

Abstract
Cells die by a variety of mechanisms. Terminally differentiated cells such as neurones die in a variety of disorders, in part, via parthanatos, a process dependent on the activity of poly (ADP-ribose)-polymerase (PARP). Parthanatos does not require the mediation of caspases for its execution, but is clearly mechanistically dependent on the nuclear translocation of the mitochondrial-associated apoptosis-inducing factor (AIF). The nuclear translocation of this otherwise beneficial mitochondrial protein, occasioned by poly (ADP-ribose) (PAR) produced through PARP overactivation, causes large-scale DNA fragmentation and chromatin condensation, leading to cell death. This review describes the multistep course of parthanatos and its dependence on PAR signalling and nuclear AIF translocation. The review also discusses potential targets in the parthanatos cascade as promising avenues for the development of novel, disease-modifying, therapeutic agents.
LINKED ARTICLES
This article is part of a themed issue on Mitochondrial Pharmacology: Energy, Injury & Beyond. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2014.171.issue-8
Keywords: PARP-1, parthanatos, AIF, mitochondria, cell death, therapy
Cell death: introduction and current nomenclature
The phenomenon of cell death is a subject of intense investigation. Current research is focused on unravelling the sequence of induction of the molecular events and characteristic signatures that define a particular death type, as well as the interactions and molecular bridges linking the different death types. As a consequence, cell death nomenclature continues to grow, as more evidence comes to light regarding multiple and distinct ways by which a cell can die. To date, morphological, enzymological, functional (biochemical) and immunological features are criteria commonly used to classify cell death (Kroemer et al., 2009; Galluzzi et al., 2012). Based on morphological appearance, apoptosis and necrosis were described as the first two distinct routes of cell death. Apoptosis, a term now more than four decades old (Kerr et al., 1972), is an active form of cell death. It is highly regulated or programmed, which, consistent with an exceptionally choreographed nature, is highly conserved among organisms (Chew et al., 2009) and required for proper development (Lorenzo and Susin, 2007). Necrosis usually is a passive process, once thought to be mostly unregulated or random, but now considered to involve programmed components in some cases (e.g. necroptosis) (Lorenzo and Susin, 2007).
There are now many other modalities of cell death, including anoikis (Frisch and Ruoslahti, 1997), autophagic cell death, caspase-dependent intrinsic apoptosis, caspase-independent intrinsic apoptosis, regulated necrosis (some investigators prefer to use this term to describe or include caspase-independent intrinsic apoptosis, given their observation that apoptosis necessarily features early caspase activation), cornification, entosis (Mormone et al., 2006), extrinsic apoptosis by death receptors, extrinsic apoptosis by dependence receptors, mitotic catastrophe, netosis, pyroptosis and parthanatos (see Galluzzi et al. 2012). Other forms of cell death may exist as well, including ferroptosis, an iron-dependent form of non-apoptotic cell death (Dixon et al., 2012). Some of these new cell-death modalities were defined initially by morphological criteria, but biochemical/functional definitions of cell-death modalities are replacing the morphological definitions to reflect accuracy and consistency of description. It should be emphasized that there are many instances when cell death in a particular setting displays features of more than a single form of cell death. This makes it rather challenging to employ morphological criteria to describe such mixed cell-death scenarios. Thus, descriptions based on biochemical or functional characteristics are ultimately more relevant. In 2009, the Nomenclature Committee on Cell Death issued recommendations in relation to the use of these concepts and definitions (Kroemer et al., 2009), which were expanded in their more recent (2012) round of recommendations, in order to produce a systematic classification of cell death based on measurable biochemical features (Galluzzi et al., 2012).
In this review, we discuss the entire course and sequence of events of parthanatos and the key role of apoptosis-inducing factor (AIF), a mitochondrial protein whose nuclear translocation from the mitochondria sets in motion the commitment-to-death fate of cells or tissues in which the event has taken place. Parthanatos is a unique and highly choreographed form of cell death, which occurs through the overactivation of the nuclear enzyme, PARP-1, also known as poly (ADP-ribose) synthetase 1 or poly (ADP-ribose) transferase 1.
PARP-1 in cell survival and cell death
PARP-1 (EC 2.4.2.30) is the best known, most abundant and seemingly most important of an expanding family of about 17 proteins classified on the basis of protein domain homology and enzymatic function (some of them have not been fully characterized). PARP-1 helps in the regulation of cellular homeostasis and the preservation of genomic stability under physiological conditions (Smith, 2001; Hong et al., 2004; Krietsch et al., 2012). It is a 116 kDa protein with three major domains: an N-terminal domain (42 kDa) that has two zinc finger motifs and a nuclear localization sequence used for DNA-binding; a central automodification domain (16 kDa) and a C-terminal catalytic domain (55 kDa) harbouring the NAD-binding site and the poly (ADP-ribose) (PAR)-synthesizing domain (Kameshita et al., 1984; Hong et al., 2004; Sousa et al., 2012; Virag and Szabo, 2002) (Figure 1). In terms of amino acid sequence, there is nearly 92% homology between the human and mouse enzyme, evidence that PARP-1 is highly conserved in eukaryotes (Virag and Szabo, 2002). Interestingly, homology is highest for the catalytic domain that contains the PARP signature sequence (Virag and Szabo, 2002). Among a number of other possible roles, PARP-1 is known to regulate gene expression and amplification, cell differentiation, cell division, malignant transformation, DNA replication, mitochondrial function and cell death (D'Amours et al., 1999; Chiarugi, 2002; Kraus and Lis, 2003; Hong et al., 2004; Gagne et al., 2008).
Figure 1.

Primary structure of PARP-1. Primary structure of PARP-1 showing its three domains: An N-terminal DNA-binding domain containing two zinc-finger motifs and a nuclear localization sequence (NLS), a central automodification domain [with a breast cancer-associated gene 1 (BRCA1) C-terminal (BRCT) motif containing phosphorylation sites for regulating PARP-1 activity], and a C-terminal catalytic domain containing the nicotinamide adenine dinucleotide (NAD)-binding site and the poly (ADP-ribose) polymer (PAR)-synthesizing domain.
Consistent with its function in maintaining nuclear homeostasis, PARP-1 constitutes a DNA base-excision repair system by sensing DNA strand nicks and breaks and facilitating their repair through the synthesis of PAR polymer. When there is mild DNA damage, the activity of PARP-1 increases up to 500-fold and the enzyme makes use of oxidized NAD (NAD+), produced through the expenditure of ATP, to synthesize PAR polymer. More than 95% of the synthesized PAR polymer comes from PARP-1, in contrast to the very minimal contribution from other isoforms (Dawson and Dawson, 2004). About 50–200 PAR polymer residues are then attached to PARP itself (autoribosylation) and to other nuclear (acceptor) proteins (heteroribosylation) such as histones, topoisomerases I and II, DNA polymerases, DNA ligase-2, high-mobility group proteins, transcription factors, as well as many other proteins (Hong et al., 2004; Shall and de Murcia, 2000; Smulson et al., 2000). This biochemical pathway is a unique one and the synthesis and degradation of PAR has been found to occur in most mitotic and post-mitotic cells in mammalian organisms (Kanai et al., 2000; Shall and de Murcia, 2000). However, when DNA damage is profound, PARP-1 becomes excessively activated and produces long-chained, branched polymers of PAR, leading to externalization of phosphatidylserine, dissipation of the mitochondrial membrane potential, translocation of AIF from the mitochondria to the nucleus, large-scale DNA fragmentation (≈50 kb) and chromatin condensation, followed by cell death (Andrabi et al., 2008; David et al., 2009).
In the context of the CNS, which is highlighted in this review to illustrate PARP-1-mediated cell death mechanisms that are shared in most cases by non-neuronal systems, stimuli that induce pathological activation of PARP-1 in in vitro and in vivo studies include oxidative stress by reactive oxygen species (ROS), such as hydrogen peroxide (H2O2) or hydroxyl radical, nitrosative stress from NO or peroxynitrite (ONOO−), inflammation, ischaemia (or ischaemic reperfusion), hypoxia, hypoglycaemia and DNA-alkylating agents, such as N-methyl-N’-nitro-N-nitrosoguanidine (MNNG). For example, ischaemic injury induces the release and accumulation of the excitatory neurotransmitter, glutamate, which causes excessive activation of its ionotropic receptors (NMDA, AMPA and kainate receptors; receptor nomenclature follows Alexander et al., 2013). These can all cause calcium influx, but NMDA receptor activation plays a primary role, leading to the activation of neuronal NO synthase (Dawson et al., 1991; 1993,1996). Activation of endothelial NOS and inducible NOS is also important to NO production (and PARP-1 activation) in non-neuronal tissues (Virag and Szabo, 2002). ROS such as superoxide anion (O2.−) or H2O2 are also produced in tissue injury. NO is able to induce poly (ADP-ribosyl)ation (Zhang et al., 1994) but, more importantly, O2.− has the capacity to react with NO to form ONOO−, a potent inducer of DNA damage and consequently of PARP-1 activation (Zhang et al., 1994; Xia et al., 1996; Eliasson et al., 1997; Gonzalez-Zulueta et al., 1998; Wang et al., 2004; Pacher and Szabo, 2008). Excessive stimulation of glutamate receptors also results from hypoxia, hypoglycaemia and inflammation, leading to DNA damage (Stone and Addae, 2002), while oxidative stress and DNA-alkylating agents may damage DNA through other means, directly or indirectly (Fatokun et al., 2008) (Figure 2).
Figure 2.
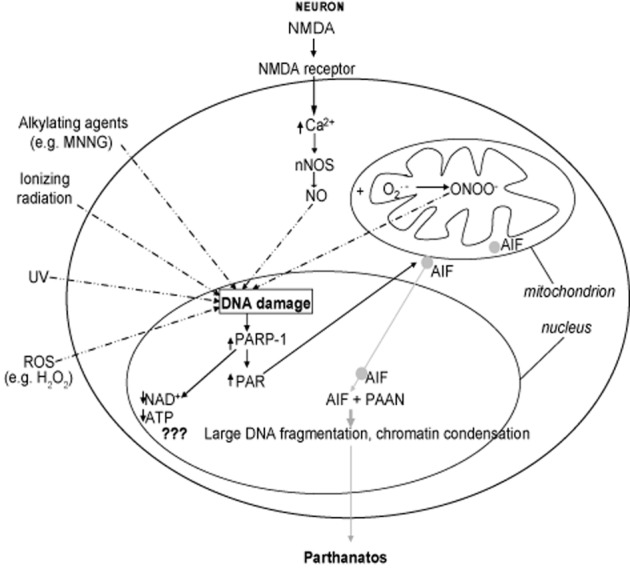
Cascade of events leading to parthanatos. Diagrammatic representation, in a typical (neuronal or non-neuronal) cell, of the multiple steps in parthanatos. In neurones, activation of the NMDA receptor leads to increased calcium influx, resulting in the activation of calcium-dependent neuronal nitric oxide synthase (nNOS) that produces NO. NO may induce DNA damage directly, but more commonly reacts with superoxide (O2._) in the mitochondria to generate peroxynitrite (ONOO−), which is a very potent inducer of DNA damage. Production of NO through activation of endothelial NOS (eNOS) or inducible NOS (iNOS) may be more important in non-neuronal cells, leading to ONOO− generation (not shown). Some stimuli can induce DNA damage directly, including reactive oxygen species (ROS), for example hydrogen peroxide (H2O2), alkylating agents [e.g. N-methyl-N’-nitro-N-nitrosoguanidine (MNNG)], UV radiation and ionizing radiation. Stimuli inducing DNA damage are shown by broken arrows. DNA damage causes PARP-1 overactivation that leads to poly (ADP-ribose) (PAR) polymer synthesis and accumulation. PARP-1 overactivation depletes cellular pool of nicotinamide adenine dinucleotide (NAD+) and ATP, but this does not seem to be the primary cause of cell death (indicated by ???). PAR polymer signals to the mitochondria and directly binds to the PAR polymer-binding site on apoptosis-inducing factor (AIF), inducing its mitochondrial release and translocation to the nucleus. Once in the nucleus, it causes large-scale (≈50 kb) DNA fragmentation and chromatin condensation through as yet unidentified parthanatos AIF-associated nuclease (PAAN). This is believed to be the cause of cell death. Events after AIF release from the mitochondria are depicted in grey, solid arrows.
Parthanatos (PARP-1-mediated cell death)
Following a relatively recent coinage, the form of cell death that occurs as a result of the overactivation of PARP-1 is referred to as ‘parthanatos,’ a portmanteau term (par-thanatos) derived from ‘par’ (for PAR polymer, synthesized following PARP-1 activation), and ‘Thanatos,’ the personification of death in Greek mythology (Andrabi et al., 2008; Harraz et al., 2008; David et al., 2009; Wang et al., 2009a). The name ‘parthanatos’ is now officially recognized and has thus entered the cell death pantheon (Galluzzi et al., 2012). As a distinct death pathway, the time course of parthanatos is associated with the biochemical events of rapid activation of PARP-1, synthesis and accumulation of PAR polymer, mitochondrial depolarization, nuclear AIF translocation (which occurs early) and, perhaps at the late stage, caspase activation, although caspase activation is not obligatory (Yu et al., 2002; 2006; Andrabi et al., 2006). It should be emphasized that, while events such as rapid activation of PARP-1, PAR synthesis and accumulation and nuclear AIF translocation are unique to parthanatos, which therefore characterize this process biochemically, other events that accompany it, such as mitochondrial depolarization, are not exclusive occurrences but shared with other forms of cell death. Some further account of the nature and sequence of this array of events has been provided by recent reviews (Andrabi et al., 2008; Wang et al., 2009a). Of particular interest is that parthanatos does not obligatorily require caspases for its execution, as it cannot be rescued by pan-caspase inhibitors, for example boc-aspartyl-fmk or Z-VAD-fmk (Yu et al., 2002). This feature, therefore, distinguishes parthanatos from caspase-dependent apoptosis that cannot proceed without the involvement of relevant caspases. Furthermore, unlike apoptosis, parthanatos does not induce the formation of apoptotic bodies, neither does it engender small-scale DNA fragmentation (Wang et al., 2009a) – it causes large-scale DNA fragmentation. Like necrosis, parthanatic death involves loss of cell membrane integrity, but, unlike it, is not accompanied by cell swelling (Yu et al., 2002; Wang et al., 2004). However, it should be emphasized that some investigators consider parthanatos and necroptosis as two subsets or examples of regulated or programmed necrosis (see Nagley et al. 2010), but even in this context, the co-involvement of PARP-1, PAR and AIF, which is lacking in any (other) forms of programmed or regulated necrosis, still distinguishes parthanatos. Major similarities and differences in biochemical, structural and other changes that occur in apoptosis, necrosis, autophagy and parthanatos are shown in Table 1 (adapted from Bredesen et al., 2006; Wang et al., 2009a; Galluzzi et al., 2012).
Table 1.
Similarities and differences between apoptosis, necrosis, autophagy and parthanatos
| Apoptosis | Necrosis | Autophagy | Parthanatos | |
|---|---|---|---|---|
| Variations/Subsets in the literature | Caspase-dependent intrinsica | Random or unregulated | Macroautophagy | |
| Caspase-independent intrinsicb | Programmed or regulated, e.g. necroptosis(some think parthanatos could also be considered a case of regulated necrosis) | Microautophagy | ||
| Extrinsic apoptosis by death receptorsc | Chaperone-mediated autophagy | |||
| Extrinsic apoptosis by dependence receptorsd | Mitophagy(but here we consider autophagy as macroautophagy) | |||
| (Biochemical) Signatures | ||||
| Mitochondrial | Caspase activation (except in b) | Loss of ultrastructure | Degradation | Depolarization |
| Mitochondrial depolarization | Swelling | Irreversible Δψm dissipation | ||
| MOMPa,c | ATP and NADH depletion | |||
| Irreversible Δψm dissipationa | AIF release | |||
| CYT c release | CYT c release | |||
| Release of IMS proteinsb | Caspase activation (late stage, non-obligatory) | |||
| Respiratory chain inhibitionb | ||||
| BID cleavagec | ||||
| PP2A activationd | ||||
| DAPK1 activationd | ||||
| Cytoplasmic | Shrinkage | Swelling (including of organelles) | Massive vacuolization | PAR polymer accumulation |
| Vacuolation | Lysosomal degradation | PAR-AIF interactions (binding) | ||
| Organellar disintegration | MAP1LC3 lipidation | Condensation | ||
| AIF translocation to the nucleus | ||||
| Nuclear | PARP cleavage | Chromatin digestion | SQSTM1 degradation | Rapid PARP-1 activation (not cleavage) |
| Chromatin condensation | DNA hydrolysis (smear) | PARP-1-mediated PAR synthesis | ||
| DNA fragmentation (small-scale, DNA ladder) | Chromatin condensation | |||
| PAAN activation (putative) | ||||
| DNA fragmentation (large-scale, ≈50 kb) | ||||
| Structural (plasma membrane) changes | Membrane integrity preserved | Loss of integrity | Double membrane-bound autophagosomes formed | Loss of integrity |
| Formation of apoptotic bodies | Blebbing | Phosphatidylserine externalization | ||
| Membrane blebbing | Cell lysis | |||
| Phosphatidylserine externalization | ||||
| Examples of trigger factors and/or conditions | Death receptor signallingc | Excitotoxicity | Amino acid starvation | Excitotoxicity |
| Dependence receptor signallingd | Ischaemia | Serum starvation | Ischaemia | |
| DNA damage | Stroke | Protein aggregates | DNA damage | |
| Trophic factor withdrawal | Reactive oxygen/nitrogen species | Stroke | ||
| Viral infections | Reactive oxygen/nitrogen species | |||
| Energy (ATP) requirement | + | − | + | − |
| (Obligatory) Caspase-dependence | +a,c,d | − | − | − |
| −b | ||||
| Inflammatory component | − | + | − | − |
| Major mediator(s) | Caspases (except in b) | Calpains, CYPD, RIP-1, RIP-3 (and PARP-1 and AIF, if parthanatos is considered regulated necrosis), etc. | ATG5, ATG6 (Beclin-1), ATG7, ATG12, VPS34, AMBRA-1 | PARP-1 |
| PAR | ||||
| AIF | ||||
| Pharmacological inhibition | Caspase inhibitors, e.g. Z-VAD-fmk (except in b) | RIP-1 inhibitors, e.g. necrostatin-1, calpain inhibitors, etc. | VPS34 inhibitors, e.g. 3-methyladenine and wortmannin | PARP-1 inhibitors, e.g. DPQ |
| Genetic inhibition (knockout/mutation, RNAi targeting) or inhibition by protein overexpression | BCL2 overexpressiona, b | Inhibition of RIP-1 or RIP-3 | Inhibition of AMBRA1, ATG5, ATG7, ATG12 or BECN1 | PARP-1 knockout, AIF down-regulation (e.g. in Harlequin mouse) |
| Inhibition of caspases (3, 8 and 9)c,d | ||||
| Inhibition of PP2Ad | ||||
| CrmA expressionc | ||||
Major similarities and differences in the biochemical, structural and other changes that occur in apoptosis, necrosis, autophagy and parthanatos as some types of cell death, as adapted from (Bredesen et al., 2006; Wang et al., 2009a; Galluzzi et al., 2012). For apoptosis, superscripts have been used where necessary to link each of its different subsets or variations to features that distinguish it. It is worthy to note that, while parthanatos is generally considered to be separate and distinct from necrosis, some investigators consider it to be a specific case of regulated necrosis, just as is necroptosis. Δψm is mitochondrial transmembrane potential.
AIF, apoptosis-inducing factor; AMBRA1, activating molecule in Beclin-1-regulated autophagy protein 1; ATG, autophagy; BCL2, B-cell lymphoma 2; BECN1, Beclin-1; CrmA, cytokine response modifier A; CYPD, cyclophilin D; CYT, cytochrome; DAPK1, death-associated protein kinase 1; DPQ, 3,4-Dihydro-5-[4-(1-piperidinyl)butoxyl]-1(2H)-isoquinolinone; IMS, intermembrane space; MAP1LC3, microtubule-associated protein 1 light chain 3; MOMP, mitochondrial outer membrane permeabilization; PAAN, parthanatos AIF-associated nuclease; PAR, poly (ADP-ribose); PP2A, protein phosphatase 2A; RIP, receptor-interacting protein; SQSTM1, sequestosome 1; VPS, vacuolar protein sorting; Z-VAD-fmk, N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone.
Extensive details of molecular mechanisms in parthanatos and the order in which they are presented in the cascade are only beginning to be unglued. It is established that factors such as the magnitude of the death-inducing stimulus, the length of exposure to such stimulus, the type and nature of cell used, and the purity of cultures are among those that can largely determine the death cascade that is activated in any experimental paradigm (Meli et al., 2004; Wang et al., 2009b). In its pure form, parthanatos has been described in several experimental cell culture and animal models that feature excessive activation of the PARP-1 enzyme, leading to severe, rather than mild-to-moderate, DNA damage. In most cases, the alkylating agent MNNG at concentrations between 50 and 500 μM applied for usually 5 to 25 min has been used as the gold standard in cell-death research to induce parthanatos in cell lines, including human cervical carcinoma HeLa cells (mostly used), CHO cells and rarely MDCK cells and the lymphoblastoid cell line TK-6 (Kim et al., 2005). This agent (MNNG) has also been used to study parthanatos in immortalized mouse embryonic fibroblasts and human fibroblasts (Yu et al., 2002; Yeh et al., 2005; David et al., 2006; Keil et al., 2006; Ethier et al., 2007; Lee et al., 2007b). Treatment of human pulmonary epithelial and vascular endothelial cells with MNNG or human umbilical vein endothelial cells with peroxide has also been found to cause activation of parthanatos (Geraets et al., 2007; Mathews and Berk, 2008). In primary cortical neuronal cultures prepared from fetal mice, NMDA (500 μM) applied briefly (5 min) robustly activates parthanatos (Yu et al., 2002; Wang et al., 2009b; Fatokun et al., 2013). Exposure to MNNG (10–300 μM, 30 min) also induced parthanatos in primary astrocyte cultures (Suzuki et al., 2010; Tang et al., 2010). It should be noted that a number of other stimuli or toxic conditions, such as H2O2, NO, ONOO− generation or oxygen-glucose deprivation, although relatively less commonly used, also induce parthanatos in a variety of cells (Moroni et al., 2001; Yu et al., 2002; Mathews and Berk, 2008; Son et al., 2009). The in vitro experimental conditions as highlighted here are known to favour parthanatos over other forms of cell death. Depending on the type, length and strength of the toxic stimulus, other forms of cell death due to DNA damage can be induced in addition to parthanatos. Thus, cell death that involves primarily parthanatos should be completely blocked by inhibitors of PARP or knockout of PARP. This is important in the design of experiments to investigate this form of cell death.
Degradation of PAR polymer by PARG
Poly (ADP-ribose) glycohydrolase (PARG) is the enzyme that regulates PAR levels by catalyzing its degradation after it is synthesized by PARP (Kameshita et al., 1984; Whitacre et al., 1995). PARG causes PAR hydrolysis to free ADP-ribose units through its endoglycosidic or exoglycosidic activity (Davidovic et al., 2001). Although encoded by a single gene, now characterized in humans and in other species (Ame et al., 1999; Winstall et al., 1999; Meyer-Ficca et al., 2004), alternatively spliced PARG isoforms have been reported (Bonicalzi et al., 2003; Haince et al., 2006; Meyer et al., 2007). Full-length PARG (110 kDa) localizes to the nucleus, but the splice variants have diverse localization patterns (Bonicalzi et al., 2003; Meyer-Ficca et al., 2004; Haince et al., 2006; Meyer et al., 2007; Burns et al., 2009). However, the relevance of each of the variants and its subcellular localization in relation to parthanatos has not been clarified. Evidence demonstrating the importance of PARG in parthanatos comes from studies involving overexpression, genetic deletion and pharmacological approaches in which PARG was found to protect against PAR-mediated cell death (Andrabi et al., 2006), H2O2-induced cell death (Blenn et al., 2006), excitotoxicity and stroke (Andrabi et al., 2006; Cozzi et al., 2006), while its deletion (PARG knockouts) increased toxicity, most probably through the accumulation of PAR (Andrabi et al., 2006). In mice, the whole-body knockout of PARG is embryonically lethal at around day E3.5 (Koh et al., 2004a), which may be due to excessive accumulation of the synthesized PAR polymer. Furthermore, trophoblasts from early PARG-knockout embryos do not survive without the PARP inhibitor, benzamide (Koh et al., 2004a). The cells were found to be extremely sensitive to MNNG and menadione, agents that activate PARP-1 excessively, leading to PAR accumulation (Koh et al., 2004a). These data show that regulation of PAR levels is critical to cell survival in parthanatos (Zhou et al., 2011).
A different enzyme, ADP-ribose-(arginine) protein hydrolase (ARH3), also has some PARG-like activity, but it does not appear that this activity plays a significant role in cell death, as ARH3 does not rescue Drosophila or mouse genetic knockouts of PARG from cell death or PAR accumulation (Hanai et al., 2004; Koh et al., 2004a).
Molecular mechanisms in parthanatos: PAR polymer as a death mediator
Quite understandably, a leading area of investigation that has generated much attention in parthanatos is the identification of the underlying molecular mechanisms, as the nature of cell death is unique and separate from those of other forms of cell death such as apoptosis and necrosis. Although there is considerable success in this area, our knowledge of molecular events in parthanatos is still not complete, and efforts are ongoing, not only to completely identify all the major players, but also to clarify the spatial and temporal relationships between such mediators.
So, how do cells die in parthanatos? It was initially assumed that the massive depletion of cellular NAD+ as a result of excessive activation of PARP-1 caused cells to commit suicide (Berger et al., 1983; Berger, 1985; Berger et al., 1985; Berger and Berger, 1986; Ha and Snyder, 1999). NAD+ is an important cofactor in glycolysis and the tricarboxylic acid cycle. The same NAD+ is required for the synthesis of PAR and, in turn, ATP is required for the synthesis of NAD+. In fact, each molecule of NAD+ requires four molecules of ATP for its synthesis. The suicide hypothesis, therefore, argues that following PARP-1 overactivation, there is excessive depletion of cytosolic and nuclear pools of NAD+, over time. Despite this observation, there is no convincing and direct evidence to date in favour of a role for energy depletion as a primary cause of parthanatos. Instead, although infarct volumes after focal ischaemic injury in PARP-1 knockout mice are smaller than in wild-type animals, cellular energy stores are not different between the two groups (Goto et al., 2002), suggesting that energy depletion is not the underlying cause of cell death in parthanatos. Moreover, in cells lacking PARG, activation of PARP-1 leads to cell death through parthanatos, in the absence of NAD+ depletion (Zhou et al., 2011).
The PAR polymer, generated when PARP-1 is overactivated, is now recognized as a key signalling molecule in the parthanatos cascade (Andrabi et al., 2006). When cells are exposed to a toxic stimulus that induces significant DNA damage, PARP-1 becomes overactivated and produces toxic levels of PAR, which translocates to the cytosol, from where it signals to AIF to effect its nuclear translocation (Andrabi et al., 2006). As AIF is a mitochondrial protein, its translocation from the mitochondria to the nucleus is a critical step that inextricably links parthanatos with the mitochondria.
PAR has been shown to interact with several proteins and its binding to these proteins might regulate (inhibit or activate) their physiological functions (Gagne et al., 2008; Krietsch et al., 2012). Evidence that PAR mediates parthanatos comes from observation of protection against cell death induced by exposure of mouse primary neurones to NMDA when cytosolic PAR was neutralized with PAR-specific antibodies (Andrabi et al., 2006). It was then established that PAR was directly toxic to neurones, as their exposure to the exogenous polymer through BioPorter-mediated delivery induced death (Andrabi et al., 2006). The BioPorter reagent (Gene Therapy Systems, San Diego, CA, USA) enables the transport of recombinant proteins, peptides or antibodies into viable cells (Zelphati et al., 2001). Toxicity increased with the dose and complexity of PAR: more death resulted from highly complex and long-chain polymers than from simple and shorter ones. This study suggests that PAR can translocate as a free polymer to induce cell death, although translocation of protein-conjugated PAR, which similarly induces cell death, cannot be ruled out and is currently the subject of active investigation. Interestingly, in a separate study, exposure to exogenous PAR was demonstrated to induce AIF translocation (Yu et al., 2006).
AIF has been identified as a PAR polymer-binding protein (Gagne et al., 2008) and a physical interaction between PAR and AIF is required in parthanatos for inducing the release of AIF from the mitochondria (Wang et al., 2011). It is interesting to note that, despite AIF being a bigger protein than cytochrome c, its translocation precedes cytochrome c release (and caspase activation) in parthanatos (Yu et al., 2002), arousing great interest in the study of its release, mitochondrial localization and translocation kinetics. Importantly, there is a small pool (30%) of AIF on the outer mitochondrial membrane (cytosolic side) (Yu et al., 2009). Using multiple biochemical and immunogold electron microscopic analyses of mouse brain mitochondria, this study showed that the localization accounted for the rapid release of a small pool of AIF, as 20% of the uncleaved AIF rapidly translocated to the nucleus and caused death following NMDA treatment (Yu et al., 2009). This implies that the release of the outer mitochondrial membrane pool of AIF is required to induce cell death in parthanatos. However, more studies are needed to clarify the different mechanisms involved in the release of the two AIF pools and their spatiotemporal regulation and interrelationship. In addition, studies are required to understand how PAR signalling affects mitochondrial (i.e. metabolic) function, integrity and maintenance.
Mitochondrial AIF is a key mediator of parthanatos
One mediator of parthanatos that has been unequivocally recognized is AIF, a mitochondrially localized flavoprotein discovered more than a decade ago (Susin et al., 1999), now reported to have five isoforms in humans (Delettre et al., 2006a,b; Loeffler et al., 2001; Lorenzo and Susin, 2007). Most death factors released from mitochondria participate in caspase-dependent cell death, whereas AIF mediates caspase-independent cell death, although some crosstalk between AIF and caspases has been proposed (Daugas et al., 2000; Arnoult et al., 2002). In retrospect, the name ‘apoptosis-inducing factor’ is somewhat a misnomer, because this protein is now known to participate in a form of cell death (parthanatos) that is distinct from apoptosis and it is possible that the involvement of AIF in caspase-dependent cell death (classical apoptosis) may be an epiphenomenon that occurs to supply caspase-activated cells with an alternative (non-caspase) pathway to death (Cregan et al., 2002; Wang et al., 2009a).
The AIF gene comprises 16 exons, is found on chromosome X (Susin et al., 1999) and is synthesized in the cytoplasm as a 67 kDa precursor, but imported into the mitochondria, where it undergoes processing to the mature 62 kDa form (Otera et al., 2005; Cao et al., 2007). AIF localizes mainly to the intermembrane space (Susin et al., 1999), although, as already mentioned, about 30% of it may loosely associate with the outer mitochondrial membrane, on the cytosolic side (Yu et al., 2009). The crystal structures of mouse and human AIFs have been solved at 2.0 (Mate et al., 2002) and 1.8 Å (Ye et al., 2002) resolutions respectively. There is a strong, positive electrostatic potential at its surface that might bind to DNA (Ye et al., 2002). There is a PAR-binding domain that is distinct from the DNA-binding domain (Wang et al., 2011). AIF has three putative functional domains: an N-terminal FAD-binding domain and a central reduced NAD (NADH)-binding domain, responsible for its oxidoreductase activity, and a C-terminal domain that is mainly responsible for its cell death (parthanatos)-mediating ability (Susin et al., 1999; Wang et al., 2009a). The oxidoreductase activity of AIF is not required for its death-inducing property (Ye et al., 2002). Although initial identification of this protein was as a cell-death mediator, subsequent attempts to understand its physiological role have revealed that, in normal conditions (when AIF is localized to the mitochondria), it supports cell survival. A very useful model to study AIF-mediated cell death is the harlequin (Hq) mouse, which has about an 80% down-regulation of AIF expression (Klein et al., 2002). This mouse shows evidence of oxidative stress and progressive degeneration of terminally differentiated cerebellar and retinal neurones (Klein et al., 2002), suggesting that AIF supports cell survival. In addition, AIF may support cortical development (Cheung et al., 2005) and may be associated with cytoprotective mechanisms in diabetes and obesity (Pospisilik et al., 2007), while deletion of AIF in muscle brings about mitochondrial dysfunction, muscle atrophy and dilated cardiomyopathy (Joza et al., 2005). Recently, when the PAR-binding site on AIF was mutated, the AIF mutant retained its NADH oxidase activity, bound FAD or DNA, and induced nuclear condensation. However, the AIF mutant was not released from the mitochondria and failed to translocate to the nucleus or mediate cell death following PARP-1 activation. This observation therefore seems to separate the role of AIF in cell metabolism (promoting survival) from its effect as a mitochondrial death effector (Wang et al., 2011). However, further studies such as the generation and use of knock-in mice are required to fully understand the effect of the AIF mutants that are incapable of binding PAR on mitochondrial (i.e. metabolic) function, integrity and maintenance.
Relating to cell death, numerous studies using diverse experimental paradigms have demonstrated convincingly that AIF translocates to the nucleus following PARP-1 overactivation caused by cellular injury (see Yu et al., 2003; Cregan et al., 2004; Virag, 2005). Exposure of cells to insults that cause DNA damage, such as NMDA excitotoxicity, oxidative or nitrosative stress and DNA alkylating agents, leads to nuclear translocation of AIF, resulting in large-scale DNA fragmentation (≈50 kb) and chromatin condensation and, eventually, cell death (Yu et al., 2002). Both NMDA and purified PAR polymer were less toxic to cortical neurones from Hq mice (Yu et al., 2006), and these mice, when exposed to excitotoxicity and stroke, had decreased lesions compared to wild-type animals (Cheung et al., 2005; Culmsee et al., 2005; Andrabi et al., 2006; Zhu et al., 2007b; Yuan et al., 2009; Wang et al., 2011). A converse experiment of virally mediated, forced AIF expression raised NMDA-induced cell death in Hq neurones to the same level as was found in wild-type neurones (Yu et al., 2006). Notably, AIF translocation from the mitochondria seems to be the commitment point for cell death in several disease states, including neurodegenerative disorders (Wang et al., 2003; Dawson and Dawson, 2004), a realization that places a premium on targeting this event in the process of translating the study of parthanatos to therapy. We need to determine whether the initial release of AIF from the mitochondria and concomitant deficits in mitochondrial function, or the large-scale fragmentation of genomic DNA that ensues after AIF enters the nucleus, is the main cause of cell death. In the nucleus, AIF binds to DNA and this step is thought to be required for the precipitation of cell death (Ye et al., 2002), although one drawback to this view comes from the realization that mouse AIF does not have recognizable structural motifs for DNA binding (Mate et al., 2002), in contrast to human AIF that does (Ye et al., 2002), thus suggesting the possibility of other mechanisms.
So far, it is not clear if AIF itself has any apparent or intrinsic endonuclease activity (Mate et al., 2002; Ye et al., 2002), raising the question of how does it induce DNA fragmentation? It has been suggested that AIF may recruit endogenous proteases or nucleases to precipitate fragmentation or, on the other hand, promote DNA vulnerability to these molecules through its interaction with DNA. Cyclophilin A (CypA) interacts with AIF, possibly forming a pro-apoptotic DNA-degradation complex and it may be a co-factor for AIF nuclear translocation and AIF-dependent chromatinolysis under conditions of cerebral hypoxia-ischaemia (Cande et al., 2004; Zhu et al., 2007a). How CypA and AIF form a pro-apoptotic DNA-degradation complex is not known, as there is no nuclease domain in CypA or AIF. Another suggested interactor with AIF is the mammalian endonuclease G (endoG). Its orthologue in Caenorhabditis elegans is CPS-6 and WAH-1, the AIF orthologue in C. elegans, associates and cooperates with CPS-6, causing DNA degradation (Wang et al., 2002). However, endoG does not seem to play a role in DNA fragmentation following parthanatos in mammals (David et al., 2006; Xu et al., 2010), suggesting that there is a parthanatos AIF-associated nuclease (PAAN) which, so far, remains to be identified in vertebrates.
Involvement of PARP in diseases affecting multiple organ systems
There is ample evidence to implicate PARP, and by extension, parthanatos, in the pathogenesis of several human diseases (see Pacher and Szabo 2008), including those that do not directly affect the nervous system. Evidence of PARP-1 activation and its pharmacological inhibition and/or genetic knockout resulting in cytoprotection has been used to demonstrate its possible or definite involvement in the pathophysiology of many diseases in a wide range of organ systems, including neurological (Endres et al., 1997; Joashi et al., 1999; LaPlaca et al., 2001) and neurodegenerative conditions (Mandir et al., 1999; Love et al., 1999a), diabetes (Yamamoto and Okamoto, 1980), colitis (Zingarelli et al., 1999), arthritis (Miesel et al., 1995), liver toxicity (Stubberfield and Cohen, 1988) and uveitis (Mabley et al., 2001) (see Virag and Szabo, 2002; Dawson and Dawson, 2004). It was in the CNS that PARP-1-dependent cell death was first detected (Zhang et al., 1994), and involvement of PARP-1, with or without the activation of parthanatos (cell death mediated by PARP-1 overactivation), has now been demonstrated, or at least suggested, in experimental models of stroke (Eliasson et al., 1997), trauma (LaPlaca et al., 2001), Alzheimer's disease (Love et al., 1999a), Parkinson's disease (Mandir et al., 1999; Outeiro et al., 2007), Huntington's disease (Vis et al., 2005), amyotrophic lateral sclerosis (Hivert et al., 1998) and spinal cord injury (Maier et al., 2007). Contributions of PARP to various dysfunctions in non-neuronal systems, such as diabetic endothelial dysfunction of the vasculature, interstitial pulmonary fibrosis and acute respiratory distress syndrome of the lung, uveitis of the eye, reperfusion injury of skeletal muscles, acetaminophen toxicity of the liver and sulfur mustard-induced vesication of the skin, are not the focus of this review but have been highlighted elsewhere (see Virag and Szabo, 2002; Szabo, 2005; de la Lastra et al., 2007). PARP has also been implicated in the ischaemic-reperfusion injury of the retina and cochlea, as well as in both haemorrhagic and endotoxin shock, and has been associated with multi-organ failure (see Virag and Szabo, 2002). Circulatory shock results in multi-organ failure and eventually death, and many changes that occur in this condition are related to oxidative and nitrosative stress in which PARP plays a major role (see Gero and Szabo, 2008)]. Furthermore, a role for PARP has been demonstrated in the development of vascular contractile failure, endothelial dysfunction, myocardial dysfunction, intestinal and pulmonary epithelial hyperpermeability and ultimately multi-organ failure (see Gero and Szabo, 2008).
It is also worthy of mention that a role for PARP has been established in cancer, which is leading the way for the development of new chemotherapeutic agents that block PARP in cancer cells to promote, rather than prevent, their death, through inhibition of DNA repair (see Peralta-Leal et al., 2008), especially when other survival mechanisms are deficient in those cells. Overall, this wide-ranging involvement of PARP has generated much research interest in PARP inhibitors, as promising therapeutic interventions in relevant conditions affecting diverse tissue types.
Therapeutic opportunities
As already highlighted, parthanatos occurs in a highly choreographed, multistep fashion and a number of steps in the cascade could therefore serve as promising therapeutic targets to develop novel compounds for use in the management of diseases associated with cell death. Current approaches to the identification of small-molecule compounds specific for inhibiting parthanatos, including high-throughput screening of relevant chemical libraries, would lead the way in rational drug design in this area. There is some progress already regarding such pharmacological interventions and PARP blockers prevent parthanatos in cell culture and animal models of diseases involving overactivation of PARP-1 (see Virag and Szabo, 2002; Gero and Szabo, 2008). It should be noted that PARP inhibitors are now being tested also in other areas of therapeutic intervention not related to cell death, although these are outside the purview of this review (Peralta-Leal et al., 2009; Virag and Szabo, 2002). Historically, first-generation PARP blockers include nicotinamide, benzamide and substituted benzamides, such as 3-aminobenzamide. Many analogues of benzamide with greater potency were later developed as second-generation PARP inhibitors, while third-generation inhibitors with greatly improved properties belong to a diverse family of chemical structures, such as derivatives of imidazopyridine, imidazoquinolinone and isoquinolindione (Eltze et al., 2008). Others being developed include benzimidazoles, pthalazinones, ideno[1,2-c]isoquinolinones and tricyclic indoles (Peralta-Leal et al., 2009). Interestingly, some natural compounds, especially flavonoids, have also been shown to inhibit PARP (Geraets et al., 2007; Weseler et al., 2009) and, very recently, 4'-methoxyflavone and 3’,4'-dimethoxyflavone, identified through a high-throughput screening campaign, were demonstrated to be neuroprotective PARP-1 inhibitors (Fatokun et al., 2013).
Some PARP inhibitors have entered clinical trials, either as single agents (e.g. KU59436, BSI-201, ABT-888, INO-1001), or in combination with other agents (e.g. INO-1001, GPI 21016) (Green and Kroemer, 2005; Jagtap and Szabo, 2005; Jagtap et al., 2005; Lorenzo and Susin, 2007; Pacher and Szabo, 2007; Peralta-Leal et al., 2009). Third-generation PARP inhibitors that also entered clinical trials include ABT-888, AG014699, AZD2281, BSI-201, CEP-8983/CEP-9722 and MK-4827, designed to compete with NAD+ at the enzyme-active site [see Rouleau et al. (2010)]. They are more potent and specific compared to earlier inhibitors (Rouleau et al., 2010). However, many of these PARP inhibitors have mainly been useful for attaining the therapeutic objective of promoting cell death, especially in cancer (Bryant et al., 2005; Farmer et al., 2005; McCabe et al., 2005; Peralta-Leal et al., 2008; 2009; Turner et al., 2008), rather than preventing cell death, as would be desirable in, for example, neurodegenerative diseases. The rationale for this use is that sustained inhibition of PARP in cancer cells promotes their death by blocking their DNA repair machinery needed for survival. In terms of efforts towards protection, INO-1001 (Inotek) entered clinical trials for myocardial ischaemia (Lorenzo and Susin, 2007). There are also ongoing efforts to develop isoform-specific PARP inhibitors (Iwashita et al., 2005; Ishida et al., 2006; Pellicciari et al., 2008; Peralta-Leal et al., 2008; Turner et al., 2008). It is anticipated that, due to their unknown potential long-term side effects, the development of PARP inhibitors for chronic neurodegenerative diseases and neuroinflammation may be more challenging (Graziani and Szabo, 2005). Conceptually, it might be much less desirable to seek to prevent cell death in the long term by blocking PARP, as doing so will occlude its beneficial effects, which are related to the maintenance of genomic stability. Nevertheless, shown in Table 2 are outcomes of the assessment of some PARP inhibitors in animal models of human neurological and neurodegenerative conditions linked to parthanatos.
Table 2.
Effects of PARP-1 inhibitors in animal models of human neurological and neurodegenerative conditions linked to parthanatos
| Relevant neurological condition(s) or neurodegenerative disease (s) | Evidence of parthanatos involvement in human disease | Animal models (rodents) | Inhibitors | Outcome of PARP-1 or parthanatos pathway inhibition in animal models/observation |
|---|---|---|---|---|
| Stroke, brain ischaemia, brain trauma (Pacher and Szabo, 2008) | PARP activated in brain sections from patients dying from stroke, brain ischaemia (from cardiac arrest) and brain trauma (Love et al., 199b; 2000) | Focal cerebral ischaemia (rat) (Strosznajder et al., 2010) | DPQ | Decrease in infarct volume (Takahashi et al., 1997) |
| 3-AB | Decrease in infarct volume after MCAO, reduction in NMDA-induced glutamate elevation, improvement in neurological outcome and motor function, marked decrease in the volume of damaged tissue (Ding et al., 2001; Lo et al., 1998; Takahashi and Greenberg, 1999; Tokime et al., 1998; Yap et al., 2008) | |||
| Cilostazol | Reduction in infarct size, nuclear AIF translocation and apoptosis after MCAO followed by reperfusion (Lee et al., 2007a) | |||
| Focal cerebral ischaemia (mouse) (Strosznajder et al., 2010) | 3-AB | Neuroprotection, decrease in infarct volume, improvement of neurological score (Couturier et al., 2003) | ||
| Global cerebral ischaemia (rat) (Strosznajder et al., 2010) | PJ34 | Inhibition of microglia/macrophage activation, decrease in CA1 neuronal death after forebrain ischaemia (Hamby et al., 2007) | ||
| Global cerebral ischaemia (gerbil) (Strosznajder et al., 2010) | DPQ | No improvement of CA1 neuronal survival after bilateral carotid artery occlusion (Moroni et al., 2001) | ||
| Benzamide | ||||
| 3-AB | Robust neuroprotection in CA1 neurones after 3-min ischaemia, reduced forebrain ischaemia (Strosznajder et al., 2003) | |||
| Neurodegenerative diseases (Pacher and Szabo, 2008) | Evidence of poly (ADP-ribosyl)ation in brain sections from patients with Alzheimer's disease, Parkinson's disease and ALS (Love et al., 1999a; Kim et al., 2003; 2004; Soos et al., 2004) | Middle cerebral artery occlusion, global brain ischaemia, ischaemic reperfusion, hypoxia-ischaemia, MPTP intoxication, hypothermia, cortical trauma (mouse, rat, gerbil, guinea pig) (Komjati et al., 2005) | 3-AB | Reduced infarct size, maintained or improved NAD, improved neurological function or status, attenuation in glutamate release, reduced neutrophil infiltration, reduced nitrosative stress, sustained ATP, decrease in water content, reduction in number of TUNEL-positive cells, decrease in immunoreactivity of PAR, CD11B, ICAM-1 and COX2, enhanced neuroprotection (in combination with melatonin), improved neuronal conductance, decrease in production of inflammatory mediator, improved survival and CNS function (Barc et al., 2001; Chiang and Lam, 2000; Couturier et al., 2003; Ding et al., 2001; Ducrocq et al., 2000; Eliasson et al., 1997; Endres et al., 1997; Goto et al., 2002; Iwashita et al., 2004; Koedel et al., 2002; Koh et al., 2004b; Lam, 1997; Lo et al., 1998; Nagayama et al., 2000; Plaschke et al., 2000; Sun and Cheng, 1998; Tabuchi et al., 2001; Tokime et al., 1998) |
| PJ34 | Reduced infarct size, improved neurological status (Abdelkarim et al., 2001; Iwashita et al., 2004) | |||
| DPQ | Reduced infarct size, reduced infarct size but not DNA damage, improved neurological status (high doses may cause protective effect to be lost) (Takahashi et al., 1997; 1999; Giovannelli et al., 2002) | |||
| Benzamide | Reduced neuronal death, reduced hyperalgesia and mechano-allodynia, reduced neuronal deficit, improved survival, maintained striatal NAD+ and ATP, protection against dopamine loss (Cosi et al., 1994; 1996; Mao et al., 1997; Meier et al., 1999) | |||
| INO-1001 | Reduced infarct volume, improved neurological outcome | |||
| Reduced immunoreactivity of PAR, NT and APP, immunoreactivity changes of AIF restored. | ||||
| Entered clinical trial for myocardial ischaemia (Komjati et al., 2004; Lorenzo and Susin, 2007) | ||||
Animal models of neurological and neurodegenerative diseases in which parthanatos is implicated and the observed effects of selected PARP-1 inhibitors, most of which are currently at the experimental stage, except INO-1001 that entered clinical trial for myocardial ischaemia. Abbreviations: 3-AB, 3-aminobenzamide; AIF, apoptosis-inducing factor; ALS, amyotrophic lateral sclerosis; APP, amyloid precursor protein; DPQ, 3,4-Dihydro-5-[4-(1-piperidinyl)butoxyl]-1(2H)-isoquinolinone; MCAO, middle cerebral artery occlusion; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; NAD+, nicotinamide adenine dinucleotide; NADP, nicotinamide adenine dinucleotide phosphate; NT, nitrotyrosine; PAR, poly (ADP-ribose) polymer; PJ34, N-(-oxo-5,6-dihydro-phenanthridin-2-yl)-N,N-dimethylacetamide. Table was adapted from references (Komjati et al., 2005; Pacher and Szabo, 2008; Strosznajder et al., 2010).
Apart from direct PARP inhibitors already mentioned, some of which entered clinical trials, most levels of potential intervention in the parthanatos cascade as discussed subsequently are still essentially at the speculative or the experimental stage, but with the hope of making it to the clinic in the future. First, identifying pharmacological or other interventions that can block the accumulation of PAR polymer or its ability to signal to the mitochondria will be a rational means of combating parthanatos, just as direct PARP inhibition, as PAR is a death signal (Andrabi et al., 2006). A novel, NMDA-induced survival protein, ‘Iduna,’ also known as RNF146, was recently shown to be a PAR-dependent E3 ubiquitin ligase (Kang et al., 2011) and, like PARG, is an endogenous inhibitor of parthanatos (Andrabi et al., 2011). Iduna was shown to protect against glutamate NMDA receptor-mediated excitotoxicity, both in vitro and in vivo, and also against stroke induced by middle cerebral artery occlusion in mice. The mechanisms of its protection are due to its E3 ubiquitin ligase activity and its ability to bind PAR. Like PARG, it acts downstream of, and does not affect, PARP-1 activity (Andrabi et al., 2011).
Manipulation of PARG, the enzyme that hydrolyses PAR polymer, also constitutes a promising means of protecting against parthanatos. Studies suggest that PARG plays an important role in cell survival by degrading PAR polymer (Koh et al., 2004a). Animals lacking PARG are dramatically sensitive to toxic insults, while mice overexpressing PARG are correspondingly resistant (Koh et al., 2004a; Andrabi et al., 2006). There is, therefore, a unique therapeutic promise from achieving an efficient and rapid clearance of synthesized PAR polymer by boosting levels of PARG.
Release and translocation of AIF, which occur downstream of PAR signalling, are another set of attractive therapeutic targets. Because the mitochondrial translocation of AIF to the nucleus seems to be the commitment point in parthanatos, this particular step appears to have a critical place among therapeutic targets in this cell-death paradigm. Developing a means of monitoring the extent of the translocation (response) and relating this response to the degree of insult applied (concentration) will allow the generation of quantitative, concentration-response style data that should furnish excellent pharmacology for development of therapeutic agents. A way to achieve this could be fluorescently tagging AIF for visualization of its nuclear translocation in live cells in real time, which has been demonstrated (Landshamer et al., 2008), and then achieving the ability to quantify this translocation in a manner that makes the approach amenable to high-content screening platforms for drug discovery purposes to identify compounds that modulate the event.
A further step that could potentially be targeted is the interaction of AIF with the DNA. This interaction has been suggested to be obligatory for the characteristic large-scale fragmentation and chromatin condensation that occur in parthanatos. As such, blocking the AIF-DNA interaction may be a further means to inhibit parthanatos, particularly if the concomitant deficits in mitochondrial function after AIF release are not the main cause of cell death in parthanatos. Thus, identifying PAAN and developing inhibitors to PAAN may be another attractive target to block parthanatos. As noted above, studies are required to dissect the relative importance of AIF release from the mitochondria versus the large-scale DNA fragmentation in the death process. The fact that AIF interacts with several proteins (mitochondrial, cytosolic, nuclear) en route to, or in, the nucleus provides a clue that identification of such AIF-interacting proteins and the blockade (or activation, in case of death-attenuating interactions) of such interactions by therapeutic agents may, at least in part, ameliorate cell death in parthanatos. Knowing that human AIF has some distinct characteristics that distinguish it from mouse AIF (e.g. human AIF has a fold identical to that in murine AIF but a different crystal packing that underlies a continuous positively charged patch thought to guide interactions with DNA) (Sevrioukova, 2011), the availability of its crystal structure (Ye et al., 2002), alongside that of the murine AIF (Mate et al., 2002), will enable a faster translation of basic investigations to therapeutic applications for human diseases. These highlights suggest that, although much still has to be done in this area to translate bench work to the bedside, they will provide credible approaches to the identification of disease-modifying therapeutic agents and thus give at least a glimmer of hope to sufferers of debilitating conditions in which parthanatos contributes significantly to pathophysiology. Figure 3 summarizes the various levels in parthanatos that could be targeted in the development of such therapeutic agents.
Figure 3.
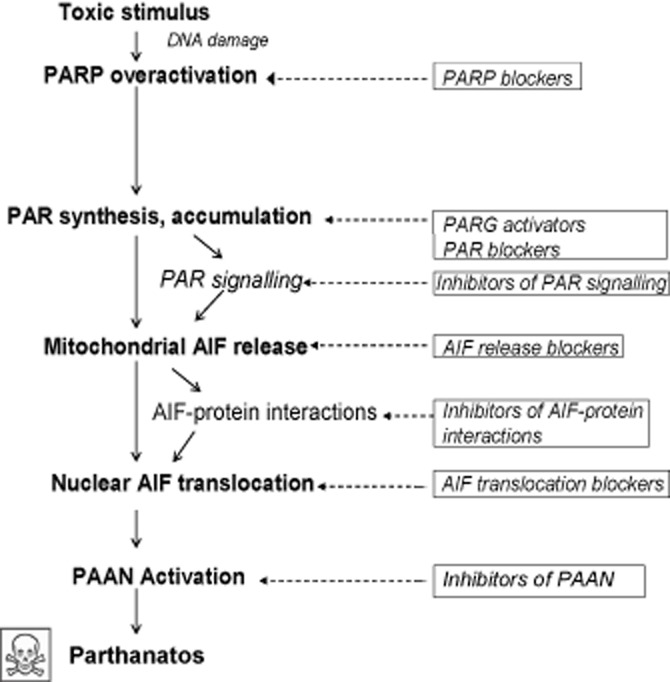
Potential therapeutic opportunities in the parthanatos cascade. Steps in the parthanatos cascade where therapeutic interventions are possible, from exposure of a cell to a toxic stimulus to the eventual cell death. Pathway progression is depicted using solid arrows. Proven or potential therapeutic interventions are in solid rectangular boxes, each linked with broken arrows to the step it modulates. To date, PARP blockers are the most advanced in development, with a number of them having entered clinical trials. The development of most other interventions is still at the experimental stage.
Summary
Parthanatos is here described as a unique, caspase-independent, cell-death pathway that is distinct from apoptosis, necrosis or other identified forms of cell death. Although largely discussed here in the context of cell death in the CNS, with relevance to neurological and neurodegenerative conditions, parthanatos is a multistep death pathway that may underlie many disease conditions, including diabetes and inflammatory disorders. Toxic stimuli that induce parthanatos activate nuclear PARP-1 excessively, causing it to synthesize a vast amount of PAR polymer. PAR polymer reaches a toxic level that translocates into the cytosol, where it constitutes a death signal through which mitochondrially localized AIF is forced out of its location and into the nucleus. Nuclear AIF translocation induced by mitochondriotoxic PAR establishes the mitochondrion as a central, indispensable player in parthanatos. AIF upon reaching the nucleus then interacts with DNA through an as-of-yet unidentified PAAN, resulting in large-scale DNA fragmentation and chromatin condensation. Parthanatic cell death is the eventual outcome. There are checkpoints in the cascade that can be usefully exploited to create therapeutic interventions to manage pathologies associated with parthanatos. However, of all these potentially druggable molecules or steps of the death cascade, the event of AIF translocation from the mitochondria to the nucleus may currently be considered a very attractive target for drug design. Reassuringly, knowledge of parthanatos continues to grow rapidly, with the hope that the uncertainties in the pathway will soon become clarified and the existing controversies fully resolved.
Acknowledgments
We apologize to colleagues whose work might not have been cited here for want of space. A. A. F. is grateful to the University of Nottingham (UK) for the award of a Nottingham Advanced Research Fellowship (NARF) and to the European Commission for the award of a Marie Curie International Incoming Fellowship (IIF) through its European Union (EU) Seventh Framework (FP7) Marie Curie People Work Programme. T. M. D. is supported by NIH/NINDS NS067525 and NS38377 and T. M. D. and V. L. D. are supported by NIDA DA000266. T. M. D. is the Leonard and Madlyn Abramson Professor of Neurodegenerative Diseases at Johns Hopkins University.
Glossary
- AIF
apoptosis-inducing factor
- ARH3
ADP-ribose-(arginine) protein hydrolase
- CypA
cyclophilin A
- endoG
endonuclease G
- Hq
harlequin
- MNNG
N-methyl-N’-nitro-N-nitrosoguanidine
- mPT
mitochondrial permeability transition
- NADH
reduced nicotinamide adenine dinucleotide
- PAAN
parthanatos AIF-associated nuclease
- PAR
poly (ADP-ribose)
- PARG
poly (ADP-ribose) glycohydrolase
- ROS
reactive oxygen species
Conflict of interest
The authors declare no conflict of interest.
References
- Abdelkarim GE, Gertz K, Harms C, Katchanov J, Dirnagl U, Szabo C, et al. Protective effects of PJ34, a novel, potent inhibitor of poly(ADP-ribose) polymerase (PARP) in in vitro and in vivo models of stroke. Int J Mol Med. 2001;7:255–260. [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: Overview. Br J Pharmacol. 2013;170:1449–1867. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ame JC, Apiou F, Jacobson EL, Jacobson MK. Assignment of the poly(ADP-ribose) glycohydrolase gene (PARG) to human chromosome 10q11.23 and mouse chromosome 14B by in situ hybridization. Cytogenet Cell Genet. 1999;85:269–270. doi: 10.1159/000015310. [DOI] [PubMed] [Google Scholar]
- Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A. 2006;103:18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SA, Dawson TM, Dawson VL. Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann N Y Acad Sci. 2008;1147:233–241. doi: 10.1196/annals.1427.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SA, Kang HC, Haince JF, Lee YI, Zhang J, Chi Z, et al. Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death. Nat Med. 2011;17:692–699. doi: 10.1038/nm.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnoult D, Parone P, Martinou JC, Antonsson B, Estaquier J, Ameisen JC. Mitochondrial release of apoptosis-inducing factor occurs downstream of cytochrome c release in response to several proapoptotic stimuli. J Cell Biol. 2002;159:923–929. doi: 10.1083/jcb.200207071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barc S, Page G, Barrier L, Piriou A, Fauconneau B. Impairment of the neuronal dopamine transporter activity in MPP(+)-treated rat was not prevented by treatments with nitric oxide synthase or poly(ADP-ribose) polymerase inhibitors. Neurosci Lett. 2001;314:82–86. doi: 10.1016/s0304-3940(01)02273-x. [DOI] [PubMed] [Google Scholar]
- Berger NA. Poly(ADP-ribose) in the cellular response to DNA damage. Radiat Res. 1985;101:4–15. [PubMed] [Google Scholar]
- Berger NA, Berger SJ. Metabolic consequences of DNA damage: the role of poly (ADP-ribose) polymerase as mediator of the suicide response. Basic Life Sci. 1986;38:357–363. doi: 10.1007/978-1-4615-9462-8_39. [DOI] [PubMed] [Google Scholar]
- Berger NA, Sims JL, Catino DM, Berger SJ. Poly(ADP-ribose) polymerase mediates the suicide response to massive DNA damage: studies in normal and DNA-repair defective cells. Princess Takamatsu Symp. 1983;13:219–226. [PubMed] [Google Scholar]
- Berger NA, Berger SJ, Catino DM, Petzold SJ, Robins RK. Modulation of nicotinamide adenine dinucleotide and poly(adenosine diphosphoribose) metabolism by the synthetic ‘C’ nucleoside analogs, tiazofurin and selenazofurin. A new strategy for cancer chemotherapy. J Clin Invest. 1985;75:702–709. doi: 10.1172/JCI111750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blenn C, Althaus FR, Malanga M. Poly(ADP-ribose) glycohydrolase silencing protects against H2O2-induced cell death. Biochem J. 2006;396:419–429. doi: 10.1042/BJ20051696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonicalzi ME, Vodenicharov M, Coulombe M, Gagne JP, Poirier GG. Alteration of poly(ADP-ribose) glycohydrolase nucleocytoplasmic shuttling characteristics upon cleavage by apoptotic proteases. Biol Cell. 2003;95:635–644. doi: 10.1016/j.biolcel.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature. 2006;443:796–802. doi: 10.1038/nature05293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- Burns DM, Ying W, Kauppinen TM, Zhu K, Swanson RA. Selective down-regulation of nuclear poly(ADP-ribose) glycohydrolase. PLoS ONE. 2009;4:e4896. doi: 10.1371/journal.pone.0004896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cande C, Vahsen N, Kouranti I, Schmitt E, Daugas E, Spahr C, et al. AIF and cyclophilin A cooperate in apoptosis-associated chromatinolysis. Oncogene. 2004;23:1514–1521. doi: 10.1038/sj.onc.1207279. [DOI] [PubMed] [Google Scholar]
- Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin XM, et al. Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J Neurosci. 2007;27:9278–9293. doi: 10.1523/JNEUROSCI.2826-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EC, Melanson-Drapeau L, Cregan SP, Vanderluit JL, Ferguson KL, McIntosh WC, et al. Apoptosis-inducing factor is a key factor in neuronal cell death propagated by BAX-dependent and BAX-independent mechanisms. J Neurosci. 2005;25:1324–1334. doi: 10.1523/JNEUROSCI.4261-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew SK, Chen P, Link N, Galindo KA, Pogue K, Abrams JM. Genome-wide silencing in Drosophila captures conserved apoptotic effectors. Nature. 2009;460:123–127. doi: 10.1038/nature08087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang SK, Lam TT. Post-treatment at 12 or 18 hours with 3-aminobenzamide ameliorates retinal ischemia-reperfusion damage. Invest Ophthalmol Vis Sci. 2000;41:3210–3214. [PubMed] [Google Scholar]
- Chiarugi A. Poly(ADP-ribose) polymerase: killer or conspirator? The ‘suicide hypothesis’ revisited. Trends Pharmacol Sci. 2002;23:122–129. doi: 10.1016/S0165-6147(00)01902-7. [DOI] [PubMed] [Google Scholar]
- Cosi C, Suzuki H, Milani D, Facci L, Menegazzi M, Vantini G, et al. Poly(ADP-ribose) polymerase: early involvement in glutamate-induced neurotoxicity in cultured cerebellar granule cells. J Neurosci Res. 1994;39:38–46. doi: 10.1002/jnr.490390106. [DOI] [PubMed] [Google Scholar]
- Cosi C, Colpaert F, Koek W, Degryse A, Marien M. Poly(ADP-ribose) polymerase inhibitors protect against MPTP-induced depletions of striatal dopamine and cortical noradrenaline in C57B1/6 mice. Brain Res. 1996;729:264–269. [PubMed] [Google Scholar]
- Couturier JY, Ding-Zhou L, Croci N, Plotkine M, Margaill I. 3-Aminobenzamide reduces brain infarction and neutrophil infiltration after transient focal cerebral ischemia in mice. Exp Neurol. 2003;184:973–980. doi: 10.1016/S0014-4886(03)00367-4. [DOI] [PubMed] [Google Scholar]
- Cozzi A, Cipriani G, Fossati S, Faraco G, Formentini L, Min W, et al. Poly(ADP-ribose) accumulation and enhancement of postischemic brain damage in 110-kDa poly(ADP-ribose) glycohydrolase null mice. J Cereb Blood Flow Metab. 2006;26:684–695. doi: 10.1038/sj.jcbfm.9600222. [DOI] [PubMed] [Google Scholar]
- Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, et al. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol. 2002;158:507–517. doi: 10.1083/jcb.200202130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan SP, Dawson VL, Slack RS. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene. 2004;23:2785–2796. doi: 10.1038/sj.onc.1207517. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Zhu C, Landshamer S, Becattini B, Wagner E, Pellecchia M, et al. Apoptosis-inducing factor triggered by poly(ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J Neurosci. 2005;25:10262–10272. doi: 10.1523/JNEUROSCI.2818-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amours D, Desnoyers S, D'Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342(Pt 2):249–268. [PMC free article] [PubMed] [Google Scholar]
- Daugas E, Susin SA, Zamzami N, Ferri KF, Irinopoulou T, Larochette N, et al. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 2000;14:729–739. [PubMed] [Google Scholar]
- David KK, Sasaki M, Yu SW, Dawson TM, Dawson VL. EndoG is dispensable in embryogenesis and apoptosis. Cell Death Differ. 2006;13:1147–1155. doi: 10.1038/sj.cdd.4401787. [DOI] [PubMed] [Google Scholar]
- David KK, Andrabi SA, Dawson TM, Dawson VL. Parthanatos, a messenger of death. Front Biosci. 2009;14:1116–1128. doi: 10.2741/3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidovic L, Vodenicharov M, Affar EB, Poirier GG. Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp Cell Res. 2001;268:7–13. doi: 10.1006/excr.2001.5263. [DOI] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM. Deadly conversations: nuclear-mitochondrial cross-talk. J Bioenerg Biomembr. 2004;36:287–294. doi: 10.1023/B:JOBB.0000041755.22613.8d. [DOI] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci U S A. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM, Bartley DA, Uhl GR, Snyder SH. Mechanisms of nitric oxide-mediated neurotoxicity in primary brain cultures. J Neurosci. 1993;13:2651–2661. doi: 10.1523/JNEUROSCI.13-06-02651.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson VL, Kizushi VM, Huang PL, Snyder SH, Dawson TM. Resistance to neurotoxicity in cortical cultures from neuronal nitric oxide synthase-deficient mice. J Neurosci. 1996;16:2479–2487. doi: 10.1523/JNEUROSCI.16-08-02479.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delettre C, Yuste VJ, Moubarak RS, Bras M, Lesbordes-Brion JC, Petres S, et al. AIFsh, a novel apoptosis-inducing factor (AIF) pro-apoptotic isoform with potential pathological relevance in human cancer. J Biol Chem. 2006a;281:6413–6427. doi: 10.1074/jbc.M509884200. [DOI] [PubMed] [Google Scholar]
- Delettre C, Yuste VJ, Moubarak RS, Bras M, Robert N, Susin SA. Identification and characterization of AIFsh2, a mitochondrial apoptosis-inducing factor (AIF) isoform with NADH oxidase activity. J Biol Chem. 2006b;281:18507–18518. doi: 10.1074/jbc.M601751200. [DOI] [PubMed] [Google Scholar]
- Ding Y, Zhou Y, Lai Q, Li J, Gordon V, Diaz FG. Long-term neuroprotective effect of inhibiting poly(ADP-ribose) polymerase in rats with middle cerebral artery occlusion using a behavioral assessment. Brain Res. 2001;915:210–217. doi: 10.1016/s0006-8993(01)02852-9. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducrocq S, Benjelloun N, Plotkine M, Ben-Ari Y, Charriaut-Marlangue C. Poly(ADP-ribose) synthase inhibition reduces ischemic injury and inflammation in neonatal rat brain. J Neurochem. 2000;74:2504–2511. doi: 10.1046/j.1471-4159.2000.0742504.x. [DOI] [PubMed] [Google Scholar]
- Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- Eltze T, Boer R, Wagner T, Weinbrenner S, McDonald MC, Thiemermann C, et al. Imidazoquinolinone, imidazopyridine, and isoquinolindione derivatives as novel and potent inhibitors of the poly(ADP-ribose) polymerase (PARP): a comparison with standard PARP inhibitors. Mol Pharmacol. 2008;74:1587–1598. doi: 10.1124/mol.108.048751. [DOI] [PubMed] [Google Scholar]
- Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA. Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J Cereb Blood Flow Metab. 1997;17:1143–1151. doi: 10.1097/00004647-199711000-00002. [DOI] [PubMed] [Google Scholar]
- Ethier C, Labelle Y, Poirier GG. PARP-1-induced cell death through inhibition of the MEK/ERK pathway in MNNG-treated HeLa cells. Apoptosis. 2007;12:2037–2049. doi: 10.1007/s10495-007-0127-z. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Fatokun AA, Stone TW, Smith RA. Oxidative stress in neurodegeneration and available means of protection. Front Biosci. 2008;13:3288–3311. doi: 10.2741/2926. [DOI] [PubMed] [Google Scholar]
- Fatokun AA, Liu JO, Dawson VL, Dawson TM. Identification through high-throughput screening of 4'-methoxyflavone and 3’,4'-dimethoxyflavone as novel neuroprotective inhibitors of parthanatos. Br J Pharmacol. 2013;169:1263–1278. doi: 10.1111/bph.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Ruoslahti E. Integrins and anoikis. Curr Opin Cell Biol. 1997;9:701–706. doi: 10.1016/s0955-0674(97)80124-x. [DOI] [PubMed] [Google Scholar]
- Gagne JP, Isabelle M, Lo KS, Bourassa S, Hendzel MJ, Dawson VL, et al. Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res. 2008;36:6959–6976. doi: 10.1093/nar/gkn771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraets L, Moonen HJ, Brauers K, Gottschalk RW, Wouters EF, Bast A, et al. Flavone as PARP-1 inhibitor: its effect on lipopolysaccharide induced gene-expression. Eur J Pharmacol. 2007;573:241–248. doi: 10.1016/j.ejphar.2007.07.013. [DOI] [PubMed] [Google Scholar]
- Gero D, Szabo C. Poly(ADP-ribose) polymerase: a new therapeutic target? Curr Opin Anaesthesiol. 2008;21:111–121. doi: 10.1097/ACO.0b013e3282f63c15. [DOI] [PubMed] [Google Scholar]
- Giovannelli L, Cozzi A, Guarnieri I, Dolara P, Moroni F. Comet assay as a novel approach for studying DNA damage in focal cerebral ischemia: differential effects of NMDA receptor antagonists and poly(ADP-ribose) polymerase inhibitors. J Cereb Blood Flow Metab. 2002;22:697–704. doi: 10.1097/00004647-200206000-00008. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Zulueta M, Ensz LM, Mukhina G, Lebovitz RM, Zwacka RM, Engelhardt JF, et al. Manganese superoxide dismutase protects nNOS neurons from NMDA and nitric oxide-mediated neurotoxicity. J Neurosci. 1998;18:2040–2055. doi: 10.1523/JNEUROSCI.18-06-02040.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto S, Xue R, Sugo N, Sawada M, Blizzard KK, Poitras MF, et al. Poly(ADP-ribose) polymerase impairs early and long-term experimental stroke recovery. Stroke. 2002;33:1101–1106. doi: 10.1161/01.str.0000014203.65693.1e. [DOI] [PubMed] [Google Scholar]
- Graziani G, Szabo C. Clinical perspectives of PARP inhibitors. Pharmacol Res. 2005;52:109–118. doi: 10.1016/j.phrs.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G. Pharmacological manipulation of cell death: clinical applications in sight? J Clin Invest. 2005;115:2610–2617. doi: 10.1172/JCI26321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha HC, Snyder SH. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci U S A. 1999;96:13978–13982. doi: 10.1073/pnas.96.24.13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haince JF, Ouellet ME, McDonald D, Hendzel MJ, Poirier GG. Dynamic relocation of poly(ADP-ribose) glycohydrolase isoforms during radiation-induced DNA damage. Biochim Biophys Acta. 2006;1763:226–237. doi: 10.1016/j.bbamcr.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Hamby AM, Suh SW, Kauppinen TM, Swanson RA. Use of a poly(ADP-ribose) polymerase inhibitor to suppress inflammation and neuronal death after cerebral ischemia-reperfusion. Stroke. 2007;38(2 Suppl):632–636. doi: 10.1161/01.STR.0000250742.61241.79. [DOI] [PubMed] [Google Scholar]
- Hanai S, Kanai M, Ohashi S, Okamoto K, Yamada M, Takahashi H, et al. Loss of poly(ADP-ribose) glycohydrolase causes progressive neurodegeneration in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2004;101:82–86. doi: 10.1073/pnas.2237114100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harraz MM, Dawson TM, Dawson VL. Advances in neuronal cell death 2007. Stroke. 2008;39:286–288. doi: 10.1161/STROKEAHA.107.511857. [DOI] [PubMed] [Google Scholar]
- Hivert B, Cerruti C, Camu W. Hydrogen peroxide-induced motoneuron apoptosis is prevented by poly ADP ribosyl synthetase inhibitors. Neuroreport. 1998;9:1835–1838. doi: 10.1097/00001756-199806010-00031. [DOI] [PubMed] [Google Scholar]
- Hong SJ, Dawson TM, Dawson VL. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol Sci. 2004;25:259–264. doi: 10.1016/j.tips.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Ishida J, Yamamoto H, Kido Y, Kamijo K, Murano K, Miyake H, et al. Discovery of potent and selective PARP-1 and PARP-2 inhibitors: SBDD analysis via a combination of X-ray structural study and homology modeling. Bioorg Med Chem. 2006;14:1378–1390. doi: 10.1016/j.bmc.2005.09.061. [DOI] [PubMed] [Google Scholar]
- Iwashita A, Tojo N, Matsuura S, Yamazaki S, Kamijo K, Ishida J, et al. A novel and potent poly(ADP-ribose) polymerase-1 inhibitor, FR247304 (5-chloro-2-[3-(4-phenyl-3,6-dihydro-1(2H)-pyridinyl)propyl]-4(3H)-quinazo linone), attenuates neuronal damage in in vitro and in vivo models of cerebral ischemia. J Pharmacol Exp Ther. 2004;310:425–436. doi: 10.1124/jpet.104.066944. [DOI] [PubMed] [Google Scholar]
- Iwashita A, Hattori K, Yamamoto H, Ishida J, Kido Y, Kamijo K, et al. Discovery of quinazolinone and quinoxaline derivatives as potent and selective poly(ADP-ribose) polymerase-1/2 inhibitors. FEBS Lett. 2005;579:1389–1393. doi: 10.1016/j.febslet.2005.01.036. [DOI] [PubMed] [Google Scholar]
- Jagtap P, Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- Jagtap PG, Baloglu E, Southan GJ, Mabley JG, Li H, Zhou J, et al. Discovery of potent poly(ADP-ribose) polymerase-1 inhibitors from the modification of indeno[1,2-c]isoquinolinone. J Med Chem. 2005;48:5100–5103. doi: 10.1021/jm0502891. [DOI] [PubMed] [Google Scholar]
- Joashi UC, Greenwood K, Taylor DL, Kozma M, Mazarakis ND, Edwards AD, et al. Poly(ADP ribose) polymerase cleavage precedes neuronal death in the hippocampus and cerebellum following injury to the developing rat forebrain. Eur J Neurosci. 1999;11:91–100. doi: 10.1046/j.1460-9568.1999.00409.x. [DOI] [PubMed] [Google Scholar]
- Joza N, Oudit GY, Brown D, Benit P, Kassiri Z, Vahsen N, et al. Muscle-specific loss of apoptosis-inducing factor leads to mitochondrial dysfunction, skeletal muscle atrophy, and dilated cardiomyopathy. Mol Cell Biol. 2005;25:10261–10272. doi: 10.1128/MCB.25.23.10261-10272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameshita I, Matsuda Z, Taniguchi T, Shizuta Y. Poly (ADP-Ribose) synthetase. Separation and identification of three proteolytic fragments as the substrate-binding domain, the DNA-binding domain, and the automodification domain. J Biol Chem. 1984;259:4770–4776. [PubMed] [Google Scholar]
- Kanai M, Uchida M, Hanai S, Uematsu N, Uchida K, Miwa M. Poly(ADP-ribose) polymerase localizes to the centrosomes and chromosomes. Biochem Biophys Res Commun. 2000;278:385–389. doi: 10.1006/bbrc.2000.3801. [DOI] [PubMed] [Google Scholar]
- Kang HC, Lee YI, Shin JH, Andrabi SA, Chi Z, Gagne JP, et al. Iduna is a poly(ADP-ribose) (PAR)-dependent E3 ubiquitin ligase that regulates DNA damage. Proc Natl Acad Sci U S A. 2011;108:14103–14108. doi: 10.1073/pnas.1108799108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keil C, Grobe T, Oei SL. MNNG-induced cell death is controlled by interactions between PARP-1, poly(ADP-ribose) glycohydrolase, and XRCC1. J Biol Chem. 2006;281:34394–34405. doi: 10.1074/jbc.M606470200. [DOI] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Henkel JS, Beers DR, Sengun IS, Simpson EP, Goodman JC, et al. PARP expression is increased in astrocytes but decreased in motor neurons in the spinal cord of sporadic ALS patients. J Neuropathol Exp Neurol. 2003;62:88–103. doi: 10.1093/jnen/62.1.88. [DOI] [PubMed] [Google Scholar]
- Kim SH, Engelhardt JI, Henkel JS, Siklos L, Soos J, Goodman C, et al. Widespread increased expression of the DNA repair enzyme PARP in brain in ALS. Neurology. 2004;62:319–322. doi: 10.1212/01.wnl.0000103291.04985.dc. [DOI] [PubMed] [Google Scholar]
- Kim WJ, Beardsley DI, Adamson AW, Brown KD. The monofunctional alkylating agent N-methyl-N'-nitro-N-nitrosoguanidine triggers apoptosis through p53-dependent and-independent pathways. Toxicol Appl Pharmacol. 2005;202:84–98. doi: 10.1016/j.taap.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, et al. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367–374. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- Koedel U, Winkler F, Angele B, Fontana A, Pfister HW. Meningitis-associated central nervous system complications are mediated by the activation of poly(ADP-ribose) polymerase. J Cereb Blood Flow Metab. 2002;22:39–49. doi: 10.1097/00004647-200201000-00005. [DOI] [PubMed] [Google Scholar]
- Koh DW, Lawler AM, Poitras MF, Sasaki M, Wattler S, Nehls MC, et al. Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc Natl Acad Sci U S A. 2004a;101:17699–17704. doi: 10.1073/pnas.0406182101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh SH, Park Y, Song CW, Kim JG, Kim K, Kim J, et al. The effect of PARP inhibitor on ischaemic cell death, its related inflammation and survival signals. Eur J Neurosci. 2004b;20:1461–1472. doi: 10.1111/j.1460-9568.2004.03632.x. [DOI] [PubMed] [Google Scholar]
- Komjati K, Mabley JG, Virag L, Southan GJ, Salzman AL, Szabo C. Poly(ADP-ribose) polymerase inhibition protect neurons and the white matter and regulates the translocation of apoptosis-inducing factor in stroke. Int J Mol Med. 2004;13:373–382. [PubMed] [Google Scholar]
- Komjati K, Besson VC, Szabo C. Poly (adp-ribose) polymerase inhibitors as potential therapeutic agents in stroke and neurotrauma. Curr Drug Targets CNS Neurol Disord. 2005;4:179–194. doi: 10.2174/1568007053544138. [DOI] [PubMed] [Google Scholar]
- Kraus WL, Lis JT. PARP goes transcription. Cell. 2003;113:677–683. doi: 10.1016/s0092-8674(03)00433-1. [DOI] [PubMed] [Google Scholar]
- Krietsch J, Rouleau M, Pic E, Ethier C, Dawson TM, Dawson VL, et al. Reprogramming cellular events by poly(ADP-ribose)-binding proteins. Mol Aspects Med. 2012;34:1066–1087. doi: 10.1016/j.mam.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam TT. The effect of 3-aminobenzamide, an inhibitor of poly-ADP-ribose polymerase, on ischemia/reperfusion damage in rat retina. Res Commun Mol Pathol Pharmacol. 1997;95:241–252. [PubMed] [Google Scholar]
- Landshamer S, Hoehn M, Barth N, Duvezin-Caubet S, Schwake G, Tobaben S, et al. Bid-induced release of AIF from mitochondria causes immediate neuronal cell death. Cell Death Differ. 2008;15:1553–1563. doi: 10.1038/cdd.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPlaca MC, Zhang J, Raghupathi R, Li JH, Smith F, Bareyre FM, et al. Pharmacologic inhibition of poly(ADP-ribose) polymerase is neuroprotective following traumatic brain injury in rats. J Neurotrauma. 2001;18:369–376. doi: 10.1089/089771501750170912. [DOI] [PubMed] [Google Scholar]
- de la Lastra CA, Villegas I, Sanchez-Fidalgo S. Poly(ADP-ribose) polymerase inhibitors: new pharmacological functions and potential clinical implications. Curr Pharm Des. 2007;13:933–962. doi: 10.2174/138161207780414241. [DOI] [PubMed] [Google Scholar]
- Lee JH, Park SY, Shin HK, Kim CD, Lee WS, Hong KW. Poly(ADP-ribose) polymerase inhibition by cilostazol is implicated in the neuroprotective effect against focal cerebral ischemic infarct in rat. Brain Res. 2007a;1152:182–190. doi: 10.1016/j.brainres.2007.03.035. [DOI] [PubMed] [Google Scholar]
- Lee MW, Kim WJ, Beardsley DI, Brown KD. N-methyl-N'-nitro-N-nitrosoguanidine activates multiple cell death mechanisms in human fibroblasts. DNA Cell Biol. 2007b;26:683–694. doi: 10.1089/dna.2007.0594. [DOI] [PubMed] [Google Scholar]
- Lo EH, Bosque-Hamilton P, Meng W. Inhibition of poly(ADP-ribose) polymerase: reduction of ischemic injury and attenuation of N-methyl-D-aspartate-induced neurotransmitter dysregulation. Stroke. 1998;29:830–836. doi: 10.1161/01.str.29.4.830. [DOI] [PubMed] [Google Scholar]
- Loeffler M, Daugas E, Susin SA, Zamzami N, Metivier D, Nieminen AL, et al. Dominant cell death induction by extramitochondrially targeted apoptosis-inducing factor. FASEB J. 2001;15:758–767. doi: 10.1096/fj.00-0388com. [DOI] [PubMed] [Google Scholar]
- Lorenzo HK, Susin SA. Therapeutic potential of AIF-mediated caspase-independent programmed cell death. Drug Resist Updat. 2007;10:235–255. doi: 10.1016/j.drup.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Love S, Barber R, Wilcock GK. Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer's disease. Brain. 1999a;122(Pt 2):247–253. doi: 10.1093/brain/122.2.247. [DOI] [PubMed] [Google Scholar]
- Love S, Barber R, Wilcock GK. Neuronal accumulation of poly(ADP-ribose) after brain ischaemia. Neuropathol Appl Neurobiol. 1999b;25:98–103. doi: 10.1046/j.1365-2990.1999.00179.x. [DOI] [PubMed] [Google Scholar]
- Love S, Barber R, Wilcock GK. Neuronal death in brain infarcts in man. Neuropathol Appl Neurobiol. 2000;26:55–66. doi: 10.1046/j.1365-2990.2000.00218.x. [DOI] [PubMed] [Google Scholar]
- Mabley JG, Jagtap P, Perretti M, Getting SJ, Salzman AL, Virag L, et al. Anti-inflammatory effects of a novel, potent inhibitor of poly (ADP-ribose) polymerase. Inflamm Res. 2001;50:561–569. doi: 10.1007/PL00000234. [DOI] [PubMed] [Google Scholar]
- McCabe N, Lord CJ, Tutt AN, Martin NM, Smith GC, Ashworth A. BRCA2-deficient CAPAN-1 cells are extremely sensitive to the inhibition of Poly (ADP-Ribose) polymerase: an issue of potency. Cancer Biol Ther. 2005;4:934–936. doi: 10.4161/cbt.4.9.2141. [DOI] [PubMed] [Google Scholar]
- Maier C, Scheuerle A, Hauser B, Schelzig H, Szabo C, Radermacher P, et al. The selective poly(ADP)ribose-polymerase 1 inhibitor INO1001 reduces spinal cord injury during porcine aortic cross-clamping-induced ischemia/reperfusion injury. Intensive Care Med. 2007;33:845–850. doi: 10.1007/s00134-007-0585-3. [DOI] [PubMed] [Google Scholar]
- Mandir AS, Przedborski S, Jackson-Lewis V, Wang ZQ, Simbulan-Rosenthal CM, Smulson ME, et al. Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc Natl Acad Sci U S A. 1999;96:5774–5779. doi: 10.1073/pnas.96.10.5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Price DD, Zhu J, Lu J, Mayer DJ. The inhibition of nitric oxide-activated poly(ADP-ribose) synthetase attenuates transsynaptic alteration of spinal cord dorsal horn neurons and neuropathic pain in the rat. Pain. 1997;72:355–366. doi: 10.1016/s0304-3959(97)00063-8. [DOI] [PubMed] [Google Scholar]
- Mate MJ, Ortiz-Lombardia M, Boitel B, Haouz A, Tello D, Susin SA, et al. The crystal structure of the mouse apoptosis-inducing factor AIF. Nat Struct Biol. 2002;9:442–446. doi: 10.1038/nsb793. [DOI] [PubMed] [Google Scholar]
- Mathews MT, Berk BC. PARP-1 inhibition prevents oxidative and nitrosative stress-induced endothelial cell death via transactivation of the VEGF receptor 2. Arterioscler Thromb Vasc Biol. 2008;28:711–717. doi: 10.1161/ATVBAHA.107.156406. [DOI] [PubMed] [Google Scholar]
- Meier HL, Ballough GP, Forster JS, Filbert MG. Benzamide, a poly(ADP-ribose) polymerase inhibitor, is neuroprotective against soman-induced seizure-related brain damage. Ann N Y Acad Sci. 1999;890:330–335. doi: 10.1111/j.1749-6632.1999.tb08010.x. [DOI] [PubMed] [Google Scholar]
- Meli E, Pangallo M, Picca R, Baronti R, Moroni F, Pellegrini-Giampietro DE. Differential role of poly(ADP-ribose) polymerase-1in apoptotic and necrotic neuronal death induced by mild or intense NMDA exposure in vitro. Mol Cell Neurosci. 2004;25:172–180. doi: 10.1016/j.mcn.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Meyer RG, Meyer-Ficca ML, Whatcott CJ, Jacobson EL, Jacobson MK. Two small enzyme isoforms mediate mammalian mitochondrial poly(ADP-ribose) glycohydrolase (PARG) activity. Exp Cell Res. 2007;313:2920–2936. doi: 10.1016/j.yexcr.2007.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Ficca ML, Meyer RG, Coyle DL, Jacobson EL, Jacobson MK. Human poly(ADP-ribose) glycohydrolase is expressed in alternative splice variants yielding isoforms that localize to different cell compartments. Exp Cell Res. 2004;297:521–532. doi: 10.1016/j.yexcr.2004.03.050. [DOI] [PubMed] [Google Scholar]
- Miesel R, Kurpisz M, Kroger H. Modulation of inflammatory arthritis by inhibition of poly(ADP ribose) polymerase. Inflammation. 1995;19:379–387. doi: 10.1007/BF01534394. [DOI] [PubMed] [Google Scholar]
- Mormone E, Matarrese P, Tinari A, Cannella M, Maglione V, Farrace MG, et al. Genotype-dependent priming to self-and xeno-cannibalism in heterozygous and homozygous lymphoblasts from patients with Huntington's disease. J Neurochem. 2006;98:1090–1099. doi: 10.1111/j.1471-4159.2006.03998.x. [DOI] [PubMed] [Google Scholar]
- Moroni F, Meli E, Peruginelli F, Chiarugi A, Cozzi A, Picca R, et al. Poly(ADP-ribose) polymerase inhibitors attenuate necrotic but not apoptotic neuronal death in experimental models of cerebral ischemia. Cell Death Differ. 2001;8:921–932. doi: 10.1038/sj.cdd.4400884. [DOI] [PubMed] [Google Scholar]
- Nagayama T, Simon RP, Chen D, Henshall DC, Pei W, Stetler RA, et al. Activation of poly(ADP-ribose) polymerase in the rat hippocampus may contribute to cellular recovery following sublethal transient global ischemia. J Neurochem. 2000;74:1636–1645. doi: 10.1046/j.1471-4159.2000.0741636.x. [DOI] [PubMed] [Google Scholar]
- Nagley P, Higgins GC, Atkin JD, Beart PM. Multifaceted deaths orchestrated by mitochondria in neurones. Biochim Biophys Acta. 2010;1802:167–185. doi: 10.1016/j.bbadis.2009.09.004. [DOI] [PubMed] [Google Scholar]
- Otera H, Ohsakaya S, Nagaura Z, Ishihara N, Mihara K. Export of mitochondrial AIF in response to proapoptotic stimuli depends on processing at the intermembrane space. EMBO J. 2005;24:1375–1386. doi: 10.1038/sj.emboj.7600614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro TF, Grammatopoulos TN, Altmann S, Amore A, Standaert DG, Hyman BT, et al. Pharmacological inhibition of PARP-1 reduces alpha-synuclein-and MPP+-induced cytotoxicity in Parkinson's disease in vitro models. Biochem Biophys Res Commun. 2007;357:596–602. doi: 10.1016/j.bbrc.2007.03.163. [DOI] [PubMed] [Google Scholar]
- Pacher P, Szabo C. Role of poly(ADP-ribose) polymerase 1 (PARP-1) in cardiovascular diseases: the therapeutic potential of PARP inhibitors. Cardiovasc Drug Rev. 2007;25:235–260. doi: 10.1111/j.1527-3466.2007.00018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Szabo C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am J Pathol. 2008;173:2–13. doi: 10.2353/ajpath.2008.080019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicciari R, Camaioni E, Costantino G, Formentini L, Sabbatini P, Venturoni F, et al. On the way to selective PARP-2 inhibitors. Design, synthesis, and preliminary evaluation of a series of isoquinolinone derivatives. ChemMedChem. 2008;3:914–923. doi: 10.1002/cmdc.200800010. [DOI] [PubMed] [Google Scholar]
- Peralta-Leal A, Rodriguez MI, Oliver FJ. Poly(ADP-ribose)polymerase-1 (PARP-1) in carcinogenesis: potential role of PARP inhibitors in cancer treatment. Clin Transl Oncol. 2008;10:318–323. doi: 10.1007/s12094-008-0207-8. [DOI] [PubMed] [Google Scholar]
- Peralta-Leal A, Rodriguez-Vargas JM, Aguilar-Quesada R, Rodriguez MI, Linares JL, de Almodovar MR, et al. PARP inhibitors: new partners in the therapy of cancer and inflammatory diseases. Free Radic Biol Med. 2009;47:13–26. doi: 10.1016/j.freeradbiomed.2009.04.008. [DOI] [PubMed] [Google Scholar]
- Plaschke K, Kopitz J, Weigand MA, Martin E, Bardenheuer HJ. The neuroprotective effect of cerebral poly(ADP-ribose)polymerase inhibition in a rat model of global ischemia. Neurosci Lett. 2000;284:109–112. doi: 10.1016/s0304-3940(00)00988-5. [DOI] [PubMed] [Google Scholar]
- Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, et al. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell. 2007;131:476–491. doi: 10.1016/j.cell.2007.08.047. [DOI] [PubMed] [Google Scholar]
- Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10:293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevrioukova IF. Apoptosis-inducing factor: structure, function, and redox regulation. Antioxid Redox Signal. 2011;14:2545–2579. doi: 10.1089/ars.2010.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shall S, de Murcia G. Poly(ADP-ribose) polymerase-1: what have we learned from the deficient mouse model? Mutat Res. 2000;460:1–15. doi: 10.1016/s0921-8777(00)00016-1. [DOI] [PubMed] [Google Scholar]
- Smith S. The world according to PARP. Trends Biochem Sci. 2001;26:174–179. doi: 10.1016/s0968-0004(00)01780-1. [DOI] [PubMed] [Google Scholar]
- Smulson ME, Simbulan-Rosenthal CM, Boulares AH, Yakovlev A, Stoica B, Iyer S, et al. Roles of poly(ADP-ribosyl)ation and PARP in apoptosis, DNA repair, genomic stability and functions of p53 and E2F-1. Adv Enzyme Regul. 2000;40:183–215. doi: 10.1016/s0065-2571(99)00024-2. [DOI] [PubMed] [Google Scholar]
- Son YO, Kook SH, Jang YS, Shi X, Lee JC. Critical role of poly(ADP-ribose) polymerase-1 in modulating the mode of cell death caused by continuous oxidative stress. J Cell Biochem. 2009;108:989–997. doi: 10.1002/jcb.22332. [DOI] [PubMed] [Google Scholar]
- Soos J, Engelhardt JI, Siklos L, Havas L, Majtenyi K. The expression of PARP, NF-kappa B and parvalbumin is increased in Parkinson disease. Neuroreport. 2004;15:1715–1718. doi: 10.1097/01.wnr.0000136175.51954.ce. [DOI] [PubMed] [Google Scholar]
- Sousa FG, Matuo R, Soares DG, Escargueil AE, Henriques JA, Larsen AK, et al. PARPs and the DNA damage response. Carcinogenesis. 2012;33:1433–1440. doi: 10.1093/carcin/bgs132. [DOI] [PubMed] [Google Scholar]
- Stone TW, Addae JI. The pharmacological manipulation of glutamate receptors and neuroprotection. Eur J Pharmacol. 2002;447:285–296. doi: 10.1016/s0014-2999(02)01851-4. [DOI] [PubMed] [Google Scholar]
- Strosznajder RP, Gadamski R, Czapski GA, Jesko H, Strosznajder JB. Poly(ADP-ribose) polymerase during reperfusion after transient forebrain ischemia: its role in brain edema and cell death. J Mol Neurosci. 2003;20:61–72. doi: 10.1385/JMN:20:1:61. [DOI] [PubMed] [Google Scholar]
- Strosznajder RP, Czubowicz K, Jesko H, Strosznajder JB. Poly(ADP-ribose) metabolism in brain and its role in ischemia pathology. Mol Neurobiol. 2010;41:187–196. doi: 10.1007/s12035-010-8124-6. [DOI] [PubMed] [Google Scholar]
- Stubberfield CR, Cohen GM. NAD+ depletion and cytotoxicity in isolated hepatocytes. Biochem Pharmacol. 1988;37:3967–3974. doi: 10.1016/0006-2952(88)90081-0. [DOI] [PubMed] [Google Scholar]
- Sun AY, Cheng JS. Neuroprotective effects of poly (ADP-ribose) polymerase inhibitors in transient focal cerebral ischemia of rats. Zhongguo Yao Li Xue Bao. 1998;19:104–108. [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Suzuki E, Okuda H, Nishida K, Fujimoto S, Nagasawa K. Protective effect of nicotinamide against poly(ADP-ribose) polymerase-1-mediated astrocyte death depends on its transporter-mediated uptake. Life Sci. 2010;86:676–682. doi: 10.1016/j.lfs.2010.02.019. [DOI] [PubMed] [Google Scholar]
- Szabo C. Roles of poly(ADP-ribose) polymerase activation in the pathogenesis of diabetes mellitus and its complications. Pharmacol Res. 2005;52:60–71. doi: 10.1016/j.phrs.2005.02.015. [DOI] [PubMed] [Google Scholar]
- Tabuchi K, Ito Z, Tsuji S, Nakagawa A, Serizawa F, Hara A, et al. Poly(adenosine diphosphate-ribose) synthetase inhibitor 3-aminobenzamide alleviates cochlear dysfunction induced by transient ischemia. Ann Otol Rhinol Laryngol. 2001;110:118–121. doi: 10.1177/000348940111000205. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Greenberg JH. The effect of reperfusion on neuroprotection using an inhibitor of poly(ADP-ribose) polymerase. Neuroreport. 1999;10:2017–2022. doi: 10.1097/00001756-199907130-00005. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Greenberg JH, Jackson P, Maclin K, Zhang J. Neuroprotective effects of inhibiting poly(ADP-ribose) synthetase on focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1997;17:1137–1142. doi: 10.1097/00004647-199711000-00001. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Pieper AA, Croul SE, Zhang J, Snyder SH, Greenberg JH. Post-treatment with an inhibitor of poly(ADP-ribose) polymerase attenuates cerebral damage in focal ischemia. Brain Res. 1999;829:46–54. doi: 10.1016/s0006-8993(99)01335-9. [DOI] [PubMed] [Google Scholar]
- Tang KS, Suh SW, Alano CC, Shao Z, Hunt WT, Swanson RA, et al. Astrocytic poly(ADP-ribose) polymerase-1 activation leads to bioenergetic depletion and inhibition of glutamate uptake capacity. Glia. 2010;58:446–457. doi: 10.1002/glia.20936. [DOI] [PubMed] [Google Scholar]
- Tokime T, Nozaki K, Sugino T, Kikuchi H, Hashimoto N, Ueda K. Enhanced poly(ADP-ribosyl)ation after focal ischemia in rat brain. J Cereb Blood Flow Metab. 1998;18:991–997. doi: 10.1097/00004647-199809000-00008. [DOI] [PubMed] [Google Scholar]
- Turner NC, Lord CJ, Iorns E, Brough R, Swift S, Elliott R, et al. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 2008;27:1368–1377. doi: 10.1038/emboj.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virag L. Structure and function of poly(ADP-ribose) polymerase-1: role in oxidative stress-related pathologies. Curr Vasc Pharmacol. 2005;3:209–214. doi: 10.2174/1570161054368625. [DOI] [PubMed] [Google Scholar]
- Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- Vis JC, Schipper E, de Boer-van Huizen RT, Verbeek MM, de Waal RM, Wesseling P, et al. Expression pattern of apoptosis-related markers in Huntington's disease. Acta Neuropathol. 2005;109:321–328. doi: 10.1007/s00401-004-0957-5. [DOI] [PubMed] [Google Scholar]
- Wang H, Shimoji M, Yu SW, Dawson TM, Dawson VL. Apoptosis inducing factor and PARP-mediated injury in the MPTP mouse model of Parkinson's disease. Ann N Y Acad Sci. 2003;991:132–139. doi: 10.1111/j.1749-6632.2003.tb07471.x. [DOI] [PubMed] [Google Scholar]
- Wang H, Yu SW, Koh DW, Lew J, Coombs C, Bowers W, et al. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J Neurosci. 2004;24:10963–10973. doi: 10.1523/JNEUROSCI.3461-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yang C, Chai J, Shi Y, Xue D. Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science. 2002;298:1587–1592. doi: 10.1126/science.1076194. [DOI] [PubMed] [Google Scholar]
- Wang Y, Dawson VL, Dawson TM. Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp Neurol. 2009a;218:193–202. doi: 10.1016/j.expneurol.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kim NS, Li X, Greer PA, Koehler RC, Dawson VL, et al. Calpain activation is not required for AIF translocation in PARP-1-dependent cell death (parthanatos) J Neurochem. 2009b;110:687–696. doi: 10.1111/j.1471-4159.2009.06167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kim NS, Haince JF, Kang HC, David KK, Andrabi SA, et al. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos) Sci Signal. 2011;4:ra20. doi: 10.1126/scisignal.2000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weseler AR, Geraets L, Moonen HJ, Manders RJ, van Loon LJ, Pennings HJ, et al. Poly (ADP-ribose) polymerase-1-inhibiting flavonoids attenuate cytokine release in blood from male patients with chronic obstructive pulmonary disease or type 2 diabetes. J Nutr. 2009;139:952–957. doi: 10.3945/jn.108.102756. [DOI] [PubMed] [Google Scholar]
- Whitacre CM, Hashimoto H, Tsai ML, Chatterjee S, Berger SJ, Berger NA. Involvement of NAD-poly(ADP-ribose) metabolism in p53 regulation and its consequences. Cancer Res. 1995;55:3697–3701. [PubMed] [Google Scholar]
- Winstall E, Affar EB, Shah R, Bourassa S, Scovassi AI, Poirier GG. Poly(ADP-ribose) glycohydrolase is present and active in mammalian cells as a 110-kDa protein. Exp Cell Res. 1999;246:395–398. doi: 10.1006/excr.1998.4321. [DOI] [PubMed] [Google Scholar]
- Xia Y, Dawson VL, Dawson TM, Snyder SH, Zweier JL. Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. Proc Natl Acad Sci U S A. 1996;93:6770–6774. doi: 10.1073/pnas.93.13.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Zhang J, David KK, Yang ZJ, Li X, Dawson TM, et al. Endonuclease G does not play an obligatory role in poly(ADP-ribose) polymerase-dependent cell death after transient focal cerebral ischemia. Am J Physiol Regul Integr Comp Physiol. 2010;299:R215–R221. doi: 10.1152/ajpregu.00747.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Okamoto H. Protection by picolinamide, a novel inhibitor of poly (ADP-ribose) synthetase, against both streptozotocin-induced depression of proinsulin synthesis and reduction of NAD content in pancreatic islets. Biochem Biophys Res Commun. 1980;95:474–481. doi: 10.1016/0006-291x(80)90762-7. [DOI] [PubMed] [Google Scholar]
- Yap E, Tan WL, Ng I, Ng YK. Combinatorial-approached neuroprotection using pan-caspase inhibitor and poly (ADP-ribose) polymerase (PARP) inhibitor following experimental stroke in rats; is there additional benefit? Brain Res. 2008;1195:130–138. doi: 10.1016/j.brainres.2007.12.024. [DOI] [PubMed] [Google Scholar]
- Ye H, Cande C, Stephanou NC, Jiang S, Gurbuxani S, Larochette N, et al. DNA binding is required for the apoptogenic action of apoptosis inducing factor. Nat Struct Biol. 2002;9:680–684. doi: 10.1038/nsb836. [DOI] [PubMed] [Google Scholar]
- Yeh TY, Sbodio JI, Nguyen MT, Meyer TN, Lee RM, Chi NW. Tankyrase-1 overexpression reduces genotoxin-induced cell death by inhibiting PARP1. Mol Cell Biochem. 2005;276:183–192. doi: 10.1007/s11010-005-4059-z. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Dawson TM, Dawson VL. Poly(ADP-ribose) polymerase-1 and apoptosis inducing factor in neurotoxicity. Neurobiol Dis. 2003;14:303–317. doi: 10.1016/j.nbd.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, et al. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci U S A. 2006;103:18314–18319. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SW, Wang Y, Frydenlund DS, Ottersen OP, Dawson VL, Dawson TM. Outer mitochondrial membrane localization of apoptosis-inducing factor: mechanistic implications for release. ASN Neuro. 2009;1:e00021. doi: 10.1042/AN20090046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Siegel C, Zeng Z, Li J, Liu F, McCullough LD. Sex differences in the response to activation of the poly (ADP-ribose) polymerase pathway after experimental stroke. Exp Neurol. 2009;217:210–218. doi: 10.1016/j.expneurol.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelphati O, Wang Y, Kitada S, Reed JC, Felgner PL, Corbeil J. Intracellular delivery of proteins with a new lipid-mediated delivery system. J Biol Chem. 2001;276:35103–35110. doi: 10.1074/jbc.M104920200. [DOI] [PubMed] [Google Scholar]
- Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Feng X, Koh DW. Activation of cell death mediated by apoptosis-inducing factor due to the absence of poly(ADP-ribose) glycohydrolase. Biochemistry. 2011;50:2850–2859. doi: 10.1021/bi101829r. [DOI] [PubMed] [Google Scholar]
- Zhu C, Wang X, Deinum J, Huang Z, Gao J, Modjtahedi N, et al. Cyclophilin A participates in the nuclear translocation of apoptosis-inducing factor in neurons after cerebral hypoxia-ischemia. J Exp Med. 2007a;204:1741–1748. doi: 10.1084/jem.20070193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Wang X, Huang Z, Qiu L, Xu F, Vahsen N, et al. Apoptosis-inducing factor is a major contributor to neuronal loss induced by neonatal cerebral hypoxia-ischemia. Cell Death Differ. 2007b;14:775–784. doi: 10.1038/sj.cdd.4402053. [DOI] [PubMed] [Google Scholar]
- Zingarelli B, Szabo C, Salzman AL. Blockade of Poly(ADP-ribose) synthetase inhibits neutrophil recruitment, oxidant generation, and mucosal injury in murine colitis. Gastroenterology. 1999;116:335–345. doi: 10.1016/s0016-5085(99)70130-7. [DOI] [PubMed] [Google Scholar]