka T, Yoshida S, Kawaguchi J, Fujita Y, Ozeki Y, Hara A, Yoshimura C, Furukawa H, Haruyama H, Ichikawa K, Yamashita M, Matsuoka T, Iijima Y. 2004. Identification of 2′-phosphodiesterase, which plays a role in the 2-5A system regulated by interferon. J Biol Chem 279(36):37832–37841 [DOI] [PubMed] [Google Scholar]
- Lai WS, Carballo E, Strum JR, Kennington EA, Phillips RS, Blackshear PJ. 1999. Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol Cell Biol 19(6):4311–4323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai WS, Stumpo DJ, Blackshear PJ. 1990. Rapid insulin-stimulated accumulation of an mRNA encoding a proline-rich protein. J Biol Chem 265(27):16556–16563 [PubMed] [Google Scholar]
- Le Roy F, Bisbal C, Silhol M, Martinand C, Lebleu B, Salehzada T. 2001. The 2-5A/RNase L/RNase L inhibitor (RLI) [correction of (RNI)] pathway regulates mitochondrial mRNAs stability in interferon alpha-treated H9 cells. J Biol Chem 276(51):48473–48482 [DOI] [PubMed] [Google Scholar]
- Le Roy F, Salehzada T, Bisbal C, Dougherty JP, Peltz SW. 2005. A newly discovered function for RNase L in regulating translation termination. Nat Struct Mol Biol 12(6):505–512 [DOI] [PubMed] [Google Scholar]
- Lee H, Komano J, Saitoh Y, Yamaoka S, Kozaki T, Misawa T, Takahama M, Satoh T, Takeuchi O, Yamamoto N, Matsuura Y, Saitoh T, Akira S. 2013a. Zinc-finger antiviral protein mediates retinoic acid inducible gene I-like receptor-independent antiviral response to murine leukemia virus. Proc Natl Acad Sci USA 110(30):12379–12384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TY, Ezelle HJ, Venkataraman T, Lapidus RG, Scheibner KA, Hassel BA. 2013b. Regulation of human RNase-L by the miR-29 family reveals a novel oncogenic role in chronic myelogenous leukemia. J Interferon Cytokine Res 33(1):34–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XL, Andersen JB, Ezelle HJ, Wilson GM, Hassel BA. 2007. Post-transcriptional regulation of RNase-L expression is mediated by the 3′-untranslated region of its mRNA. J Biol Chem 282(11):7950–7960 [DOI] [PubMed] [Google Scholar]
- Li XL, Blackford JA, Hassel BA. 1998. RNase L mediates the antiviral effect of interferon through a selective reduction in viral RNA during encephalomyocarditis virus infection. J Virol 72(4):2752–2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XL, Blackford JA, Judge CS, Liu M, Xiao W, Kalvakolanu DV, Hassel BA. 2000. RNase-L-dependent destabilization of interferon-induced mRNAs. A role for the 2-5A system in attenuation of the interferon response. J Biol Chem 275(12):8880–8888 [DOI] [PubMed] [Google Scholar]
- Li XL, Ezelle HJ, Kang TJ, Zhang L, Shirey KA, Harro J, Hasday JD, Mohapatra SK, Crasta OR, Vogel SN, Cross AS, Hassel BA. 2008. An essential role for the antiviral endoribonuclease, RNase-L, in antibacterial immunity. Proc Natl Acad Sci USA 105(52):20816–20821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu LQ, Ilaria R, Jr., Kingsley PD, Iwama A, van Etten RA, Palis J, Zhang DE. 1999. A novel ubiquitin-specific protease, UBP43, cloned from leukemia fusion protein AML1-ETO-expressing mice, functions in hematopoietic cell differentiation. Mol Cell Biol 19(4):3029–3038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long TM, Chakrabarti A, Ezelle HJ, Brennan-Laun SE, Raufman JP, Polyakova I, Silverman RH, Hassel BA. 2013. RNase-L deficiency exacerbates experimental colitis and colitis-associated cancer. Inflamm Bowel Dis 19(6):1295–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahat DB, Brennan-Laun SE, Fialcowitz-White EJ, Kishor A, Ross CR, Pozharskaya T, Rawn JD, Blackshear PJ, Hassel BA, Wilson GM. 2012. Coordinated expression of tristetraprolin post-transcriptionally attenuates mitogenic induction of the oncogenic Ser/Thr kinase Pim-1. Plos One 7(3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra RK, Mcmillan NAJ, Desai S, Mcswiggen J, Hovanessian AG, Sen G, Williams BRG, Silverman RH. 1994. Hiv-1 Tar RNA has an intrinsic ability to activate interferon-inducible enzymes. Virology 204(2):823–827 [DOI] [PubMed] [Google Scholar]
- Malathi K, Dong B, Gale M, Jr., Silverman RH. 2007. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448(7155):816–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malathi K, Paranjape JM, Bulanova E, Shim M, Guenther-Johnson JM, Faber PW, Eling TE, Williams BR, Silverman RH. 2005. A transcriptional signaling pathway in the IFN system mediated by 2′-5′-oligoadenylate activation of RNase L. Proc Natl Acad Sci USA 102(41):14533–14538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malathi K, Saito T, Crochet N, Barton DJ, Gale M, Silverman RH. 2010. RNase L releases a small RNA from HCV RNA that refolds into a potent PAMP. RNA-a Publication of the RNA Society 16(11):2108–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal S, Abebe F, Chaudhary J. 2011. 2′-5′ oligoadenylate synthetase 1 polymorphism is associated with prostate cancer. Cancer 117(24):5509–5518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazan-Mamczarz K, Galban S, de Silanes IL, Martindale JL, Atasoy U, Keene JD, Gorospe M. 2003. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc Natl Acad Sci USA 100(14):8354–8359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinaro RJ, Jha BK, Malathi K, Varambally S, Chinnaiyan AM, Silverman RH. 2006. Selection and cloning of poly(rC)-binding protein 2 and Raf kinase inhibitor protein RNA activators of 2′,5′-oligoadenylate synthetase from prostate cancer cells. Nucleic Acids Res 34(22):6684–6695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K, Wall R. 1991. Interleukin-6 signals activating junB and TIS11 gene transcription in a B-cell hybridoma. Mol Cell Biol 11(3):1409–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi M, Yoshimura A, Ishida N, Ueno Y, Kitade Y. 2004. Contribution of Tyr712 and Phe716 to the activity of human RNase l. European Journal of Biochemistry 271(13):2737–2744 [DOI] [PubMed] [Google Scholar]
- Nilsen TW, Baglioni C. 1979. Mechanism for discrimination between viral and host mRNA in interferon-treated cells. Proc Natl Acad Sci USA 76(6):2600–2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW, Maroney PA, Baglioni C. 1981. Double-stranded RNA causes synthesis of 2′,5′-oligo(A) and degradation of messenger RNA in interferon-treated cells. J Biol Chem 256(15):7806–7811 [PubMed] [Google Scholar]
- Nilsen TW, Maroney PA, Baglioni C. 1982. Synthesis of (2′-5′)oligoadenylate and activation of an endoribonuclease in interferon-treated HeLa cells infected with reovirus. J Virol 42(3):1039–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurenberg E, Tampe R. 2013. Tying up loose ends: ribosome recycling in eukaryotes and archaea. Trends Biochem Sci 38(2):64–74 [DOI] [PubMed] [Google Scholar]
- Ogawa K, Chen F, Kim YJ, Chen Y. 2003. Transcriptional regulation of tristetraprolin by transforming growth factor-beta in human T cells. J Biol Chem 278(32):30373–30381 [DOI] [PubMed] [Google Scholar]
- Pandey M, Rath PC. 2004. Expression of interferon-inducible recombinant human RNase L causes RNA degradation and inhibition of cell growth in Escherichia coli. Biochem Biophys Res Commun 317(2):586–597 [DOI] [PubMed] [Google Scholar]
- Paschoud S, Dogar AM, Kuntz C, Grisoni-Neupert B, Richman L, Kuhn LC. 2006. Destabilization of interleukin-6 mRNA requires a putative RNA stem-loop structure, an AU-rich element, and the RNA-binding protein AUF1. Mol Cell Biol 26(22):8228–8241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paukku K, Backlund M, De Boer RA, Kalkkinen N, Kontula KK, Lehtonen JYA. 2012. Regulation of AT1R expression through HuR by insulin. Nucleic Acids Res 40(12):5250–5261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisarev AV, Skabkin MA, Pisareva VP, Skabkina OV, Rakotondrafara AM, Hentze MW, Hellen CU, Pestova TV. 2010. The role of ABCE1 in eukaryotic posttermination ribosomal recycling. Mol Cell 37(2):196–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisareva VP, Skabkin MA, Hellen CU, Pestova TV, Pisarev AV. 2011. Dissociation by Pelota, Hbs1 and ABCE1 of mammalian vacant 80S ribosomes and stalled elongation complexes. EMBO J 30(9):1804–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putland RA, Sassinis TA, Harvey JS, Diamond P, Coles LS, Brown CY, Goodall GJ. 2002. RNA destabilization by the granulocyte colony-stimulating factor stem-loop destabilizing element involves a single stem-loop that promotes deadenylation. Mol Cell Biol 22(6):1664–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi MY, Wang ZZ, Zhang Z, Shao Q, Zeng A, Li XQ, Li WQ, Wang C, Tian FJ, Li Q, Zou J, Qin YW, Brewer G, Huang S, Jing Q. 2012. AU-rich-element-dependent translation repression requires the cooperation of tristetraprolin and RCK/P54. Mol Cell Biol 32(5):913–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CR, Brennan-Laun SE, Wilson GM. 2012. Tristetraprolin: Roles in cancer and senescence. Ageing Res Rev 11(4):473–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross J. 1995. mRNA stability in mammalian cells. Microbiol Rev 59(3):423–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusch L, Dong B, Silverman RH. 2001. Monitoring activation of ribonuclease L by 2′,5′-oligoadenylates using purified recombinant enzyme and intact malignant glioma cells. Methods Enzymol 342:10–20 [DOI] [PubMed] [Google Scholar]
- Sadler AJ, Williams BR. 2008. Interferon-inducible antiviral effectors. Nat Rev Immunol 8(7):559–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehzada T, Cambier L, Vu Thi N, Manchon L, Regnier L, Bisbal C. 2009. Endoribonuclease L (RNase L) regulates the myogenic and adipogenic potential of myogenic cells. PLoS One 4(10):e7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehzada T, Silhol M, Lebleu B, Bisbal C. 1991. Polyclonal antibodies against Rnase-L - subcellular-localization of this enzyme in mouse cells. J Biol Chem 266(9):5808–5813 [PubMed] [Google Scholar]
- Sawaoka H, Dixon DA, Oates JA, Boutaud O. 2003. Tristetraprolin binds to the 3′-untranslated region of cyclooxygenase-2 mRNA. A polyadenylation variant in a cancer cell line lacks the binding site. J Biol Chem 278(16):13928–13935 [DOI] [PubMed] [Google Scholar]
- Shetzline SE, Suhadolnik RJ. 2001. Characterization of a 2′,5′-oligoadenylate (2-5A)-dependent 37-kDa RNase L. Azido photoaffinity labeling and 2-5A-dependent activation. (vol 276, pg 23707, 2001). J Biol Chem 276(34):32392–32392 [DOI] [PubMed] [Google Scholar]
- Silverman RH, Skehel JJ, James TC, Wreschner DH, Kerr IM. 1983. rRNA cleavage as an index of ppp(A2′p)nA activity in interferon-treated encephalomyocarditis virus-infected cells. J Virol 46(3):1051–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G, Glynn SA, Komaroff AL, Mikovits JA, Tobler LH, Hackett J, Tang N, Switzer WM, Heneine W, Hewlett IK, Zhao J, Lo SC, Alter HJ, Linnen JM, Gao K, Coffin JM, Kearney MF, Ruscetti FW, Pfost MA, Bethel J, Kleinman S, Holmberg JA, Busch MP, Blood XMRV Scientific Research Working Group (SRWG) 2011 Failure to confirm XMRV/MLVs in the blood of patients with chronic fatigue syndrome: a multi-laboratory study. Science. 2011334(6057):814–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N, Dever TE. 2003. Eukaryotic translation initiation factors and regulators. Curr Opin Struct Biol 13(1):56–63 [DOI] [PubMed] [Google Scholar]
- Stark GR, Kerr IM, Williams BRG, Silverman RH, Schreiber RD. 1998. How cells respond to interferons. Annu Rev Biochem 67:227–264 [DOI] [PubMed] [Google Scholar]
- Sukumar NA. 1991. Regulation of 2-5A dependent RNase at the level of its phosphorylation In Department of Pathology Uniformed Services University of the Health Sciences122 [Google Scholar]
- Suswam E, Li Y, Zhang X, Gillespie GY, Li X, Shacka JJ, Lu L, Zheng L, King PH. 2008. Tristetraprolin down-regulates interleukin-8 and vascular endothelial growth factor in malignant glioma cells. Cancer Res 68(3):674–682 [DOI] [PubMed] [Google Scholar]
- Suzuki K, Nakajima H, Ikeda K, Maezawa Y, Suto A, Takatori H, Saito Y, Iwamoto I. 2003. IL-4-Stat6 signaling induces tristetraprolin expression and inhibits TNF-alpha production in mast cells. J Exp Med 198(11):1717–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Nakanishi M, Kusakabe Y, Goto Y, Kitade Y, Nakamura KT. 2004. Structural basis for recognition of 2′,5′-linked oligoadenylates by human ribonuclease L. EMBO J 23(20):3929–3938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang QQ, Lane MD. 2000. Role of C/EBP homologous protein (CHOP-10) in the programmed activation of CCAAT/enhancer-binding protein-beta during adipogenesis. Proc Natl Acad Sci USA 97(23):12446–12450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiedje C, Ronkina N, Tehrani M, Dhamija S, Laass K, Holtmann H, Kotlyarov A, Gaestel M. 2012. The p38/MK2-driven exchange between tristetraprolin and HuR regulates AU-rich element-dependent translation. PLoS Genet 8(9):e1002977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tnani M, Aliau S, Bayard B. 1998. Localization of a molecular form of interferon-regulated RNase L in the cytoskeleton. J Interferon Cytokine Res 18(6):361–368 [DOI] [PubMed] [Google Scholar]
- Tomecki R, Dziembowski A. 2010. Novel endoribonucleases as central players in various pathways of eukaryotic RNA metabolism. Rna-a Publication of the Rna Society 16(9):1692–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojanowicz B, Dralle H, Hoang-Vu C. 2011. AUF1 and HuR: possible implications of mRNA stability in thyroid function and disorders. Thyroid Res 4Suppl 1:S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubeda M, Schmitt-Ney M, Ferrer J, Habener JF. 1999. CHOP/GADD153 and methionyl-tRNA synthetase (MetRS) genes overlap in a conserved region that controls mRNA stability. Biochem Biophys Res Commun 262(1):31–38 [DOI] [PubMed] [Google Scholar]
- Uchida N, Hoshino S, Imataka H, Sonenberg N, Katada T. 2002. A novel role of the mammalian GSPT/eRF3 associating with poly(A)-binding protein in Cap/Poly(A)-dependent translation. J Biol Chem 277(52):50286–50292 [DOI] [PubMed] [Google Scholar]
- Uehata T, Akira S. 2013. mRNA degradation by the endoribonuclease Regnase-1/ZC3H12a/MCPIP-1. Biochim Biophys Acta 1829(6–7):708–713 [DOI] [PubMed] [Google Scholar]
- Varnum BC, Lim RW, Sukhatme VP, Herschman HR. 1989. Nucleotide sequence of a cDNA encoding TIS11, a message induced in Swiss 3T3 cells by the tumor promoter tetradecanoyl phorbol acetate. Oncogene 4(1):119–120 [PubMed] [Google Scholar]
- Wang WG, Furneaux H, Cheng HM, Caldwell MC, Hutter D, Liu YS, Holbrook N, Gorospe M. 2000. HuR regulates p21 mRNA stabilization by UV light. Mol Cell Biol 20(3):760–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BR. 2001. Signal integration via PKR. Sci STKE 2001(89):re2. [DOI] [PubMed] [Google Scholar]
- Worthington M, Rowlett R, Pelo J, Harp M, Chrestensen C, Schroeder M, Sturgill TW. 2005. Regulation of TNF expression at the post-transcriptional level: Polya binding protein inhibits tristetraprolin-mediated degradation of TNF Rna via a PABP-TTP complex. Gastroenterology 128(4):A218 [Google Scholar]
- Wreschner DH, James TC, Silverman RH, Kerr IM. 1981a. Ribosomal RNA cleavage, nuclease activation and 2–5A(ppp(A2′p)nA) in interferon-treated cells. Nucleic Acids Res 9(7):1571–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wreschner DH, McCauley JW, Skehel JJ, Kerr IM. 1981b. Interferon action–sequence specificity of the ppp(A2′p)nA-dependent ribonuclease. Nature 289(5796):414–417 [DOI] [PubMed] [Google Scholar]
- Xiang Y, Wang ZF, Murakami J, Plummer S, Klein EA, Carpten JD, Trent JM, Isaacs WB, Casey G, Silverman RH. 2003. Effects of RNase L mutations associated with prostate cancer on apoptosis induced by 2′,5′-oligoadenylates. Cancer Res 63(20):6795–6801 [PubMed] [Google Scholar]
- Yang F, Schoenberg DR. 2004. Endonuclease-mediated mRNA decay involves the selective targeting of PMR1 to polyribosome-bound substrate mRNA. Mol Cell 14(4):435–45 [DOI] [PubMed] [Google Scholar]
- Ye S, Guo Q, Tang JP, Yang CD, Shen N, Chen SL. 2007. Could 2′ 5′-oligoadenylate synthetase isoforms be biomarkers to differentiate between disease flare and infection in lupus patients? A pilot study. Clinical Rheumatology 26(2):186–190 [DOI] [PubMed] [Google Scholar]
- Yin T, Yang YC. 1993. Protein tyrosine phosphorylation and activation of primary response genes by interleukin 11 in B9-TY1 cells. Cell Growth Differ 4(7):603–609 [PubMed] [Google Scholar]
- Zhou A, Hassel BA, Silverman RH. 1993. Expression cloning of 2-5A-dependent RNAase: a uniquely regulated mediator of interferon action. Cell 72(5):753–765 [DOI] [PubMed] [Google Scholar]
- Zhou A, Paranjape J, Brown TL, Nie H, Naik S, Dong B, Chang A, Trapp B, Fairchild R, Colmenares C, Silverman RH. 1997. Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J 16(21):6355–6363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou AM, Molinaro RJ, Malathi K, Stlverman RH. 2005. Mapping of the human RNASEL promoter and expression in cancer and normal cells. J Interferon Cytokine Res 25(10):595–603 [DOI] [PubMed] [Google Scholar]
- Zhou JH, Liu CY, Back SH, Clark RL, Peisach D, Xu ZH, Kaufman RJ. 2006. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc Natl Acad Sci USA 103(39):14343–14348 [DOI] [PMC free article] [PubMed] [Google Scholar]