

Abstract
BACKGROUND AND PURPOSE
Dynamin-related protein 1 (Drp1) mediates mitochondrial fission and is thought to promote Bax/Bak-induced cytochrome c release during apoptosis. Conformationally active Bax, Bak and Bax/Bak-activating BH3-only proteins, such as Bim, are restrained by anti-apoptotic Bcl-2 proteins in cells that are ‘primed for death’. Inhibition of Bcl-2/Bcl-xL/Bcl-w by the antagonist ABT-737 causes rapid apoptosis of primed cells. Hence, we determined whether Drp1 is required for cytochrome c release, respiratory alterations and apoptosis of cells that are already primed for death.
EXPERIMENTAL APPROACH
We tested the Drp1 inhibitor mdivi-1 for inhibition of cytochrome c release in MCF10A cells primed by Bcl-2 overexpression. We measured ATP synthesis-dependent,-independent and cytochrome c-limited maximal oxygen consumption rates (OCRs) and cell death of immortalized wild-type (WT) and Drp1 knockout (KO) mouse embryonic fibroblasts (MEFs) treated with ABT-737.
KEY RESULTS
Mdivi-1 failed to attenuate ABT-737-induced cytochrome c release. ABT-737 decreased maximal OCR measured in the presence of uncoupler in both WT and Drp1 KO MEF, consistent with respiratory impairment due to release of cytochrome c. However, Drp1 KO MEF were slightly less sensitive to this ABT-737-induced respiratory inhibition compared with WT, and were resistant to an initial ABT-737-induced increase in ATP synthesis-independent O2 consumption. Nevertheless, caspase-dependent cell death was not reduced. Pro-apoptotic Bax was unaltered, whereas Bak was up-regulated in Drp1 KO MEF.
CONCLUSIONS AND IMPLICATIONS
The findings indicate that once fibroblast cells are primed for death, Drp1 is not required for apoptosis. However, Drp1 may contribute to ABT-737-induced respiratory changes and the kinetics of cytochrome c release.
LINKED ARTICLES
This article is part of a themed issue on Mitochondrial Pharmacology: Energy, Injury & Beyond. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2014.171.issue-8
Keywords: primed for death, fission, Bax, Bak, Bim, Bcl-xL, oxygen, apoptosis, mitochondria, caspases
Introduction
BH3-only pro-apoptotic molecules such as Bim and Bid cause oligomerization of Bax and its close homologue Bak (Wei et al., 2000; Polster et al., 2001; Gavathiotis et al., 2010). Bax/Bak oligomerization leads to pore formation in the mitochondrial outer membrane, releasing cytochrome c and other intermembrane space proteins which then initiate apoptosis (Polster and Fiskum, 2004). Although the exact nature of the outer membrane channel resulting from Bax/Bak activation is not known, evidence has been obtained suggesting that the pore is formed by lipid or by a combination of lipid and protein (Hardwick and Polster, 2002; Kuwana et al., 2002; Polster et al., 2003; Terrones et al., 2004).
Bax co-localizes with the mitochondrial fission factor dynamin-related protein 1 (Drp1) at mitochondrial membrane scission sites during apoptosis (Karbowski et al., 2002). Drp1-dependent mitochondrial fragmentation temporally coincides with cytochrome c release (Frank et al., 2001). In vitro experiments suggest that Drp1 facilitates Bax oligomerization and pore formation by promoting formation of phospholipid membrane hemifission or hemifusion intermediates (Montessuit et al., 2010). Although Bax can permeabilize synthetic liposomes in the absence of mitochondrial proteins (Kuwana et al., 2002; Polster et al., 2003), Drp1 or other mitochondrial proteins augment Bax-induced mitochondrial outer membrane permeabilization (Cassidy-Stone et al., 2008; Montessuit et al., 2010; Kushnareva et al., 2012), and the putative Drp1 antagonist mdivi-1 blocks Bax-induced membrane poration (Cassidy-Stone et al., 2008; Kushnareva et al., 2012). Mitochondrial fission can be uncoupled from cytochrome c release, suggesting that Drp1 can independently promote mitochondrial fragmentation and Bax-dependent cytochrome c efflux (Parone et al., 2006; Cassidy-Stone et al., 2008; Sheridan et al., 2008; Montessuit et al., 2010).
Development of the high-affinity Bcl-2/Bcl-xL/Bcl-w antagonist ABT-737 (Oltersdorf et al., 2005) led to the recognition that some cells exhibit a primed for death state characterized by an absolute dependence on anti-apoptotic Bcl-2 proteins for survival (Certo et al., 2006; van Delft et al., 2006). In most healthy cells, Bax and BH3-only proteins are primarily non-mitochondrial and only accumulate at the mitochondrial outer membrane in response to cellular stress. However, in cells primed for death, Bax and BH3-only proteins such as Bim often constitutively localize to mitochondria where they are held in check by anti-apoptotic Bcl-2 family proteins (Konopleva et al., 2006; Del Gaizo Moore et al., 2007; Tagscherer et al., 2008; Merino et al., 2012). When bound to anti-apoptotic Bcl-2 or Bcl-xL, Bax or Bak adopt an active (or partially active) configuration despite being restrained (Dlugosz et al., 2006; Billen et al., 2008; Edlich et al., 2011; Llambi et al., 2011). Whether Drp1 still promotes pore formation by Bax/Bak in cells already in a primed for death state has yet to be clarified. In primary cerebellar granule neurons, overexpression of dominant negative Drp1 or knockdown of Bax by small-interfering RNA (siRNA) inhibited ABT-737-induced cell death (Young et al., 2007), indicating that Drp1 is an essential participant in the Bax-induced death process even in primed cells. In contrast, Drp1 deficiency did not prevent the apoptosis of mouse embryonic fibroblasts (MEFs) in response to staurosporine, etoposide or UV irradiation (Ishihara et al., 2009; Wakabayashi et al., 2009) despite its reliance on the Bax/Bak-dependent intrinsic mitochondrial pathway.
The primary goal of this study was to evaluate whether Drp1 is required for the cytochrome c release, associated respiratory alterations and death of cells exhibiting a primed state. We exploited two models: (i) MCF10A human mammary epithelial cells in which a primed for death state was induced by stable Bcl-2 overexpression and (ii) spontaneously immortalized MEF cells, which exhibited cell death priming following extended serial passage. Mitochondrial cytochrome c release in cells was assessed as an impairment of maximal O2 consumption rate (OCR) using our recently developed bioenergetics-based profiling technique (Clerc et al., 2012). Because ABT-737 also specifically elevates ATP synthesis-independent respiration in primed Bcl-2 overexpressing MCF10A cells (Clerc et al., 2012), we additionally assessed ATP synthesis-independent respiration. This respiration, measured in the presence of the ATP synthase inhibitor oligomycin, is largely due to mitochondrial inner membrane proton leak and reactive oxygen species production.
Overall, the results indicated that Drp1 was not required for the ABT-737-induced apoptotic cytochrome c release in cells exhibiting a primed for death state in the two models employed. However, the Drp1 knockout (KO) MEF were slightly resistant to ABT-737-induced cytochrome c release compared with wild-type (WT) cells, as well as to an initial ABT-737-mediated elevation in ATP synthesis-independent oxygen consumption. Unexpectedly, Drp1 KO MEF displayed an up-regulation of pro-apoptotic Bak, indicating that changes in mitochondrial proteins in Drp1 KO MEF are not restricted to Drp1.
Methods
Cell culture
WT and Drp1 KO MEF (Wakabayashi et al., 2009) became spontaneously immortalized following extended (>30 times) serial passage (Kageyama et al., 2012). These cells were cultured in Iscove's modified Dulbecco's medium supplemented with 10% FBS and 100 μg·mL−1 primocin (InvivoGen, San Diego, CA, USA). The creation of MCF10A cells stably transfected with a vector overexpressing Bcl-2 was as previously described (Martin and Leder, 2001). These cells were cultured in a 1:1 mixture of F12 medium and DMEM (DMEM-F12) supplemented with hydrocortisone (0.5 μg·mL−1), insulin (10 μg·mL−1), EGF (20 ng·mL−1), 5% horse serum, penicillin (100 IU·mL−1) and streptomycin (100 μg·mL−1). Immortalized WT and Bax/Bak double KO (DKO) MEF were generously provided by Drs Tullia Lindsten and Craig Thompson (University of Pennsylvania, Philadelphia, PA, USA) and were cultured in DMEM supplemented with 10% FBS, L-glutamine (2 mM), penicillin (100 IU·mL−1) and streptomycin (100 μg·mL−1) (Zong et al., 2001). All cells were maintained in a humidified atmosphere of 95% air/5% CO2 at 37°C and passaged one to two times weekly.
XF24 microplate-based respirometry
An XF24 extracellular flux analyser (Seahorse Bioscience, Billerica, MA, USA) was used to measure OCR from intact and permeabilized cells as previously described (Wu et al., 2007; Clerc et al., 2012; Clerc and Polster, 2012). MCF10A Bcl-2 cells were plated at a density of 6 × 104 cells per well, and WT and Drp1 KO cells were plated at a density of 2 × 104 to 4 × 104 cells per well to achieve ∼90% confluence at the time of assay (16–24 h after plating). All comparisons were made with cells at a similar density at the time of assay. XF24 assay medium consisted of 120 mM NaCl, 3.5 mM KCl, 1.3 mM CaCl2, 0.4 mM KH2PO4, 1 mM MgCl2, 5 mM HEPES, 15 mM glucose and 4 mg·mL−1 fatty acid-free BSA, pH 7.4. For experiments with permeabilized MCF10A cells, 1.3 mM CaCl2 was replaced with 1.86 mM CaCl2 plus 5 mM EGTA to yield a low Ca2+ (∼100 nM) assay medium that approximates cytoplasmic [Ca2+] and prevents mitochondrial Ca2+ overload (Abramov and Duchen, 2008). For experiments in which WT and Drp1 KO MEF were acutely permeabilized, cells were assayed in normal assay medium. EGTA (5 mM), diluted from a pH-adjusted 500 mM stock, was then included in the saponin permeabilization solution to reduce calcium to cytoplasmic levels. The XF24 assay medium with increased buffering capacity (20 mM HEPES) was used for experiments with permeabilized cells to help neutralize protons released by the binding of Ca2+ to EGTA (Patton et al., 2004).
Immunocytochemistry and fluorescence microscopy
MCF10A Bcl-2 overexpressing cells were plated at 5 × 105 cells per well (4.2 cm2) in 2-well Nunc™ Lab-Tek™ chambered #1.0 borosilicate coverglass slides (ThermoScientific, Waltham, MA, USA) and allowed to proliferate overnight. The next day, cells were treated with mdivi-1 (100 μM) or DMSO vehicle for 3 h in XF24 assay medium, fixed in 4% formaldehyde for 20 min, washed with 1X PBS and stored in 1X PBS overnight. On the following day, cells were permeabilized in 0.15% Triton-X (Sigma-Aldrich, St. Louis, MO, USA) for 20 min, washed with 1X PBS and then blocked for 45 min with 7.5% BSA in 1X PBS. Cells were then incubated with rabbit polyclonal anti-Tom20 (1:2000) and mouse monoclonal anti-Drp1 (1:200) for 90 min, followed by incubation with Alexa Fluor 594 goat anti-rabbit (1:250; emission 620 nm) and Alexa Fluor 488 goat anti-mouse (1:250; emission 520 nm; Life Technologies, Grand Island, NY, USA). All procedures were carried out at room temperature. Cells were imaged with a Zeiss LSM510 confocal microscope system equipped with a 100× oil objective and Axiovision release 4.8 analysis software (Zeiss Microimaging, Oberkochen, Germany).
Cell death measurements
Drp1 KO MEF or WT control cells were treated with ABT-737 or vehicle (DMSO) for 4 h in XF24 assay medium, and cell death was quantified using the cytotoxicity detection kit (Roche Applied Science, Indianapolis, IN, USA) according to the instructions of the manufacturer. This assay measures cell death as a percentage of activity of the cytoplasmic enzyme LDH detected in the medium relative to the total measured in the medium plus cell lysate.
Protein detection by immunoblot
Cells were lysed in RIPA buffer consisting of 150 mM NaCl, 50 mM Tris, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS and protease inhibitor cocktail set III (EMD Biosciences, San Diego, CA, USA), pH 7.4. WT and Drp1 KO cell lysates (55–65 μg) were loaded on NuPAGE Novex 4–12% Bis-Tris gradient gels (Invitrogen, Carlsbad, CA, USA). SDS-PAGE and immunodetection for Drp1 (1:1000), Bax (1:1000), Bak (1:500), cytochrome c (1:1000) and β-actin (1:2000) were performed as described previously (Polster et al., 2001). Densitometric quantification of protein levels was conducted using National Institutes of Health Image J software (Bethesda, MD, USA).
Data analysis and statistics
Data are expressed as mean ± SD for representative respirometry data and as mean ± SEM for histograms compiled from three to five independent experiments. anova was used to determine statistical significance at P < 0.05, with Tukey's post hoc analysis employed for pairwise comparisons. anova with repeated measures was used to analyse data with multiple time points. Independent sample t-tests were run to compare WT and Drp1 KO at individual drug doses and for densitometry data. Statistical analyses were carried out using IBM SPSS statistics software (Armonk, NY, USA).
Drugs and reagents
ABT-737 (C42H45ClN6O5S2) was obtained from ChemieTek (Indianapolis, IN, USA). Mdivi-1 [3-(2,4-dichloro-5-methoxyphenyl)-2,3-dihydro-2-thioxo-4(1H)-quinazolinone] was purchased from Enzo Life Sciences (Farmingdale, NY, USA). Drp1 and cytochrome c mouse monoclonal antibodies were from BD Biosciences (San Jose, CA, USA; catalogue #611113 and #556433 respectively). Bax NT and Bak NT rabbit polyclonal antibodies were from EMD Millipore (Billerica, MA, USA; catalogue #06-499 and #06-536 respectively). β-Actin mouse monoclonal antibody was obtained from Sigma-Aldrich (catalogue #A5316). Tom20 rabbit polyclonal antibody was from Santa Cruz Biotechnology (catalogue #sc-11415; Dallas, TX, USA). Alexa Fluor secondary antibodies were from Life Technologies. Cell culture products were from Invitrogen. Other reagents were purchased from Sigma-Aldrich unless otherwise indicated.
Results
Mdivi-1 fails to impair ABT-737-induced cytochrome c release in primed MCF10A Bcl-2 overexpressing cells
Stable Bcl-2 overexpression primes MCF10A mammary epithelial cells for death (Clerc et al., 2012). Inhibition of Bcl-2 by ABT-737 induces rapid and complete Bax/Bak-dependent cytochrome c release from MCF10A Bcl-2 overexpressing mitochondria, whereas mitochondria within MCF10A control-transfected cells are impervious to ABT-737 (Clerc et al., 2012). Utilizing this primed for death model, we tested the ability of the quinazolinone derivative mdivi-1 to block ABT-737-triggered cytochrome c release over the same concentration range reported to inhibit Drp1-mediated mitochondrial fission in cells or Bax/Bak-induced cytochrome c release from isolated mitochondria (Cassidy-Stone et al., 2008). Oxygen consumption was monitored as an indicator of cytochrome c release. Maximal OCR is a sensitive indicator of cytochrome c release because cytochrome c is required for electron transfer between complex III and complex IV (Nicholls and Ferguson, 2002). MCF10A Bcl-2 overexpressing cells were permeabilized by saponin, a cholesterol-removing agent that when carefully titrated selectively affects the plasma membrane without disrupting mitochondrial membranes (Fiskum et al., 1980; Clerc et al., 2012; Clerc and Polster, 2012). Prior studies multiplexing quantitative cytochrome c elisa to MCF10A respiration measurements established that saponin does not compromise mitochondrial integrity while also validating respirometry as a precise indicator of cytochrome c release (Clerc et al., 2012). Mdivi-1 or vehicle were added together with saponin and the mitochondrial complex II substrate succinate, followed by exposure of mitochondria within permeabilized cells to ABT-737. ADP-stimulated oxygen consumption was stable in the presence of mdivi-1 (100 μM) or vehicle but was nearly abolished following 20 min of 10 μM ABT-737 treatment (Figure 1). Mdivi-1 failed to alter the rate or extent of respiratory decline induced by ABT-737 (Figure 1). Similar results were obtained using 50 or 75 μM mdivi-1 (data not shown). Exogenous purified cytochrome c reversed the respiratory decline both in the absence and in the presence of mdivi-1 (Figure 1), confirming that impaired respiration was due to cytochrome c release and that mdivi-1 did not cause cytochrome c-independent inhibition of complex II-dependent respiration.
Figure 1.

The Drp1 inhibitor mdivi-1 does not impair ABT-737-induced cytochrome c release. MCF10A Bcl-2 overexpressing cells were exposed to the plasma membrane-permeabilizing agent saponin (10 μg·mL−1) plus succinate (5 mM), rotenone (0.5 μM), ADP (1 mM) and K2HPO4 (3.6 mM) in the absence or presence of mdivi-1 (100 μM, first arrow). ABT-737 (ABT; 10 μM) or vehicle control (con; second arrow), cyt c (100 μM) or con (third arrow) and finally sodium azide (5 mM, fourth arrow) were subsequently injected. Results are mean ± SD from one experiment in triplicate and are representative of three independent experiments. OCR is baseline normalized to the point before saponin addition. In some cases the error bars are smaller than the symbol size.
Immunocytochemical staining verified that Drp1 was at least partly localized to mitochondria in MCF10A Bcl-2 overexpressing cells both in the absence (Figure 2A) and in the presence (Figure 2B) of mdivi-1 treatment. Mitochondria were already fairly elongated in the Bcl-2 overexpressing cells, and mitochondrial morphology was not noticeably altered by a 3 h treatment with mdivi-1 (Figure 2). Thus, we cannot exclude the possibility that mdivi-1 does not inhibit Drp1 as effectively in MCF10A Bcl-2 overexpressing cells as it does in other cell types.
Figure 2.
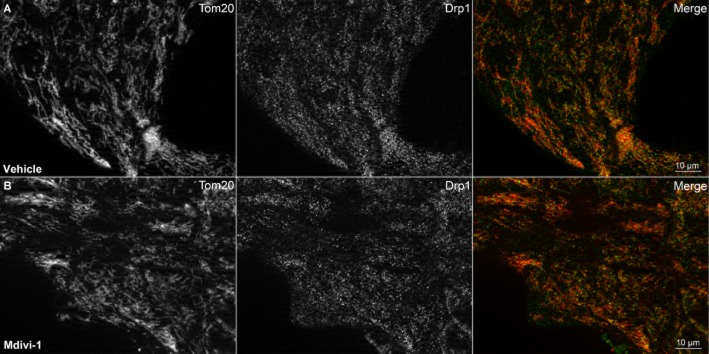
Drp1 is partly localized to mitochondria in MCF10A Bcl-2 overexpressing cells. Mitochondrial morphology and Drp1 subcellular localization were visualized by immunocytochemical staining for the mitochondrial outer membrane protein Tom20 (first column) and Drp1 (second column) in DMSO vehicle-treated (A) and mdivi-1 (100 μM)-treated (B) cells. Co-localization of Tom20 (red) and Drp1 (green) is shown in the third column (merge).
Drp1 deficiency delays but does not prevent ABT-737-induced respiratory impairment caused by cytochrome c release in MEF
Suppression of maximal respiration by ABT-737 is also a sensitive indicator of cytochrome c release in intact cells and does not occur in immortalized Bax/Bak DKO MEF (Clerc et al., 2012). Thus, we employed bioenergetics-based profiling, our recently described method characterized by adding Bcl-2 antagonist prior to the sequential addition of the ATP synthase inhibitor oligomycin and the uncoupler FCCP (Clerc et al., 2012), to determine whether genetic Drp1 deficiency confers resistance to ABT-737. Drp1 KO MEF and WT control MEF became spontaneously immortalized after extended serial passage. Both WT (Figure 3A, B and E) and Drp1 KO MEF (Figure 3C–E) exhibited dose-dependent attenuation of maximal OCR by ABT-737, with a significant loss of uncoupled OCR apparent at 0.1 μM ABT-737 in WT cells and at 0.5 μM ABT-737 in Drp1 KO cells. Considerable variability in both the maximal OCR relative to baseline under control conditions and OCR sensitivity to ABT-737 was observed in WT and Drp1 KO MEF over time in culture. Representative experiments are displayed for WT and Drp1 KO MEF at multiple passages to illustrate the range of responses with passage number. Overall, Drp1 KO MEF cells were significantly less sensitive in the extent of OCR impairment over the range of 0.5–5.0 μM ABT-737 but not at 0.1 or 10 μM ABT-737. Cytochrome c content did not differ between WT and Drp1 KO MEF (Figure 3F), eliminating increased cytochrome c content as a potential explanation for the resistance of Drp1 KO cells to ABT-737-induced respiratory inhibition. Drp1 deficiency was confirmed by immunoblot (Figure 3G).
Figure 3.
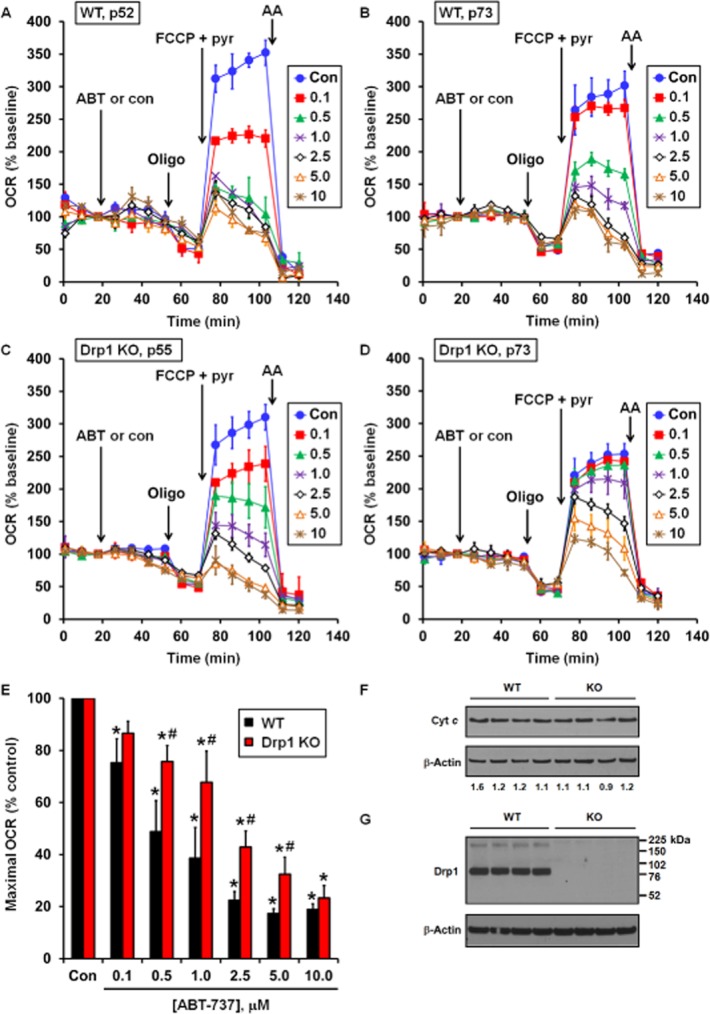
ABT-737 induces dose-dependent impairment of maximal OCR in immortalized WT and Drp1 KO MEF cells. (A–D) Representative bioenergetic profiles of WT (A, passage 52; B, passage 73) and Drp1 KO (C, passage 55; D, passage 73) cells treated with vehicle (con) or ABT-737 (ABT), oligomycin (oligo; 0.3 μg·mL−1), FCCP (3 μM) and antimycin A (AA; 1 μM) as indicated. Optimal oligomycin and FCCP concentrations were determined by titration for each cell type. Pyruvate (pyr; 10 mM) was added in combination with FCCP to ensure that substrate supply was not rate limiting for maximal OCR. Numbers in legends correspond to ABT-737 concentration in μM. Representative traces are means from individual experiments performed in triplicate. OCR is baseline normalized to the point before vehicle or ABT-737 addition. (E) Maximal OCR following addition of ABT-737 as percentage vehicle control. Maximal OCR was calculated as the uncoupled OCR just before AA addition minus AA-insensitive OCR. Results are mean ± SEM of five independent experiments with one to three technical replicates per experiment. *P < 0.05 for ABT-737 treated relative to control treated; #P < 0.05 for Drp1 KO relative to WT. (F) Immunodetection for cytochrome c and (G) for Drp1 in WT and Drp1 KO MEF, with β-actin as a loading control. Numbers in (F) are band densities normalized to β-actin.
To verify that respiratory inhibition in both WT and Drp1 KO cells was due to cytochrome c release, MEF were treated with ABT-737 (10 μM) for 1 h followed by acute permeabilization in the presence of mitochondrial substrate, FCCP and the absence or presence of exogenous cytochrome c. ABT-737-impaired respiration was fully rescued by cytochrome c in both WT (Figure 4A) and Drp1 KO MEF (Figure 4B), confirming that ABT-737-triggered changes in OCR are a specific measure of mitochondrial cytochrome c loss. In addition, the extent of respiratory impairment was significantly less in Drp1 KO cells at 5 min (Figure 4C) but not at 10 min (Figure 4D) following FCCP addition, indicating that Drp1 deficiency alters the kinetics but not the ultimate extent of cytochrome c release.
Figure 4.

ABT-737-impaired maximal O2 consumption is restored by exogenous cytochrome c in both WT and Drp1 KO cells. WT (A) or Drp1 KO (B) MEF cells were exposed to ABT-737 (10 μM) or vehicle (con) for 1 h, followed by acute plasma membrane permeabilization by saponin (sap; 7.5 μg·mL−1) in the presence of the complex II substrate succinate (5 mM), the complex I inhibitor rotenone (0.5 μM), the uncoupler FCCP (5 μM) and the absence or presence of cytochrome c (cyt c; 100 μM). Results in (A) and (B) are mean ± SD for representative experiments performed in triplicate. OCR is baseline normalized to the point before vehicle or ABT-737 addition. In some cases, the error bars are smaller than the symbol size. (C) Quantification of the maximal OCR at the initial measurement point after saponin permeabilization as a percentage of control (no ABT-737 or cyt c). (D) Quantification of the maximal OCR at the second measurement point after permeabilization as a percentage of control. Results in (C) and (D) are mean ± SEM of three (WT) or four (Drp1 KO) independent experiments with two to three replicates per experiment. *P < 0.05 relative to the control treatment. #P < 0.05 for Drp1 KO relative to WT.
Drp1 deficiency protects against a small initial ABT-737-induced increase in ATP synthesis-independent oxygen consumption
The ATP synthase inhibitor oligomycin was added in the bioenergetic profiling experiments depicted in Figure 3 to investigate changes in ATP synthesis-independent OCR. ATP synthesis-independent OCR is primarily due to proton leak across the mitochondrial inner membrane and reactive oxygen species production. Variability in oligomycin-insensitive OCR was high but did not differ significantly between WT and Drp1 KO MEF (35.6 ± 14.0% of basal OCR vs. 46.1 ± 5.9% of basal OCR, respectively, mean ± SD, n = 5). ABT-737 induced a small but significant increase in oligomycin-insensitive OCR in WT cells (Figure 5A and C) that was significantly attenuated by Drp1 deficiency (Figure 5B and C). Evaluation of bioenergetic characteristics of WT versus Drp1 KO cells in the absence of ABT-737 treatment will be reported elsewhere (P. Clerc et al., in preparation).
Figure 5.
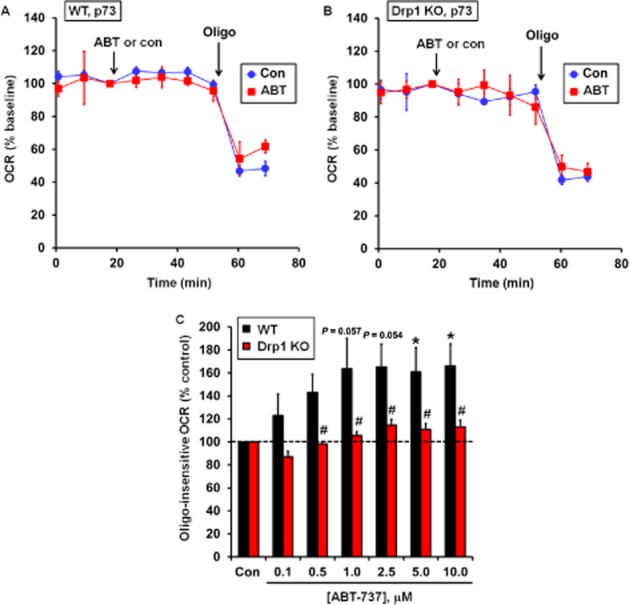
Drp1 KO cells are resistant to an ABT-737-induced increase in ATP synthesis-independent O2 consumption. Representative experiments performed in triplicate are shown for WT (A) and Drp1 KO cells (B) treated with vehicle (con) or 1 μM ABT-737 (ABT) followed by oligomycin (oligo; 0.3 μg·mL−1). OCR is baseline normalized to the point before vehicle or ABT-737 addition. (C) Oligo-insensitive OCR following addition of ABT-737 as percentage vehicle control. Oligo-insensitive OCR was calculated as a percentage of the vehicle control at the second measurement point after oligo addition in (A) and (B). Results in (C) are mean ± SEM of five independent experiments with one to three technical replicates per experiment. *P < 0.05 for ABT-737 treated relative to control treated; #P < 0.05 for Drp1 KO relative to WT. The P-values for 1.0 and 2.5 μM ABT-737-treated WT MEF relative to control treated cells were 0.057 and 0.054 respectively.
Drp1 KO MEF are not protected from ABT-737-induced caspase-dependent cell death
Next, we tested whether Drp1 KO MEF are resistant to ABT-737-induced cell death. A 4 h treatment with ABT-737 induced cell death in both WT and Drp1 KO MEF, as measured by release of the cytoplasmic enzyme LDH (Figure 6). The extent of LDH release was not significantly different between WT and Drp1 KO MEF at 0.5, 1.0 or 10.0 μM ABT-737. LDH release was completely blocked by the caspase inhibitor Q-VD in both cell types (Figure 6), indicating that death occurred by a caspase-dependent apoptotic mechanism. WT and Drp1 KO MEF did not differ in the extent of LDH release induced by saponin (Figure 6), indicating that the releasable LDH pool was similar in both cell types.
Figure 6.
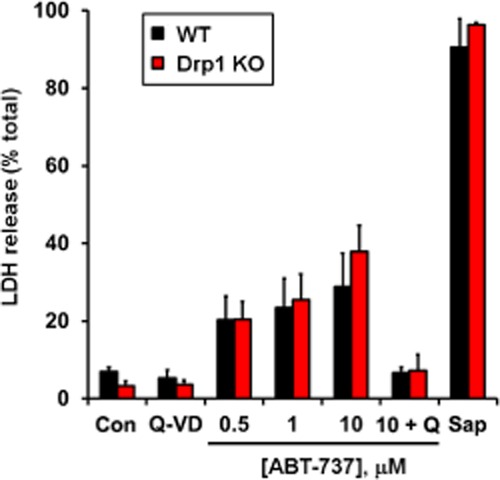
Drp1 deficiency does not attenuate rapid, ABT-737-induced caspase-dependent cell death. WT or Drp1 KO MEF cells were treated with vehicle control (con), ABT-737 (0.5, 1.0 or 10 μM), Q-VD (20 μM), ABT-737 (10 μM) + Q-VD (20 μM) or saponin (100 μg·mL−1) for 4 h. Cell death is expressed as percentage release of the cytoplasmic enzyme LDH. Results are mean ± SEM of three independent experiments performed in triplicate.
Pro-apoptotic Bak is up-regulated in Drp1 KO MEF
Finally, because sensitivity to ABT-737 can be influenced by Bcl-2 family protein expression levels, we surveyed the expression of the key pro-apoptotic death effectors Bax and Bak in WT and Drp1 KO MEF. Drp1 KO MEF displayed no difference compared with WT in the expression of Bax (Figure 7A and C). In contrast, ∼28 kDa Bak and a ∼76 kDa band detected by the Bak antibody were significantly elevated in Drp1 KO MEF (Figure 7B and C). The 28 kDa band was absent in immortalized Bax/Bak DKO MEF (Figure 7D), confirming its identity as Bak, whereas the 76 kDa band was present, indicating that it was the result of non-specific immunoreactivity. Bax identity was also confirmed using Bax/Bak DKO cells (data not shown).
Figure 7.
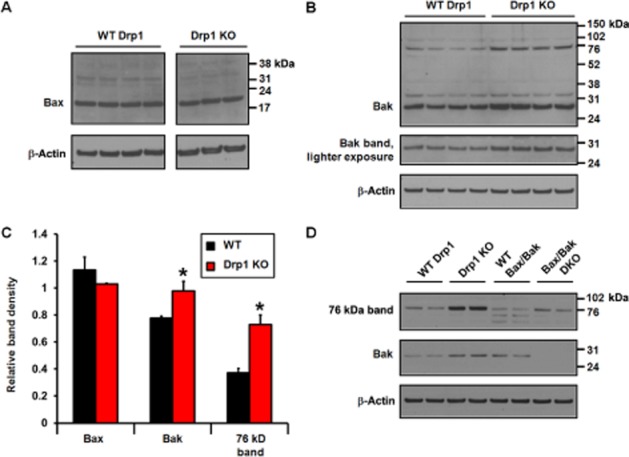
Pro-apoptotic Bak but not Bax is up-regulated in Drp1 KO MEF. Bax (A) and Bak (B) were detected by immunoblot in WT and Drp1 KO MEF, with immunodetection for β-actin as a control for loading. In (C), band densities for Bax, Bak and the ∼76 kDa Bak antibody immunoreactive band were quantified by densitometry and normalized to β-actin. Results are mean ± SEM, n = 3–4. *P < 0.05 for Drp1 KO relative to WT. In (D), Bak antibody immunoreactive bands in WT and Drp1 KO MEF were compared with those detected in WT and Bax/Bak DKO MEF.
Discussion and conclusions
Bax and Bak are considered essential gatekeepers of the mitochondrial pathway of apoptosis (Wei et al., 2001). The role of Drp1 in mitochondrial outer membrane permeabilization, cytochrome c release and apoptosis is more controversial. We evaluated the contribution of Drp1 to these phenomena in ‘primed for death’ cells that absolutely depend on anti-apoptotic Bcl-2 proteins to suppress death signals, reasoning that once cells are primed, the apoptotic function of Drp1 may no longer be required.
We found that although Drp1 localized to MCF10A Bcl-2 mitochondria, the putative Drp1 inhibitor mdivi-1 exhibited no ability to block ABT-737-induced cytochrome c release from primed MCF10A Bcl-2 overexpressing cells at the same or higher concentrations reported to inhibit Bid/Bax-induced membrane permeabilization (Cassidy-Stone et al., 2008; Kushnareva et al., 2012). Assuming mdivi-1 attenuates Drp1 activity as effectively in MCF10A Bcl-2 cells as it does in numerous other cell types, this suggests that mdivi-1 inhibits Bax/Bak at an activation stage that is already bypassed in primed MCF10A Bcl-2 cells. Anti-apoptotic Bcl-2 family proteins forestall Bax/Bak activation by binding direct activators such as Bid and Bim, termed MODE 1 inhibition, or by sequestering the Bax/Bak death effectors themselves, termed MODE 2 inhibition (Llambi et al., 2011). During MODE 2 inhibition, Bax and Bak exhibit conformational alterations consistent with partial activation, including Bax membrane insertion (Llambi et al., 2011). Thus, it is possible that Bax and Bak are already in an activated state when bound to anti-apoptotic Bcl-2 in primed MCF10A Bcl-2 cells, eliminating the need for the membrane-altering functions of Drp1.
Kushnareva et al. (2012) observed inhibition of Bax-induced membrane permeabilization by mdivi-1 even in the absence of detectable Drp1, making it unclear whether Drp1 is the relevant mdivi-1 target in its blockade of cytochrome c efflux. To more specifically examine the role of Drp1 versus mitochondrial mdivi-1 targets in Bax/Bak-mediated mitochondrial outer membrane permeabilization, we compared the effects of ABT-737 on WT and Drp1 KO MEF. Dose-dependent loss of maximal OCR due to the disappearance of mitochondrial cytochrome c electron transfer activity was observed in Drp1 KO MEF at similar ABT-737 concentrations to those attenuating maximal OCR in WT cells. This finding indicates that Drp1 is dispensable for cytochrome c release in MEF. The extent of maximal OCR decline was less in Drp1 KO MEF over a range of ABT-737 concentrations compared with WT cells although cytochrome c content was similar, suggesting that the kinetics of cytochrome c release are likely impaired. This delay was more clearly observed in ABT-737-treated Drp1 KO MEF permeabilized by saponin in the absence and presence of cytochrome c, where a difference in cytochrome c-sensitive respiratory impairment was initially observed relative to WT cells that disappeared after an additional 5 min of incubation.
Our findings suggesting that Drp1 regulates the kinetics of cytochrome c release in immortalized MEF are consistent with reports that Drp1 regulates the mitochondrial efflux of cytochrome c through the remodelling of mitochondrial cristae structure (Germain et al., 2005; Ban-Ishihara et al., 2013). Notably, in several reports the release of the intermembrane space protein Smac/DIABLO, in contrast to cytochrome c, was not modulated by Drp1 (Parone et al., 2006; Estaquier and Arnoult, 2007; Ishihara et al., 2009). Thus, Drp1 may separately promote apoptosis through mobilization of intracristal cytochrome c pools and via acceleration of mitochondrial outer membrane pore formation. A limitation of our study is that we did not directly monitor the release of cytochrome c and Smac/DIABLO in response to ABT-737, making it impossible to distinguish between these two possible functions of Drp1. Nevertheless, we observed an interesting difference between the two primed for death models employed, with no effect of mdivi-1 on cytochrome c release in the MCF10A Bcl-2 overexpression model and an apparent delay of cytochrome c release by Drp1 deficiency in the spontaneously immortalized MEF model. A potential explanation for this difference is that pro-apoptotic Bax and Bak are primarily held in check by MODE 2 inhibition in MCF10A Bcl-2 overexpressing cells but by MODE 1 inhibition (or by a mixture of the two) in immortalized MEF. Drp1 may be required for the Bax/Bak activation step triggered by the liberation of BH3-only molecules using ABT-737 (repression of MODE 1) but not for the subsequent pore formation that occurs after repression of Bcl-2 bound to already activated Bax/Bak (repression of MODE 2). Consistent with this possibility, overexpression of dominant negative Drp1 was found to impair Bax membrane insertion (Brooks et al., 2011), an activation step already bypassed in MODE 2-inhibited primed cells (Llambi et al., 2011). A goal of future work will be to elucidate whether the existence of MODE 1 versus MODE 2 priming can explain inconsistencies in regulation of cytochrome c release by Drp1.
In addition to controversy surrounding the importance of Drp1 to Bax/Bak-induced cytochrome c release, discrepancies in the ability of Drp1 deficiency to attenuate apoptosis have also been documented. Interestingly, Drp1 was required for developmental apoptosis during neural tube formation in vivo but not for the apoptosis of MEF in response to staurosporine, etoposide or UV irradiation (Wakabayashi et al., 2009). Here, we found that spontaneously immortalized MEF generated from the same Drp1 KO animals showed no resistance to ABT-737-induced cell death. In contrast to our results obtained with MEF, albeit in a different cell type, siRNA knockdown of Drp1 protected cerebellar granule neurons from ABT-737-induced apoptosis, measured at the same time point as in our study. A pitfall of using constitutive genetic manipulations to evaluate protein function is the propensity for secondary changes in the proteome. In this study, we identified up-regulation of pro-apoptotic Bak and a 76 kDa protein recognized by the Bak antibody in Drp1 KO MEF. Interestingly, the unidentified 76 kDa protein that was elevated in Drp1 KO MEF was also higher in apoptosis-deficient Bax/Bak DKO MEF compared with WT. It is possible that up-regulation of Bak or another protein was able to compensate for loss of Drp1 function in our model but did not occur during developmental neural tube apoptosis in vivo or following short-term Drp1 knockdown in cerebellar granule cells. The discovery of elevated Bak in Drp1 KO cells warrants a more complete examination of Bcl-2 family protein alterations resulting from Drp1 deficiency, as well as experiments to decipher how these changes impact mitochondrial morphology and apoptosis sensitivity. In addition to its apoptotic function, Bak was implicated in mitochondrial fragmentation during apoptosis (Brooks et al., 2007) and therefore may also influence mitochondrial dynamics in the context of Drp1 deficiency.
Finally, we observed a small but significant increase in ATP synthesis-independent oxygen consumption in WT MEF that was mitigated by Drp1 deficiency. Bax-dependent mitochondrial cytochrome c efflux increases reactive oxygen species production (Starkov et al., 2002). The delay in cytochrome c release in Drp1 KO cells relative to WT may explain the reduction of ATP synthesis-independent oxygen consumption in Drp1 KO cells if the change in oxygen consumption is due to reactive oxygen species generation. Alternatively, Drp1 deletion may prevent increased mitochondrial inner membrane proton leak occurring downstream of Bax/Bak activation (Scorrano et al., 2002), perhaps associated with Drp1-dependent mitochondrial cristae remodelling (Germain et al., 2005).
The primary conclusion of our study is that Drp1 is not required for cytochrome c release and apoptosis of cells that are already primed for death. However, under some contexts Drp1 contributes to the kinetics of cytochrome c release and regulates alterations in respiration, probably as a consequence of its effect on cytochrome c efflux. An up-regulation of Bak was identified in Drp1 KO cells for the first time, highlighting the importance of investigating changes in potentially compensatory apoptotic pathways that may shape conclusions on the role of Drp1 in apoptosis. Examination of Drp1 deficiency in the absence of changes in Bcl-2 family proteins, as well as a more detailed investigation of Bax/Bak activation status in primed cells will ultimately be necessary to completely unravel how Drp1 participates in the cytochrome c release and death pathways.
Acknowledgments
This work was supported by NIH/NINDS grant NS064978 to B. M. P. and by NIH/NIGMS grant GM089853 to H. S.
Glossary
- DKO
double knockout
- Drp1
dynamin-related protein 1
- KO
knockout
- MEF
mouse embryonic fibroblasts
- OCR
oxygen consumption rate
- WT
wild type
Conflict of interest
B. M. P. has consulted for Seahorse Bioscience, manufacturer of equipment used to conduct experiments described in this article.
References
- Abramov AY, Duchen MR. Mechanisms underlying the loss of mitochondrial membrane potential in glutamate excitotoxicity. Biochim Biophys Acta. 2008;1777:953–964. doi: 10.1016/j.bbabio.2008.04.017. [DOI] [PubMed] [Google Scholar]
- Ban-Ishihara R, Ishihara T, Sasaki N, Mihara K, Ishihara N. Dynamics of nucleoid structure regulated by mitochondrial fission contributes to cristae reformation and release of cytochrome c. Proc Natl Acad Sci U S A. 2013;110:11863–11868. doi: 10.1073/pnas.1301951110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billen LP, Kokoski CL, Lovell JF, Leber B, Andrews DW. Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol. 2008;6:e147. doi: 10.1371/journal.pbio.0060147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks C, Wei Q, Feng L, Dong G, Tao Y, Mei L, et al. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci U S A. 2007;104:11649–11654. doi: 10.1073/pnas.0703976104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks C, Cho SG, Wang CY, Yang T, Dong Z. Fragmented mitochondria are sensitized to Bax insertion and activation during apoptosis. Am J Physiol Cell Physiol. 2011;300:C447–C455. doi: 10.1152/ajpcell.00402.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Clerc P, Polster BM. Investigation of mitochondrial dysfunction by sequential microplate-based respiration measurements from intact and permeabilized neurons. PLoS ONE. 2012;7:e34465. doi: 10.1371/journal.pone.0034465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerc P, Carey GB, Mehrabian Z, Wei M, Hwang H, Girnun GD, et al. Rapid detection of an ABT-737-sensitive primed for death state in cells using microplate-based respirometry. PLoS ONE. 2012;7:e42487. doi: 10.1371/journal.pone.0042487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117:112–121. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dlugosz PJ, Billen LP, Annis MG, Zhu W, Zhang Z, Lin J, et al. Bcl-2 changes conformation to inhibit Bax oligomerization. EMBO J. 2006;25:2287–2296. doi: 10.1038/sj.emboj.7601126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C, et al. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell. 2011;145:104–116. doi: 10.1016/j.cell.2011.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estaquier J, Arnoult D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 2007;14:1086–1094. doi: 10.1038/sj.cdd.4402107. [DOI] [PubMed] [Google Scholar]
- Fiskum G, Craig SW, Decker GL, Lehninger AL. The cytoskeleton of digitonin-treated rat hepatocytes. Proc Natl Acad Sci U S A. 1980;77:3430–3434. doi: 10.1073/pnas.77.6.3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, et al. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- Gavathiotis E, Reyna DE, Davis ML, Bird GH, Walensky LD. BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol Cell. 2010;40:481–492. doi: 10.1016/j.molcel.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain M, Mathai JP, McBride HM, Shore GC. Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. EMBO J. 2005;24:1546–1556. doi: 10.1038/sj.emboj.7600592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick JM, Polster BM. Bax, along with lipid conspirators, allows cytochrome c to escape mitochondria. Mol Cell. 2002;10:963–965. doi: 10.1016/s1097-2765(02)00751-7. [DOI] [PubMed] [Google Scholar]
- Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- Kageyama Y, Zhang Z, Roda R, Fukaya M, Wakabayashi J, Wakabayashi N, et al. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J Cell Biol. 2012;197:535–551. doi: 10.1083/jcb.201110034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, et al. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002;159:931–938. doi: 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–388. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Kushnareva Y, Andreyev AY, Kuwana T, Newmeyer DD. Bax activation initiates the assembly of a multimeric catalyst that facilitates Bax pore formation in mitochondrial outer membranes. PLoS Biol. 2012;10:e1001394. doi: 10.1371/journal.pbio.1001394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- Llambi F, Moldoveanu T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL, et al. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell. 2011;44:517–531. doi: 10.1016/j.molcel.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SS, Leder P. Human MCF10A mammary epithelial cells undergo apoptosis following actin depolymerization that is independent of attachment and rescued by Bcl-2. Mol Cell Biol. 2001;21:6529–6536. doi: 10.1128/MCB.21.19.6529-6536.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino D, Khaw SL, Glaser SP, Anderson DJ, Belmont LD, Wong C, et al. Bcl-2, Bcl-xL and Bcl-w are not equivalent targets of ABT-737 and Navitoclax (ABT-263) in lymphoid and leukemic cells. Blood. 2012;119:5807–5816. doi: 10.1182/blood-2011-12-400929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, et al. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell. 2010;142:889–901. doi: 10.1016/j.cell.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Ferguson SJ. Bioenergetics 3. London: Academic Press; 2002. [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Parone PA, James DI, Da CS, Mattenberger Y, Donze O, Barja F, et al. Inhibiting the mitochondrial fission machinery does not prevent Bax/Bak-dependent apoptosis. Mol Cell Biol. 2006;26:7397–7408. doi: 10.1128/MCB.02282-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton C, Thompson S, Epel D. Some precautions in using chelators to buffer metals in biological solutions. Cell Calcium. 2004;35:427–431. doi: 10.1016/j.ceca.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Polster BM, Fiskum G. Mitochondrial mechanisms of neural cell apoptosis. J Neurochem. 2004;90:1281–1289. doi: 10.1111/j.1471-4159.2004.02572.x. [DOI] [PubMed] [Google Scholar]
- Polster BM, Kinnally KW, Fiskum G. BH3 death domain peptide induces cell type-selective mitochondrial outer membrane permeability. J Biol Chem. 2001;276:37887–37894. doi: 10.1074/jbc.M104552200. [DOI] [PubMed] [Google Scholar]
- Polster BM, Basanez G, Young M, Suzuki M, Fiskum G. Inhibition of Bax-induced cytochrome c release from neural cell and brain mitochondria by dibucaine and propranolol. J Neurosci. 2003;23:2735–2743. doi: 10.1523/JNEUROSCI.23-07-02735.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, et al. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- Sheridan C, Delivani P, Cullen SP, Martin SJ. Bax-or Bak-induced mitochondrial fission can be uncoupled from cytochrome C release. Mol Cell. 2008;31:570–585. doi: 10.1016/j.molcel.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Starkov AA, Polster BM, Fiskum G. Regulation of hydrogen peroxide production by brain mitochondria by calcium and Bax. J Neurochem. 2002;83:220–228. doi: 10.1046/j.1471-4159.2002.01153.x. [DOI] [PubMed] [Google Scholar]
- Tagscherer KE, Fassl A, Campos B, Farhadi M, Kraemer A, Bock BC, et al. Apoptosis-based treatment of glioblastomas with ABT-737, a novel small molecule inhibitor of Bcl-2 family proteins. Oncogene. 2008;27:6646–6656. doi: 10.1038/onc.2008.259. [DOI] [PubMed] [Google Scholar]
- Terrones O, Antonsson B, Yamaguchi H, Wang HG, Liu J, Lee RM, et al. Lipidic pore formation by the concerted action of proapoptotic BAX and tBID. J Biol Chem. 2004;279:30081–30091. doi: 10.1074/jbc.M313420200. [DOI] [PubMed] [Google Scholar]
- Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, et al. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. 2009;186:805–816. doi: 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14:2060–2071. [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292:C125–C136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- Young KW, Pinon LG, Dhiraj D, Twiddy D, Macfarlane M, Hickman J, et al. Mitochondrial fragmentation and neuronal cell death in response to the Bcl-2/Bcl-x(L)/Bcl-w antagonist ABT-737. Neuropharmacology. 2010;58:1258–1267. doi: 10.1016/j.neuropharm.2010.03.008. [DOI] [PubMed] [Google Scholar]
- Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 2001;15:1481–1486. doi: 10.1101/gad.897601. [DOI] [PMC free article] [PubMed] [Google Scholar]