

Abstract
Progressive mitochondrial dysfunction is linked with the onset of many age-related pathologies and neurological disorders. Mitochondrial damage can come in many forms and be induced by a variety of cellular insults. To preserve organelle function during biogenesis or times of stress, multiple surveillance systems work to ensure the persistence of a functional mitochondrial network. This review provides an overview of these processes, which collectively contribute to the maintenance of a healthy mitochondrial population, which is critical for cell physiology and survival.
Linked Articles
This article is part of a themed issue on Mitochondrial Pharmacology: Energy, Injury & Beyond. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2014.171.issue-8
Keywords: mitochondria, protease, mitochondrial morphology, protein quality control, neurodegenerative disease, apoptosis
Mitochondria
Mitochondria are double membrane-bound organelles that play a central role in cellular metabolism and ultimately cellular survival. The most notable function attributed to the organelle is ATP generation through respiration, resulting in the famous textbook description of mitochondria as the ‘powerhouse of the cell’. The workhorse of this ATP-producing compartment is the protein complement that resides within it. This army of workers, which can amount to 1000–1500 proteins, ensures all mitochondrial functions are executed to support organelle integrity and function. Thus, maintenance of the mitochondrial proteome is of high importance in the context of cellular survival. Consequently, a landscape of hierarchical systems of quality control surveillance mechanisms is in place at both the organelle and cellular level (Anand et al., 2013; Voos, 2013). The first line of defence is at the molecular level and includes a conserved repertoire of chaperones and proteases that serve to uphold mitochondrial protein homeostasis (Baker et al., 2011; Tatsuta and Langer, 2007). In addition to the quality control system localized in mitochondria, there is also a role for the ubiquitin-proteasome system (UPS) in the turnover of mitochondrial proteins (Livnat-Levanon and Glickman, 2011; Taylor and Rutter, 2011). At the organelle level, mitochondria are highly dynamic and constantly undergo fission and fusion events (Westermann, 2010; Chan, 2012; Elgass et al., 2013). The dynamic nature of the organelle offers an additional line of defence and regulates mitochondrial function by enabling mitochondrial recruitment to subcellular compartments, content exchange between mitochondria and changes in mitochondrial morphology. Finally, at the cellular level the quality of the mitochondrial population is maintained by mitophagy, a form of autophagy for selective removal of damaged mitochondria (Twig et al., 2008; Osellame et al., 2012; Youle and van der Bliek, 2012). Crosstalk between these processes ensures the persistence of a healthy mitochondrial population and ultimately cellular survival. Here, we will describe these surveillance mechanisms and how their interplay promotes mitochondrial health. We will also look at the failure of these systems and the effect this has on human disease.
Quality control at the molecular level – maintaining the mitochondrial proteome
Mitochondrial function and cellular metabolism is dependent on the maintenance of the mitochondrial proteome. It is critical to ensure that the mitochondrial proteome is adjusted depending on cellular demands and this is achieved by protein import and protein quality control. These events encompass protein synthesis, trafficking, import and folding and ultimately protein turnover. Although mitochondrial protein import and quality control may have been traditionally investigated independently, the overlap between the two is now exceedingly clear and mitochondrial protein homeostasis cannot be sustained without either of these processes.
Protein import is absolutely fundamental to mitochondrial function as it facilitates entry of nearly all the protein complement of the organelle (Neupert and Herrmann, 2007; Chacinska et al., 2009; Endo and Yamano, 2009). Import failure can result in mitochondrial injury and dysfunction due to the accumulation of immature precursors that would be prone to misfolding and aggregation either within the organelle or the cytosol. Mitochondrial protein import is more complicated than most eukaryotic trafficking systems due to the fact that (i) the organelle has a complex architecture, which consists of four subcompartments: the outer membrane, intermembrane space, inner membrane and matrix, and (ii) the mitochondrial proteome is encoded within two genomes, the mitochondrial and nuclear genomes (Sickmann et al., 2003; Dolezal et al., 2006). The mitochondrial genome encodes only a handful of proteins belonging to the respiratory chain, thus, the remaining proteins are all encoded within the nucleus and must be imported into mitochondria. Advances in the last decade, particularly due to the application of proteomic approaches, have significantly extended our understanding of the machineries cooperating in mitochondrial protein import (Sickmann et al., 2003; Prokisch et al., 2004; Pagliarini et al., 2008). These and previous discoveries have provided us with the current picture, which suggests there are numerous translocation and assembly machineries within mitochondria (Figure 1). We will describe these here briefly (for more details see Koehler, 2000; Neupert and Herrmann, 2007; Becker et al., 2009; Chacinska et al., 2009; Endo and Yamano, 2009; Gebert et al., 2011; Riemer et al., 2011; Dudek et al., 2013; Varabyova et al., 2013).
Figure 1.
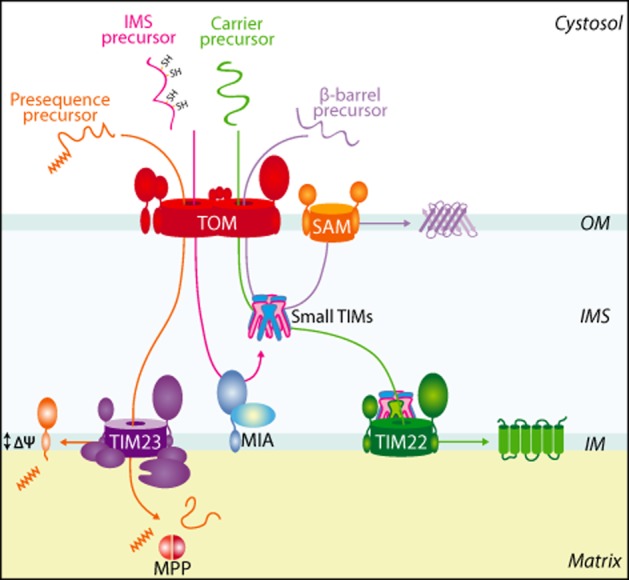
Different import pathways and machineries for precursor translocation into mitochondria. Mitochondrial precursor proteins begin their journey in the cytosol and are delivered to the outer mitochondrial (OM) TOM complex. Precursor proteins containing a positively charged N-terminal presequence (depicted in orange) are then delivered to the translocase of the inner membrane 23 (TIM23) complex for their translocation into or across the inner mitochondrial membrane in a membrane potential (Δψ)-dependent manner. Following import, the N-terminal presequence is proteolytically removed by the MPP. Import of intermembrane space precursor (IMS) proteins that contain characteristic cysteine residues (depicted in pink), such as the small TIM chaperones, is coupled to their oxidative folding by exploiting the MIA machinery. Hydrophobic membrane proteins belonging to the carrier family (depicted in green) are guided by small TIM chaperones to the TIM22 complex for inner membrane (IM) integration, or in the case of β-barrel precursor proteins (depicted in purple) to the sorting and assembly machinery (SAM) complex in the outer membrane of mitochondria.
The translocase of the outer membrane (TOM) complex provides the first line of contact at mitochondria for nearly all nuclear-encoded precursor proteins making the journey to the organelle (Figure 1). The central subunit of the TOM complex is Tom40, a β-barrel and channel-forming protein that allows precursor proteins to translocate from the cytosol into the mitochondria. This transport is assisted by several other TOM subunits that regulate the architecture and function of the TOM translocase (Neupert and Herrmann, 2007; Becker et al., 2009; Chacinska et al., 2009). Following translocation across the outer membrane via the TOM complex specific targeting signals found within a mitochondrial precursor define the biogenesis pathway it will follow. The classical import pathway into mitochondria is governed by the well-defined positively charged targeting signal known as a presequence (Neupert and Herrmann, 2007; Chacinska et al., 2009; Mokranjac and Neupert, 2010; Dudek et al., 2013). Presequence containing precursors are delivered to the inner membrane TIM23 (translocase of the inner membrane 23) complex for translocation through into the mitochondrial matrix, or in some cases lateral release into the inner membrane (Neupert and Herrmann, 2007; Chacinska et al., 2009; Mokranjac and Neupert, 2010; Dudek et al., 2013) (Figure 1). Many proteins residing in the inner membrane, intermembrane space and outer membrane of mitochondria contain non-cleavable targeting signals and employ different biogenesis pathways. Hydrophobic inner membrane metabolite carriers are inserted into the inner membrane by an alternative translocase, the TIM22 complex (Figure 1) (Koehler, 2000; Rehling et al., 2004; Chacinska et al., 2009). The TIM22 complex facilitates the insertion of carrier proteins into the inner membrane in a membrane potential-dependent manner (Koehler, 2000; Rehling et al., 2004; Chacinska et al., 2009). Alternatively, hydrophobic precursors of the outer membrane belonging to the β-barrel proteins require the action of the sorting and assembly machinery complex of the outer membrane for their successful integration and assembly into functional complexes (Figure 1) (Becker et al., 2008; 2009; Chacinska et al., 2009). Both carrier and β-barrel proteins are chaperoned through the aqueous intermembrane space by the ATP-independent chaperone family, the small TIMs. Finally, many intermembrane space proteins are cysteine-rich proteins that exploit an oxidative folding machinery of the intermembrane space, known as (mitochondrial intermembrane space assembly (MIA) (Figure 1) (Riemer et al., 2011; Stojanovski et al., 2012). The MIA machinery consists of the central receptor and oxidoreductase Mia40 and the sulfhydryl oxidase Erv1 and couples the processes of protein translocation into the intermembrane space with protein oxidation. Mia40 engages with its substrates by transient disulphide intermediates and in doing so facilitates oxidation of these substrates (Chacinska et al., 2004; Naoe et al., 2004; Terziyska et al., 2005).
Once protein import into the mitochondria has been achieved, mitochondrial precursors are then able to undergo maturation by folding and in many cases, assembly into multimeric complexes. This is where the action of the mitochondrial protein quality control machinery comes into play (Figure 2). Protein quality control comprises all reactions that deal with newly imported polypeptides or misfolded polypeptides, with respect to their stabilization, folding or refolding or complete removal by proteolysis. The mitochondrial protein quality control machinery consists of an elaborate network of molecular chaperones and protein degradation factors, which continually monitor and maintain the integrity of the proteome (Baker and Haynes, 2011; Baker et al., 2011; Varabyova et al., 2013; Voos, 2013). In the case of the mitochondrial matrix, a string of classical chaperones and co-chaperones are present to assist in protein folding and to maintain these proteins in a proper conformation (Figure 2) (Voos, 2013). Chaperones belonging to the Hsp60, Hsp70 and Hsp100 (Hsp78 in mitochondria) families play an important role in mitochondrial protein homeostasis. Hsp60 and Hsp70 mainly facilitate protein translocation and folding reactions (Voos, 2013), whereas Hsp78 is required at elevated temperatures, conferring compartment-specific thermotolerance to yeast mitochondria and is necessary for the resolubilization of aggregated proteins in vivo (Schmitt et al., 1996; Neupert and Herrmann, 2007). As mentioned above, proteins residing in the mitochondrial matrix or the inner membrane are guided by the presence of an N-terminal presequence. This presequence must be removed once it has fulfilled its function of delivering the protein to mitochondria. This cleavage event is performed by the mitochondrial processing peptidase (MPP) (Hawlitschek et al., 1988). Two additional proteases, Icp55 and Oct1, have been shown to play a role in further processing steps post MPP cleavage for a defined subset of proteins (Vogtle et al., 2009; 2011). In the event of protein misfolding, soluble protease systems of the mitochondrial matrix, including the peptidases Lon and ClpXP, extend the protein quality control capacity of the mitochondrial matrix (Fischer et al., 2012; Voos, 2013). Lon is a major protein quality control protease for soluble matrix proteins in both yeast (known as Pim1) and mammalian cells. Lon degrades endogenous mitochondrial proteins due to destabilizing mutations (Bateman et al., 2002) or oxidative modifications (Bota and Davies, 2002) that induce conformational changes in the proteins. In the case of ClpXP, it has been demonstrated in Caenorhabditis elegans that ClpP, which is the proteolytic component of ClpXP machine, in conjunction with the matrix peptide exporter HAF-1 are essential upstream components for the induction of the mitochondrial unfolded protein response (UPR) (Haynes et al., 2007; 2010). The mitochondrial UPR is a stress response that activates transcription of nuclear-encoded mitochondrial chaperone genes to promote protein homeostasis within the organelle. The peptides generated from the ClpP-dependent degradation of accumulated unfolded matrix proteins are believed to cross the inner membrane via HAF-1 and then cross the porous outer membrane into the cytosol. Once in the cytosol, it is suspected that the peptides themselves or some additional activity of HAF-1 are necessary for mitochondrial UPR signalling (Haynes and Ron, 2010).
Figure 2.
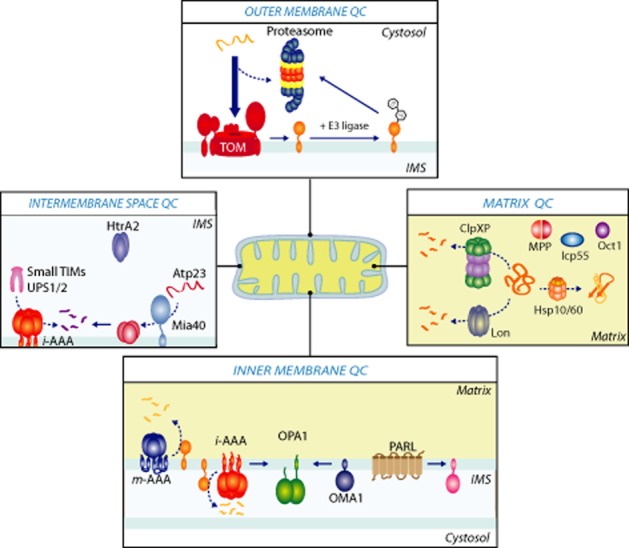
Mitochondrial protein quality control. Each mitochondrial compartment has its own quality control machinery. Outer mitochondrial membrane proteins can be ubiquitinated by E3 ligases at the mitochondria surface and are degraded through the ubiquitin/26S proteasome system. Premature precursors in the cytosol can also be targets for the proteasome, prior to being imported into mitochondria. In the mitochondrial matrix, chaperones belonging to Hsp60 and Hsp70 families facilitate protein translocation and folding reactions. Precursor processing such as removal of the N-terminal presequence is mediated by the MPP. Two additional proteases, Icp55 and Oct1, play a role in processing steps post MPP cleavage. Damaged proteins within the mitochondrial matrix are preferentially degraded by the Lon protease and the ClpXP complex. Protein quality control in the inner membrane is primarily monitored by two AAA proteolytic complexes, the i-AAA protease and m-AAA protease with their respective active sites facing the intermembrane space (i-AAA) or matrix (m-AAA). The i-AAA protease in conjunction with the inner membrane metallopeptidase OMA1 regulates mitochondrial morphology by proteolytic processing of the dynamin-like GTPase OPA1. The rhomboid serine protease, PARL, mediates cleavage of outer membrane kinase Pink1 in healthy mitochondria. Finally, in the intermembrane space, the oxidoreductase Mia40 can serve as a chaperone for the import of substrates such as the metallopeptidase Atp23. The i-AAA protease and Atp23 can degrade intermembrane space proteins, such as Ups1, whereas the i-AAA alone is responsible for the turnover of Ups2 and the small TIM chaperones, Tim9 and Tim10. Finally, HtrA2 encodes a serine protease and has been implicated in both apoptosis and Parkinson's disease.
Protein quality control in the mitochondrial inner membrane is primarily monitored by two AAA proteolytic complexes, the i-AAA protease and m-AAA protease, which are oriented with their respective active sites facing the intermembrane space (i-AAA) or matrix (m-AAA) (Figure 2) (Leonhard et al., 1996; Tatsuta and Langer, 2009; Voos et al., 2013). The i-AAA protease is a homo-oligomeric machine composed of a single protein subunit known as Yme1 (yeast mitochondrial escape protein 1) in both yeast and humans (known as YME1L). Conversely, the m-AAA protease is hetero-oligomeric complex and is composed of the Yta10 and Yta12 subunits in yeast, while mammalian mitochondria incorporate different isoenzymes of m-AAA proteases (paraplegin, AFG3L1 and AFG3L2 subunits) that assemble into homo-or hetero-oligomeric complexes (Koppen et al., 2007). The substrate repertoire of these inner membrane machines includes non-assembled inner membrane proteins that are peripherally associated or integrated into the inner membrane (Leonhard et al., 1996; Arlt et al., 1998; Korbel et al., 2004; Stiburek et al., 2012). In yeast, it has been shown that the m-AAA protease has an additional role in the processing of the mitochondrial subunit Mrpl32, which is required for the assembly of mitochondrial ribosomes (Nolden et al., 2005; Bonn et al., 2011). A number of additional proteases are localized to the mitochondrial inner membrane, such as Atp23, a conserved metallopeptidase that associates peripherally with the inner membrane. In yeast mitochondria, Atp23 acts as a peptidase for the maturation and assembly of the F1FO-ATP synthase subunit Atp6 (Osman et al., 2007; Zeng et al., 2007). Another protease that resides in the inner membrane is the metallopeptidase Oma1, which in yeast has overlapping activity with the m-AAA protease acting as a quality control enzyme (Kaser et al., 2003; Khalimonchuk et al., 2012). Interestingly, mammalian OMA1 has evolved an additional function as a stress-inducible protease that controls mitochondrial morphology through the regulated processing of the fusion mediator, OPA1 (discussed in section on Mitochondrial Fusion) (Ehses et al., 2009; Head et al., 2009).
The mechanisms and machineries that govern protein quality control within the intermembrane space remain less defined. The intermembrane space does contain the small TIM chaperones, which transport hydrophobic membrane proteins passing through this aqueous environment to one of the mitochondrial membranes (Koehler, 2000; Chacinska et al., 2009). However, the role of chaperones in the folding and assembly of intermembrane space proteins themselves remains more elusive. Interestingly, the intermembrane space oxidoreductase Mia40 (described above) was also recently shown to also act as chaperone and facilitate protein folding in a redox-independent manner (Weckbecker et al., 2012), thus suggesting a broader role for this protein in quality control within the intermembrane space (Figure 2). The inner membrane localized i-AAA protease is responsible for the degradation of the intrinsically unstable intermembrane space proteins, Ups1 and Ups2, which are involved in maintenance of mitochondrial phospholipid levels (Potting et al., 2010). Additionally, Yme1 was recently shown to be responsible for the degradation of misfolded and non-assembled small TIM proteins (Baker et al., 2012) (Figure 2). Ups1 is also degraded by Atp23, along with Yme1 (Potting et al., 2010). These recent discoveries have contributed to unravelling protein quality control mechanisms within the intermembrane space. In higher eukaryotes, an additional protease is found within the intermembrane space of mitochondria, the serine protease HtrA2/Omi (Suzuki et al., 2004). The bacterial homolog of HtrA2, called DegP mediates protein quality control within the periplasm (Kim and Kim, 2005), which is analogous to the mitochondrial intermembrane space. However, a defined role in intermembrane space quality control has not as yet been assigned to HtrA2.
With regards to the outer membrane, given the apparent absence of proteases in this compartment, it remained enigmatic if and how outer membrane proteins were proteolytically regulated. Current research places the UPS at the forefront of regulating the mitochondrial outer membrane proteome (Figure 2) (Karbowski and Youle, 2011; Livnat-Levanon and Glickman, 2011). The outer membrane of mitochondria is an intriguing environment, as it houses proteins with very distinct topologies ranging from β-barrel proteins, single spanning membrane proteins with α-helical transmembrane domains and multispanning membrane proteins. Many of these proteins have roles in essential mitochondrial pathways, for instance proteins involved in organelle morphology, protein import into mitochondria and apoptosis. Several lines of evidence make a strong case for UPS involvement in regulating the outer membrane proteome: (i) several ubiquitin ligases have been established as residents of the outer membrane, including MARCH-V/MITOL (Nakamura et al., 2006; Yonashiro et al., 2006; Karbowski et al., 2007; Sugiura et al., 2013), MULAN (Li et al., 2008) and Mdm30 (Fritz et al., 2003; Escobar-Henriques et al., 2006); (ii) ubiquitin-conjugated proteins can be detected in purified mitochondria by mass spectrometry (Peng et al., 2003; Jeon et al., 2007); (iii) cells treated with proteasomal inhibitors display an increase in ubiquitinated mitochondrial proteins (Margineantu et al., 2007); and (iv) studies have identified specific mitochondrial substrates of the UPS, such as Fzo1/Mfn1/Mfn2, outer membrane proteins involved in mitochondrial fusion (discussed below) (Gegg et al., 2010; Anton et al., 2011; Chan et al., 2011) and the β-barrel protein VDAC1 (Narendra et al., 2010b).
Surprisingly, the UPS has also been suggested to be involved in regulating the degradation of proteins from other mitochondrial compartments, including the matrix (OSCP) (Margineantu et al., 2007), the inner membrane (UCP2 and UCP3) (Azzu and Brand, 2010) and the intermembrane space (Endo G) (Radke et al., 2008). Recently, Bragoszewski et al. (2013) reported on a role for the proteasome in the early biogenesis of intermembrane space precursor proteins. The study revealed that intermembrane space proteins that utilize the MIA pathway can be ubiquitinated and degraded by the proteasome before they even reach the mitochondria (Bragoszewski et al., 2013). Thus, by regulating the fate of premature precursors in the cytosol, the UPS can in essence control the proteome of the intermembrane space. All of these independent lines of evidence make a strong case for a role of the UPS in maintaining mitochondrial protein homeostasis. However, the mechanistic details of the signalling network and regulatory events that take place await clarification.
Quality control at the organellar level – maintaining a healthy mitochondrial network
When the molecular pathways that regulate the mitochondrial proteome become overwhelmed, the dynamic nature of mitochondria can assist to ensure that the whole organelle is not at risk of damage. Mitochondria undergo constant changes in number, size and positioning within the cell, often reflecting the cellular energetic requirements (Bereiter-Hahn and Voth, 1994). The importance of mitochondrial fusion and fission events in the regulation of organelle morphology had been evident for some time (Bereiter-Hahn and Voth, 1994). However, the significance of these events in maintaining mitochondrial function in a physiological context remained unclear. The biological relevance of the pathways that mediate organelle dynamics is substantiated by the number of human diseases that have now been attributed to mutations in the genes encoding components that govern mitochondrial morphology (Alexander et al., 2000; Delettre et al., 2000; Baxter et al., 2002; Zuchner et al., 2004; Shirendeb et al., 2011; Song et al., 2011). Thus, mitochondrial dynamics and the maintenance of a dynamic mitochondrial population underpin cellular survival.
Mitochondrial fission
The process of organelle division or fission is essential since mitochondria cannot be created de novo. Mitochondrial fission ensures that a full complement of mitochondria is inherited by daughter cells following mitosis (Yaffe, 1999). Additionally, mitochondrial fission may serve to segregate damaged mitochondria, thus preserving the health of the entire network (Twig et al., 2008). Both mitochondrial fusion and fission events are mediated by large GTPases of the dynamin family (Otsuga et al., 1998; Bleazard et al., 1999; Alexander et al., 2000; Wong et al., 2000; Chen et al., 2003; Sesaki et al., 2003). Mitochondrial fission is mediated by the cytosolic dynamin-related protein, Drp1, which needs to translocate to mitochondria in order to sever the organelle (Figure 3). Drp1 translocates to points of mitochondrial constriction that represent future sites of scission and assembles into ring-shaped oligomers (Smirnova et al., 2001; Legesse-Miller et al., 2003; Ingerman et al., 2005). By utilizing GTP hydrolysis as a driving force, Drp1 severs both the outer and inner mitochondrial membranes, resulting in mitochondrial fission (Lackner et al., 2009; Mears et al., 2011). The true physiological significance of Drp1 has been highlighted by the identification of a patient with a dominant negative allele of Drp1 (Waterham et al., 2007). The patient died at 37 days and displayed poor brain development and optic atrophy. In accordance with such observations, a mouse model lacking Drp1 results in embryonic lethality (Ishihara et al., 2009; Wakabayashi et al., 2009).
Figure 3.
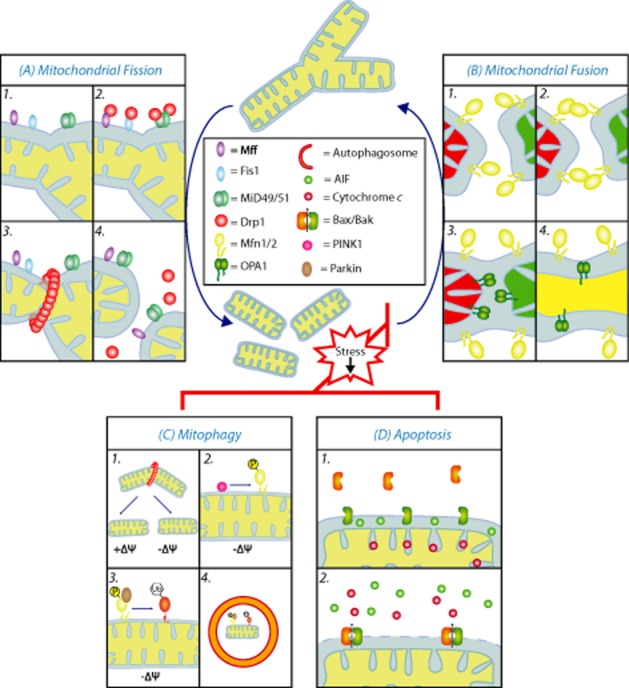
Mitochondrial morphology mediators in mammalian cells. (A) Mitochondrial fission requires recruitment of the cytosolic Drp1 to the outer membrane. Mitochondrial fission factor (Mff), Fis1 and Mitochondrial dynamics proteins of 49 and 51 kDa (MiD49 and MiD51) have been proposed to act in recruitment and assembly of Drp1 at the outer mitochondrial membrane. In the presence of GTP, Drp1 forms rings around mitochondrial tubules at sites of scission. GTP hydrolysis causes constriction of the Drp1 rings and facilitates scission of the mitochondrion. (B) Mitochondrial fusion relies on two machineries located within each of the mitochondrial membranes. Outer membrane fusion is mediated by Mfn1 and Mfn2. Mfn1/2 homo and heterodimers assemble in trans and form an organelle tethering complex. Inner membrane fusion is mediated by the inner membrane GTPase Optic atrophy 1, Opa1. Fusion of both the outer and inner membrane is coordinated and normally occurs simultaneously. (C) Under conditions of stress, such as mitochondrial depolarization, mitochondrial fusion is blocked and mitochondrial fission continues unopposed causing a fragmentation of the network. This reduces the mitochondria to a size appropriate for subsequent engulfment by an autophagosome. In dysfunctional mitochondria, such as depolarized mitochondria, PINK1 accumulates at the outer membrane and flags the damaged mitochondria. PINK1 phosphorylates Mfn2 and this stimulates recruitment of Parkin from the cytosol to the impaired mitochondria, where it ubiquitinates outer membrane proteins, which results in the elimination of the unwanted mitochondria. Δψ, membrane potential; P, phosphorylation; Ub, Ubiquitination. (D) Induction of the intrinsic pathway of apoptosis results in the recruitment of cytosolic Bax to mitochondria. Bax and Bak oligomerize in the outer membrane and induce MOMP, which permits release of cytochrome c and other mitochondrial intermembrane space proteins such as the apoptosis inducing factor (AIF). A number of components of the fission and fusion machinery including OPA1, Fis1, Drp1, Mfn1 and Mfn2 have been directly implicated in the regulation of apoptosis.
How Drp1 is recruited to the surface of mitochondria has been a contentious issue. Post-translational modification of Drp1 (discussed below) as well as several receptor proteins seem to be important in determining its recruitment and activity (Figure 3). In yeast, Dnm1p (the Drp1 equivalent) is recruited to the outer membrane by the receptor protein, Fis1p (Mozdy et al., 2000). The yeast system also involves two additional adaptor proteins Mdv1p and Caf4p, which helps to recruit and assemble Dnm1p into helical structures (Tieu et al., 2002; Cerveny and Jensen, 2003; Griffin et al., 2005; Koirala et al., 2010). The situation in higher eukaryotes is more complicated. Firstly, higher eukaryotes have no obvious Mdv1 homologue, but they do have a Fis1 homologue (James et al., 2003; Yoon et al., 2003; Stojanovski et al., 2004). The function of human Fis1 has however been brought into question by genetic data showing that Fis1 is not required for fission (Otera et al., 2010). A number of alternative outer membrane receptors have been proposed for Drp1 in higher eukaryotes including Mff, MiD49 and MiD51/MIEF1 (Gandre-Babbe and van der Bliek, 2008; Otera et al., 2010; Palmer et al., 2011; Zhao et al., 2011). Knockdown and overexpression studies of Mff, MiD49 and MiD51/Mief1 have been shown independently to influence mitochondrial fission events, suggesting that there are multiple ways to control fission (Otera et al., 2010; Palmer et al., 2011; Zhao et al., 2011). Recent work has suggested that Fis1, Mff, MiD49 and MiD51/MIEF1 act in a partially redundant manner to promote Drp1-dependent fission (Koirala et al., 2013; Loson et al., 2013). It remains to be dissected how all of these receptor proteins act in the same pathway to modulate the recruitment and activity of Drp1.
The steady-state levels and cycling activity of Drp1 are regulated by various post-translational modifications, including ubiquitination (Nakamura et al., 2006; Yonashiro et al., 2006; Karbowski et al., 2007; Horn et al., 2011; Wang et al., 2011b), sumoylation (Harder et al., 2004; Wasiak et al., 2007; Braschi et al., 2009; Figueroa-Romero et al., 2009; Zunino et al., 2009), S-nitrosylation (Cho et al., 2009; Nakamura et al., 2010) and phosphorylation (Chang and Blackstone, 2007; Cribbs and Strack, 2007; Taguchi et al., 2007; Cereghetti et al., 2008; Han et al., 2008). For example, sumoylation of Drp1 by the SUMO E3 ligase, MAPL in conjunction with the SUMO E2 ligase Ubc9, serves to enhance Drp1 stability and mitochondrial fission (Harder et al., 2004). Ubiquitination of Drp1 is mediated by the E3 ubiquitin ligase MARCH-V/MITOL; however, this does not lead to the degradation of Drp1, but rather serves to regulate the activity of the protein and regulates formation of fission complexes at the outer membrane (Karbowski et al., 2007). Phosphorylation of Drp1 at Ser637 by cAMP-dependent PKA has been shown to result in impaired mitochondrial fission (Chang and Blackstone, 2007; Cribbs and Strack, 2007), while S-nitrosylation of Drp1 at Cys644 by treatment of cells with nitric oxide increases GTPase the activity and dimerization of Drp1 (Cho et al., 2009). These modifications are testament to the central role of Drp1 in organelle dynamics.
Mitochondrial fusion
During mitochondrial fusion, the outer and inner membranes, which delineate the organelle, combine with the corresponding membranes on an adjacent mitochondrion. This results in mixing of the membranes, the contents of the intermembrane space and the matrix and allows mixing of the protein and mtDNA content (Figure 3). Mitochondrial content mixing helps to ease organelle stress by mixing the contents of partially damaged mitochondria as a form of complementation. Unlike mitochondrial fission, which appears to rely on a single set of machinery at the outer membrane, mitochondrial fusion relies on two sets, located within each of the mitochondrial membranes. The key proteins involved in fusion are the outer membrane GTPases mitofusins, Mfn1 and Mfn2 (Fzo1 in yeast) and the inner membrane GTPase. Optic atrophy 1, (Opa1; Mgm1p in yeast) (Figure 3). Fusion of both the outer and inner membrane is coordinated and normally occurs simultaneously to ensure that mitochondrial compartmentalization is maintained. Mouse knockout studies have provided strong evidence to suggest that both Mfn1 and Mfn2 are essential for mitochondrial fusion (Chen et al., 2003; 2005). Cells lacking either Mfn1 or Mfn2 display a highly fragmented mitochondrial network and fusion assays indicate a great reduction in the levels of mitochondrial fusion in knockout mouse embryonic fibroblasts of either Mfn1 or Mfn2 alone (Chen et al., 2003) and a complete loss of fusion in cells lacking both Mfn1 and Mfn2 (Koshiba et al., 2004; Chen et al., 2005). Interestingly, Mfn1 and Mfn2 are required on adjacent mitochondria to mediate membrane fusion. Both in vivo and in vitro membrane fusion assays have suggested that mitochondria lacking the mitofusins do not fuse with wild-type mitochondria (Koshiba et al., 2004; Meeusen et al., 2004; Song et al., 2009).
OPA1 is a dynamin-like protein associated within the inner membrane and is involved in mitochondrial cristae remodelling and inner membrane fusion (Wong et al., 2003; Ishihara et al., 2004). OPA1 has been identified as the gene mutated in the most common form of dominant optic atrophy, a disease in which retinal ganglion cells degenerate and cause atrophy of the optic nerve (Alexander et al., 2000; Delettre et al., 2000). Loss of OPA1 results in a loss of inner membrane fusion, while outer membrane fusion can still proceed (Song et al., 2009), indicating that the mitofusins and OPA1 act in distinct steps during the fusion process. OPA1 is anchored into the inner membrane by a single transmembrane domain with the bulk of the protein facing the intermembrane space (Wong et al., 2003; Frezza et al., 2006; Ishihara et al., 2006). The protein has a fascinating biogenesis pathway and due to differential RNA splicing and post-translational processing, several isoforms of the protein can exist within mitochondria. These isoforms are described as long (L) and short (S) and the balance between L-OPA1 and S-OPA1 is required for membrane fusion and the maintenance of steady-state mitochondrial morphology (Song et al., 2007). There are eight mRNA splice forms (Delettre et al., 2001) and following subsequent proteolytic processing within mitochondria, OPA1 isoforms migrate as a complex mixture of at least five species, designated (a–e). The species denoted as a and b are thought to be a mixture of long isoforms of OPA1, whereas the shorter species (c–e) are thought to result from additional proteolytic processing within mitochondria (Ishihara et al., 2006). All mRNA splice forms of OPA1 encode a protease processing site in exon 5 (denoted the S1 cleavage site) and some contain an additional site (denoted the S2 cleavage site) in the alternative exon 5b (Ishihara et al., 2006). The mRNA splice forms 1 and 2 encode only the S1 cleavage site and thus only yield a single long and a single short form, while mRNA splice forms 4 and 7 encode sites S1 and S2 and therefore yield two short forms in addition to a single long form.
Because mitochondrial fusion, and ultimately mitochondrial morphology, is so intimately linked to the proper ratio of long and short OPA1 isoforms, the proteolytic processing of OPA1 is highly regulated. The mitochondrial inner membrane YME1L (i-AAA protease) and OMA1 have emerged as the most likely candidates for mediating the proteolytic processing of OPA1. Depletion of YME1L decreases processing of OPA1 at the S2 site and in accordance with this cells that have reduced levels of YME1L display a fragmented mitochondrial network (Griparic et al., 2007). OMA1 is a zinc metallopeptidase localized to the inner membrane and is necessary for S1 cleavage of OPA1. OMA1 mediates both constitutive and stress-induced cleavage of OPA1 at the S1 cleavage site (Ehses et al., 2009; Quiros et al., 2012). Cellular insults that result in mitochondrial dysfunction influence such processing events. Cellular stress in the form of a dissipated membrane potential, loss of mtDNA, altered ATP levels and apoptosis have all been shown to affect OPA1 processing (Ishihara et al., 2006; Duvezin-Caubet et al., 2007).
In yeast, the outer membrane protein Ugo1 is an additional fusion mediator and has a proposed role of stabilizing Fzo1p and Mgm1p to facilitate lipid mixing following mitochondrial tethering (Sesaki and Jensen, 2001; Sesaki et al., 2003; Wong et al., 2003). However, a mammalian homologue of Ugo1 awaits identification. Interestingly, post-translation modifications are also involved in the regulation of the fusion machinery, although probably not to the extent observed with Drp1. The protein levels of Fzo1p are regulated by Mdm30p, a cytosolic mitochondrially associated F-box protein (Escobar-Henriques et al., 2006; Cohen et al., 2008). Recently, it was shown that two independent pathways reversibly ubiquitinate yeast Fzo1p. Ubiquitination in one pathway by Mdm30 leads to an activation of Fzo1p for fusion (Anton et al., 2013). Alternatively, in the second pathway ubiquitination by an as yet unidentified E3 ligase marks Fzo1p for degradation by the proteasome. These two pathways cause modification of different lysine residues within the protein thereby distinguishing the events of protein activation and degradation. Interestingly, Anton et al. (2013) revealed that both Fzo1p ubiquitination pathways are reversible and specific deubiquitylases for each pathway were identified. Ubp12 was shown to act on Fzo1 oligomers and selectively remove the activating ubiquitin chains (Anton et al., 2013). Conversely, Ubp2 was shown to remove destabilizing ubiquitin chains that were attached to Fzo1p independently of Mdm30 (Anton et al., 2013). Thus, the selective ubiquitination and deubiquitination of Fzo1p represents a unique mechanism for regulation of mitochondrial fusion. Although there is no apparent Mdm30p homologue in higher eukaryotes, the human orthologues of Fzo1p, Mfn1 and Mfn2, are degraded by the UPS. Both Mfn1 and Mfn2 have shown to be targets for an E3-ubiquitin ligase known as Parkin (described in section on Quality Control at the Cellular Level) (Tanaka et al., 2010; Chan et al., 2011).
Mitochondrial transport and distribution
Mitochondrial morphology and distribution are intimately linked, with changes in mitochondrial fission and fusion affecting not only mitochondrial shape but also distribution throughout the cell (Chen et al., 2003; 2007,; Li et al., 2004; Verstreken et al., 2005). The reliance of the cell on correct mitochondrial distribution is most evident in neuronal cells, with efficient transport of mitochondria to synapses shown to be important for the regulation of synaptic activity and transmission of synaptic signals (Li et al., 2004; Verstreken et al., 2005). The responsive nature of mitochondria to the energy demands of the cell was further demonstrated recently with alterations to mitochondrial motility observed following increased axonal electrical activity (Ohno et al., 2011), potentially due to changes in localized ATP and/or metabolite concentration. Fragmented mitochondria formed following loss of Mfn2 exhibit a loss of motility (Chen et al., 2003) with reduced dendritic spine formation observed in Purkinje cells from Mfn2 mutant mice (Chen et al., 2007), while loss of Drp1 function has been described to reduce synaptic mitochondria (Verstreken et al., 2005).
Mammalian and yeast cells have evolved separate mechanisms for mitochondrial distribution, primarily using different cytoskeletal scaffolds for transport. In yeast, mitochondria predominantly utilize the actin cytoskeleton to facilitate movement (Okamoto and Shaw, 2005; Anesti and Scorrano, 2006; Boldogh and Pon, 2007; Frederick and Shaw, 2007). In mammalian cells, mitochondria principally rely on their association with microtubules for movement through association with motor proteins (Pilling et al., 2006); however, a role for actin in mitochondrial transport and/or docking in mammalian cells has been proposed (Chada and Hollenbeck, 2004; Lee and Peng, 2008). The motor proteins kinesin and dynein are mechanochemical motors that associate with microtubules, transporting mitochondria throughout the cell in an anterograde and retrograde manner respectively (Nangaku et al., 1994; Tanaka et al., 1998; Zhao et al., 2001; Vale, 2003; Pilling et al., 2006). The kinesin-like protein KLP6 has been shown to be important for the maintenance of mitochondrial motility (Tanaka et al., 2011), confirming the role of kinesin in mitochondrial anterograde transport. Cytoplasmic dynein transports mitochondria in a retrograde motion towards the nucleus (Pilling et al., 2006), with a direct interaction between mitochondria and dynein confirmed recently (van Spronsen et al., 2013).
The adaptor molecule Milton is a mitochondrial protein that interacts with kinesin-1, mediating microtubule-based transport of mitochondria, with two isoforms Milton1 and Milton2 present in mammalian cells (Cox and Spradling, 2006; Koutsopoulos et al., 2010). Milton recruits kinesin-1 in axonal cells by binding to the heavy chain and activating anterograde transport of mitochondria (Glater et al., 2006). Milton associates with a mitochondrial Rho-GTPase (Miro) that mediates mitochondrial transport through the sensing of local calcium concentration (Fransson et al., 2003; 2006; Glater et al., 2006; Saotome et al., 2008; Russo et al., 2009). While association of Milton with dynein has long been suggested, it was only recently demonstrated that Milton2 preferentially interacts with the retrograde motor dynein in axonal cells as part of the Milton/Miro complex (van Spronsen et al., 2013). Docking of mitochondria to microtubules may also be regulated by syntaphilin (SNPH), where overexpression of SNPH results in extended docking of axonal mitochondria to microtubules (Kang et al., 2008).
Quality control at the cellular level – maintaining a healthy mitochondrial population by mitophagy
When entire segments of mitochondria become damaged, they can be selectively removed by mitophagy, a quality control process for disposal of mitochondria via the autophagic pathway. When molecular quality control at the organellar level fails, elimination of the damaged organelles is essential. Interestingly, the process of mitophagy has been linked to mitochondrial dynamics demonstrating that the hierarchical systems of quality control that oversee organelle function are intimately linked. Transport of mitochondria back to the cell body has also been suggested to be a mechanism to recycle old and damaged organelles, with mitochondria of low membrane potential recycled back from the cell periphery (Miller and Sheetz, 2004); however, this mechanism is still under debate, with a subsequent study reporting no difference in the membrane potential of mitochondrial cellular populations (Verburg and Hollenbeck, 2008). It has been proposed that fragmentation of the mitochondrial network facilitates elimination via mitophagy (Narendra et al., 2008; Twig et al., 2008); however, fragmentation alone is not sufficient to stimulate mitophagy indicating that additional mechanisms are involved.
The serine/threonine kinase PINK1 and the E3 ubiquitin ligase Parkin have been implicated in a pathway to degrade dysfunctional mitochondria in higher eukaryotes (Narendra et al., 2010a; Youle and Narendra, 2011). Both the PINK1 and PARKIN genes have been found mutated in some cases of familial Parkinson's disease, providing evidence that mitochondrial dysfunction is an underlying feature of Parkinson's disease (Kitada et al., 1998; Valente et al., 2004). In healthy mitochondria, the levels of PINK1 are constitutively suppressed by import into the inner membrane and proteolytic cleavage by the rhomboid protease PARL (Jin et al., 2010). In dysfunctional mitochondria, such as depolarized mitochondria, PINK1 accumulates at the outer membrane and serves to flag the damaged mitochondria for removal. PINK1 stabilization in the outer membrane stimulates recruitment of Parkin from the cytosol to the impaired mitochondria, where it ubiquitinates outer membrane proteins, which results in the elimination of the unwanted mitochondria (Narendra et al., 2010a). Interestingly, Chen and Dorn (2013) recently revealed that phosphorylation of the fusion mediator Mfn2 by PINK1 causes Parkin recruitment to mitochondria. This suggests that the Mfn2 could serve as the receptor for Parkin at the outer membrane (Chen and Dorn, 2013).
Until recently, the best understood Parkin substrates were Mfn1 and Mfn2, and two proteins that are involved in mitochondrial trafficking Miro1/2 (Poole et al., 2008; Gegg et al., 2010; Tanaka et al., 2010; Chan et al., 2011). Parkin-dependent proteasomal turnover of these outer membrane GTPases alters mitochondrial fission and fusion cycles and mitochondrial trafficking. This acts to segregate the dysfunctional mitochondria and preventing their mixing with healthy populations. Furthermore, by blocking fusion through the degradation of the mitofusins, mitochondrial fission would take place unopposed leading to fragmentation of the damaged mitochondrial network, reducing the mitochondria to size more readily engulfed by autophagosomes. Recently, exciting work by Sarraf et al. (2013) determined the Parkin-dependent ubiquitylome in response to mitochondrial depolarization. The study revealed hundreds of dynamically regulated ubiquitination sites on mitochondrial outer membrane and cytosolic proteins. These included proteins involved in: protein import into mitochondria (TOMM70 and TOM22); mitochondrial morphology (Mfn1 and Fis1); metabolite exchange across the outer membrane (VDAC1, VDAC2 and VDAC3); apoptosis (Bax); and a large repertoire of proteasomal proteins (Sarraf et al., 2013). Thus, Parkin can be involved in the regulation of many cellular processes and the ubiquitylome indicates that Parkin dramatically alters the ubiquitylation status of the mitochondrial proteome.
Interestingly, yeast cells do not have PINK1 and Parkin equivalents; however, several lines of evidence suggest that damaged mitochondria in yeast also undergo elimination by mitophagy. For instance, depletion of Mdm35 causes loss of the inner membrane potential, mitochondrial swelling and fragmentation, which leads to elimination of these abnormal mitochondria by mitophagy (Nowikovsky et al., 2007). To identify proteins acting in the removal of mitochondria from yeast cells, two groups performed a genome wide screen of non-essential gene deletions strains for yeast mutants defective in selective mitochondrial degradation (Kanki et al., 2009; Okamoto et al., 2009). Among the >30 genes identified in both of these screens, the YIL146C gene, which encodes the Atg32 protein was looked into further. Deletion of Atg32 was shown to completely inhibit mitophagy, while other processes such as non-selective authophagy, pexophagy and the cytoplasm-to-vacuole targeting pathway remained unaffected. Atg32 is anchored in the mitochondrial outer membrane via a C-terminal transmembrane domain and uses Atg11 as an adaptor for cargo recognition (Kanki et al., 2009; Okamoto et al., 2009). The interaction between Atg11 and Atg32 increases upon conditions of nitrogen starvation that can induce mitophagy. Interestingly, one of the screens that identified Atg32, also uncovered DNM1, the yeast equivalent of Drp1 (Kanki et al., 2009), which is in agreement with previous findings that suggest mitochondrial fragmentation is a prerequisite for mitophagy (Twig et al., 2008). Interestingly, carbonyl cyanide 3-chlorophenylhydrazone (CCCP) treatment induces mitophagy in mammalian cells (Narendra et al., 2008), but not in yeast cells (Kanki et al., 2009). This implies that the induction of mitophagy in yeast may require additional proteins and/or signalling events in addition to mitochondrial depolarization.
Mitochondrial dynamics and cell death
If all quality control measures fail and mitochondrial dysfunction cannot be alleviated by any of the measures described so far, the cell would face death through apoptosis. The intrinsic pathway or mitochondrial pathway of cell death depends on the release of cytochrome c and other proapoptotic molecules from the intermembrane space of mitochondria (Figure 3). Interestingly, mitochondrial fusion and fission events have been shown to participate in the intrinsic cell death pathway although, as yet, the reason for this is still not well understood. In cellular models of apoptosis, mitochondria undergo increased fission and dramatically fragment due to increased Drp1 recruitment near the time of cytochrome c release (Frank et al., 2001). Drp1 inhibition can prevent mitochondrial fission and reduce cytochrome c release (Frank et al., 2001). The commitment stage in the intrinsic pathway is mitochondrial outer membrane permeabilization (MOMP), which is mediated by the cohort of Bcl-2 proteins (Youle and Strasser, 2008). This represents the point of no return in the cells' commitment to death and it is believed that Drp1 might work with the two proapoptotic Bcl-2 proteins, Bak and Bax to permeabilize the outer membrane (Frank et al., 2001; Wasiak et al., 2007; Suen et al., 2008). A critical event in the induction of MOMP by Bax and Bak is the oligomerization of the proteins in the outer membrane and it has been proposed that Drp1 can facilitate Bax oligomerization during apoptosis (Montessuit et al., 2010). Furthermore, depletion of the fission mediators Mff and Fis1 has also been shown to reduce apoptosis (Lee et al., 2004; Gandre-Babbe and van der Bliek, 2008). This suggests that fission of the mitochondrial network is a requirement for intrinsic cell death, or that the mitochondrial fission machinery is hijacked to participate in additional functions that contribute to the execution of the cell.
Given that mitochondrial fusion and fission events are opposing and the role of mitochondrial fission in promoting cell death, it is plausible that mitochondrial fusion should have a protective role in cells. Indeed, mitochondrial fusion does have a protective effect on cells and activation of apoptosis appears to block mitochondrial fusion (Olichon et al., 2003; Lee et al., 2004; Sugioka et al., 2004). However, the exact roles of mitochondrial fusion and fission in cell death are not so black and white and there are opposing findings. For instance, Mfn2 has been shown to be a determinant of oxidative stress-mediated heart muscle cell apoptosis (Shen et al., 2007) contrary to the protective effects detailed for fusion mediators. Additionally, Drp1 knockout mice have no defect in apoptosis (Wakabayashi et al., 2009), suggesting that Bax/Bak mediated MOMP can proceed in the absence of Drp1. Clearly, more studies are needed to tease out the details of this exciting link between the mitochondrial morphology regulators and apoptosis.
An additional role for mitochondrial fusion in quality control was recently uncovered and termed stress-induced mitochondrial hyperfusion (Tondera et al., 2009). This has been suggested to be a prosurvival mechanism that prevents apoptosis and maintains mitochondrial function during cell stress (Tondera et al., 2009; Gomes et al., 2011). Supporting this notion, many cell lines display an interconnected and elongated mitochondrial network in response to insults that induce stress, such as UV irradiation and nutrient starvation (Tondera et al., 2009; Gomes et al., 2011). Mitochondrial hyperfusion is dependent on the fusion mediators Mfn1 and OPA1 and an additional protein known as Stomatin-like protein 2 (Tondera et al., 2009). Cells unable to mount this response display greater sensitivity to the cellular insult and cell stress and undergo apoptosis more readily. However, the true physiological significance of this response needs to be clarified.
Mitochondrial quality control in neurodegenerative disorders
Thus far, we described a hierarchy of quality control systems put in place to sustain the mitochondrial proteome and mitochondrial integrity and function. These include: (i) quality control at the molecular level, involving mitochondrial chaperones and proteases and the UPS; (ii) quality control at the organelle level focusing on mitochondrial dynamics and distribution and the ability of the organelle to undergo fusion and fission and be transported within the cell and (iii) quality control at the cellular level involving removal of damaged organelles by mitophagy and ultimately removal of the cell through apoptosis. Although each system is distinct, there is a significant amount of crosstalk in order to ensure the persistence of a healthy mitochondrial population. What is evident, is that dysfunction at any level within the quality control hierarchy can result in neurological insult and disease. Indeed, numerous neurological disorders have been linked to proteins involved in maintaining mitochondrial quality control. These are listed in Table 1 and some have been selected for further discussion below.
Table 1.
Neurological disorders and mitochondrial association
| Disease | Protein/localization | Protein function | Pathogenesis and mitochondrial association |
|---|---|---|---|
| Alzheimer's disease (AD) | Amyloid precursor protein (APP)/cytosol | Synaptic formation and repair; cell signalling; cell adhesion; iron export, hormonal regulation | APP can accumulate in the TOMM40 pore1, resulting in mitochondrial dysfunction. Oxidative stress is a primary event in the progression of AD.2 Mitochondrial dynamics have been found to be altered in neurons or fibroblasts in AD patients and animal models.3 The disease is characterized by the presence of senile plaques, filamentous aggregates of amyloid beta peptide4 and neurofibrillary tangles mainly made up the mitochondrial-associated protein Tau.5 This is also accompanied by marked neuronal cell death.6 |
| Amyotrophic lateral sclerosis (ALS) | SOD1/cytosol and IMS | Detoxification of superoxide free radicals | Mutant SOD1 may be imported into mitochondria where it can interfere with the apoptotic machinery7, MPTP8, activate apoptosis9, protein import10, the redox balance11, mitochondrial dynamics and axonal transport.12,13 ALS is characterized by progressive loss of motor neurons followed by muscle weakness, paralysis and eventually death.14 |
| Charcot–Marie–Tooth neuropathy (CMT) | Mitofusin2 (Mfn2)/MOM GDAP1/MOM | Mitochondrial outer membrane fusion Maintenance of mitochondrial morphology | Deletion of MFN2 or loss of function mutants result in impaired mitochondrial fusion15 and most likely perturbs mitochondrial ER contacts.16 MFN2 mutations cause CMT2A, which is characterized by progressive distal limb muscle weakness and/or atrophy, stepping gait, distal sensory loss and mobility impairment.17 The function of GDAP1 still remains unclear; however, mutations in the GDAP1 gene result in CMT4A and CMT2K.18 |
| Dominant optic atrophy (DOA) | OPA1/MIM | Mitochondrial inner membrane fusion | Pathogenic mutations in OPA1 that cause DOA have been shown to impair mitochondrial morphology, mitochondrial ultrastructure, mitochondrial fusion and oxidative phosphorylation in fibroblasts.19 The disease affects the retinal ganglion cell layer and their axons that form the optic nerve.20 This results in visual impairment of the patient with a highly variable severity of the clinical presentation.21 |
| Parkinson's disease (PD) | DJ-1/Cytosol HtrA2/IMS Parkin/Cytosol PARL/MIM PINK1/MIM and MOM | Transcriptional regulation; anti-oxidative stress reaction; mitochondrial regulation Apoptotic regulation E3 Ubiquitin ligase; mitochondrial quality control; mitophagy PINK1 processing; HtrA2 processing Respiratory chain function; mtDNA maintenance; mitophagy | DJ-1 deficiency results in impaired mitochondrial dynamics and increased levels of oxidative stress.22 Mutations of HtrA2 seem to result in increased mitochondrial dysfunction and pathogenic mutations have been detected in various populations.23,24,25,26,27 Parkin is required for the clearance of dysfunctional mitochondria.28 Pathogenic mutations in the PARKIN gene disrupt important interactions intramolecular interactions and the catalytic environment of the RING2 domain.29 Autocatalytic processing of PARL regulates its activity and mutations in this processing site have been identified in patients with PD.30 Mutations in the PINK1 gene result in altered mitochondrial calcium buffering capacity and impaired respiration31, which can be rescued by supplementing with vitamin K2.32 PD is the most prevalent movement disorder in modern society. The motor symptoms arise due to degeneration of the dopaminergic neurons in the substantia nigra pars compacta.33 |
| Hereditary spastic paraplegia (HSP) | Paraplegin/MIM Hsp60/MM | Degradation of misfolded proteins; protein processing Protein folding; heat stress response | Mutations in the paraplegin gene linked to HSP display impaired proteolytic activity.34 Mutations in HSP60 associated with HSP result in impaired chaperonin function.35 HSP is characterized by progressive weakness and spasticity of the lower limbs.36 The phenotypes are often variable and arise due to retrograde degeneration of cortical motor neuron axons.37 |
| Huntington's disease (HD) | Huntingtin/cytosol and nucleus | Vesicular and organellar trafficking; transcriptional regulation | Pathogenic forms of Huntingtin have been shown to reduce PGC-1α activity by direct binding and reducing its expression.38,39 Huntingtin can bind the fission regulator Drp1 to promote mitochondrial fission and interfere with axonal transport of mitochondria.40,41 Huntington's disease results from expanded CAG nucleotide repeats encoding for a polyglutamine stretch in the Huntingtin gene.42 The disease is characterized by progressive motor impairment, personality changes, psychiatric illness and intellectual decline.43 |
| Spinocerebellar ataxia (SCA28) | AFG3L2/MIM | Degradation of misfolded proteins; ribosome biogenesis | Heterozygous mutations in the AFG3L2 gene that result in dominant SCA28 alter the proteolytic competence of the m-AAA protease and induce defective cytochrome c activity.44 Autosomal dominant forms of SCA are neurological disorders characterized by imbalance, progressive gait and limb ataxia and dysarthria caused by degeneration of the cerebellum.45,46 |
ER, endoplasmic reticulum; IMS, intermembrane space; Mfn, Mitofusin; MIM, mitochondrial inner membrane; MM, mitochondrial matrix; MOM, mitochondrial outer membrane; MPTP, mitochondrial permeability transition pore; SCA, spinocerebellar ataxia.
Devi et al. (2006);
Bonda et al. (2010);
Zhu et al. (2013);
Duyckaerts et al. (2009);
Grundke-Iqbal et al. (1986);
Gschwind and Huber (1995);
Pasinelli et al. (2004);
Martin et al. (2009);
Pasinelli et al. (2000);
Li et al. (2010);
Ferri et al. (2006);
Ferri et al. (2010);
De Vos et al. (2007);
Rowland (1984);
Ishihara et al. (2004);
de Brito and Scorrano (2008);
Zuchner et al. (2006);
Cassereau et al. (2011);
Zanna et al. (2008);
Alexander et al. (2000);
Delettre et al. (2001);
Irrcher et al. (2010);
Strauss et al. (2005);
Simon-Sanchez and Singleton (2008);
Kruger et al. (2011);
Wang et al. (2011a);
Lin et al. (2011);
Narendra et al. (2010a);
Spratt et al. (2013);
Shi et al. (2011);
Gandhi et al. (2009);
Vos et al. (2012);
Chinta and Andersen (2005);
Bonn et al. (2010);
Bross et al. (2008);
Reid (1997);
Harding (1993);
Chaturvedi et al. (2009);
Cui et al. (2006);
Song et al. (2011);
Reddy et al. (2009);
Huntington's Disease Collaborative Research Group (1993);
Martin and Gusella (1986);
Di Bella et al. (2010);
Schols et al. (2004);
Koeppen (2005).
A number of neurodegenerative disorders have been linked to molecular quality control systems. Disease mutations in the SPG7 gene, which encodes the m-AAA subunit paraplegin, were described 15 years ago (Casari et al., 1998), and were found in patients with an autosomal recessive form of hereditary spastic paraplegia. More recently, mutations in the gene of the other m-AAA subunit AFGL32 were described in one form of spinocerebellar ataxia (Cagnoli et al., 2010; Di Bella et al., 2010). Mouse models have demonstrated that the intermembrane space peptidase HtrA2 may be linked to Parkinson's disease (Vande Walle et al., 2008; Clausen et al., 2011). Mouse models using mutated or targeted deletion of HTRA2 presented phenotypes reminiscent of Parkinson's disease and display early onset neurodegeneration due to mitochondrial dysfunction (Jones et al., 2003; Martins et al., 2004). The mitochondrial chaperonin Hsp60 mediates protein folding in the mitochondrial matrix and is encoded by the HSPD1 gene. Mutations in this gene have been associated with hereditary spastic paraplegia and (Hansen et al., 2002) and Pelizaeus–Merzbacher-like disease (Magen et al., 2008).
Several human diseases are caused by mutation in genes encoding proteins critical for mitochondrial fusion or fission. In terms of mitochondrial fusion, heterozygous mutations in the OPA1 gene cause dominant optic atrophy, and heterozygous mutations in MFN2 gene cause the peripheral neuropathy Charcot–Marie–Tooth type 2A (Alexander et al., 2000; Delettre et al., 2000; Zuchner et al., 2004). In terms of mitochondrial fission, as explained earlier, a patient with a dominant negative allele of Drp1 (Waterham et al., 2007) has been described and has been linked to neonatal lethality with multisystem failure. Interestingly, mitochondrial dynamics have been implicated in other neurological disorders, including Parkinson's, Alzheimer's and Huntington's disease. As noted earlier, mutations in PINK1 and PARKIN lead to early onset autosomal recessive Parkinson's disease (Kitada et al., 1998; Valente et al., 2004). Drosophila lacking PINK1 or Parkin display swollen and defective mitochondria, which leads to muscle and neuron degeneration, which is reminiscent of Parkinson's disease (Poole et al., 2008; Yang et al., 2008). However, deficiency of Pink1 in mammalian cells leads to mitochondrial fragmentation (Exner et al., 2007; Dagda et al., 2009). It is important to note that the role of PINK1 and Parkin and mitochondrial dysfunction in Parkinson's disease has been limited by the cellular models exploited. Investigations have primarily been limited to cell culture models and mitochondrial dysfunction initiated by depolarization using CCCP, typically at high concentrations that would depolarize, and thus damage the whole mitochondrial network. This is perhaps not entirely representative of a cellular situation where presumably only small sections of the mitochondrial network would be damaged. Thus, the relevance of these observations and how the PINK1/Parkin pathway relates to mitochondrial function and the onset of Parkinson's disease remain to be explored. Recently a mouse model, referred to as the MitoPark model, assessed mitochondrial morphology in dopaminergic neurons in response to respiratory chain deficiency and was found to induce mitochondrial fragmentation and formation of large cytoplasmic bodies derived from mitochondria (Sterky et al., 2011). Interestingly, the dysfunctional dopaminergic neurons did not recruit Parkin to mitochondria in vivo and mitochondrial clearance and neurodegeneration were not affected by the absence of Parkin (Sterky et al., 2011).
Studies have also described a role for the regulation of mitochondrial morphology in Alzheimer's disease. One theory for the onset of Alzheimer's disease is that aggregation of the amyloid-β-peptide (Aβ) (product generated due to processing of the amyloid precursor protein) is the executioner of the disease resulting in cellular plaques and neurofibrillary tangles. Aβ has been shown to accumulate at mitochondria (Caspersen et al., 2005; Manczak et al., 2006) and studies have suggested that it can interact with Drp1 (Manczak et al., 2011; Manczak and Reddy, 2012). In both fibroblasts and neurons from patients with Alzheimer's disease, Drp1 protein levels are decreased (Wang et al., 2008; 2009,). Surprisingly, the levels of OPA1, Mfn1 and Mfn2 are reduced, while Fis1 expression is increased in these cells (Wang et al., 2009). It is believed that the altered expression of these morphology regulators will affect the distribution of the organelle leading to a loss of synaptic activity. Drp1 has also been shown to bind to mutant Huntington protein, which results in increased GTPase activity, oligomerization and increased dephosphorylation of Drp1 (Costa et al., 2010; Song et al., 2011). Defects in transport and morphology in cells expressing mutant Huntington can be rescued by expression of the GTPase dominant negative mutant of Drp1, known as Drp1K38A (Song et al., 2011) suggesting that Drp1 has a key role in the mitochondrial dysfunction observed in Huntington's disease. All of the findings reiterate the central role of mitochondrial dynamics and mitochondrial health in neuronal health and organism development; however, whether the mitochondrial morphology machinery can be exploited in a therapeutic manner remains to be determined.
Perspective
Mitochondrial function is central to cellular survival and consequently mitochondrial dysfunction is attributed to a myriad of diseases. In this review, we have summarized the hierarchy of systems that exist to ensure the persistence of a healthy mitochondrial population. Even though at first glance these systems may appear as distinct, it is evident that there is a significant level of crosstalk and that these events are integrated into a cellular network aimed at keeping mitochondria healthy. For instance, mitophagy depends on mitochondrial fission, which serves to reduce the size of damaged mitochondria for efficient engulfment by an autophagosome. There is also significant crosstalk between autophagy and apoptosis (Maiuri et al., 2007). For instance, mitophagy progression is influenced by mitochondrial permeability transition and can inhibited by inhibitors like cyclosporine A or overexpression of the anti-apoptotic protein Bcl2 (Xue et al., 2001; Rodriguez-Enriquez et al., 2006), suggesting common steps in these two physiologically distinct processes. Future discoveries on the molecular mechanisms and molecular connections between all of these pathways will yield important insights into cross-regulation and interdependence. Understanding how molecular, organellar and cellular events contribute to mitochondrial quality control is essential not only to understand the biology that regulates this amazing organelle, but to understand how these events underpin health and how their downfall can lead to disease.
Acknowledgments
The Stojanovski laboratory is currently funded by the National Health and Medical Research Council.
Glossary
- CCCP
carbonyl cyanide 3-chlorophenylhydrazone
- MIA
mitochondrial intermembrane space assembly
- MOMP
mitochondrial outer membrane permeablization
- MPP
mitochondrial processing peptidase
- mtDNA
mitochondrial DNA
- TOM
translocase of the outer membrane
- TIM
translocase of the inner membrane
- UPR
unfolded protein response
- UPS
ubiquitin-proteasome system
Conflict of interest
None.
References
- Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211–215. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- Anand R, Langer T, Baker MJ. Proteolytic control of mitochondrial function and morphogenesis. Biochim Biophys Acta. 2013;1833:195–204. doi: 10.1016/j.bbamcr.2012.06.025. [DOI] [PubMed] [Google Scholar]
- Anesti V, Scorrano L. The relationship between mitochondrial shape and function and the cytoskeleton. Biochim Biophys Acta. 2006;1757:692–699. doi: 10.1016/j.bbabio.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Anton F, Fres JM, Schauss A, Pinson B, Praefcke GJ, Langer T, et al. Ugo1 and Mdm30 act sequentially during Fzo1-mediated mitochondrial outer membrane fusion. J Cell Sci. 2011;124(Pt 7):1126–1135. doi: 10.1242/jcs.073080. [DOI] [PubMed] [Google Scholar]
- Anton F, Dittmar G, Langer T, Escobar-Henriques M. Two deubiquitylases act on mitofusin and regulate mitochondrial fusion along independent pathways. Mol Cell. 2013;49:487–498. doi: 10.1016/j.molcel.2012.12.003. [DOI] [PubMed] [Google Scholar]
- Arlt H, Steglich G, Perryman R, Guiard B, Neupert W, Langer T. The formation of respiratory chain complexes in mitochondria is under the proteolytic control of the m-AAA protease. EMBO J. 1998;17:4837–4847. doi: 10.1093/emboj/17.16.4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzu V, Brand MD. Degradation of an intramitochondrial protein by the cytosolic proteasome. J Cell Sci. 2010;123(Pt 4):578–585. doi: 10.1242/jcs.060004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BM, Haynes CM. Mitochondrial protein quality control during biogenesis and aging. Trends Biochem Sci. 2011;36:254–261. doi: 10.1016/j.tibs.2011.01.004. [DOI] [PubMed] [Google Scholar]
- Baker MJ, Tatsuta T, Langer T. Quality control of mitochondrial proteostasis. Cold Spring Harb Perspect Biol. 2011;3:a007559. doi: 10.1101/cshperspect.a007559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker MJ, Mooga VP, Guiard B, Langer T, Ryan MT, Stojanovski D. Impaired folding of the mitochondrial small TIM chaperones induces clearance by the i-AAA protease. J Mol Biol. 2012;424:227–239. doi: 10.1016/j.jmb.2012.09.019. [DOI] [PubMed] [Google Scholar]
- Bateman JM, Iacovino M, Perlman PS, Butow RA. Mitochondrial DNA instability mutants of the bifunctional protein Ilv5p have altered organization in mitochondria and are targeted for degradation by Hsp78 and the Pim1p protease. J Biol Chem. 2002;277:47946–47953. doi: 10.1074/jbc.M209071200. [DOI] [PubMed] [Google Scholar]
- Baxter RV, Ben Othmane K, Rochelle JM, Stajich JE, Hulette C, Dew-Knight S, et al. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat Genet. 2002;30:21–22. doi: 10.1038/ng796. [DOI] [PubMed] [Google Scholar]
- Becker T, Vogtle FN, Stojanovski D, Meisinger C. Sorting and assembly of mitochondrial outer membrane proteins. Biochim Biophys Acta. 2008;1777:557–563. doi: 10.1016/j.bbabio.2008.03.017. [DOI] [PubMed] [Google Scholar]
- Becker T, Gebert M, Pfanner N, van der Laan M. Biogenesis of mitochondrial membrane proteins. Curr Opin Cell Biol. 2009;21:484–493. doi: 10.1016/j.ceb.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Bereiter-Hahn J, Voth M. Dynamics of mitochondria in living cells: shape changes, dislocations, fusion, and fission of mitochondria. Microsc Res Tech. 1994;27:198–219. doi: 10.1002/jemt.1070270303. [DOI] [PubMed] [Google Scholar]
- Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q, et al. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat Cell Biol. 1999;1:298–304. doi: 10.1038/13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldogh IR, Pon LA. Mitochondria on the move. Trends Cell Biol. 2007;17:502–510. doi: 10.1016/j.tcb.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Bonda DJ, Wang X, Perry G, Nunomura A, Tabaton M, Zhu X, et al. Oxidative stress in Alzheimer disease: a possibility for prevention. Neuropharmacology. 2010;59:290–294. doi: 10.1016/j.neuropharm.2010.04.005. [DOI] [PubMed] [Google Scholar]
- Bonn F, Pantakani K, Shoukier M, Langer T, Mannan AU. Functional evaluation of paraplegin mutations by a yeast complementation assay. Hum Mutat. 2010;31:617–621. doi: 10.1002/humu.21226. [DOI] [PubMed] [Google Scholar]
- Bonn F, Tatsuta T, Petrungaro C, Riemer J, Langer T. Presequence-dependent folding ensures MrpL32 processing by the m-AAA protease in mitochondria. EMBO J. 2011;30:2545–2556. doi: 10.1038/emboj.2011.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bota DA, Davies KJ. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat Cell Biol. 2002;4:674–680. doi: 10.1038/ncb836. [DOI] [PubMed] [Google Scholar]
- Bragoszewski P, Gornicka A, Sztolsztener ME, Chacinska A. The ubiquitin-proteasome system regulates mitochondrial intermembrane space proteins. Mol Cell Biol. 2013;33:2136–2148. doi: 10.1128/MCB.01579-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braschi E, Zunino R, McBride HM. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 2009;10:748–754. doi: 10.1038/embor.2009.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- Bross P, Naundrup S, Hansen J, Nielsen MN, Christensen JH, Kruhoffer M, et al. The Hsp60-(p.V98I) mutation associated with hereditary spastic paraplegia SPG13 compromises chaperonin function both in vitro and in vivo. J Biol Chem. 2008;283:15694–15700. doi: 10.1074/jbc.M800548200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagnoli C, Stevanin G, Brussino A, Barberis M, Mancini C, Margolis RL, et al. Missense mutations in the AFG3L2 proteolytic domain account for approximately 1.5% of European autosomal dominant cerebellar ataxias. Hum Mutat. 2010;31:1117–1124. doi: 10.1002/humu.21342. [DOI] [PubMed] [Google Scholar]
- Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–983. doi: 10.1016/s0092-8674(00)81203-9. [DOI] [PubMed] [Google Scholar]
- Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, et al. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- Cassereau J, Chevrollier A, Gueguen N, Desquiret V, Verny C, Nicolas G, et al. Mitochondrial dysfunction and pathophysiology of Charcot-Marie-Tooth disease involving GDAP1 mutations. Exp Neurol. 2011;227:31–41. doi: 10.1016/j.expneurol.2010.09.006. [DOI] [PubMed] [Google Scholar]
- Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, et al. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci U S A. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerveny KL, Jensen RE. The WD-repeats of Net2p interact with Dnm1p and Fis1p to regulate division of mitochondria. Mol Biol Cell. 2003;14:4126–4139. doi: 10.1091/mbc.E03-02-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacinska A, Pfannschmidt S, Wiedemann N, Kozjak V, Sanjuan Szklarz LK, Schulze-Specking A, et al. Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. EMBO J. 2004;23:3735–3746. doi: 10.1038/sj.emboj.7600389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138:628–644. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chada SR, Hollenbeck PJ. Nerve growth factor signaling regulates motility and docking of axonal mitochondria. Curr Biol. 2004;14:1272–1276. doi: 10.1016/j.cub.2004.07.027. [DOI] [PubMed] [Google Scholar]
- Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, et al. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–1737. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CR, Blackstone C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem. 2007;282:21583–21587. doi: 10.1074/jbc.C700083200. [DOI] [PubMed] [Google Scholar]
- Chaturvedi RK, Adhihetty P, Shukla S, Hennessy T, Calingasan N, Yang L, et al. Impaired PGC-1alpha function in muscle in Huntington's disease. Hum Mol Genet. 2009;18:3048–3065. doi: 10.1093/hmg/ddp243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Chen Y, Dorn GW., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta SJ, Andersen JK. Dopaminergic neurons. Int J Biochem Cell Biol. 2005;37:942–946. doi: 10.1016/j.biocel.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, et al. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen T, Kaiser M, Huber R, Ehrmann M. HTRA proteases: regulated proteolysis in protein quality control. Nat Rev Mol Cell Biol. 2011;12:152–162. doi: 10.1038/nrm3065. [DOI] [PubMed] [Google Scholar]
- Cohen MM, Leboucher GP, Livnat-Levanon N, Glickman MH, Weissman AM. Ubiquitin-proteasome-dependent degradation of a mitofusin, a critical regulator of mitochondrial fusion. Mol Biol Cell. 2008;19:2457–2464. doi: 10.1091/mbc.E08-02-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa V, Giacomello M, Hudec R, Lopreiato R, Ermak G, Lim D, et al. Mitochondrial fission and cristae disruption increase the response of cell models of Huntington's disease to apoptotic stimuli. EMBO Mol Med. 2010;2:490–503. doi: 10.1002/emmm.201000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox RT, Spradling AC. Milton controls the early acquisition of mitochondria by Drosophila oocytes. Development. 2006;133:3371–3377. doi: 10.1242/dev.02514. [DOI] [PubMed] [Google Scholar]
- Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8:939–944. doi: 10.1038/sj.embor.7401062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Dagda RK, Cherra SJ, 3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem. 2009;284:13843–13855. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet. 2007;16:2720–2728. doi: 10.1093/hmg/ddm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- Delettre C, Griffoin JM, Kaplan J, Dollfus H, Lorenz B, Faivre L, et al. Mutation spectrum and splicing variants in the OPA1 gene. Hum Genet. 2001;109:584–591. doi: 10.1007/s00439-001-0633-y. [DOI] [PubMed] [Google Scholar]
- Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006;26:9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bella D, Lazzaro F, Brusco A, Plumari M, Battaglia G, Pastore A, et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet. 2010;42:313–321. doi: 10.1038/ng.544. [DOI] [PubMed] [Google Scholar]
- Dolezal P, Likic V, Tachezy J, Lithgow T. Evolution of the molecular machines for protein import into mitochondria. Science. 2006;313:314–318. doi: 10.1126/science.1127895. [DOI] [PubMed] [Google Scholar]
- Dudek J, Rehling P, van der Laan M. Mitochondrial protein import: common principles and physiological networks. Biochim Biophys Acta. 2013;1833:274–285. doi: 10.1016/j.bbamcr.2012.05.028. [DOI] [PubMed] [Google Scholar]
- Duvezin-Caubet S, Koppen M, Wagener J, Zick M, Israel L, Bernacchia A, et al. OPA1 processing reconstituted in yeast depends on the subunit composition of the m-AAA protease in mitochondria. Mol Biol Cell. 2007;18:3582–3590. doi: 10.1091/mbc.E07-02-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol (Berl) 2009;118:5–36. doi: 10.1007/s00401-009-0532-1. [DOI] [PubMed] [Google Scholar]
- Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, Tondera D, et al. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol. 2009;187:1023–1036. doi: 10.1083/jcb.200906084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgass K, Pakay J, Ryan MT, Palmer CS. Recent advances into the understanding of mitochondrial fission. Biochim Biophys Acta. 2013;1833:150–161. doi: 10.1016/j.bbamcr.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Endo T, Yamano K. Multiple pathways for mitochondrial protein traffic. Biol Chem. 2009;390:723–730. doi: 10.1515/BC.2009.087. [DOI] [PubMed] [Google Scholar]
- Escobar-Henriques M, Westermann B, Langer T. Regulation of mitochondrial fusion by the F-box protein Mdm30 involves proteasome-independent turnover of Fzo1. J Cell Biol. 2006;173:645–650. doi: 10.1083/jcb.200512079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exner N, Treske B, Paquet D, Holmstrom K, Schiesling C, Gispert S, et al. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci. 2007;27:12413–12418. doi: 10.1523/JNEUROSCI.0719-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri A, Cozzolino M, Crosio C, Nencini M, Casciati A, Gralla EB, et al. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc Natl Acad Sci U S A. 2006;103:13860–13865. doi: 10.1073/pnas.0605814103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri A, Fiorenzo P, Nencini M, Cozzolino M, Pesaresi MG, Valle C, et al. Glutaredoxin 2 prevents aggregation of mutant SOD1 in mitochondria and abolishes its toxicity. Hum Mol Genet. 2010;19:4529–4542. doi: 10.1093/hmg/ddq383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa-Romero C, Iniguez-Lluhi JA, Stadler J, Chang CR, Arnoult D, Keller PJ, et al. SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. FASEB J. 2009;23:3917–3927. doi: 10.1096/fj.09-136630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer F, Hamann A, Osiewacz HD. Mitochondrial quality control: an integrated network of pathways. Trends Biochem Sci. 2012;37:284–292. doi: 10.1016/j.tibs.2012.02.004. [DOI] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, et al. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- Fransson A, Ruusala A, Aspenstrom P. Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J Biol Chem. 2003;278:6495–6502. doi: 10.1074/jbc.M208609200. [DOI] [PubMed] [Google Scholar]
- Fransson S, Ruusala A, Aspenstrom P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun. 2006;344:500–510. doi: 10.1016/j.bbrc.2006.03.163. [DOI] [PubMed] [Google Scholar]
- Frederick RL, Shaw JM. Moving mitochondria: establishing distribution of an essential organelle. Traffic. 2007;8:1668–1675. doi: 10.1111/j.1600-0854.2007.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Fritz S, Weinbach N, Westermann B. Mdm30 is an F-box protein required for maintenance of fusion-competent mitochondria in yeast. Mol Biol Cell. 2003;14:2303–2313. doi: 10.1091/mbc.E02-12-0831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, et al. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell. 2009;33:627–638. doi: 10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. 2008;19:2402–2412. doi: 10.1091/mbc.E07-12-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebert N, Ryan MT, Pfanner N, Wiedemann N, Stojanovski D. Mitochondrial protein import machineries and lipids: a functional connection. Biochim Biophys Acta. 2011;1808:1002–1011. doi: 10.1016/j.bbamem.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19:4861–4870. doi: 10.1093/hmg/ddq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol. 2006;173:545–557. doi: 10.1083/jcb.200601067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin EE, Graumann J, Chan DC. The WD40 protein Caf4p is a component of the mitochondrial fission machinery and recruits Dnm1p to mitochondria. J Cell Biol. 2005;170:237–248. doi: 10.1083/jcb.200503148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griparic L, Kanazawa T, van der Bliek AM. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol. 2007;178:757–764. doi: 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986;261:6084–6089. [PubMed] [Google Scholar]
- Gschwind M, Huber G. Apoptotic cell death induced by beta-amyloid 1–42 peptide is cell type dependent. J Neurochem. 1995;65:292–300. doi: 10.1046/j.1471-4159.1995.65010292.x. [DOI] [PubMed] [Google Scholar]
- Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y, Tomizawa K, et al. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol. 2008;182:573–585. doi: 10.1083/jcb.200802164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JJ, Durr A, Cournu-Rebeix I, Georgopoulos C, Ang D, Nielsen MN, et al. Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am J Hum Genet. 2002;70:1328–1332. doi: 10.1086/339935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder Z, Zunino R, McBride H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol. 2004;14:340–345. doi: 10.1016/j.cub.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Harding AE. Hereditary spastic paraplegias. Semin Neurol. 1993;13:333–336. doi: 10.1055/s-2008-1041143. [DOI] [PubMed] [Google Scholar]
- Hawlitschek G, Schneider H, Schmidt B, Tropschug M, Hartl FU, Neupert W. Mitochondrial protein import: identification of processing peptidase and of PEP, a processing enhancing protein. Cell. 1988;53:795–806. doi: 10.1016/0092-8674(88)90096-7. [DOI] [PubMed] [Google Scholar]
- Haynes CM, Ron D. The mitochondrial UPR – protecting organelle protein homeostasis. J Cell Sci. 2010;123(Pt 22):3849–3855. doi: 10.1242/jcs.075119. [DOI] [PubMed] [Google Scholar]
- Haynes CM, Petrova K, Benedetti C, Yang Y, Ron D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell. 2007;13:467–480. doi: 10.1016/j.devcel.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Haynes CM, Yang Y, Blais SP, Neubert TA, Ron D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell. 2010;37:529–540. doi: 10.1016/j.molcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol. 2009;187:959–966. doi: 10.1083/jcb.200906083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn SR, Thomenius MJ, Johnson ES, Freel CD, Wu JQ, Coloff JL, et al. Regulation of mitochondrial morphology by APC/CCdh1-mediated control of Drp1 stability. Mol Biol Cell. 2011;22:1207–1216. doi: 10.1091/mbc.E10-07-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Ingerman E, Perkins EM, Marino M, Mears JA, McCaffery JM, Hinshaw JE, et al. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol. 2005;170:1021–1027. doi: 10.1083/jcb.200506078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irrcher I, Aleyasin H, Seifert EL, Hewitt SJ, Chhabra S, Phillips M, et al. Loss of the Parkinson's disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum Mol Genet. 2010;19:3734–3746. doi: 10.1093/hmg/ddq288. [DOI] [PubMed] [Google Scholar]
- Ishihara N, Eura Y, Mihara K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci. 2004;117(Pt 26):6535–6546. doi: 10.1242/jcs.01565. [DOI] [PubMed] [Google Scholar]
- Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25:2966–2977. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem. 2003;278:36373–36379. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- Jeon HB, Choi ES, Yoon JH, Hwang JH, Chang JW, Lee EK, et al. A proteomics approach to identify the ubiquitinated proteins in mouse heart. Biochem Biophys Res Commun. 2007;357:731–736. doi: 10.1016/j.bbrc.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JM, Datta P, Srinivasula SM, Ji W, Gupta S, Zhang Z, et al. Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature. 2003;425:721–727. doi: 10.1038/nature02052. [DOI] [PubMed] [Google Scholar]
- Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, et al. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008;132:137–148. doi: 10.1016/j.cell.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki T, Wang K, Baba M, Bartholomew CR, Lynch-Day MA, Du Z, et al. A genomic screen for yeast mutants defective in selective mitochondria autophagy. Mol Biol Cell. 2009;20:4730–4738. doi: 10.1091/mbc.E09-03-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Youle RJ. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr Opin Cell Biol. 2011;23:476–482. doi: 10.1016/j.ceb.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Neutzner A, Youle RJ. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol. 2007;178:71–84. doi: 10.1083/jcb.200611064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaser M, Kambacheld M, Kisters-Woike B, Langer T. Oma1, a novel membrane-bound metallopeptidase in mitochondria with activities overlapping with the m-AAA protease. J Biol Chem. 2003;278:46414–46423. doi: 10.1074/jbc.M305584200. [DOI] [PubMed] [Google Scholar]
- Khalimonchuk O, Jeong MY, Watts T, Ferris E, Winge DR. Selective Oma1 protease-mediated proteolysis of Cox1 subunit of cytochrome oxidase in assembly mutants. J Biol Chem. 2012;287:7289–7300. doi: 10.1074/jbc.M111.313148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DY, Kim KK. Structure and function of HtrA family proteins, the key players in protein quality control. J Biochem Mol Biol. 2005;38:266–274. doi: 10.5483/bmbrep.2005.38.3.266. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Koehler CM. Protein translocation pathways of the mitochondrion. FEBS Lett. 2000;476:27–31. doi: 10.1016/s0014-5793(00)01664-1. [DOI] [PubMed] [Google Scholar]
- Koeppen AH. The pathogenesis of spinocerebellar ataxia. Cerebellum. 2005;4:62–73. doi: 10.1080/14734220510007950. [DOI] [PubMed] [Google Scholar]
- Koirala S, Bui HT, Schubert HL, Eckert DM, Hill CP, Kay MS, et al. Molecular architecture of a dynamin adaptor: implications for assembly of mitochondrial fission complexes. J Cell Biol. 2010;191:1127–1139. doi: 10.1083/jcb.201005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koirala S, Guo Q, Kalia R, Bui HT, Eckert DM, Frost A, et al. Interchangeable adaptors regulate mitochondrial dynamin assembly for membrane scission. Proc Natl Acad Sci U S A. 2013;110:E1342–E1351. doi: 10.1073/pnas.1300855110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppen M, Metodiev MD, Casari G, Rugarli EI, Langer T. Variable and tissue-specific subunit composition of mitochondrial m-AAA protease complexes linked to hereditary spastic paraplegia. Mol Cell Biol. 2007;27:758–767. doi: 10.1128/MCB.01470-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel D, Wurth S, Kaser M, Langer T. Membrane protein turnover by the m-AAA protease in mitochondria depends on the transmembrane domains of its subunits. EMBO Rep. 2004;5:698–703. doi: 10.1038/sj.embor.7400186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–862. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- Koutsopoulos OS, Laine D, Osellame L, Chudakov DM, Parton RG, Frazier AE, et al. Human Miltons associate with mitochondria and induce microtubule-dependent remodeling of mitochondrial networks. Biochim Biophys Acta. 2010;1803:564–574. doi: 10.1016/j.bbamcr.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Kruger R, Sharma M, Riess O, Gasser T, Van Broeckhoven C, Theuns J, et al. A large-scale genetic association study to evaluate the contribution of Omi/HtrA2 (PARK13) to Parkinson's disease. Neurobiol Aging. 2011;32:548.e9–548.e18. doi: 10.1016/j.neurobiolaging.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner LL, Horner JS, Nunnari J. Mechanistic analysis of a dynamin effector. Science. 2009;325:874–877. doi: 10.1126/science.1176921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CW, Peng HB. The function of mitochondria in presynaptic development at the neuromuscular junction. Mol Biol Cell. 2008;19:150–158. doi: 10.1091/mbc.E07-05-0515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–5011. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legesse-Miller A, Massol RH, Kirchhausen T. Constriction and Dnm1p recruitment are distinct processes in mitochondrial fission. Mol Biol Cell. 2003;14:1953–1963. doi: 10.1091/mbc.E02-10-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhard K, Herrmann JM, Stuart RA, Mannhaupt G, Neupert W, Langer T. AAA proteases with catalytic sites on opposite membrane surfaces comprise a proteolytic system for the ATP-dependent degradation of inner membrane proteins in mitochondria. EMBO J. 1996;15:4218–4229. [PMC free article] [PubMed] [Google Scholar]
- Li Q, Vande Velde C, Israelson A, Xie J, Bailey AO, Dong MQ, et al. ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc Natl Acad Sci U S A. 2010;107:21146–21151. doi: 10.1073/pnas.1014862107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Bengtson MH, Ulbrich A, Matsuda A, Reddy VA, Orth A, et al. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle's dynamics and signaling. PLoS One. 2008;3:e1487. doi: 10.1371/journal.pone.0001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Lin CH, Chen ML, Chen GS, Tai CH, Wu RM. Novel variant Pro143Ala in HTRA2 contributes to Parkinson's disease by inducing hyperphosphorylation of HTRA2 protein in mitochondria. Hum Genet. 2011;130:817–827. doi: 10.1007/s00439-011-1041-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livnat-Levanon N, Glickman MH. Ubiquitin-proteasome system and mitochondria – reciprocity. Biochim Biophys Acta. 2011;1809:80–87. doi: 10.1016/j.bbagrm.2010.07.005. [DOI] [PubMed] [Google Scholar]
- Loson OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013;24:659–667. doi: 10.1091/mbc.E12-10-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magen D, Georgopoulos C, Bross P, Ang D, Segev Y, Goldsher D, et al. Mitochondrial hsp60 chaperonopathy causes an autosomal-recessive neurodegenerative disorder linked to brain hypomyelination and leukodystrophy. Am J Hum Genet. 2008;83:30–42. doi: 10.1016/j.ajhg.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- Manczak M, Reddy PH. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer's disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet. 2012;21:2538–2547. doi: 10.1093/hmg/dds072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet. 2011;20:2495–2509. doi: 10.1093/hmg/ddr139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margineantu DH, Emerson CB, Diaz D, Hockenbery DM. Hsp90 inhibition decreases mitochondrial protein turnover. PLoS One. 2007;2:e1066. doi: 10.1371/journal.pone.0001066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JB, Gusella JF. Huntington's disease. Pathogenesis and management. N Engl J Med. 1986;315:1267–1276. doi: 10.1056/NEJM198611133152006. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Gertz B, Pan Y, Price AC, Molkentin JD, Chang Q. The mitochondrial permeability transition pore in motor neurons: involvement in the pathobiology of ALS mice. Exp Neurol. 2009;218:333–346. doi: 10.1016/j.expneurol.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins LM, Morrison A, Klupsch K, Fedele V, Moisoi N, Teismann P, et al. Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol Cell Biol. 2004;24:9848–9862. doi: 10.1128/MCB.24.22.9848-9862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears JA, Lackner LL, Fang S, Ingerman E, Nunnari J, Hinshaw JE. Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat Struct Mol Biol. 2011;18:20–26. doi: 10.1038/nsmb.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeusen S, McCaffery JM, Nunnari J. Mitochondrial fusion intermediates revealed in vitro. Science. 2004;305:1747–1752. doi: 10.1126/science.1100612. [DOI] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP. Axonal mitochondrial transport and potential are correlated. J Cell Sci. 2004;117(Pt 13):2791–2804. doi: 10.1242/jcs.01130. [DOI] [PubMed] [Google Scholar]
- Mokranjac D, Neupert W. The many faces of the mitochondrial TIM23 complex. Biochim Biophys Acta. 2010;1797:1045–1054. doi: 10.1016/j.bbabio.2010.01.026. [DOI] [PubMed] [Google Scholar]
- Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, et al. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell. 2010;142:889–901. doi: 10.1016/j.cell.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozdy AD, McCaffery JM, Shaw JM. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol. 2000;151:367–380. doi: 10.1083/jcb.151.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Kimura Y, Tokuda M, Honda S, Hirose S. MARCH-V is a novel mitofusin 2-and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 2006;7:1019–1022. doi: 10.1038/sj.embor.7400790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Cieplak P, Cho DH, Godzik A, Lipton SA. S-nitrosylation of Drp1 links excessive mitochondrial fission to neuronal injury in neurodegeneration. Mitochondrion. 2010;10:573–578. doi: 10.1016/j.mito.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nangaku M, Sato-Yoshitake R, Okada Y, Noda Y, Takemura R, Yamazaki H, et al. KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell. 1994;79:1209–1220. doi: 10.1016/0092-8674(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Naoe M, Ohwa Y, Ishikawa D, Ohshima C, Nishikawa S, Yamamoto H, et al. Identification of Tim40 that mediates protein sorting to the mitochondrial intermembrane space. J Biol Chem. 2004;279:47815–47821. doi: 10.1074/jbc.M410272200. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010b;6:1090–1106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010a;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neupert W, Herrmann JM. Translocation of proteins into mitochondria. Annu Rev Biochem. 2007;76:723–749. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- Nolden M, Ehses S, Koppen M, Bernacchia A, Rugarli EI, Langer T. The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell. 2005;123:277–289. doi: 10.1016/j.cell.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Nowikovsky K, Reipert S, Devenish RJ, Schweyen RJ. Mdm38 protein depletion causes loss of mitochondrial K+/H+ exchange activity, osmotic swelling and mitophagy. Cell Death Differ. 2007;14:1647–1656. doi: 10.1038/sj.cdd.4402167. [DOI] [PubMed] [Google Scholar]
- Ohno N, Kidd GJ, Mahad D, Kiryu-Seo S, Avishai A, Komuro H, et al. Myelination and axonal electrical activity modulate the distribution and motility of mitochondria at CNS nodes of Ranvier. J Neurosci. 2011;31:7249–7258. doi: 10.1523/JNEUROSCI.0095-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Shaw JM. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu Rev Genet. 2005;39:503–536. doi: 10.1146/annurev.genet.38.072902.093019. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell. 2009;17:87–97. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003;278:7743–7746. doi: 10.1074/jbc.C200677200. [DOI] [PubMed] [Google Scholar]
- Osellame LD, Blacker TS, Duchen MR. Cellular and molecular mechanisms of mitochondrial function. Best Pract Res Clin Endocrinol Metab. 2012;26:711–723. doi: 10.1016/j.beem.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman C, Wilmes C, Tatsuta T, Langer T. Prohibitins interact genetically with Atp23, a novel processing peptidase and chaperone for the F1Fo-ATP synthase. Mol Biol Cell. 2007;18:627–635. doi: 10.1091/mbc.E06-09-0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191:1141–1158. doi: 10.1083/jcb.201007152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuga D, Keegan BR, Brisch E, Thatcher JW, Hermann GJ, Bleazard W, et al. The dynamin-related GTPase, Dnm1p, controls mitochondrial morphology in yeast. J Cell Biol. 1998;143:333–349. doi: 10.1083/jcb.143.2.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011;12:565–573. doi: 10.1038/embor.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasinelli P, Houseweart MK, Brown RH, Jr, Cleveland DW. Caspase-1 and-3 are sequentially activated in motor neuron death in Cu,Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2000;97:13901–13906. doi: 10.1073/pnas.240305897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, et al. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43:19–30. doi: 10.1016/j.neuron.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, et al. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol. 2003;21:921–926. doi: 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- Pilling AD, Horiuchi D, Lively CM, Saxton WM. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell. 2006;17:2057–2068. doi: 10.1091/mbc.E05-06-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potting C, Wilmes C, Engmann T, Osman C, Langer T. Regulation of mitochondrial phospholipids by Ups1/PRELI-like proteins depends on proteolysis and Mdm35. EMBO J. 2010;29:2888–2898. doi: 10.1038/emboj.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokisch H, Scharfe C, Camp DG, 2nd, Xiao W, David L, Andreoli C, et al. Integrative analysis of the mitochondrial proteome in yeast. PLoS Biol. 2004;2:e160. doi: 10.1371/journal.pbio.0020160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiros PM, Ramsay AJ, Sala D, Fernandez-Vizarra E, Rodriguez F, Peinado JR, et al. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J. 2012;31:2117–2133. doi: 10.1038/emboj.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radke S, Chander H, Schafer P, Meiss G, Kruger R, Schulz JB, et al. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J Biol Chem. 2008;283:12681–12685. doi: 10.1074/jbc.C800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, Mao P, Manczak M. Mitochondrial structural and functional dynamics in Huntington's disease. Brain Res Rev. 2009;61:33–48. doi: 10.1016/j.brainresrev.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehling P, Brandner K, Pfanner N. Mitochondrial import and the twin-pore translocase. Nat Rev Mol Cell Biol. 2004;5:519–530. doi: 10.1038/nrm1426. [DOI] [PubMed] [Google Scholar]
- Reid E. Pure hereditary spastic paraplegia. J Med Genet. 1997;34:499–503. doi: 10.1136/jmg.34.6.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemer J, Fischer M, Herrmann JM. Oxidation-driven protein import into mitochondria: insights and blind spots. Biochim Biophys Acta. 2011;1808:981–989. doi: 10.1016/j.bbamem.2010.06.003. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Enriquez S, Kim I, Currin RT, Lemasters JJ. Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy. 2006;2:39–46. doi: 10.4161/auto.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland LP. Looking for the cause of amyotrophic lateral sclerosis. N Engl J Med. 1984;311:979–981. doi: 10.1056/NEJM198410113111509. [DOI] [PubMed] [Google Scholar]
- Russo GJ, Louie K, Wellington A, Macleod GT, Hu F, Panchumarthi S, et al. Drosophila Miro is required for both anterograde and retrograde axonal mitochondrial transport. J Neurosci. 2009;29:5443–5455. doi: 10.1523/JNEUROSCI.5417-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, et al. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A. 2008;105:20728–20733. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–376. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt M, Neupert W, Langer T. The molecular chaperone Hsp78 confers compartment-specific thermotolerance to mitochondria. J Cell Biol. 1996;134:1375–1386. doi: 10.1083/jcb.134.6.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schols L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3:291–304. doi: 10.1016/S1474-4422(04)00737-9. [DOI] [PubMed] [Google Scholar]
- Sesaki H, Jensen RE. UGO1 encodes an outer membrane protein required for mitochondrial fusion. J Cell Biol. 2001;152:1123–1134. doi: 10.1083/jcb.152.6.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesaki H, Southard SM, Yaffe MP, Jensen RE. Mgm1p, a dynamin-related GTPase, is essential for fusion of the mitochondrial outer membrane. Mol Biol Cell. 2003;14:2342–2356. doi: 10.1091/mbc.E02-12-0788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen T, Zheng M, Cao C, Chen C, Tang J, Zhang W, et al. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J Biol Chem. 2007;282:23354–23361. doi: 10.1074/jbc.M702657200. [DOI] [PubMed] [Google Scholar]
- Shi G, Lee JR, Grimes DA, Racacho L, Ye D, Yang H, et al. Functional alteration of PARL contributes to mitochondrial dysregulation in Parkinson's disease. Hum Mol Genet. 2011;20:1966–1974. doi: 10.1093/hmg/ddr077. [DOI] [PubMed] [Google Scholar]
- Shirendeb U, Reddy AP, Manczak M, Calkins MJ, Mao P, Tagle DA, et al. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington's disease: implications for selective neuronal damage. Hum Mol Genet. 2011;20:1438–1455. doi: 10.1093/hmg/ddr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmann A, Reinders J, Wagner Y, Joppich C, Zahedi R, Meyer HE, et al. The proteome of Saccharomyces cerevisiae mitochondria. Proc Natl Acad Sci U S A. 2003;100:13207–13212. doi: 10.1073/pnas.2135385100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon-Sanchez J, Singleton AB. Sequencing analysis of OMI/HTRA2 shows previously reported pathogenic mutations in neurologically normal controls. Hum Mol Genet. 2008;17:1988–1993. doi: 10.1093/hmg/ddn096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Chen J, Petrilli A, Liot G, Klinglmayr E, Zhou Y, et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat Med. 2011;17:377–382. doi: 10.1038/nm.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell. 2009;20:3525–3532. doi: 10.1091/mbc.E09-03-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratt DE, Martinez-Torres RJ, Noh YJ, Mercier P, Manczyk N, Barber KR, et al. A molecular explanation for the recessive nature of parkin-linked Parkinson's disease. Nat Commun. 2013;4:1983. doi: 10.1038/ncomms2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Spronsen M, Mikhaylova M, Lipka J, Schlager MA, van den Heuvel DJ, Kuijpers M, et al. TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron. 2013;77:485–502. doi: 10.1016/j.neuron.2012.11.027. [DOI] [PubMed] [Google Scholar]
- Sterky FH, Lee S, Wibom R, Olson L, Larsson NG. Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc Natl Acad Sci U S A. 2011;108:12937–12942. doi: 10.1073/pnas.1103295108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiburek L, Cesnekova J, Kostkova O, Fornuskova D, Vinsova K, Wenchich L, et al. YME1L controls the accumulation of respiratory chain subunits and is required for apoptotic resistance, cristae morphogenesis, and cell proliferation. Mol Biol Cell. 2012;23:1010–1023. doi: 10.1091/mbc.E11-08-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojanovski D, Koutsopoulos OS, Okamoto K, Ryan MT. Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J Cell Sci. 2004;117(Pt 7):1201–1210. doi: 10.1242/jcs.01058. [DOI] [PubMed] [Google Scholar]
- Stojanovski D, Bragoszewski P, Chacinska A. The MIA pathway: a tight bond between protein transport and oxidative folding in mitochondria. Biochim Biophys Acta. 2012;1823:1142–1150. doi: 10.1016/j.bbamcr.2012.04.014. [DOI] [PubMed] [Google Scholar]
- Strauss KM, Martins LM, Plun-Favreau H, Marx FP, Kautzmann S, Berg D, et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson's disease. Hum Mol Genet. 2005;14:2099–2111. doi: 10.1093/hmg/ddi215. [DOI] [PubMed] [Google Scholar]
- Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev. 2008;22:1577–1590. doi: 10.1101/gad.1658508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugioka R, Shimizu S, Tsujimoto Y. Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J Biol Chem. 2004;279:52726–52734. doi: 10.1074/jbc.M408910200. [DOI] [PubMed] [Google Scholar]
- Sugiura A, Nagashima S, Tokuyama T, Amo T, Matsuki Y, Ishido S, et al. MITOL Regulates Endoplasmic Reticulum-Mitochondria Contacts via Mitofusin2. Mol Cell. 2013;51:20–34. doi: 10.1016/j.molcel.2013.04.023. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Takahashi-Niki K, Akagi T, Hashikawa T, Takahashi R. Mitochondrial protease Omi/HtrA2 enhances caspase activation through multiple pathways. Cell Death Differ. 2004;11:208–216. doi: 10.1038/sj.cdd.4401343. [DOI] [PubMed] [Google Scholar]
- Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Sugiura Y, Ichishita R, Mihara K, Oka T. KLP6: a newly identified kinesin that regulates the morphology and transport of mitochondria in neuronal cells. J Cell Sci. 2011;124(Pt 14):2457–2465. doi: 10.1242/jcs.086470. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, et al. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. 1998;93:1147–1158. doi: 10.1016/s0092-8674(00)81459-2. [DOI] [PubMed] [Google Scholar]
- Tatsuta T, Langer T. Studying proteolysis within mitochondria. Methods Mol Biol. 2007;372:343–360. doi: 10.1007/978-1-59745-365-3_25. [DOI] [PubMed] [Google Scholar]
- Tatsuta T, Langer T. AAA proteases in mitochondria: diverse functions of membrane-bound proteolytic machines. Res Microbiol. 2009;160:711–717. doi: 10.1016/j.resmic.2009.09.005. [DOI] [PubMed] [Google Scholar]
- Taylor EB, Rutter J. Mitochondrial quality control by the ubiquitin-proteasome system. Biochem Soc Trans. 2011;39:1509–1513. doi: 10.1042/BST0391509. [DOI] [PubMed] [Google Scholar]
- Terziyska N, Lutz T, Kozany C, Mokranjac D, Mesecke N, Neupert W, et al. Mia40, a novel factor for protein import into the intermembrane space of mitochondria is able to bind metal ions. FEBS Lett. 2005;579:179–184. doi: 10.1016/j.febslet.2004.11.072. [DOI] [PubMed] [Google Scholar]
- Tieu Q, Okreglak V, Naylor K, Nunnari J. The WD repeat protein, Mdv1p, functions as a molecular adaptor by interacting with Dnm1p and Fis1p during mitochondrial fission. J Cell Biol. 2002;158:445–452. doi: 10.1083/jcb.200205031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009;28:1589–1600. doi: 10.1038/emboj.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD. The molecular motor toolbox for intracellular transport. Cell. 2003;112:467–480. doi: 10.1016/s0092-8674(03)00111-9. [DOI] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Vande Walle L, Lamkanfi M, Vandenabeele P. The mitochondrial serine protease HtrA2/Omi: an overview. Cell Death Differ. 2008;15:453–460. doi: 10.1038/sj.cdd.4402291. [DOI] [PubMed] [Google Scholar]
- Varabyova A, Stojanovski D, Chacinska A. Mitochondrial protein homeostasis. IUBMB Life. 2013;65:191–201. doi: 10.1002/iub.1122. [DOI] [PubMed] [Google Scholar]
- Verburg J, Hollenbeck PJ. Mitochondrial membrane potential in axons increases with local nerve growth factor or semaphorin signaling. J Neurosci. 2008;28:8306–8315. doi: 10.1523/JNEUROSCI.2614-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–378. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Vogtle FN, Wortelkamp S, Zahedi RP, Becker D, Leidhold C, Gevaert K, et al. Global analysis of the mitochondrial N-proteome identifies a processing peptidase critical for protein stability. Cell. 2009;139:428–439. doi: 10.1016/j.cell.2009.07.045. [DOI] [PubMed] [Google Scholar]
- Vogtle FN, Prinz C, Kellermann J, Lottspeich F, Pfanner N, Meisinger C. Mitochondrial protein turnover: role of the precursor intermediate peptidase Oct1 in protein stabilization. Mol Biol Cell. 2011;22:2135–2143. doi: 10.1091/mbc.E11-02-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voos W. Chaperone-protease networks in mitochondrial protein homeostasis. Biochim Biophys Acta. 2013;1833:388–399. doi: 10.1016/j.bbamcr.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Voos W, Ward LA, Truscott KN. The role of AAA+ proteases in mitochondrial protein biogenesis, homeostasis and activity control. Subcell Biochem. 2013;66:223–263. doi: 10.1007/978-94-007-5940-4_9. [DOI] [PubMed] [Google Scholar]
- Vos M, Esposito G, Edirisinghe JN, Vilain S, Haddad DM, Slabbaert JR, et al. Vitamin K2 is a mitochondrial electron carrier that rescues pink1 deficiency. Science. 2012;336:1306–1310. doi: 10.1126/science.1218632. [DOI] [PubMed] [Google Scholar]
- Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, et al. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. 2009;186:805–816. doi: 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CY, Xu Q, Weng L, Zhang Q, Zhang HN, Guo JF, et al. Genetic variations of Omi/HTRA2 in Chinese patients with Parkinson's disease. Brain Res. 2011a;1385:293–297. doi: 10.1016/j.brainres.2011.02.037. [DOI] [PubMed] [Google Scholar]
- Wang H, Song P, Du L, Tian W, Yue W, Liu M, et al. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J Biol Chem. 2011b;286:11649–11658. doi: 10.1074/jbc.M110.144238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, et al. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A. 2008;105:19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, et al. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasiak S, Zunino R, McBride HM. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol. 2007;177:439–450. doi: 10.1083/jcb.200610042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wanders RJ, Leonard JV. A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med. 2007;356:1736–1741. doi: 10.1056/NEJMoa064436. [DOI] [PubMed] [Google Scholar]
- Weckbecker D, Longen S, Riemer J, Herrmann JM. Atp23 biogenesis reveals a chaperone-like folding activity of Mia40 in the IMS of mitochondria. EMBO J. 2012;31:4348–4358. doi: 10.1038/emboj.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B. Mitochondrial dynamics in model organisms: what yeasts, worms and flies have taught us about fusion and fission of mitochondria. Semin Cell Dev Biol. 2010;21:542–549. doi: 10.1016/j.semcdb.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Wong ED, Wagner JA, Gorsich SW, McCaffery JM, Shaw JM, Nunnari J. The dynamin-related GTPase, Mgm1p, is an intermembrane space protein required for maintenance of fusion competent mitochondria. J Cell Biol. 2000;151:341–352. doi: 10.1083/jcb.151.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ED, Wagner JA, Scott SV, Okreglak V, Holewinske TJ, Cassidy-Stone A, et al. The intramitochondrial dynamin-related GTPase, Mgm1p, is a component of a protein complex that mediates mitochondrial fusion. J Cell Biol. 2003;160:303–311. doi: 10.1083/jcb.200209015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L, Fletcher GC, Tolkovsky AM. Mitochondria are selectively eliminated from eukaryotic cells after blockade of caspases during apoptosis. Curr Biol. 2001;11:361–365. doi: 10.1016/s0960-9822(01)00100-2. [DOI] [PubMed] [Google Scholar]
- Yaffe MP. Dynamic mitochondria. Nat Cell Biol. 1999;1:E149–E150. doi: 10.1038/14101. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, et al. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A. 2008;105:7070–7075. doi: 10.1073/pnas.0711845105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonashiro R, Ishido S, Kyo S, Fukuda T, Goto E, Matsuki Y, et al. A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J. 2006;25:3618–3626. doi: 10.1038/sj.emboj.7601249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. 2003;23:5409–5420. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Zanna C, Ghelli A, Porcelli AM, Karbowski M, Youle RJ, Schimpf S, et al. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain. 2008;131(Pt 2):352–367. doi: 10.1093/brain/awm335. [DOI] [PubMed] [Google Scholar]
- Zeng X, Neupert W, Tzagoloff A. The metalloprotease encoded by ATP23 has a dual function in processing and assembly of subunit 6 of mitochondrial ATPase. Mol Biol Cell. 2007;18:617–626. doi: 10.1091/mbc.E06-09-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Takita J, Tanaka Y, Setou M, Nakagawa T, Takeda S, et al. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell. 2001;105:587–597. doi: 10.1016/s0092-8674(01)00363-4. [DOI] [PubMed] [Google Scholar]
- Zhao J, Liu T, Jin S, Wang X, Qu M, Uhlen P, et al. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011;30:2762–2778. doi: 10.1038/emboj.2011.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Perry G, Smith MA, Wang X. Abnormal mitochondrial dynamics in the pathogenesis of Alzheimer's disease. J Alzheimers Dis. 2013;33(Suppl 1):S253–S262. doi: 10.3233/JAD-2012-129005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- Zuchner S, De Jonghe P, Jordanova A, Claeys KG, Guergueltcheva V, Cherninkova S, et al. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann Neurol. 2006;59:276–281. doi: 10.1002/ana.20797. [DOI] [PubMed] [Google Scholar]
- Zunino R, Braschi E, Xu L, McBride HM. Translocation of SenP5 from the nucleoli to the mitochondria modulates DRP1-dependent fission during mitosis. J Biol Chem. 2009;284:17783–17795. doi: 10.1074/jbc.M901902200. [DOI] [PMC free article] [PubMed] [Google Scholar]