

Abstract
The autophagic-lysosomal degradation pathway is critical for cardiac homeostasis, and defects in this pathway are associated with development of cardiomyopathy. Autophagy is responsible for the normal turnover of organelles and long-lived proteins. Autophagy is also rapidly up-regulated in response to stress, where it rapidly clears dysfunctional organelles and cytotoxic protein aggregates in the cell. Autophagy is also important in clearing dysfunctional mitochondria before they can cause harm to the cell. This quality control mechanism is particularly important in cardiac myocytes, which contain a very high volume of mitochondria. The degradation of proteins and organelles also generates free fatty acids and amino acids, which help maintain energy levels in myocytes during stress conditions. Increases in autophagy have been observed in various cardiovascular diseases, but a major question that remains to be answered is whether enhanced autophagy is an adaptive or maladaptive response to stress. This review discusses the regulation and role of autophagy in the myocardium under baseline conditions and in various aetiologies of heart disease. It also discusses whether this pathway represents a new therapeutic target to treat or prevent cardiovascular disease and the concerns associated with modulating autophagy.
Linked Articles
This article is part of a themed issue on Mitochondrial Pharmacology: Energy, Injury & Beyond. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2014.171.issue-8
Keywords: autophagy, BNIP3, heart, mitochondria, mitophagy, Parkin
Introduction
Autophagy is an evolutionarily conserved process concerned with the degradation of long-lived proteins and damaged or excess organelles. During this process, a double-membrane autophagosome sequesters cytoplasmic material and subsequently fuses with a lysosome, thereby degrading the contents (Choi et al., 2013). This constitutive process was originally believed to be a non-selective mechanism for removal of cytosolic material. However, it is now known that autophagosomes can specifically target bacteria (Knodler and Celli, 2011), protein aggregates (Tannous et al., 2008) and organelles such as peroxisomes (Iwata et al., 2006), endoplasmic reticulum (Bernales et al., 2007) and mitochondria (Quinsay et al., 2010) in different mammalian cells. The autophagic-lysosomal pathway is essential for normal cardiac function, and a disruption in this pathway has severe consequences for the heart. Disruption leads to accumulation of protein aggregates and dysfunctional organelles, resulting in cellular dysfunction and heart failure (Nishino et al., 2000; Tanaka et al., 2000; Nakai et al., 2007). During stress, this pathway is quickly activated and removes cytotoxic protein aggregates and damaged organelles (Hamacher-Brady et al., 2006; Nakai et al., 2007; Tannous et al., 2008; Kubli et al., 2013). Autophagy also serves to preserve energy status during energy deprivation. The degradation of proteins and organelles generates fatty acids and amino acids that are used for ATP production, thereby promoting cardiomyocyte survival (Kanamori et al., 2009; 2011,).
Increases in autophagy have been observed in various cardiovascular diseases and a major question that remains to be answered is whether autophagy is an adaptive or maladaptive response to stress. Although cardiac autophagy is a critical quality control mechanism, increased autophagy can have deleterious effects on the heart under some circumstances. Excessive activation of autophagy may deplete essential molecules and organelles responsible for cellular survival. Thus, tightly controlled regulation of autophagy is important for maintaining cardiac homeostasis, and dysregulation of this pathway resulting in either excessive or insufficient autophagy will have deleterious effects. This review discusses the regulation and role of autophagy and mitochondrial autophagy (mitophagy) in the myocardium. It also addresses the therapeutic potential of this pathway and concerns associated with modulating autophagy.
Overview of autophagy
Formation of the autophagosome begins with the creation of a precursor membrane, or phagophore (Figure 1). Lipid sources for the phagophore membrane are still unclear, but mounting evidence suggests that the endoplasmic reticulum (Hayashi-Nishino et al., 2009), plasma membrane (Ravikumar et al., 2010) and mitochondria (Hailey et al., 2010) are all likely sources. Formation of the autophagosome is regulated by Atg proteins (see Mizushima et al., 2011). Initiation of autophagy begins with Beclin 1/Atg6 forming a complex with vacuolar protein sorting (VPS) 34 and VPS15, resulting in activation of this class III PI3K complex and nucleation of the isolation membrane or phagophore (Kihara et al., 2001; Zeng et al., 2006). Elongation of the phagophore involves two ubiquitin-like conjugation systems, Atg12-Atg5 and Atg8/LC3 (microtubule-associated protein 1 light chain 3). The resulting mature LC3-II resides on the autophagosome membrane and interacts with specific adapter proteins or receptors that are present on the cargo to be degraded (Geisler et al., 2010; Novak et al., 2010). Finally, the autophagosome fuses with a lysosome, and the contents are degraded in the resulting autophagolysosome by acid hydrolase enzymes (Choi et al., 2013). The term autophagic flux is used to describe the dynamic process of autophagosome synthesis, sequestration of material, delivery of autophagic cargo to the lysosome and degradation of autophagic cargo inside the lysosome. Assessing autophagic flux experimentally is a more accurate indicator of autophagic activity in cells and tissues than measurements of autophagosome numbers. An increase in the number of autophagosomes at a given time does not always correspond to increased autophagic activity. It can represent either induction of autophagy or defects in the fusion between autophagosomes and lysosomes.
Figure 1.
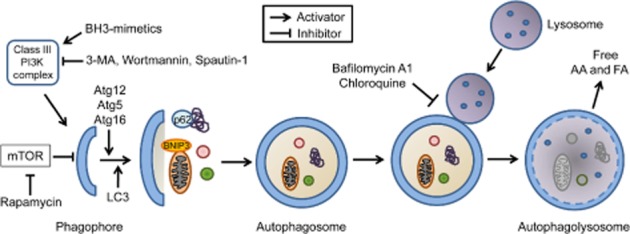
Autophagic flux. Autophagy is initiated upon inhibition of mTOR and activation of a class III PI3K complex, which consists in part of Beclin 1 and VPS34. Elongation of the phagophore and maturation of the autophagosome requires two ubiquitin-like conjugation systems, Atg12 and Atg8/LC3. The forming autophagosomal membrane sequesters material in the cytosol that has been marked for degradation by proteins such as BNIP3 and p62. The mature autophagosome then fuses with a lysosome to form the autophagolysosome. The cargo and the inner membrane of the autophagosome are degraded by lysosomal hydrolases. The degradation generates free amino acids (AA) and fatty acids (FA) that are transported into the cytosol.
The BCL-2 proteins are well-known regulators of mitochondria-mediated cell death. However, it is now clear that these proteins can also regulate autophagy in cells. Anti-apoptotic BCL-2 and BCL-XL inhibit autophagy by interacting with Beclin 1 (Pattingre et al., 2005; Maiuri et al., 2007b). The pro-apoptotic BH3-only protein BIM has also been reported to inhibit autophagy by interacting with Beclin 1 (Luo et al., 2012), demonstrating that inhibition of autophagy via Beclin 1 is not limited to anti-apoptotic BCL-2 proteins. Other pro-apoptotic BH3-only proteins induce autophagy by disrupting the interaction between BCL-2/BCL-XL and Beclin 1, thereby allowing Beclin 1 to initiate nucleation of the phagophore (Maiuri et al., 2007a; Zhang et al., 2008). The BH3-only proteins BCL2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3) and BNIP3L/NIX can also act as autophagy receptors on organelles that need to be cleared by autophagosomes (Schwarten et al., 2009; Novak et al., 2010; Hanna et al., 2012).
Mitochondrial autophagy
Cells use autophagy to selectively degrade targets such as mitochondria. Removal of mitochondria via autophagy, termed mitophagy, is essential for cellular homeostasis and is particularly important in cells with a high density of mitochondria such as myocytes. Mitophagy selectively removes dysfunctional mitochondria, enriching for healthy organelles (Twig et al., 2008; Suen et al., 2010). Although mitochondria are critical in providing myocytes with ATP to sustain contraction, they are also a source of reactive oxygen species (ROS). Functional mitochondria produce some ROS as a by-product of ATP production by the electron transport chain. However, damaged mitochondria can produce up to 10-fold more ROS than healthy mitochondria, exceeding the cell's antioxidant capacity. ROS can damage mitochondrial proteins and DNA, resulting in more dysfunctional mitochondria, further continuing the cycle (Baines, 2010). Additionally, mitochondria are important in cell death signaling. Permeabilization of the outer mitochondrial membrane leads to release of cytochrome c and other pro-apoptotic factors, while opening of the mitochondrial permeability transition pore (mPTP) and collapse of the proton gradient results in necrosis (Baines, 2010). Thus, swift yet selective removal of damaged mitochondria by mitophagy is essential for cell survival. However, excessive activation of mitophagy can lead to complete clearance of mitochondria in cells (Narendra et al., 2008). Because myocytes are highly dependent on mitochondria for energy to sustain contraction, rates of clearance that exceeds synthesis of new mitochondria can be detrimental to myocytes.
One of the pathways important for specificity in mitophagy is controlled by Parkin and phosphatase and tensin homologue-induced putative kinase 1 (PINK1; Figure 2A). Parkin is an E3 ubiquitin ligase that is cytosolic under normal conditions. Upon mitochondrial membrane depolarization, it quickly translocates to mitochondria where it ubiquitinates proteins in the outer membrane, thereby labelling them for removal (Narendra et al., 2008). Currently, there are four known substrates for Parkin at the mitochondria: VDAC1 (Geisler et al., 2010), mitofusins 1 and 2 (Mfn1/2; Gegg et al., 2010; Poole et al., 2010) and MIRO-1 (Wang et al., 2011). Mfn2 also acts as a mitochondrial receptor for Parkin (Chen and Dorn, 2013). Loss of Mfn2 prevents Parkin translocation to depolarized mitochondria and results in accumulation of dysfunctional mitochondria (Chen and Dorn, 2013). The adaptor protein p62/SQSTM1 (p62) subsequently binds to the ubiquitinated proteins on the mitochondrion (Geisler et al., 2010) and to LC3 on the autophagosome (Pankiv et al., 2007). In this way, it is postulated that p62 anchors the autophagosome to the target mitochondrion. However, whether p62 is an essential component of Parkin-mediated mitophagy is still not clear. One study found that p62 knockdown significantly inhibits mitophagy (Geisler et al., 2010), while another study found no noticeable effect on mitophagy upon p62 knockdown (Narendra et al., 2010a). It would be dangerous for the cell to rely on only one adaptor protein for mitophagy and it is likely that there exist additional currently unidentified mitophagy adaptor proteins. A possible candidate is NBR1, which has been found to bind ubiquitinated proteins where it acts as an autophagy adaptor (Kirkin et al., 2009).
Figure 2.
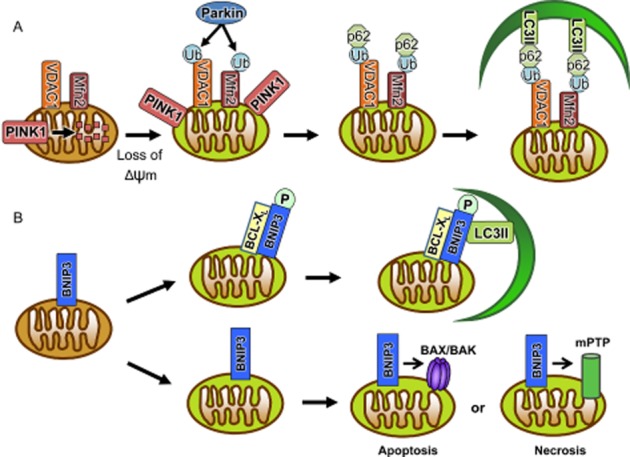
Mitophagy pathways. (A) Loss of Δψm results in accumulation of PINK1 on the outer mitochondrial membrane, leading to Parkin recruitment. Parkin then ubiquitinates mitochondrial membrane proteins. The adaptor protein p62 binds to ubiquitin and recruits LC3-II located on the autophagosome membrane. (B) Phosphorylation of BNIP3 leads to binding to LC3-II directly on the autophagosome. Binding of the anti-apoptotic BCL-XL to BNIP3 positively regulates the interaction between BNIP3 and LC3-II. Alternatively, BNIP3 can induce cell death via activation of BAX/BAK and opening of the mitochondrial permeability transition pore.
PINK1 is a serine/threonine kinase that serves to communicate the collapse of the mitochondrial membrane potential (Δψm) to Parkin. Normally, PINK1 is rapidly imported into the mitochondrial matrix where it is cleaved by mitochondrial proteases (Jin et al., 2010). As a result, there are low levels of PINK1 on the mitochondria under normal conditions. However, upon collapse of the Δψm, PINK1 import and degradation is halted, resulting in the accumulation of PINK1 on the outer mitochondrial membrane (Narendra et al., 2010b). Studies have suggested that this accumulation is essential for Parkin translocation to the mitochondria, although the exact mechanism is unclear. Some suggest that PINK1 interacts directly with Parkin, physically anchoring Parkin to mitochondria (Xiong et al., 2009). Others propose that Parkin is activated by direct phosphorylation by PINK1 (Sha et al., 2010) or that PINK1 phosphorylates Parkin substrates on mitochondria (Wang et al., 2011; Chen and Dorn, 2013). For example, PINK1 phosphorylation of Mfn2 is necessary for Parkin ubiquitination of Mfn2, and is also a prerequisite for Parkin translocation (Chen and Dorn, 2013). Wang et al. (2011) found that phosphorylation of mitochondrial MIRO-1 by PINK1 leads to Parkin-dependent degradation of MIRO-1 (Wang et al., 2011). MIRO-1 is a component of the primary motor/adaptor complex for mitochondrial trafficking, and loss of MIRO-1 on the mitochondrial membrane detaches the mitochondrion from the kinesin network, preventing mitochondrial movement. This may serve to quarantine damaged mitochondria to limit cellular damage, as well as serving as a signal for the removal of the mitochondrion by mitophagy.
Regulation of mitophagy by BNIP3 and BNIP3L/NIX
The BH3-only proteins BNIP3 and BNIP3L/NIX are pro-apoptotic members of the BCL-2 family. Both activate cell death via activation of BAX/BAK and opening of the mPTP (Vande et al., 2000; Regula et al., 2002; Kubli et al., 2007; Chen et al., 2010). However, recent studies have found that these proteins are also potent inducers of autophagy in cells and that this function is independent their pro-cell death activity (Figure 2B; Daido et al., 2004; Schweers et al., 2007; Sandoval et al., 2008; Rikka et al., 2011). In fact, both BNIP3 and NIX function as autophagy receptors on mitochondria. The LC3-interacting region (LIR) on their N-termini is essential for mitochondrial clearance (Schwarten et al., 2009; Novak et al., 2010; Hanna et al., 2012; Zhu et al., 2013). For instance, NIX interacts with the autophagy proteins LC3A and GABA-associated protein (GABARAP) on the autophagosome, and mutations of critical residues in its LIR disrupts the Nix-GABARAP/LC3A interaction and inhibits mitochondrial clearance (Schwarten et al., 2009; Novak et al., 2010).
It is still unclear how the pro-death versus mitophagy functions of NIX and BNIP3 are differentially regulated. However, a recent study found that phosphorylation serves an important role in determining whether BNIP3 will induce cell death or autophagy. This study found that phosphorylation of serine residues flanking the LIR in BNIP3 promotes the interaction between BNIP3 and LC3B (Zhu et al., 2013). Interestingly, this study also reported that anti-apoptotic BCL-XL enhances BNIP3-mediated mitophagy. This suggests that different post-translational modifications of BNIP3 and NIX might serve to specify whether they will promote mitophagy or cell death. The interaction with other proteins such as BCL-XL might also serve to promote the pro-autophagy function of these proteins. A limitation of this study is that they did not investigate whether mutating the phosphorylation sites has any effect on the pro-death activity of BNIP3. Further, the kinase responsible for phosphorylating these residues in BNIP3 remains to be identified. Interestingly, only one of the serine residues identified to be important for regulating BNIP3-mediated mitophagy is conserved in NIX (Novak et al., 2010). Hence, it will be important to determine whether phosphorylation of NIX also plays an important role in determining whether NIX is to induce autophagy or cell death.
Autophagy in the heart
A functional autophagic-lysosomal pathway is important for cardiac homeostasis. For instance, conditional deletion of Atg5 in the adult mouse heart results in accumulation of damaged mitochondria and rapid cardiac dysfunction (Nakai et al., 2007). This indicates that autophagy occurs constitutively in the adult heart and is an important quality control mechanism to maintain myocyte health. In contrast, mice with cardiac-specific deficiency of Atg5 during cardiogenesis have no cardiac defect under baseline conditions into adulthood (Nakai et al., 2007). This therefore suggests the activation of another pathway that can compensate for the lack of autophagy early on. In contrast, when autophagy is abruptly disrupted in the adult heart, there is no time for a compensatory pathway(s) to be activated. In addition, Danon disease is caused by mutations in the gene encoding for LAMP-2, a lysosomal protein that is important for the fusion of lysosomes with autophagosomes. Thus, the fusion between autophagosomes and lysosomes are much slower in these patients and leads to accumulation of autophagosomes in the cytosol (Nishino et al., 2000; Tanaka et al., 2000). These patients develop a lethal cardiomyopathy, suggesting that the accumulation of autophagosomes containing undigested cargo is detrimental to myocytes. Recently, Thomas et al., (2013) demonstrated that cardiac specific deletion of MCL-1, an anti-apoptotic BCL-2 protein, results in disruption of autophagy and rapid onset of heart failure (Thomas et al., 2013). Overall, these studies demonstrate that baseline autophagy is an important cellular quality control mechanism.
Autophagy in myocardial infarction (MI) and ischaemia/reperfusion (I/R)
Autophagy is activated during both the acute and chronic stages of a MI. Studies have found that the enhanced autophagy serves to limit injury and remodelling of the heart after the infarct. For instance, inhibition of autophagic flux using bafilomycin A1 significantly aggravates post-infarction cardiac dysfunction and remodelling, whereas enhancing autophagy with rapamycin reduces cardiac dysfunction and remodelling (Kanamori et al., 2011). This study also found that inhibition of autophagy reduced the ATP content after the infarct, whereas enhancing autophagy before the infarct increased myocardial levels of amino acids and ATP. Moreover, mitophagy also occurs in response to MI (Hoshino et al., 2012; Kubli et al., 2013). Parkin-deficient mice have reduced mitophagy and accumulate dysfunctional mitochondria after the infarction, which leads to increased remodelling and reduced survival (Kubli et al., 2013). In addition, Hoshino et al. recently reported that the tumour suppressor p53 contributes to pathological remodelling after an infarction by inhibiting the clearance of damaged mitochondria. Not surprisingly, p53-/- mice have reduced myocyte apoptosis and reduced remodelling after an infarction. Interestingly, this study identified p53 to be a negative regulator of BNIP3-mediated mitophagy and concluded that the inhibition of mitophagy leads to accumulation of damaged mitochondria that activate apoptosis after the infarction (Hoshino et al., 2012). It is well established that p53 can induce apoptosis via regulation of gene expression and by direct action at the mitochondria. However, this study did not examine whether any other modes of p53-mediated cell death are activated after the infarction. Collectively, these studies suggest that enhanced autophagy protects against progression of post-infarction cardiac remodelling by clearing dysfunctional mitochondria and preserving ATP levels, implying that augmenting autophagy could be a therapeutic strategy.
Studies agree that myocardial I/R increases autophagy, but whether autophagy is protective or detrimental to the myocytes is still under debate. There are also conflicting reports as to whether autophagic flux is impaired during I/R. Hamacher-Brady et al. initially reported that autophagic flux was suppressed during ischaemia in a cardiac cell line and that autophagic flux only partially recovered during reperfusion (Hamacher-Brady et al., 2006). Nevertheless, they found that enhanced autophagy reduced cell death after simulated I/R, demonstrating that the enhanced autophagy served as an important defence against I/R injury. Shortly thereafter, another study reported that activation of autophagy via the AMP-induced PK pathway during ischaemia was protective, whereas reperfusion resulted in up-regulation of Beclin 1 and excessive levels of autophagy (Matsui et al., 2007). This study found that activation of autophagy and cardiac injury during the reperfusion phase was significantly attenuated in Beclin 1+/− mice. In a later study, this group reported that I/R increased autophagic flux and that oxidative stress played an important role in mediating autophagy and myocardial injury during I/R (Hariharan et al., 2011). More recently, another study reported that I/R is associated with impaired autophagic flux during the reperfusion phase. In contrast to the study by Hariharan et al., they found that increased oxidative stress contributes to impaired flux by reducing LAMP-2 protein levels (Ma et al., 2012b). As LAMP-2 is essential for fusion between autophagosomes and lysosomes, the autophagosomes start to accumulate. More surprising was their finding that increased levels of Beclin 1 leads to impaired, rather than increased, flux (Ma et al., 2012b). It had been assumed that enhanced levels of Beclin 1 would lead to excessive and detrimental levels of autophagy (Matsui et al., 2007; Zhu et al., 2007). However, the study by Ma et al. challenges this concept. In fact, their study suggests that a negative feedback mechanism exists to prevent excessive Beclin 1-mediated autophagy and/or that Beclin 1 has other currently unknown functions in the cell that can regulate myocyte viability. The possibility that Beclin 1 has additional autophagy-independent functions in cells is also supported by the embryonic lethal phenotype observed in Beclin 1-/- mice (Yue et al., 2003). In contrast, deletion of autophagy genes, Atg5 or Atg7, do not result in lethality during embryogenesis (Kuma et al., 2004; Komatsu et al., 2005).
It is unclear why there is such a discrepancy between studies on autophagy using the I/R model. One possibility is the differences in the length of both the ischaemia and the reperfusion phase between the different studies. Clearly, prolonged ischaemia will have different effects on autophagy due to increased necrosis and ATP depletion, which will activate many other pathways in the cell and induce an inflammatory response. Additional studies using more consistent models are needed to resolve when autophagic flux is impaired during I/R and when autophagy is protective or detrimental during the I/R.
Autophagy in pressure overload
Haemodynamic stress is also associated with activation of the autophagic-lysosomal pathway in the heart (Nakai et al., 2007; Zhu et al., 2007; Cao et al., 2011; Sun et al., 2013), but the functional role of autophagy under these conditions is still controversial. Nakai et al. reported that activation of autophagy plays a beneficial role in the heart in response to pressure overload (Nakai et al., 2007). They found that autophagy-deficient mice show severe cardiac dysfunction and left ventricular dilatation as early as one week after aortic constriction (Nakai et al., 2007). Interestingly, they found that heart-to-body weight ratio and myocyte cross-sectional area increase to a similar degree in wild type and Atg5-deficient mice after aortic constriction, suggesting that the heart failure is not due to failure to undergo cardiac hypertrophy. As autophagy is a mechanism for maintaining energy homeostasis, it is also possible that it compensates for increased energy demand during remodelling. Moreover, cathepsin-L is a key lysosomal protease in the heart, and loss of this protein leads to reduced lysosomal activity and accumulation of autophagosomes. Mice with cathepsin-L deficiency develop exacerbated cardiac hypertrophy and cardiac dysfunction following pressure overload compared with wild-type mice (Sun et al., 2013).
In contrast, other studies have found that activation of myocyte autophagy is required for hypertrophic growth of the heart in response to haemodynamic stress, and that the enhanced autophagy plays a maladaptive role (Zhu et al., 2007; Cao et al., 2011). Zhu et al. found that autophagy was rapidly increased after aortic banding and remained elevated for at least 2 weeks (Zhu et al., 2007). Moreover, Beclin 1+/- mice have decreased myocyte autophagy and diminished pathological remodelling induced by pressure overload. Conversely, Beclin 1 overexpression in the heart increases autophagic activity and pathological remodelling, implicating Beclin 1-mediated autophagy in the pathogenesis of heart failure. One important difference between the two contradictory studies by Nakai et al. and Zhu et al. is that genetic deletion of Atg5 leads to complete disruption of autophagy, whereas heterozygous disruption of Beclin 1 does not completely disrupt autophagy. Thus, these results suggest that low-grade autophagy is protective during transverse aortic constriction, whereas complete disruption or excessive levels of autophagy are detrimental. In addition, because autophagy-mediated protein and organelle degradation would be expected to reduce cell growth, it is surprising that enhanced autophagy, such as is observed in the Beclin 1 transgenic mice, is associated with increased cardiac hypertrophy. Therefore, it is likely that the functional role of autophagy under these conditions is to provide energy and nutrients for the cell rather than regulating size.
Defects in the mitophagy pathway also lead to development of cardiac hypertrophy. PINK1-/- mice develop left ventricular dysfunction and evidence of pathological cardiac hypertrophy as early as 2 months of age (Billia et al., 2011). Although PINK1 is upstream of Parkin in the mitophagy pathway, Parkin-/- mice have no baseline phenotype and do not develop cardiac hypertrophy with age (Kubli et al., 2013). There are several possible reasons for this discrepancy. First, although PINK1 and Parkin act in the same pathway to regulate mitophagy, PINK1 might have other functions that are important for cardiac homeostasis. Second, it is possible that there are other E3 ubiquitin ligases that compensate for lack of Parkin.
Autophagy and cardiac unloading
Although the heart undergoes hypertrophy in response to mechanical load, the hypertrophy can regress upon unloading. Recent studies have identified a critical role for autophagy in mediating regression of cardiac hypertrophy during unloading (Cao et al., 2013; Hariharan et al., 2013). Cao et al. recently reported that activation of the transcription factor FoxO3 and up-regulation of its downstream target BNIP3 are increased by mechanical unloading (Cao et al., 2013). Additionally, transgenic mice overexpressing FoxO3 in the heart develop severe cardiac atrophy and have early mortality. FoxO3-mediated cardiomyocyte atrophy and autophagy require BNIP3 activation, but the early mortality phenotype involves mechanisms independent of BNIP3, autophagy and myocardial atrophy. The cardiac atrophy caused by FoxO3 overexpression is not reversed in Beclin 1+/- mice, suggesting that low-level autophagy is sufficient to mediate the atrophy induced by FoxO3.
Another study found that unloading by removing the constriction after aortic banding in mice is accompanied by up-regulation of FoxO1 (Hariharan et al., 2013). Transgenic mice with cardiac-specific overexpression of FoxO1 have smaller hearts and enhanced autophagy. In contrast to the study by Cao et al., this study found that Beclin 1+/− mice have reduced reversal of the cardiac hypertrophy after removing the constriction. It is very likely that this disparity is due to differences in models to study ventricular unloading and atrophy. Studies using FoxO3 transgenic mice will be limited to pathways activated by the transgene, whereas the surgical model will activate a more complex network of pathways. Exactly how BNIP3 and activation of autophagy and mitophagy contribute to regression of cardiac hypertrophy remain to be elucidated.
Autophagy and anthracyclines
Anthracyclines such as doxorubicin are effective anti-neoplastic drugs used in the treatment of a broad range of human malignancies. Unfortunately, their clinical use is limited by their dose-dependent cardiotoxicity, which can lead to heart failure. Anthracylines can interfere with many different intracellular processes, and it has been difficult to determine the underlying mechanisms of acute and chronic cardiotoxicity. Autophagy is enhanced by doxorubicin but its functional role appears to be influenced by factors such as dose and length of treatment. In a rat model of chronic doxorubicin cardiotoxicity, increased autophagy contributed to loss of myocytes and the pathogenesis of cardiomyopathy (Lu et al., 2009). This study found that treatment with 3-methyladenine (3-MA), an inhibitor of the Beclin 1 PI3K complex, reduces autophagy and improves cardiac function, suggesting that autophagy exerted a detrimental effect in chronic doxorubicin cardiotoxicity. However, the selectivity of 3-MA as an autophagy inhibitor in vivo is unclear, and this study also found that 3-MA reduces mitochondrial injury (Lu et al., 2009). Although it is very possible that excessive autophagy contributes to loss of myocytes, it is less clear how inhibition of autophagy preserves mitochondrial function. Thus, it cannot be ruled out that 3-MA affected additional pathways in this study. Chronic doxorubicin is a more clinically relevant model because patients receive multiple doses of the drug over time. Therefore, additional studies are necessary to determine the functional role of autophagy in chronic models of anthracyline-mediated cardiomyopathy.
In contrast, autophagy has been linked with protection against acute doxorubicin cardiotoxicity (Kawaguchi et al., 2012). Fasting is a potent inducer of autophagy in vivo (Kanamori et al., 2009), and fasting of mice prior to doxorubicin exposure preserves ATP levels and improves cardiac function compared with fed mice. This protection is likely due to clearance of old organelles that might be more sensitive to damage, as well as the generation of additional amino acids and fatty acids to preserve energy levels during stress. Doxorubicin is also known to induce mitochondrial damage, and it was recently reported that doxorubicin exposure attenuates mitophagy in myocytes. This study found that cytosolic p53 binds to Parkin and prevents its translocation to damaged mitochondria and their subsequent clearance by mitophagy in doxorubicin-treated hearts (Hoshino et al., 2013). Thus, this suggests that doxorubicin treatment can both activate autophagy and impair mitochondrial clearance.
BNIP3 and NIX as regulators of cell death and mitophagy in the myocardium
BNIP3 and NIX play dual roles in both mitophagy and cell death in the heart, but how the cell switches between these different functions is not clear. Both proteins have been implicated in the progression of cardiovascular disease, attributed to their role as pro-apoptotic BH3-only proteins (Regula et al., 2002; Yussman et al., 2002; Diwan et al., 2007; 2008,; Hamacher-Brady et al., 2007). Interestingly, NIX appears to have much stronger pro-death activity than BNIP3 in the heart. Transgenic overexpression of NIX in mouse hearts leads to loss of myocytes and death within 2 weeks of birth (Yussman et al., 2002). In contrast, although cardiac-specific BNIP3 transgenic mice develop progressive left ventricular dilation and cardiac dysfunction, they are viable for at least 40 weeks (Diwan et al., 2007). These studies did not assess whether autophagy and mitochondrial clearance are enhanced in these hearts, so it is unclear if excessive mitophagy contributes to the cardiac phenotype in these mice.
Studies of NIX and BNIP3 single-and double-knockout (DKO) mice have confirmed the importance of BNIP3 and NIX in regulating mitochondrial turnover in the myocardium. Both Bnip3-and Nix-deficient mice accumulate dysfunctional mitochondria in the heart with age, which correlates with development of cardiac dysfunction (Dorn, 2010). Interestingly, Bnip3/Nix DKO mice accumulate dysfunctional mitochondria at a much faster rate, suggesting that these proteins have overlapping function as mitophagy receptors. Although NIX is also a regulator of mitophagy (Schweers et al., 2007), most studies in the heart have focused on BNIP3-mediated mitophagy. As discussed in previous sections, BNIP3-mediated autophagy contributes to cardiac atrophy during unloading (Cao et al., 2013), and is inhibited in a p53-dependent manner after an infarction (Hoshino et al., 2012). Other studies have reported that increased autophagy protects against BNIP3-mediated cell death (Hamacher-Brady et al., 2007; Ma et al., 2012a).
It has been assumed that the clearance of dysfunctional mitochondria in response to BNIP3 in part functions to protect the cell against activation of cell death by these mitochondria. However, recent studies suggest that BNIP3-mediated mitophagy might also be involved in regulating energy homeostasis (Glick et al., 2012; Jamart et al., 2013; Lira et al., 2013). Autophagy is rapidly activated in response to increased energy demand during exercise (He et al., 2012), and studies have found that BNIP3 is up-regulated in skeletal muscle in response to exercise (Jamart et al., 2013; Lira et al., 2013). In the liver, BNIP3 is also rapidly up-regulated in response to fasting and plays an important role in regulating the balance between lipid synthesis and fatty acid oxidation (Glick et al., 2012). Thus, another possibility is that BNIP3-mediated mitophagy also serves to preserve energy status by generating amino acids and fatty acids by degrading mitochondria that are not generating ATP as efficiently as other healthy mitochondria. These mitochondria are selectively marked for degradation by BNIP3 and used for recycling under conditions of nutrient deprivation.
Therapeutic strategies and challenges
Pharmacological approaches to up-regulate or inhibit autophagy are currently receiving significant attention. For example, enhancing autophagy may have therapeutic benefits after MI, whereas reducing autophagy may be a strategy to limit pathological remodelling of the heart in response to haemodynamic stress. However, before autophagy can be targeted therapeutically, it is necessary to determine which step(s) in the autophagic-lysosomal pathway should be targeted to achieve maximum efficacy. Potential steps that can be targeted are (1) autophagosome formation, as with rapamycin to increase, or with 3-MA to block formation; (2) the fusion between autophagosomes and lysosomes, as with bafilomycin A1 or chloroquine, (3) the lysosomal activity or (4) the clearance of protein aggregates or organelles by targeting receptors or adapter proteins such as BNIP3/NIX or p62 (Figure 1). It will also be critical to understand the role of autophagy in the different cardiovascular aetiologies. For instance, if autophagic flux is blocked, as has been reported in myocardial I/R (Hamacher-Brady et al., 2006; Ma et al., 2012a) and Danon disease (Nishino et al., 2000; Tanaka et al., 2000), then a therapeutic strategy, which increases autophagosome formation will not be beneficial. Rather, increased autophagosome formation may result in their accumulation in the cytosol, which could be harmful to the cell. In addition, constitutive activation of autophagy may be associated with potential unwanted side effects. First, chronic or excessive autophagy can lead to degradation of too many organelles and critical proteins and lead to cell death. Thus, a major challenge will be how to accurately monitor autophagic flux in patients' hearts. Second, there is evidence that autophagy suppresses tumour formation. Beclin 1 deficiency is associated with enhanced susceptibility to breast, ovarian and prostate cancer in humans (Liang et al., 1999), and increased spontaneous malignancies in mice (Qu et al., 2003; Yue et al., 2003). Thus, suppression of autophagy in patients with cardiovascular disease might increase the risk of developing certain cancers.
A more attractive strategy is to enhance or block the specific targeting of damaged organelles such as mitochondria. For instance, drugs that interfere with the LIR of BNIP3 and NIX could potentially be used to block specific degradation of mitochondria, and may therefore be efficacious to prevent excessive cardiac atrophy following mechanical unloading. Likewise, BNIP3 may be targeted for activation in order to up-regulate mitophagy to enhance recovery following myocardial I/R. However, it may be difficult to differentially control the autophagy-promoting functions of BNIP3 without unintentionally activating its pro-apoptotic properties. Therefore, specific activation of the PINK1/Parkin pathway may have fewer off-target effects. However, more research is necessary to elucidate the exact mechanisms of PINK1/Parkin activation before the therapeutic potential of this pathway can be evaluated.
Conclusions
Overall, studies support that constitutive autophagy is essential for controlling protein quality, and maintaining cellular function and survival in the heart. A major question that remains unanswered is whether enhanced autophagy in the heart in various diseases is an adaptive or maladaptive response. It appears that the levels and duration of autophagy determine whether it is protective or detrimental to the myocytes, where too little or too much autophagy in response to stress might be detrimental. To date, studies on autophagy and mitophagy have focused on cardiac myocytes. However, it is very likely that autophagy is important in other cells found in the heart such as fibroblasts and endothelial cells. Mice with global deletion of Parkin are more sensitive to MI, which was mainly attributed to the impaired mitophagy and accumulation dysfunctional mitochondria in myocytes (Kubli et al., 2013). However, these mice also had very thin scars after the infarctions compared with wild-type mice, suggesting that fibroblast function might also be impaired in the Parkin-/- mice. This was not explored in this study. Thus, these issues need to be resolved before autophagy and mitophagy can safely be targeted therapeutically in the heart.
Whether the autophagy pathway represents a useful target in the treatment of cardiovascular disease will ultimately need to be addressed by conducting clinical trials in patients. Preclinical experiments and mouse models are useful in establishing the role of autophagy in certain diseases such as MI where all studies to date have reported a beneficial role for autophagy. However, there are still conflicting reports regarding the role of autophagy in myocardial I/R injury and haemodynamic stress. These discrepancies are likely to be due to differences in the experimental set-up such as length of the ischaemia and/or reperfusion, or the severity of the constriction in models of pressure overload. There is also a need to develop additional specific modulators of autophagy that can be used to further dissect the steps in the process, and assess their effects of cell survival and disease. More selective drugs will also limit the influences of autophagy-independent effects. Overall, pharmacological modulation of different steps in the autophagy pathway represents a promising future therapeutic strategy. However, additional understanding of this process and identification of proteins involved in regulating the pathway are still needed before this becomes a possibility.
Acknowledgments
Å. B. Gustafsson is supported by the National Institutes of Health grants R01HL087023, R01HL101217, and R01HL092136.
Glossary
- 3-MA
3-methyladenine
- BNIP3
BCL2/adenovirus E1B 19-kDa protein-interacting protein 3
- I/R
ischaemia/reperfusion
- LC3
microtubule-associated protein 1 light chain 3
- LIR
LC3-interacting region
- Mfn1/2
mitofusin-1/2
- Δψm
mitochondrial membrane potential
- MI
myocardial infarction
- mPTP
mitochondria permeability transition pore
- PINK1
phosphatase and tensin homologue-induced putative kinase 1
- VPS
vacuolar protein sorting
Conflict of interest
None.
References
- Baines CP. The cardiac mitochondrion: nexus of stress. Annu Rev Physiol. 2010;72:61–80. doi: 10.1146/annurev-physiol-021909-135929. [DOI] [PubMed] [Google Scholar]
- Bernales S, Schuck S, Walter P. ER-phagy: selective autophagy of the endoplasmic reticulum. Autophagy. 2007;3:285–287. doi: 10.4161/auto.3930. [DOI] [PubMed] [Google Scholar]
- Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci U S A. 2011;108:9572–9577. doi: 10.1073/pnas.1106291108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, et al. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A. 2011;108:4123–4128. doi: 10.1073/pnas.1015081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao DJ, Jiang N, Blagg A, Johnstone JL, Gondalia R, Oh M, et al. Mechanical unloading activates FoxO3 to trigger Bnip3-dependent cardiomyocyte atrophy. J Am Heart Assoc. 2013;2:e000016. doi: 10.1161/JAHA.113.000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dorn GW., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Lewis W, Diwan A, Cheng EH, Matkovich SJ, Dorn GW., 2nd Dual autonomous mitochondrial cell death pathways are activated by Nix/BNip3L and induce cardiomyopathy. Proc Natl Acad Sci U S A. 2010;107:9035–9042. doi: 10.1073/pnas.0914013107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:1845–1846. doi: 10.1056/NEJMc1303158. [DOI] [PubMed] [Google Scholar]
- Daido S, Kanzawa T, Yamamoto A, Takeuchi H, Kondo Y, Kondo S. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004;64:4286–4293. doi: 10.1158/0008-5472.CAN-03-3084. [DOI] [PubMed] [Google Scholar]
- Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, et al. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest. 2007;117:2825–2833. doi: 10.1172/JCI32490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwan A, Wansapura J, Syed FM, Matkovich SJ, Lorenz JN, Dorn GW., 2nd Nix-mediated apoptosis links myocardial fibrosis, cardiac remodeling, and hypertrophy decompensation. Circulation. 2008;117:396–404. doi: 10.1161/CIRCULATIONAHA.107.727073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn GW., 2nd Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors. J Cardiovasc Transl Res. 2010;3:374–383. doi: 10.1007/s12265-010-9174-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19:4861–4870. doi: 10.1093/hmg/ddq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Glick D, Zhang W, Beaton M, Marsboom G, Gruber M, Simon MC, et al. BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol Cell Biol. 2012;32:2570–2584. doi: 10.1128/MCB.00167-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–29787. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, et al. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14:146–157. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with BNIP3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287:19094–19104. doi: 10.1074/jbc.M111.322933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariharan N, Zhai P, Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal. 2011;14:2179–2190. doi: 10.1089/ars.2010.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariharan N, Ikeda Y, Hong C, Alcendor RR, Usui S, Gao S, et al. Autophagy plays an essential role in mediating regression of hypertrophy during unloading of the heart. Plos ONE. 2013;8:e51632. doi: 10.1371/journal.pone.0051632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11:1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481:511–515. doi: 10.1038/nature10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino A, Matoba S, Iwai-Kanai E, Nakamura H, Kimata M, Nakaoka M, et al. p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. J Mol Cell Cardiol. 2012;52:175–184. doi: 10.1016/j.yjmcc.2011.10.008. [DOI] [PubMed] [Google Scholar]
- Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, et al. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun. 2013;4:2308. doi: 10.1038/ncomms3308. [DOI] [PubMed] [Google Scholar]
- Iwata J, Ezaki J, Komatsu M, Yokota S, Ueno T, Tanida I, et al. Excess peroxisomes are degraded by autophagic machinery in mammals. J Biol Chem. 2006;281:4035–4041. doi: 10.1074/jbc.M512283200. [DOI] [PubMed] [Google Scholar]
- Jamart C, Naslain D, Gilson H, Francaux M. Higher activation of autophagy in skeletal muscle of mice during endurance exercise in the fasted state. Am J Physiol Endocrinol Metab. 2013;305:E964–E974. doi: 10.1152/ajpendo.00270.2013. [DOI] [PubMed] [Google Scholar]
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamori H, Takemura G, Maruyama R, Goto K, Tsujimoto A, Ogino A, et al. Functional significance and morphological characterization of starvation-induced autophagy in the adult heart. Am J Pathol. 2009;174:1705–1714. doi: 10.2353/ajpath.2009.080875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamori H, Takemura G, Goto K, Maruyama R, Tsujimoto A, Ogino A, et al. The role of autophagy emerging in postinfarction cardiac remodelling. Cardiovasc Res. 2011;91:330–339. doi: 10.1093/cvr/cvr073. [DOI] [PubMed] [Google Scholar]
- Kawaguchi T, Takemura G, Kanamori H, Takeyama T, Watanabe T, Morishita K, et al. Prior starvation mitigates acute doxorubicin cardiotoxicity through restoration of autophagy in affected cardiomyocytes. Cardiovasc Res. 2012;96:456–465. doi: 10.1093/cvr/cvs282. [DOI] [PubMed] [Google Scholar]
- Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001;2:330–335. doi: 10.1093/embo-reports/kve061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505–516. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- Knodler LA, Celli J. Eating the strangers within: host control of intracellular bacteria via xenophagy. Cell Microbiol. 2011;13:1319–1327. doi: 10.1111/j.1462-5822.2011.01632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Ycaza JE, Gustafsson AB. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem J. 2007;405:407–415. doi: 10.1042/BJ20070319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. 2013;288:915–926. doi: 10.1074/jbc.M112.411363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- Lira VA, Okutsu M, Zhang M, Greene NP, Laker RC, Breen DS, et al. Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J. 2013;27:4184–4193. doi: 10.1096/fj.13-228486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Wu W, Yan J, Li X, Yu H, Yu X. Adriamycin-induced autophagic cardiomyocyte death plays a pathogenic role in a rat model of heart failure. Int J Cardiol. 2009;134:82–90. doi: 10.1016/j.ijcard.2008.01.043. [DOI] [PubMed] [Google Scholar]
- Luo S, Garcia-Arencibia M, Zhao R, Puri C, Toh PP, Sadiq O, et al. Bim inhibits autophagy by recruiting Beclin 1 to microtubules. Mol Cell. 2012;47:359–370. doi: 10.1016/j.molcel.2012.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Godar RJ, Liu H, Diwan A. Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death. Autophagy. 2012a;8:297–309. doi: 10.4161/auto.18658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, et al. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012b;125:3170–3181. doi: 10.1161/CIRCULATIONAHA.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuri MC, Criollo A, Tasdemir E, Vicencio JM, Tajeddine N, Hickman JA, et al. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L) Autophagy. 2007a;3:374–376. doi: 10.4161/auto.4237. [DOI] [PubMed] [Google Scholar]
- Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007b;26:2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010a;6:1090–1106. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. Plos Biol. 2010b;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease) Nature. 2000;406:906–910. doi: 10.1038/35022604. [DOI] [PubMed] [Google Scholar]
- Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. Plos ONE. 2010;5:e10054. doi: 10.1371/journal.pone.0010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinsay MN, Thomas RL, Lee Y, Gustafsson AB. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy. 2010;6:17–24. doi: 10.4161/auto.6.7.13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010;12:747–757. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regula KM, Ens K, Kirshenbaum LA. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res. 2002;91:226–231. doi: 10.1161/01.res.0000029232.42227.16. [DOI] [PubMed] [Google Scholar]
- Rikka S, Quinsay MN, Thomas RL, Kubli DA, Zhang X, Murphy AN, et al. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ. 2011;18:721–731. doi: 10.1038/cdd.2010.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarten M, Mohrluder J, Ma P, Stoldt M, Thielmann Y, Stangler T, et al. Nix directly binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy. 2009;5:690–698. doi: 10.4161/auto.5.5.8494. [DOI] [PubMed] [Google Scholar]
- Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104:19500–19505. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha D, Chin LS, Li L. Phosphorylation of parkin by Parkinson disease-linked kinase PINK1 activates parkin E3 ligase function and NF-kappaB signaling. Hum Mol Genet. 2010;19:352–363. doi: 10.1093/hmg/ddp501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci U S A. 2010;107:11835–11840. doi: 10.1073/pnas.0914569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Ouzounian M, de Couto G, Chen M, Yan R, Fukuoka M, et al. Cathepsin-L ameliorates cardiac hypertrophy through activation of the autophagy-lysosomal dependent protein processing pathways. J Am Heart Assoc. 2013;2:e000191. doi: 10.1161/JAHA.113.000191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lullmann-Rauch R, et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. 2000;406:902–906. doi: 10.1038/35022595. [DOI] [PubMed] [Google Scholar]
- Tannous P, Zhu H, Nemchenko A, Berry JM, Johnstone JL, Shelton JM, et al. Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation. 2008;117:3070–3078. doi: 10.1161/CIRCULATIONAHA.107.763870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB, et al. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 2013;27:1365–1377. doi: 10.1101/gad.215871.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande VC, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, et al. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000;20:5454–5468. doi: 10.1128/mcb.20.15.5454-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H, Wang D, Chen L, Choo YS, Ma H, Tang C, et al. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J Clin Invest. 2009;119:650–660. doi: 10.1172/JCI37617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yussman MG, Toyokawa T, Odley A, Lynch RA, Wu G, Colbert MC, et al. Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat Med. 2002;8:725–730. doi: 10.1038/nm719. [DOI] [PubMed] [Google Scholar]
- Zeng X, Overmeyer JH, Maltese WA. Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J Cell Sci. 2006;119:259–270. doi: 10.1242/jcs.02735. [DOI] [PubMed] [Google Scholar]
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Massen S, Terenzio M, Lang V, Chen-Lindner S, Eils R, et al. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem. 2013;288:1099–1113. doi: 10.1074/jbc.M112.399345. [DOI] [PMC free article] [PubMed] [Google Scholar]