

Abstract
The mitochondrion plays an important role in the production of energy as ATP, the regulation of cell viability and apoptosis, and the biosynthesis of major structural and regulatory molecules, such as lipids. During ATP production, reactive oxygen species are generated that alter the intracellular redox state and activate apoptosis. Mitochondrial dysfunction is a well-recognized component of the pathogenesis of diseases such as cancer. Understanding mitochondrial function, and how this is dysregulated in disease, offers the opportunity for the development of drug molecules to specifically target such defects. Altered energy metabolism in cancer, in which ATP production occurs largely by glycolysis, rather than by oxidative phosphorylation, is attributable in part to the up-regulation of cell survival signalling cascades. These pathways also regulate the balance between pro-and anti-apoptotic factors that may determine the rate of cell death and proliferation. A number of anti-cancer drugs have been developed that target these factors and one of the most promising groups of agents in this regard are the lipid-based molecules that act directly or indirectly at the mitochondrion. These molecules have emerged in part from an understanding of the mitochondrial actions of naturally occurring fatty acids. Some of these agents have already entered clinical trials because they specifically target known mitochondrial defects in the cancer cell.
LINKED ARTICLES
This article is part of a themed issue on Mitochondrial Pharmacology: Energy, Injury & Beyond. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2014.171.issue-8
Keywords: N-acylethanolamines, cancer cell, ether phospholipids, fatty acid biotransformation, free fatty acids, intrinsic pathway of apoptosis, mitochondrial ATP production, polyunsaturated fatty acid epoxides, reactive oxygen species
Mitochondrial function: introduction
The mitochondrion has a number of critical homeostatic functions, including the production of energy as ATP, the control of apoptotic cell death and viability, and the biosynthesis of molecules such as steroids and lipids that perform regulatory and structural roles in cells (Kroemer et al., 2007). Mitochondria are also the major source of reactive oxygen species (ROS) that determine the intracellular redox state and modulate cell proliferation and apoptosis. It is increasingly recognized that disease processes, such as tumourigenesis and metabolic syndrome, are associated with mitochondrial dysfunction (Kroemer et al., 2007). A detailed understanding of how the regulation of mitochondrial function is altered during cancer progression may provide opportunities for drug design strategies that target underlying defects in disease.
Mitochondrial energy production and altered ATP generation in cancer cells
The role of the mitochondrion in energy metabolism is well established and glucose is the primary fuel molecule utilized by the cell in ATP production (Kroemer et al., 2007). The initial glycolytic step occurs in the cytoplasm of the cell and generates pyruvate that enters the mitochondrion where it is converted to citrate; together, these two reactions produce four molecules of ATP from each molecule of glucose. In addition, four large multi-protein respiratory complexes in the inner mitochondrial membrane work in concert to generate a much larger number of ATPs by oxidative phosphorylation (∼28–32 per glucose molecule). Simultaneously, this builds up the proton gradient across the inner and outer mitochondrial membranes, which drives many mitochondrial processes.
Fats and proteins may also be utilized by the mitochondrion to produce ATP (Kroemer et al., 2007). Triglyceride esters in adipose tissue are hydrolysed to free fatty acids that undergo mitochondrial β-oxidation to acetyl-CoA units that are then able to enter the citric acid cycle and generate ATP. The oxidative deamination of amino acids produces up to 15% of total metabolic energy in animals. In the initial phase, amino acids are converted to a series of intermediary molecules – pyruvate, α-ketoglutarate, succinyl-CoA, fumarate, oxaloacetate, acetyl-CoA and acetoacetate – that may then enter the citric acid cycle and generate ATP.
Whereas normal cells generate much of their ATP by oxidative phosphorylation, aggressive cancer cells exhibit pronounced bioenergetic differences and overproduce lactate even under normoxic conditions by ‘aerobic glycolysis’ (Rossignol et al., 2004). This occurs because most of the pyruvate formed by glycolysis is unable to enter the mitochondrion, and is instead converted to lactate by cytosolic lactate dehydrogenase.
Mitochondria and ROS production: activation of the intrinsic pathway of apoptosis
The ROS H2O2, superoxide (O2−) and hydroxyl radical (OH−) are generated by mitochondrial respiratory complexes during uncoupled substrate turnover in the process of ATP formation (Cadenas and Davies, 2000; Hanahan and Weinberg, 2011). Premature leakage of electrons from respiratory complexes, rather than coupled transfer during ATP synthesis, leads to superoxide formation (Skulachev, 1998; Di Paola and Lorusso, 2006; Pike et al., 2011). The mitochondrion is not only the main site of ROS generation; it is also their primary target. The generation of ROS has several consequences in cells, including direct peroxidative damage to membranes and modification of DNA bases that may initiate mutagenesis (Larsen et al., 2005; Nathan and Cunningham-Bussel, 2013). ROS also modulate cellular redox homeostasis (Higdon et al., 2012) and decrease the concentration of thiol-containing species, such as glutathione, which are cytoprotective (Mari et al., 2009). Indeed, glutathione depletion decreases the integrity and activities of mitochondrial respiratory complexes, which compromises ATP production and cell viability (Merad-Boudia et al., 1998). More recently, it has been recognized that ROS are also able to activate the JNK (Trachootham et al., 2006) and p38 MAPK (Ito et al., 2006) signalling pathways that may trigger apoptotic cell death.
Apoptosis is a coordinated cell death programme that is important in normal tissues (Fulda and Debatin, 2006). There are two major pathways of apoptosis: the extrinsic and intrinsic pathways. In the extrinsic pathway, tumour necrosis factor family receptors at the plasma membrane are activated by FasL and related ligands and modulate intracellular signalling pathways leading to cell deletion. The intrinsic, or mitochondrial, pathway of apoptosis is activated intracellularly by ROS, the inhibition of pro-survival signalling cascades or by major cellular stresses, including DNA damage from exposure to cytotoxic anti-cancer drugs.
The relative ratio of pro-and anti-apoptotic Bcl-2 family proteins is a determinant of the response of cells to apoptotic stimuli (Fulda and Debatin, 2006). Thus, pro-apoptotic Bcl-2 proteins, such as Bax, Bak and Bid, form dimers that destabilize the outer mitochondrial membrane to apoptotic stimuli by forming channels that allow mitochondrial factors to exit, whereas anti-apoptotic members such as Bcl-2, Bcl-XL and Bcl-w stabilize the membrane. Thus, cytochrome c, Smac/Diablo or HtrA2/Omi are released to the cytosol, which triggers the commitment step of the mitochondrial apoptotic cascade (Kluck et al., 1997; Yang et al., 2003). The intrinsic and extrinsic apoptotic pathways converge on the executioner cysteine-aspartic acid proteases (caspases)-3 and-7, which lyse a number of cellular protein targets, including the DNA-repair protein poly (ADP ribose) polymerase (PARP); inactivation of the latter promotes apoptosis (Fulda and Debatin, 2006). However, release of Smac/Diablo or HtrA2/Omi from the mitochondrion does not directly activate caspase; rather these factors block the action of the protein inhibitor of apoptosis that normally acts to suppress caspases. Finally, there is also a caspase-independent mitochondrial pathway in which apoptosis-inducing factor and endonuclease G are released to the cytosol (Susin et al., 1999). The translocation of these factors leads to DNA fragmentation, which is the hallmark of apoptosis.
Mitochondrial permeability is controlled by the permeability transition pore complex, which regulates the flow of small molecules across the mitochondrial membrane. Pore opening occurs in response to elevated Ca2+ concentrations, increased ROS and depleted adenine nucleotides (Kroemer et al., 2007). Permeability transition pore opening in turn dissipates the proton gradient, uncouples oxidative phosphorylation and decreases ATP formation. In consequence, antioxidant molecules such as glutathione exit the mitochondrion, which further decreases the capacity to detoxify locally generated ROS (Mari et al., 2009). The release of cytochrome c from the mitochondrion sets in train the activation of apoptotic death.
The permeability transition pore is a complex arrangement of proteins including the adenine nucleotide translocator, which exchanges ATP and ADP (Marzo et al., 1998a; Brenner et al., 2000), the voltage-dependent anion channel (also termed porin), the soluble mitochondrial matrix protein cyclophilin D (Woodfield et al., 1998), a number of Bcl-2 family proteins (Marzo et al., 1998b; Shimizu et al., 1999), the peripheral benzodiazepine receptor and several proteins that regulate energy metabolism (e.g. hexokinase II and creatine kinase; Marzo et al., 1998a). Several genes encoding components of the mitochondrial permeability transition pore including the peripheral benzodiazepine receptor, the associated protein Prax-1 and the energy-metabolizing enzyme creatine kinase, are overexpressed in some tumours (Kanazawa et al., 1998; Venturini et al., 1998; Galiegue et al., 1999).
While the pro-apoptotic Bax may be down-regulated in cancer cells (Brimmell et al., 1998), Bcl-2 or its anti-apoptotic homologues are frequently overexpressed (Kroemer et al., 2007). This shifts the balance in the cancer cell towards the prevention of apoptosis. Thus, an important adaptation is that the PI3K/Akt signalling cascade inhibits pro-apoptotic Bcl-2 factors and positively regulates the anti-apoptotic factor Bcl-2 (Skorski et al., 1997; Pugazhenthi et al., 2000). As described above, this shift in Bcl-2 factor composition stabilizes the outer mitochondrial membrane. PI3K/Akt has additional survival actions in tumour cells in that it is able to impair signalling by the pro-apoptotic JNK MAPK, enhance glucose uptake by transporters and activate glycolytic enzymes, such as hexokinase (Skorski et al., 1997; Pugazhenthi et al., 2000; Kroemer et al., 2007) These adaptations shift the capacity for energy production towards the glycolytic pathway that characterizes the cancer cell.
Biosynthetic roles of the mitochondrion in normal and cancer cells
Some important steps in lipid metabolism occur in the mitochondrion. While fatty acid synthesis occurs primarily in the cytoplasm, β-oxidation to produce ATP occurs in mitochondria. However, the mitochondrion also has a central role in the biosynthesis of phospholipids and triglycerides. The first step in the phospholipid synthesis pathway is the esterification of α-glycerol phosphate by acyl-CoA to produce lysophosphatidic and phosphatidic acids (Zborowski and Wojtczak, 1969; Bremer et al., 1976). Some phospholipids, such as phosphatidylcholine, phosphatidylserine and phosphatidylinositol, are synthesized in other organelles and transported to the mitochondrion where further biotransformation occurs. Cardiolipin, a characteristic phospholipid of the inner mitochondrial membrane, is synthesized within this organelle (Hostetler and van den Bosch, 1972).
In mammals, fatty acids are activated to acyl-CoAs on the outer mitochondrial membrane before entering the glycerolipid biosynthetic pathway via glycerol-3-phosphate acyltransferase. Highly proliferative cancer cells have an increased requirement for lipids for the assembly of cell membranes (Samudio et al., 2009; Zaugg et al., 2011). Increased expression of fatty acid synthase in tumour cells promotes formation of long-chain fatty acids and confers a growth advantage (Sabine et al., 1967; Ookhtens et al., 1984). Indeed, fatty acid synthase inhibition decreases the rate of cancer cell proliferation and promotes apoptosis, as reflected by increased caspase-3 activation, down-regulation of anti-apoptotic proteins and the release of cytochrome c (Pizer et al., 1998; De Schrijver et al., 2003).
Lipid synthesis and biotransformation in cells
Fatty acids perform structural roles in cells by modulating membrane fluidity and the arrangement of receptors and other proteins in plasma membrane lipid rafts (Spector and Yorek, 1985). Naturally occurring long-chain fatty acids of 18–22 carbons in length are stored in cell membranes as triglycerides or phospholipids. Triglycerides are composed of three fatty acid residues connected to the hydroxyl groups of glycerol via ester linkages (Figure 1A). Phosphoglycerides are the major phospholipid class, and are comprised of five components: two fatty acids, glycerol, a phosphate and an alcohol (Figure 1B). Subclasses are designated according to the alcohol group; thus phosphatidylcholines contain the choline group (Figure 1C). Other common alcohols include serine, ethanolamine and inositol. Phosphatidates share this same basic structure but lack the alcohol group, and are intermediates in the biosynthesis of many phosphoglycerides. The glycerol group may also be substituted with sphingosine (2-amino-4-octadecene-1,3-diol), giving rise to sphingomyelins.
Figure 1.
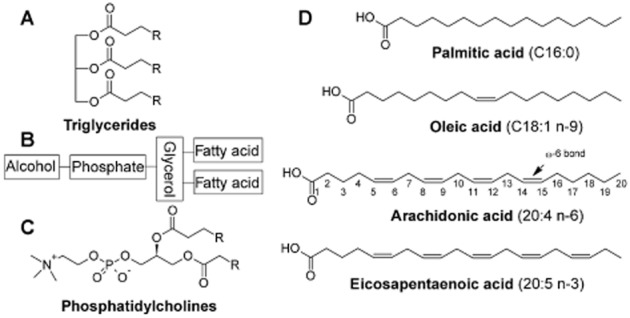
Chemical structures of fatty acids, triglycerides and phospholipids. (A) general structure of triglycerides; (B) general structure of phospholipids; (C) phosphatidylcholine; (D) important dietary fatty acids with carbon numbering.
Constituent fatty acids in membranes are either saturated [contain no carbon-carbon double bonds, as exemplified by palmitic acid (C16:0)], monounsaturated [contain only one olefinic bond; such as oleic acid (18:1 n−9)] or polyunsaturated [PUFAs that possess multiple olefinic bonds, typified by ω-6 arachidonic acid (20:4 n-6) and ω-3 eicosapentaenoic acid (20:5 n-3); Figure 1D]. The double bonds in naturally occurring unsaturated fatty acids are primarily in the cis configuration, which kinks the carbon chain and imparts greater membrane fluidity.
The carbon atoms of fatty acid chains are numbered starting from the carboxyl group and the carbon at the distal end of the molecule is the ω-carbon. Monounsaturated fatty acids and PUFA are classified by the position of the olefinic bond that is furthest from the carboxylate, and this is indicated in the lipid number notation. Thus, arachidonic acid (20:4 n-6) is a C20 fatty acid that has four olefinic bonds, with the double bond furthest from the carboxylate located six bonds from the ω-carbon (to produce a ω-6 olefinic bond).
Release of fatty acids from their esterified phospholipid forms in cell membranes is mediated by PLA2. Free PUFAs are substrates for COX, lipoxygenase (LOX) and cytochrome P450 (CYP) enzymes that generate multiple metabolites that have diverse homeostatic functions (Figure 2; Spector and Yorek, 1985; Oates et al., 1988; Oliw, 1994; Chen et al., 1995; Murray, 1999; Marden et al., 2003). Together, these enzymes produce PGs, thromboxanes, fatty acid peroxides and their downstream LTs, hydroxyfatty acids and epoxides. In comparison, saturated fatty acids primarily undergo CYP-dependent oxidation to the corresponding ω-and ω-1 hydroxy acids.
Figure 2.
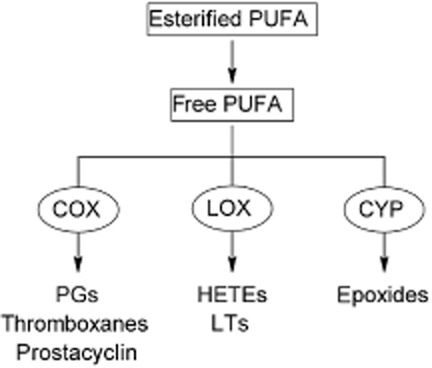
PUFA biotransformation pathways. PUFA are released from their esterified form in cell membranes to free fatty acids. The enzymatic actions of COX, LOX and CYP generate signalling molecules with diverse homeostatic actions.
Apart from free fatty acids, there is evidence that certain lipids may be present at increased levels in cancer cells. Thus, phosphocholine derivatives are increased in cancer cells and solid tumours (Ackerstaff et al., 2003) and plasma triglycerides are reportedly higher in women with invasive breast cancers (Goodwin et al., 1997). Some important enzymes involved in fatty acid biotransformation are also differentially expressed in cancers. Overexpression of COX and CYP enzymes promotes the formation of certain PG and epoxide metabolites that influence the fate of cells, including the rate of proliferation and the inhibition of apoptosis (Tsujii et al., 1997; Jiang et al., 2005). Moreover PUFA-derived metabolites modulate signalling pathways such as ERK and PI3K/Akt that have been implicated in tumourigenesis. As discussed below, inhibition of the formation of these PUFA metabolites modulates apoptosis and other aspects of tumour growth (Chen et al., 2009). Moreover, inhibition of the enzymes that mediate PUFA biotransformation has proven successful in the prevention of experimental tumour progression in vitro and in vivo (Koehne and DuBois, 2004; Chen et al., 2009). However, at present, there is a deficiency of inhibitory agents that are well tolerated over the prolonged treatment periods required for anti-cancer chemotherapy.
The potential of lipids as anti-cancer agents that act at the mitochondrion
Fatty acids are thought to uncouple oxidative phosphorylation and decrease ATP production by facilitating the leakage of protons across the lipid mitochondrial membrane. Uncoupling is greatest with C12–C16 saturated and longer cis-unsaturated fatty acids (Korshunov et al., 1998; Bernardi et al., 2002). For example, 5 μM laurate (C12-saturated) effectively inhibited H2O2 production by mitochondria (Korshunov et al., 1998). Free fatty acids also impair electron transport and activate apoptosis by releasing cytochrome c from the inner mitochondrial membrane. There is increasing evidence that several classes of naturally occurring and synthetic lipids have the potential for development as anti-cancer agents that mediate their effects, at least in part, at the mitochondrion. However, some of these lipid-mediated anti-mitochondrial actions may be indirect, by modulating intracellular signalling pathways.
Naturally occurring free fatty acids and synthetic analogues
Saturated fatty acids
In the non-ionized state, medium-and long-chain fatty acids readily penetrate the mitochondrial membrane (McLaughlin and Dilger, 1980; Gutknecht, 1988; Kamp and Hamilton, 1992). Butyric acid and similar short-chain fatty acids induce cell cycle arrest and apoptosis by dissipating the mitochondrial membrane potential (ΔΨ), leading to growth arrest and apoptosis in colon carcinoma cells in vitro, as demonstrated by activation of caspase-3 (Heerdt et al., 1997). In a small structure-activity study, the C4-butyric acid inhibited the growth of the human colonic adenocarcinoma cell lines HT-29, Colo-320 and SW-948 (IC50s 0.55–2.28 mM), while C3-propionic acid was less potent and C5-valeric and C2-acetic acids were ineffective (Figure 3, Table 1; Milovic et al., 2000). The apoptotic mechanism of butyric acid in colon cancer cells is mediated at least in part by inducing overexpression of the pro-apoptotic protein Bak, which alters the Bak/Bcl-2 ratio (Hague et al., 1997; Ruemmele et al., 1999), but not by modulating ROS or ATP production (Heerdt et al., 1997). Bromo-analogues of butyric and propionic acids (IC50s 0.13–0.39 mM) were several-fold more potent pro-apoptotic agents than butyrate, but also produced some cytotoxicity, so cautious development of these molecules as potential mitochondrial targeted agents is warranted (Milovic et al., 2000).
Figure 3.
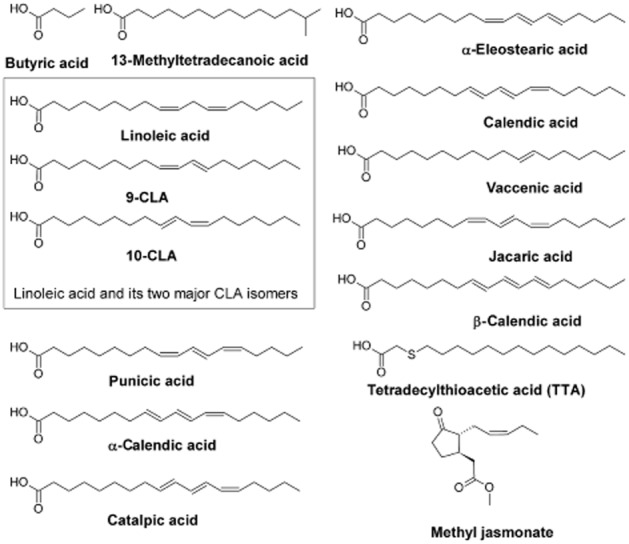
Naturally occurring fatty acids and analogues with anti-cancer activity.
Table 1.
Naturally occurring and synthetic fatty acids and other lipids: mitochondrial actions of potential value in cancer chemotherapy
| Fatty acid derivative | Mitochondrial actions | Pathways effected | Function | References |
|---|---|---|---|---|
| Butyric acid | Dissipation of the mitochondrial membrane potential (ΔΨ) | Caspase-3 activation | ↑Apoptosis Growth arrest | Heerdt et al. (1998) Milovic et al. (2000) |
| Bromo-analogues of butyric and propionic acids | ↑ROS, DNA damage | ↑Apoptosis | Milovic et al. (2000) | |
| Palmitic acid | ↓Mitochondrial membrane ΔΨ, cytochrome c release | ↑Apoptosis ↑Cell death | Merrill and Jones (1990) Schlame et al. (2000) Ostrander et al. (2001) | |
| 13-Methyltetradecanoic acid | Disrupted mitochondrial integrity and caused dysfunction, cytochrome c release | ↑Bax, ↓Bcl-2 Caspase-3 activation ↓pAkt ↑p38 and JNK MAPK | ↑Apoptosis Growth inhibition | Yang et al. (2000) Wongtangtintharn et al. (2005) Lin et al. (2012) |
| Arachidonic acid-derived PGE2 | ↑EGFR/ERK MAPK ↑PI3K/Akt | ↑Survival ↑Proliferation | Chen et al. (2009) | |
| Eicosapentaenoic acid (EPA) | ↑ATP glycolysis, ↑ROS, ↓mitochondrial membrane ΔΨ | Caspase-3 activation PKC inhibition, ↓Bcl-2 | ↑Apoptosis Growth inhibition | Denys et al. (2005) Colquhoun (2009) |
| EPA-derived epoxides | p38 MAPK | ↑Apoptosis Growth inhibition | Cui et al. (2011) | |
| Docosahexaenoic acid | PKC inhibition, ↓Bcl-2 | ↑Apoptosis | Denys et al. (2005) | |
| CLA | ↓DNA synthesis | ↓Bcl-2, ↓PI3K/Akt | ↑Apoptosis ↓Proliferation | Ip et al. (1999) Ip et al. (2000) Kim et al. (2002) |
| Jacaric acid | ↑ROS | ↓Bcl-2, caspase-3,-8 and-9 activation, PARP cleavage | ↑Apoptosis (intrinsic and/or extrinsic pathways) | Shinohara et al. (2012) Gasmi and Sanderson (2013) |
| α-Eleostearic acid | ↓Mitochondrial membrane ΔΨ, ↑DNA fragmentation ↑lipid peroxidation | ↑Apoptosis | Tsuzuki et al. (2004), Grossmann et al. (2009) | |
| Vaccenic acid | ↑DNA fragmentation | Growth inhibition ↓Cytosolic glutathione ↑Cell death | Miller et al. (2003) | |
| Punicic acid | Disrupted the mitochondrial membrane ΔΨ, ↑DNA fragmentation, ↑lipid peroxidation | Caspase-3 and-9 activation ↓Bcl-2 (decreased Bcl-2:Bax ratio) ↓pAkt | ↑Apoptosis | Grossmann et al. (2010), Gasmi and Sanderson (2010) |
| Tetradecylthioacetic acid | ↑Mitochondrial proliferation, ↑oxidative stress, ↑ROS | Caspase-3 activation PARP cleavage | ↑Apoptosis ↓Proliferation | Tronstad et al. (2001) Lin et al. (2012) Tronstad et al. (2003) |
| Jasmonates | Cytochrome c release, membrane depolarization, swelling of mitochondria, ↓ATP | ↑Cell death | Fingrut and Flescher (2002) Rotem et al. (2005) Fingrut et al. (2005) Heyfets and Flescher (2007) Goldin et al. (2007) | |
| EETs | ↑EGFR/ERK ↑PI3K/Akt | ↑Survival ↑Proliferation | Chen et al. (2009) | |
| HETEs and downstream LTs | Inhibition of 5-LOX depleted mitochondrial glutathione and ↑lipid peroxidation | ↑apoptotic Bcl-2 family proteins | ↑Survival Enhanced growth | Ghosh and Myers (1998) Avis et al. (2001) Hoque et al. (2005) |
| Short chain ceramides | Cytochrome c release, ↓mitochondrial membrane ΔΨ, ↑ROS | Caspase-3 activation, ↓Bcl-2: Bax ratio, ↑p38 and JNK MAPK phosphorylation, PARP | ↑Apoptosis | Fillet et al. (2003) Flowers et al. (2012) |
| Anandamide and N-acylethanolamines | ↑Permeability of inner mitochondrial membrane, ↑↓lipid peroxidation*, energetic and permeability transition alterations, Ca2+ overload and PTP modulation | ↑↓Apoptosis ↑↓proliferation | Epps et al. (1982) Gulaya et al. (1993) Schwarz et al. (1994) De Petrocellis et al. (1998) Maccarrone et al. (2000) Sarker et al. (2000) Wasilewski et al. (2004) | |
| Cardiolipin | Involved in ROS-promoted cytochrome c release | Membrane targeting of Bid, activation of Bax | ↑Apoptosis | Kuwana et al. (2002) |
| Perifosine | ↑Cytochrome c release, mitochondrial oxidative phosphorylation disruption, induction of permeability transition | Akt inhibition Caspase-3,-8 and-9 activation ↑JNK MAPK translocation | ↑Apoptosis | Kondapaka et al. (2003) Nieto-Miguel et al. (2006) Chiarini et al. (2008) Burgeiro et al. (2013) |
| Edelfosine | Mitochondrial dysfunction via multiple mechanisms Altered mitochondrial membrane ΔΨ, ↑DNA fragmentation, ↑ROS | Caspase-3 activation PARP | ↑Apoptosis | Cabaner et al. (1999) Gajate et al. (2000) Mollinedo et al. (2011) |
| Isopropylaminopropanol derivatives (C16:0 and C18:1) | Inhibition of multiple pro-survival kinases | Growth inhibition | Cao et al. (2013) |
Dependent on fatty acid chain length and saturation.
Longer chain saturated fatty acids also act at the mitochondrion to activate apoptosis. Palmitic acid (C16:0) decreased ΔΨ and effected cytochrome c release, which induced the proteolysis of PARP and the fragmentation of DNA (de Pablo et al., 1999). This may also be a process of endogenous importance because long-chain fatty acids can be generated intracellularly by activation of PLA2. In addition, such free fatty acids may also accumulate in mitochondria following exposure of cells to stimuli such as oxidative stress or increased Ca2+ concentrations (Broekemeier and Pfeiffer, 1995).
13-Methyltetradecanoic acid is an iso-C15 branched-chain saturated fatty acid that has been found to disrupt mitochondrial integrity and induce apoptosis in a wide range of cancer cell lines (Figure 3, Table 2003; Yang et al., 2000; Wongtangtintharn et al., 2005; Lin et al., 2012). Induction of apoptosis by 13-methyltetradecanoic acid was rapid and was detected after only 2 h of treatment over the concentration range 0.04–0.35 mM (Lin et al., 2012). In some cell types, apoptosis appeared to be a caspase-independent pathway but, in human bladder cancer cells, 13-methyltetradecanoic acid down-regulated Bcl-2, up-regulated Bax, promoted mitochondrial dysfunction and cytochrome c release, and activated caspases (Lin et al., 2012). 13-Methyltetradecanoic acid also inhibited the PI3K/Akt survival cascade and activated the pro-apoptotic p38 and JNK MAPK pathways (Lin et al., 2012).
In vivo growth of the prostate carcinoma cell line DU 145 and hepatocarcinoma-derived LCI-D35 cells after orthotopic implantation of tumour xenografts into nude mice was also inhibited by 13-methyltetradecanoic acid (35–105 mg kg−1 day−1). Apoptosis was induced without evidence of major toxicity, which suggests that 13-methyltetradecanoic acid could be a potential candidate for chemotherapy of human cancers (Yang et al., 2000).
Unsaturated fatty acids
Naturally occurring PUFAs also modulate cell proliferation and apoptosis. The ω-3 PUFA eicosapentaenoic acid (20:5 n-3) is incorporated into mitochondrial phospholipids, which has a number of consequences for mitochondrial function, including decreased mitochondrial membrane potential and ATP production, increased ROS generation and increased apoptosis (Colquhoun, 2009). ω-6 PUFA also have the potential to activate apoptosis in human cancer cell lines by promoting lipid peroxidation (Cao et al., 2000).
Conjugated linoleic acid (CLA) is a mixture of geometric isomers of linoleic acid (Figure 3) that decreases the viability of various cancer cell types, including those of the skin, forestomach, mammary gland and colon. CLA (32 μM), but not linoleic acid, inhibited growth of rat mammary epithelial cell organoids that was mediated both by a decrease in DNA synthesis and increased apoptosis (Ip et al., 1999). In vivo activity was also noted and feeding CLA to rats that harboured premalignant lesions induced apoptosis in a mammary tumour cell line, as determined by a decrease in expression of the anti-apoptotic bcl-2 (Table 2003; Ip et al., 2000). Cho et al. (2003) showed that CLA inhibited the proliferation of HT-29 human colorectal cancer cell line by activating apoptosis, due in part to inhibition of PI3K/Akt signalling. Similarly, the 10-trans,12-cis CLA isomer (5 μM) inhibited the proliferation of Caco-2 colon carcinoma cells, and enhanced apoptosis in premalignant lesions, but not in normal cells (Kim et al., 2002).
Other types of naturally occurring conjugated fatty acids that have pro-apoptotic actions include CLAs such as α-eleostearic acid (9-cis,11-trans,13-trans-18:3) from bitter gourd oil and calendic acid (8-trans,10-trans-12-cis-18:3) from pot marigold. α-Eleostearic acid was quite potent relative to CLA isomers against tumour cells in vitro (Tsuzuki et al., 2004). α-Eleostearic acid (40 μM) promoted lipid peroxidation and apoptosis in MDA-MB-231-derived cell lines as evidenced by a loss of mitochondrial membrane potential and the release of apoptotic factors from the mitochondrion. When treated with α-eleostearic acid in the presence of α-tocotrienol (20 μM), growth inhibition and apoptosis did not occur, thus providing further support for involvement of ROS-mediated lipid peroxidation in the apoptotic mechanism (Grossmann et al., 2009).
The monounsaturated fatty acid vaccenic acid (11-trans-18:1 n-7) decreased cell growth, induced DNA fragmentation and depleted cytosolic glutathione levels; these findings are consistent with activation of the intrinsic pathway of apoptosis by lipid peroxides (Miller et al., 2003). Punicic acid (18:3 n-5) is an ω-5 long-chain PUFA found in pomegranate seed oil. In MDA-MB-231 and MDA-ERα7 cells, punicic acid disrupted the mitochondrial membrane potential and induced apoptosis, apparently also by a prooxidant mechanism (Grossmann et al., 2010). DNA fragmentation was observed after 24 h treatment of cells at concentrations in the range 10–100 μM (Gasmi and Sanderson, 2010).
Jacaric acid is a linolenic acid isomer obtained from jacaranda that has a conjugated triene system and elicits potent anti-tumour effects both in vitro and in vivo in nude mice into which DLD-1 cells had been xeno-transplanted (Shinohara et al., 2012). When compared with natural conjugated linolenic acids in DLD-1 adenocarcinoma cells, the anti-tumour effects of jacaric acid were most potent and correlated with increased ROS production. Thus, jacaric acid induced concentration-and time-dependent LNCaP cell death in part through activation of intrinsic apoptotic pathways, resulting in cleavage of caspase-3,-8 and-9, modulation of pro-and anti-apoptotic Bcl-2 family of proteins and increased cleavage of PARP-1 (Gasmi and Sanderson, 2013).
Several dietary C18 unsaturated fatty acids have been tested for cytotoxicity and induction of apoptosis in human prostate cancer cells. These included three octadecatrienoic geometric isomers (α-and β-calendic and catalpic acids) and two monounsaturated C18 fatty acids (trans-and cis-vaccenic acid) in addition to jacaric and punicic acids (Figure 3, Table 2003). Jacaric acid and four of its octadecatrienoic acid geometric isomers selectively induced apoptosis in both hormone-dependent (LNCaP) and hormone-independent (PC-3) human prostate cancer cells when tested at concentrations around 10 μM, without affecting the viability of normal human prostate epithelial cells (Gasmi and Sanderson, 2013). Together, these findings suggest that some of the pro-apoptotic actions of anti-tumour fatty acids may be cell type specific.
From the foregoing, it is evident that most studies to date have focused on ROS generation by unsaturated fatty acids as the mechanism of their pro-apoptotic action. However, there may be additional, more selective, mechanisms that could be developed in synthetic anti-cancer fatty acids. In a recent study, a series of n-3 monounsaturated fatty acids of chain length C16-C22 was synthesized and evaluated in MDA-MB-468 breast cancer cells that stably overexpressed COX-2 (Cui et al., 2012). This reflects the situation that may operate in many human cancers in which COX-2 is up-regulated. The longer chain C19-C22 analogues were found to inhibit proliferation and activate apoptosis; C16-C18 analogues were less active. PGE2 formation was decreased by the C19-C22 analogues, consistent with COX-2 inhibition, which was supported by molecular modelling that revealed effective interactions with specific amino acid residues in the COX-2 active site. Strategies of this type, in which potential fatty acid drugs target a biotransformation enzyme present in tumours, may be worthy of further consideration. Such agents may enable approaches based on COX-2 inhibition to be retained, perhaps without the toxicity associated with conventional non-steroidal anti-inflammatory drugs.
Fatty acid analogues
Tetradecylthioacetic acid (TTA) is a saturated fatty acid that has a sulphur atom inserted at the C3 position in the carbon chain. TTA (0.2–0.5 mM) decreased proliferation and induced apoptosis in a diverse range of tumour cell lines in vitro and in vivo (Tronstad et al., 2003; Iversen et al., 2006). Long-chain 3-thia-fatty acids in general also uncouple oxidative phosphorylation and dissipate the mitochondrial membrane potential (ΔΨ) by direct interaction with the adenine nucleotide translocator to open the mitochondrial permeability transition pore, which leads to decreased ATP production (Wieckowski and Wojtczak, 1998). In accord with this mechanism, TTA stimulates mitochondrial ROS production (Tronstad et al., 2001), leading to glutathione depletion, which renders mitochondria susceptible to further damage (Tronstad et al., 2003). Activation of apoptosis was indicated by the release of mitochondrial cytochrome c that enhanced caspase-3 activation and PARP cleavage.
TTA is resistant to mitochondrial β-oxidation and, compared with naturally occurring saturated fatty acids, is degraded relatively slowly to dicarboxylic acids by microsomal oxidation at the ω-carbon and sulphur atoms and by peroxisomal β-oxidation (Hvattum et al., 1991). This property could be useful in development of TTA and analogues as drugs since these molecules are likely to have superior durations of action in vivo. Indeed, a diet containing TTA increased the vascularization of colon cancer xenografts in mice and improved the survival of mice with leukaemia xenografts (Jensen et al., 2007).
Jasmonates are plant hormones that structurally resemble fatty acid esters (Figure 3). These agents interact directly with mitochondria in cancer cells to detach hexokinase-II from its location on the voltage-dependent anion channel in the mitochondrial permeability transition pore (Goldin et al., 2008). Indeed, the susceptibility in cancer cells to these molecules is dependent on the association of hexokinase with the mitochondrion. Pro-apoptotic mitochondrial actions of low millimolar concentrations of jasmonates include membrane depolarization, mitochondrial swelling, cytochrome c release and cell death; interestingly, the agents were inactive in normal cells (Table 2003; Fingrut and Flescher, 2002; Rotem et al., 2005).
Correlations have been reported between methyl jasmonate cytotoxicity in a range of cell types and the extent of ATP depletion (Goldin et al., 2007). Glucose protected against this loss of ATP, whereas the glycolysis inhibitor, 2-deoxyglucose, synergized with methyl jasmonate (Fingrut et al., 2005; Heyfets and Flescher, 2007). Similar effects have been elicited by other hexokinase-detaching agents, such as hypericin and clotrimazole, that also deplete ATP and decrease cell viability (Miccoli et al., 1998; Machida et al., 2006). These findings are consistent with the functional importance of hexokinase in glycolysis, which is a major pathway of ATP production in cancer cells.
Taken together, a number of studies have found that endogenous fatty acids, including saturated and certain unsaturated analogues, activate apoptosis via the mitochondrial intrinsic pathway and impair ATP production. Frequently, this may be due to increased ROS production, which alters mitochondrial membrane potential, but the example of jasmonate, involving hexokinase detachment, illustrates the potential for further development of mitochondrially targeted fatty acids that selectively disrupt energy metabolism in cancer cells.
Fatty acid metabolites
Several PUFA-derived metabolites have been shown to modulate signalling pathways that are implicated in tumourigenesis. Thus, inhibition of the enzymic formation of these fatty acid metabolites has been found to decrease cancer cell viability. Important metabolites that have emerged in this regard include COX-mediated PGE2, CYP-dependent epoxyeicosatrienoic acids (EETs) and certain LOX-mediated hydroxyeicosatetraenoic acids (HETEs) that inhibit apoptosis and enhance proliferation (Avis et al., 2001; Koehne and DuBois, 2004; Chen et al., 2009). An understanding of the involvement of fatty acid metabolites in the pathogenesis of cancer could open new avenues for the production of new and safer therapeutic and chemopreventive agents.
Certain fatty acid biotransformation enzymes have been detected at high level in human cancers. Thus, COX-2 and CYP2J2 are overexpressed in many invasive human cancers (Tsujii et al., 1997; Jiang et al., 2005). COX-2-derived PGE2, LOX metabolites and CYP2J2-derived EETs are implicated in aggressive tumour behaviour (Figure 4, Table 2003; Avis et al., 2001; Koehne and DuBois, 2004; Hoque et al., 2005; Jiang et al., 2005). Mechanistic information is available for some of these metabolites. Thus, PGE2 and EETs enhance tumour cell proliferation and survival by activating the proliferative EGF receptor (EGFR)/ERK MAPK and anti-apoptotic PI3K/Akt signalling pathways (Chen et al., 2009).
Figure 4.
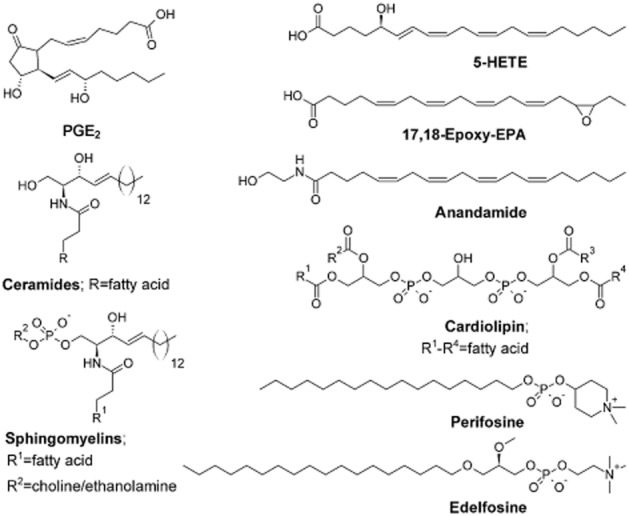
Chemical structures of important fatty acid metabolites and phospholipids, and general structures of ceramides and sphingomyelins.
Inhibition of 5-LOX by MK886 induces apoptosis in both hormone-responsive (LNCaP) and hormone-unresponsive (PC3) prostate cancer cells (Ghosh and Myers, 1998). An immediate and sustained rise in cytosolic calcium is followed by mitochondrial uncoupling and cytochrome c release (Maccarrone et al., 2001). Soon after treatment, cells underwent the mitochondrial permeability transition, followed by other apoptotic events, including externalization of phosphatidylserine and degradation of DNA (Ghosh and Myers, 1998). Cell death was prevented by direct addition of 5-HETE and analogues such as 5-HETE-lactone and 5-oxoeicosatetraenoic acid (50–500 nM; Ghosh and Myers, 1998). In breast cancer cells, inhibition of 5-LOX, but not COX, reduced growth, increased apoptosis, down-regulated bcl-2, up-regulated bax and caused G1 cell cycle arrest (Avis et al., 2001). Lipid peroxidation and the depletion of mitochondrial glutathione have been linked to the activation of apoptotic Bcl-2 proteins in these cells.
While the inhibition of biotransformation enzymes that generate lipid metabolites may be beneficial in the prevention of cancer progression, there is also increasing evidence emerging that ω-3 PUFAs undergo biotransformation to metabolites that inhibit carcinogenesis. Inhibition of PKC by the parent ω-3 PUFAs eicosapentaenoic and docosahexaenoic acids activates apoptosis by down-regulating Bcl-2 (Denys et al., 2005); COX-2-derived ω-3 PGE3 was recently implicated in anti-cancer actions of ω-3 PUFAs (Szymczak et al., 2008). Epoxides of ω-3 PUFAs have also been shown to exert growth suppressing and anti-cancer effects. The 17,18-epoxide of eicosapentaenoic acid (ω-3-epoxy-eicosatetraenoic acid), but not other regio-isomeric epoxides of eicosatetraenoic acid, was shown to inhibit cell proliferation. At physiologically relevant concentrations, ω-3-epoxy-eicosapentaenoic acid induced apoptosis and cell cycle arrest of brain microvascular endothelial bEND.3 cells through activation of growth-suppressing p38 MAPK and subsequent down-regulation of cyclin D1 (Cui et al., 2011). More recently, Zhang et al. (2013) showed that epoxygenase-mediated metabolites of the C22 ω-3 fatty acid docosahexaenoic acid exerted anti-cancer effects by suppressing vascular endothelial growth factor-mediated angiogenesis. Inhibition of angiogenesis resulted in a decrease in primary tumour growth and metastasis in vitro. By co-administration with t-AUCB, a soluble epoxide hydrolase inhibitor, the 19,20-epoxide was active in vivo, reducing tumour growth in the Lewis lung carcinoma model (Zhang et al., 2013). These actions were opposite to those of ω-6 arachidonic acid-derived EETs. However, at present, whether PUFA epoxides also interact directly with the mitochondrion, in addition to their indirect actions mediated by signalling cascades, to elicit these actions is unclear.
N-acyl lipids
Ceramides
Ceramides are a class of N-acylated sphingoid bases (Figure 4) found at high concentration in cell membranes and cytosolic organelle membranes and act as intracellular second messengers in the regulation of growth, differentiation and apotosis. Ceramide accumulation occurs after treatment of cells with apoptotic agents including chemotherapeutic agents, or after treatment with saturated fatty acids such as palmitic acid (Merrill and Jones, 1990). Direct addition of ∼1 μM ceramide to mitochondria and ceramide accumulation in cells both produced changes in the mitochondrial transmembrane potential by forming channels or targeting Bcl-2 family members that leads to translocation of cytochrome c from mitochondria to the cytoplasm, and caspase-3 activation (Garcia-Ruiz et al., 1997). Because respiratory complex III is inhibited by C2-ceramide, the proximal effects of ceramide in cells are mediated at least in part at the mitochondrion (Gudz et al., 1997). Inhibition of p38 and JNK MAPKs decreased ceramide-induced apoptosis by preventing the loss of mitochondrial transmembrane potential and inhibition of caspase activation (Chen et al., 2008). This suggests that ceramides may operate by both direct and indirect mechanisms to exert apoptotic actions at the mitochondrion.
In tumour cells, the cell-permeable shorter chain exogenous C2-and C6-ceramide analogues, but not the longer chain, naturally occurring C16-ceramide, activated intrinsic apoptotic events at concentrations ≥10 μM, including caspase-3 activation, PARP degradation and mitochondrial cytochrome c release (Fillet et al., 2003; Flowers et al., 2012). C6-ceramide increased ROS levels in MDA-MB-231 cells, shifted the Bax:Bcl-2 ratio and depolarized the mitochondrial membrane (Flowers et al., 2012). Thus, analogues containing fatty acids of medium chain length may be particularly suited to development as putative anti-cancer agents that target the mitochondrion.
N-acylethanolamines
Anandamide (Figure 4) and other N-acylethanolamines reportedly promote apoptosis and/or inhibit cell proliferation (Table 2003; Schwarz et al., 1994; De Petrocellis et al., 1998; Maccarrone et al., 2000; Sarker et al., 2000). These molecules appear to act in part by increasing the inner mitochondrial membrane permeability at concentrations up to 100 μM (Epps et al., 1982). Anandamide and N-oleoylethanolamine exerted protonophoric effects in mitochondria due to dissipation of the transmembrane potential and opening of the permeability transition pore (Wasilewski et al., 2004). Long-chain N-acylethanolamines, including anandamide, accumulate in mammalian tissues under a variety of pathological conditions (Schmid et al., 2002). Indeed, they have been detected at levels up to 500 nmol g−1 of infarcted canine myocardium. They have also been shown to inhibit the growth of various cancer cell lines in vitro. Cancer tissues usually contain substantially higher concentrations of these lipids than adjacent benign tissue, when normalized to wet weight since most tumours also contained higher levels of phospholipids.
Despite these apparent pro-apoptotic effects of N-acylethanolamines in cancer cells, there is also evidence for protective effects in cells. The long-chain N-palmitoyl-and N-stearoylethanolamine (100 μM) inhibited lipid peroxidation in hepatic mitochondria, which is consistent with membrane-protective properties (Gulaya et al., 1998). The beneficial effect of N-acylethanolamines on cell survival when oxygen availability is low depends in part on the inhibition of lipid oxidation. The effect of different N-acylethanolamines on lipid peroxidation seems to be dependent on the length of acyl chain as was found for other effects of N-acylethanolamines in the cell (Gulaya et al., 1993). These findings highlight the need for clarification of the relationships between chain length in N-acylethanolamines and their cellular actions prior to development of analogues as potential anti-cancer drugs.
Very recently, novel fatty acid derivatives of isopropylaminopropanol containing C16:0 or C18:1 substituents were prepared and were found to be effective against the growth of hepatoma cells in vitro (IC50s ∼5–10 μM) and in vivo xenografts when dosed at 25 mg kg−1 (Cao et al., 2013). These agents inhibited the activity of multiple kinases, including the prosurvival Akt, which increased caspase and PARP cleavage. It will now be of interest to explore in greater detail how these agents modulate mitochondrial activity to elicit anti-cancer actions.
Phospholipid derivatives
Cardiolipin
Cardiolipin is a structurally complex diphosphatidylglycerol lipid that is synthesized by the mitochondrion (Figure 4). Mitochondrial respiratory complexes have been shown to require cardiolipin for full function. Palmitic acid decreased the levels of cardiolipin, which is responsible for insertion and retention of cytochrome c in the inner membrane of the mitochondrion (Schlame et al., 2000; Ostrander et al., 2001). Decreased cardiolipin and altered mitochondrial function mediate palmitate-induced breast cancer cell death by promoting ROS production and release of cytochrome c by permeabilization of the outer membrane (Petrosillo et al., 2003); overexpression of the anti-apoptotic Bcl-2 family members Bcl-xL and Bcl-w blocked apoptosis (Kuwana et al., 2002).
Ether phospholipids
The ether phospholipids are a promising class of anti-tumour lipids that act in part at the mitochondrion. Ether phospholipids have one or more glycerol carbons bonded to an alkyl chain via an ether linkage, as opposed to the usual ester linkage. Ether phospholipids include miltefosine, ilmofosine, perifosine, edelfosine and erucylphosphocholine (Figure 4).
The novel alkylphospholipid analogue perifosine is a PI3K/Akt inhibitor (Table 2003; Kondapaka et al., 2003) that inhibits growth at low micromolar concentrations and activates the intrinsic pathway of apoptosis in cells as evidenced by increased caspase activity and cleavage of PARP. Activated caspase-8 cleaves Bid, which migrates to mitochondria and induces cytochrome c release (Chiarini et al., 2008). Interestingly, these agents are effective in rapidly proliferating cancer cells, but not quiescent normal cells.
Clinical evaluation of perifosine has been conducted, or is continuing, in patients with cancers of the endometrium, breast, prostate, bladder and other tissues. In 2010, perifosine was evaluated in phase II trials for metastatic colon cancer and extended the time taken for tumour progression. However, in 2013, it was announced that the drug failed trials in patients with relapsed or relapsed/refractory multiple myeloma (http://www.aezsinc.com/en/page.php?p=60&q=550, accessed 28 June 2013). When used in combination with the multikinase inhibitor sorafenib, perifosine induced intrinsic apoptosis in cells and anti-tumour effects in NOD/SCID mice with Hodgkin lymphoma cell line xenografts (Locatelli et al., 2013). In cell lines, the combination treatment inhibited MAPK, activated PI3K/Akt phosphorylation and suppressed growth; synergistic induction of mitochondrial dysfunction and cell death was noted. In in vivo xenograft studies, there was a reduction in tumour burden, increased survival time, increased apoptosis and necrosis in perifosine/sorafenib-treated animals compared with single agents. Subsequently, treatment of human leukaemia T-cells with the PI3K/Akt inhibitor perifosine and etoposide also effected synergistic induction of apoptosis by dual activation of intrinsic and extrinsic pathways (Nyåkern et al., 2006). This combination produced a twofold increase in caspase-8 activation, and a marked increase in caspase-9, caspase-3 and PARP cleavage, as well as increased Bim, Bid and Bcl-XL expression. Etoposide and perifosine induced leukaemic cell death in part by inactivation of the PI3K/Akt pathway that increased mitochondrial dysfunction. However, it is of considerable interest that ether phospholipids proved to be highly effective when used in combination with other anti-cancer agents.
Edelfosine induces changes in mitochondrial membrane permeability and inhibits mitochondrial respiration (Burgeiro et al., 2013). Edelfosine, miltefosine, erucylphosphocholine and perifosine all activate the JNK MAPK pathway, which induces apoptosis (Ruiter et al., 1999; Nieto-Miguel et al., 2006). The mechanism involves direct activation of apoptosis by phosphorylation of the anti-apoptotic Bcl-XL (Kharbanda et al., 2000; Aoki et al., 2002). Edelfosine also disrupts the mitochondrial transmembrane potential, apparently by altering mitochondrial membrane phosphocholine content (Vrablic et al., 2001), promotes the cleavage of caspase-3 and PARP, and enhances production of ROS in human leukaemic T-cells (Table 2003; Cabaner et al., 1999; Gajate et al., 2000). Edelfosine-induced apoptosis is blocked by Bcl-2 (Mollinedo et al., 1997; Ruiter et al., 2003; Hideshima et al., 2006).
New fluorescent edelfosine analogues retained the pro-apoptotic activity of the parent, and colocalized with mitochondria (Mollinedo et al., 2011). These agents induced the swelling of isolated mitochondria, consistent with an increase in mitochondrial membrane permeability. Free radical scavengers did not affect swelling, suggesting that ROS do not contribute. It was suggested that edelfosine promoted a redistribution of lipid rafts from plasma membrane to mitochondria (Mollinedo et al., 2011). In summary, ether phospholipids have potential for development as a novel class of anti-cancer agents that act in part by altering mitochondrial function.
Challenges in the development of lipid molecules as drugs
Fatty acids differ from typical drugs in that there is a large degree of molecular flexibility inherent in fatty acid alkyl chains. Although fewer than 10 rotatable bonds is seen as desirable for adequate oral bioavailability and membrane permeation (Veber et al., 2002), fatty acids possess satisfactory pharmacokinetic profiles. For example, medium-and long-chain dietary fatty acids are readily absorbed and distributed throughout all tissues of the body including the brain. Moreover, the ether phospholipid edelfosine is orally active despite possessing 27 rotatable bonds. The favourable pharmacokinetic profiles of fatty acids may be in part due to the facilitation of uptake and transport (Ramirez et al., 2001), including binding to serum albumin and incorporation into triglycerides, that distribute these essential nutrients throughout the body. However, the flexibility of fatty acids does present challenges in drug design, particularly in ligand-based approaches where the structure of the drug target is unknown. Conformational analysis is a critical step in pharmacophore and pseudo-receptor modelling, and the high number of rotatable bonds can make identification of low-energy and bioactive conformations difficult and time consuming. When the structure of the drug target is known, ligand docking approaches are straightforward, as exemplified by our recent modelling of the interactions of a series of novel ω-3 monounsaturated fatty acids in the COX-2 active site (Cui et al., 2012).
As mentioned earlier, fatty acids are subject to numerous metabolic processes mediated by COX, LOX and CYP enzymes that can present challenges for the development of lipid-derived drugs that have acceptable in vivo stability. Furthermore, lipid-derived mediators (e.g. PGs, epoxides, resolvins and others) frequently possess labile functional groups that are also readily metabolized. Improvement of the metabolic as well as chemical stability of lipid-based drugs, particularly those derived from PGs (Collins and Djuric, 1993; Das et al., 2007) and lipoxin (Duffy and Guiry, 2010), has received much attention and a number of strategies have been developed and successfully employed in drug discovery settings. These strategies commonly involve addition of functional groups to block particular metabolic processes, or bioisosteric replacement of labile functional groups with more robust equivalents. For example, incorporation of a heteroatom in alkyl chains at the position β to the carboxylate functionality can improve the duration of action in vivo, as shown by the PG analogue cicaprost (Hildebrand et al., 1989). These approaches have led to the development of numerous marketed drugs, which clearly demonstrate the potential of lipids as drugs and as lead compounds in drug discovery.
Development of new mitochondrial targeted inhibitors in cancer chemotherapy
Evidence is increasing that fatty acids and other lipids act in part by modulation of mitochondrial function. These actions may be direct, such as the detachment of hexokinase from the cancer cell mitochondrial transition pore complex or by uncoupling of respiratory complexes that produce ATP. Alternatively, some lipid agents may act indirectly by increasing ROS activity in cells or by interfering with cell signalling pathways, to perturb the balance of pro-and anti-apoptotic bcl-2 proteins that regulate mitochondrial membrane stability. It should be noted, however, that mitochondrial actions of certain lipid-based molecules may operate alongside non-mitochondrial mechanisms, including altered membrane raft composition or altered gene regulation.
A point of major interest that has emerged from cellular studies is that fatty acid derivatives often exhibit activity against cancer cells, but not normal cells. This appears to be a property that augers well for new anti-cancer drug development based on lipids. The available information does not provide a full understanding for this selectivity because the targeted pathways may be present in both cell types. It will now be of major importance to define in detail the defects present in mitochondria of cancer cells so that new drugs may be developed that have optimal potency with fewer off-target actions.
Acknowledgments
Support of research in the authors' laboratory by the Australian National Health and Medical Research Council is gratefully acknowledged.
Glossary
- Caspase
cysteine-aspartic protease
- CLA
conjugated linoleic acid
- CYP
cytochrome P450
- EET
epoxyeicosatrienoic acid
- HETE
hydroxyeicosatetraenoic acid
- LOX
lipoxygenase
- PARP
poly (ADP ribose) polymerase
- PUFA
polyunsaturated fatty acid
- ROS
reactive oxygen species
- TTA
tetradecylthioacetic acid
Conflict of interest
None.
References
- Ackerstaff E, Glunde K, Bhujwalla ZM. Choline phospholipid metabolism: a target in cancer cells? J Cell Biochem. 2003;90:525–533. doi: 10.1002/jcb.10659. [DOI] [PubMed] [Google Scholar]
- Aoki H, Kang PM, Hampe J, Yoshimura K, Noma T, Matsuzaki M, et al. Direct activation of mitochondrial apoptosis machinery by c-Jun N-terminal kinase in adult cardiac myocytes. J Biol Chem. 2002;277:10244–10250. doi: 10.1074/jbc.M112355200. [DOI] [PubMed] [Google Scholar]
- Avis I, Hong SH, Martinez A, Moody T, Choi YH, Trepel J, et al. Five-lipoxygenase inhibitors can mediate apoptosis in human breast cancer cell lines through complex eicosanoid interactions. FASEB J. 2001;15:2007–2009. doi: 10.1096/fj.00-0866fje. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Penzo D, Wojtczak L. Mitochondrial energy dissipation by fatty acids. Vitam Horm. 2002;65:97–126. doi: 10.1016/s0083-6729(02)65061-7. [DOI] [PubMed] [Google Scholar]
- Bremer J, Bjerve KS, Borrebaek B, Christiansen R. The glycerophosphate acyltransferases and their function in the metabolism of fatty acids. Mol Cell Biochem. 1976;12:113–125. doi: 10.1007/BF01731557. [DOI] [PubMed] [Google Scholar]
- Brenner C, Cardiou H, Vieira HL, Zamzami N, Marzo I, Xie Z, et al. Bcl-2 and Bax regulate the channel activity of the mitochondrial adenine nucleotide translocator. Oncogene. 2000;19:329–336. doi: 10.1038/sj.onc.1203298. [DOI] [PubMed] [Google Scholar]
- Brimmell M, Mendiola R, Mangion J, Packham G. BAX frameshift mutations in cell lines derived from human hematopoietic malignancies are associated with resistance to apoptosis and microsatellite instability. Oncogene. 1998;16:1803–1812. doi: 10.1038/sj.onc.1201704. [DOI] [PubMed] [Google Scholar]
- Broekemeier KM, Pfeiffer DR. Inhibition of the mitochondrial permeability transition by cyclosporin A during long time frame experiments: relationship between pore opening and the activity of mitochondrial phospholipases. Biochemistry. 1995;34:16440–16449. doi: 10.1021/bi00050a027. [DOI] [PubMed] [Google Scholar]
- Burgeiro A, Pereira CV, Carvalho FS, Pereira GC, Mollinedo F, Oliveira PJ. Edelfosine and perifosine disrupt hepatic mitochondrial oxidative phosphorylation and induce the permeability transition. Mitochondrion. 2013;13:25–35. doi: 10.1016/j.mito.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Cabaner C, Gajate C, Macho A, Munoz E, Modolell M, Mollinedo F. Induction of apoptosis in human mitogen-activated peripheral blood T-lymphocytes by the ether phospholipid ET-18-OCH3: involvement of the Fas receptor/ligand system. Br J Pharmacol. 1999;127:813–825. doi: 10.1038/sj.bjp.0702606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000;29:222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- Cao M, Prima V, Nelson D, Svetlov S. Composite fatty acid ether amides suppress growth of liver cancer cells in vitro and in an in vivo allograft mouse model. Cell Oncol. 2013;36:247–257. doi: 10.1007/s13402-013-0132-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Pearman AT, Zimmerman GA, McIntyre TM, Prescott SM. Intracellular unesterified arachidonic acid signals apoptosis. Proc Natl Acad Sci USA. 2000;97:11280–11285. doi: 10.1073/pnas.200367597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Li G, Liao W, Wu J, Liu L, Ma D, et al. Selective inhibitors of CYP2J2 related to terfenadine exhibit strong activity against human cancers in vitro and in vivo. J Pharmacol Exp Ther. 2009;329:908–918. doi: 10.1124/jpet.109.152017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CL, Lin CF, Chang WT, Huang WC, Teng CF, Lin YS. Ceramide induces p38 MAPK and JNK activation through a mechanism involving a thioredoxin-interacting protein-mediated pathway. Blood. 2008;111:4365–4374. doi: 10.1182/blood-2007-08-106336. [DOI] [PubMed] [Google Scholar]
- Chen J, Murray M, Liddle C, Jiang XM, Farrell GC. Down-regulation of male-specific cytochrome P450s 2C11 and 3A2 in bile duct-ligated male rats: importance to reduced hepatic content of cytochrome P450 in cholestasis. Hepatology. 1995;22:580–587. [PubMed] [Google Scholar]
- Chiarini F, Del Sole M, Mongiorgi S, Gaboardi GC, Cappellini A, Mantovani I, et al. The novel Akt inhibitor, perifosine, induces caspase-dependent apoptosis and downregulates P-glycoprotein expression in multidrug-resistant human T-acute leukemia cells by a JNK-dependent mechanism. Leukemia. 2008;22:1106–1116. doi: 10.1038/leu.2008.79. [DOI] [PubMed] [Google Scholar]
- Cho HJ, Kim WK, Kim EJ, Jung KC, Park S, Lee HS, et al. Conjugated linoleic acid inhibits cell proliferation and ErbB3 signaling in HT-29 human colon cell line. Am J Physiol Gastrointest Liver Physiol. 2003;284:G996–1005. doi: 10.1152/ajpgi.00347.2002. [DOI] [PubMed] [Google Scholar]
- Collins PW, Djuric SW. Synthesis of therapeutically useful prostaglandin and prostacyclin analogs. Chem Rev. 1993;93:1533–1564. [Google Scholar]
- Colquhoun A. Mechanisms of action of eicosapentaenoic acid in bladder cancer cells in vitro: alterations in mitochondrial metabolism, reactive oxygen species generation and apoptosis induction. J Urol. 2009;181:1885–1893. doi: 10.1016/j.juro.2008.11.092. [DOI] [PubMed] [Google Scholar]
- Cui PH, Petrovic N, Murray M. The ω-3 epoxide of eicosapentaenoic acid inhibits endothelial cell proliferation by p38 MAP kinase activation and cyclin D1/CDK4 downregulation. Br J Pharmacol. 2011;162:1143–1155. doi: 10.1111/j.1476-5381.2010.01113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui PH, Rawling T, Bourget K, Kim T, Duke CC, Doddareddy MR, et al. Antiproliferative and antimigratory actions of synthetic long chain n-3 monounsaturated fatty acids in breast cancer cells that overexpress cyclooxygenase-2. J Med Chem. 2012;55:7163–7172. doi: 10.1021/jm300673z. [DOI] [PubMed] [Google Scholar]
- Das S, Chandrasekhar S, Yadav JS, Gree R. Recent developments in the synthesis of prostaglandins and analogues. Chem Rev. 2007;107:3286–3337. doi: 10.1021/cr068365a. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Melck D, Palmisano A, Bisogno T, Laezza C, Bifulco M, et al. The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc Natl Acad Sci USA. 1998;95:8375–8380. doi: 10.1073/pnas.95.14.8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Schrijver E, Brusselmans K, Heyns W, Verhoeven G, Swinnen JV. RNA interference-mediated silencing of the fatty acid synthase gene attenuates growth and induces morphological changes and apoptosis of LNCaP prostate cancer cells. Cancer Res. 2003;63:3799–3804. [PubMed] [Google Scholar]
- Denys A, Hichami A, Khan NA. n-3 PUFAs modulate T-cell activation via protein kinase C-alpha and-epsilon and the NF-kappaB signaling pathway. J Lipid Res. 2005;46:752–758. doi: 10.1194/jlr.M400444-JLR200. [DOI] [PubMed] [Google Scholar]
- Di Paola MD, Lorusso M. Interaction of free fatty acids with mitochondria: coupling, uncoupling and permeability transition. Biochem Biophys Acta. 2006;1757:1330–1337. doi: 10.1016/j.bbabio.2006.03.024. [DOI] [PubMed] [Google Scholar]
- Duffy CD, Guiry PJ. Recent advances in the chemistry and biology of stable synthetic Lipoxin analogues. MedChemComm. 2010;1:249–265. [Google Scholar]
- Epps DE, Palmer JW, Schmid HHO, Pfeiffer DR. Inhibition of permeability-dependent Ca2+ release from mitochondria by N-acylethanolamines, a class of lipids synthesized in ischemic heart tissue. J Biol Chem. 1982;257:1383–1391. [PubMed] [Google Scholar]
- Fillet M, Bentires-Alj M, Deregowski V, Greimers R, Gielen J, Piette J, et al. Mechanisms involved in exogenous C2-and C6-ceramide-induced cancer cell toxicity. Biochem Pharmacol. 2003;65:1633–1642. doi: 10.1016/s0006-2952(03)00125-4. [DOI] [PubMed] [Google Scholar]
- Fingrut O, Flescher E. Plant stress hormones suppress the proliferation and induce apoptosis in human cancer cells. Leukemia. 2002;16:608–616. doi: 10.1038/sj.leu.2402419. [DOI] [PubMed] [Google Scholar]
- Fingrut O, Reischer D, Rotem R, Goldin N, Altboum I, Zan-Bar I, et al. Jasmonates induce nonapoptotic death in high-resistance mutant p53-expressing B-lymphoma cells. Br J Pharmacol. 2005;146:800–808. doi: 10.1038/sj.bjp.0706394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flowers M, Fabrias G, Delgado A, Casas J, Abad JL, Cabot MC. C6-ceramide and targeted inhibition of acid ceramidase induce synergistic decreases in breast cancer cell growth. Breast Cancer Res Treat. 2012;133:447–458. doi: 10.1007/s10549-011-1768-8. [DOI] [PubMed] [Google Scholar]
- Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006;25:4798–4811. doi: 10.1038/sj.onc.1209608. [DOI] [PubMed] [Google Scholar]
- Gajate C, Santos-Beneit M, Macho A, Carmen Lazaro del M, Hernandez-De Rojas A, Modolell M, et al. Involvement of mitochondria and caspase-3 in Et-18-OCH3-induced apoptosis of human leukemic cells. Int J Cancer. 2000;86:208–218. doi: 10.1002/(sici)1097-0215(20000415)86:2<208::aid-ijc10>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Galiegue S, Jbilo O, Combes T, Bribes E, Carayon P, Le Fur G, et al. Cloning and characterization of PRAX-1. A new protein that specifically interacts with the peripheral benzodiazepin receptor. J Biol Chem. 1999;274:2938–2952. doi: 10.1074/jbc.274.5.2938. [DOI] [PubMed] [Google Scholar]
- Garcia-Ruiz C, Colell A, Mari M, Morales A, Fernandez-Checa JC. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J Biol Chem. 1997;272:11369–11377. doi: 10.1074/jbc.272.17.11369. [DOI] [PubMed] [Google Scholar]
- Gasmi J, Sanderson JT. Growth inhibitory, antiandrogenic, and pro-apoptotic effects of punicic acid in LNCaP human prostate cancer cells. J Agric Food Chem. 2010;58:12149–12156. doi: 10.1021/jf103306k. [DOI] [PubMed] [Google Scholar]
- Gasmi J, Sanderson JT. Jacaric acid and its octadecatrienoic acid geoisomers induce apoptosis selectively in cancerous human prostate cells: a mechanistic and 3-D structure-activity study. Phytomedicine. 2013;20:734–742. doi: 10.1016/j.phymed.2013.01.012. [DOI] [PubMed] [Google Scholar]
- Ghosh J, Myers CE. Inhibition of arachidonate 5-lipoxygenase triggers massive apoptosis in human prostate cancer cells. Proc Natl Acad Sci USA. 1998;95:13182–13187. doi: 10.1073/pnas.95.22.13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin N, Heyfets A, Reischer D, Flescher E. Mitochondria-mediated ATP depletion by anti-cancer agents of the jasmonate family. J Bioenerg Biomemb. 2007;39:51–57. doi: 10.1007/s10863-006-9061-y. [DOI] [PubMed] [Google Scholar]
- Goldin N, Arzoine L, Heyfets A, Israelson A, Zaslavsky Z, Bravman T, et al. Methyl jasmonate binds to and detaches mitochondria-bound hexokinase. Oncogene. 2008;27:4636–4643. doi: 10.1038/onc.2008.108. [DOI] [PubMed] [Google Scholar]
- Goodwin PJ, Boyd NF, Hanna W, Hartwick W, Murray D, Qizilbash A, et al. Elevated levels of plasma triglycerides are associated with histologically defined premenopausal breast cancer risk. Nutr Cancer. 1997;27:284–292. doi: 10.1080/01635589709514539. [DOI] [PubMed] [Google Scholar]
- Grossmann ME, Mizuno NK, Dammen ML, Schuster T, Ray A, Cleary MP. Eleostearic acid inhibits breast cancer proliferation by means of an oxidation-dependent mechanism. Cancer Prev Res. 2009;2:879–886. doi: 10.1158/1940-6207.CAPR-09-0088. [DOI] [PubMed] [Google Scholar]
- Grossmann ME, Mizuno NK, Schuster T, Cleary MP. Punicic acid is an omega-5 fatty acid capable of inhibiting breast cancer proliferation. Int J Oncol. 2010;36:421–426. [PubMed] [Google Scholar]
- Gudz TI, Tserng KY, Hoppel CL. Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J Biol Chem. 1997;272:24154–24158. doi: 10.1074/jbc.272.39.24154. [DOI] [PubMed] [Google Scholar]
- Gulaya NM, Melnik AA, Balkov DI, Volkov GL, Vysotskiy MV, Vaskovsky VE. The effect of long-chain N-acylethanolamines on some membrane-associated functions of neuroblastoma C1300 N18 cells. Biochim Biophys Acta. 1993;1152:280–288. doi: 10.1016/0005-2736(93)90259-3. [DOI] [PubMed] [Google Scholar]
- Gulaya NM, Kuzmenko AI, Margitich VM, Govseeva NM, Melnichuk SD, Goridko TM, et al. Long-chain N-acylethanolamines inhibit lipid peroxidation in rat liver mitochondria under acute hypoxic hypoxia. Chem Phys Lipids. 1998;97:49–54. doi: 10.1016/s0009-3084(98)00093-0. [DOI] [PubMed] [Google Scholar]
- Gutknecht J. Proton conductance caused by long-chain fatty acids in phospholipid bilayer membranes. J Membrane Biol. 1988;106:83–93. doi: 10.1007/BF01871769. [DOI] [PubMed] [Google Scholar]
- Hague A, Diaz GD, Hicks DJ, Krajewski S, Reed JC, Paraskeva C. Bcl-2 and bak may play a pivotal role in sodium butyrate-induced apoptosis in colonic epithelial cells; however overexpression of bcl-2 does not protect against bak-mediated apoptosis. Int J Cancer. 1997;72:898–905. doi: 10.1002/(sici)1097-0215(19970904)72:5<898::aid-ijc30>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Heerdt BG, Houston MA, Augenlicht LH. Short-chain fatty acid initiated cell cycle arrest and apoptosis of colonic epithelial cells is linked to mitochondrial function. Cell Growth Differ. 1997;8:523–532. [PubMed] [Google Scholar]
- Heerdt BG, Houston MA, Anthony GM, Augenlicht LH. Mitochondrial membrane potential (ΔΨ) in the coordination of p53-independent proliferation and apoptosis pathways in human colonic carcinoma cells. Cancer Res. 1998;58:2869–2875. [PubMed] [Google Scholar]
- Heyfets A, Flescher E. Cooperative cytotoxicity of methyl jasmonate with anti-cancer drugs and 2-deoxy-D-glucose. Cancer Lett. 2007;50:300–310. doi: 10.1016/j.canlet.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Hideshima T, Catley L, Yasui H, Ishitsuka K, Raje N, Mitsiades C, et al. Perifosine, an oral bioactive novel alkylphospholipid, inhibits akt and induces in vitro and in vivo cytotoxicity in human multiple myeloma cells. Blood. 2006;107:4053–4062. doi: 10.1182/blood-2005-08-3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higdon A, Diers AR, Oh JY, Landar A, Darley-Usmar VM. Cell signalling by reactive lipid species: new concepts and molecular mechanisms. Biochem J. 2012;442:453–464. doi: 10.1042/BJ20111752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand M, Staks T, Schuett A, Matthes H. Pharmacokinetics of 3H-cicaprost in healthy volunteers. Prostaglandins. 1989;37:259–273. doi: 10.1016/0090-6980(89)90062-2. [DOI] [PubMed] [Google Scholar]
- Hoque A, Lippman SM, Wu TT, Xu Y, Liang ZD, Swisher S, et al. Increased 5-lipoxygenase expression and induction of apoptosis by its inhibitors in esophageal cancer: a potential target for prevention. Carcinogenesis. 2005;26:785–791. doi: 10.1093/carcin/bgi026. [DOI] [PubMed] [Google Scholar]
- Hostetler KY, Bosch van den H. Subcellular and submitochondrial localization of the biosynthesis of cardiolipin and related phospholipids in rat liver. Biochim Biophys Acta. 1972;260:380–386. doi: 10.1016/0005-2760(72)90052-5. [DOI] [PubMed] [Google Scholar]
- Hvattum E, Bergseth S, Pedersen CN, Bremer J, Aarsland A, Berge RK. Microsomal oxidation of dodecylthioacetic acid (a 3-thia fatty acid) in rat liver. Biochem Pharmacol. 1991;41:945–953. doi: 10.1016/0006-2952(91)90200-o. [DOI] [PubMed] [Google Scholar]
- Ip C, Ip MM, Loftus T, Shoemaker S, Shea-Eaton W. Induction of apoptosis by conjugated linoleic acid in cultured mammary tumor cells and premalignant lesions of the rat mammary gland. Cancer Epidemiol Biomarkers Prev. 2000;9:689–696. [PubMed] [Google Scholar]
- Ip MM, Masso-Welch PA, Shoemaker SF, Shea-Eaton WK, Ip C. Conjugated linoleic acid inhibits proliferation and induces apoptosis of normal rat mammary epithelial cells in primary culture. Exp Cell Res. 1999;250:22–34. doi: 10.1006/excr.1999.4499. [DOI] [PubMed] [Google Scholar]
- Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12:446–451. doi: 10.1038/nm1388. [DOI] [PubMed] [Google Scholar]
- Iversen PO, Sørensen DR, Tronstad KJ, Gudbrandsen OA, Rustan AC, Berge RK, et al. A bioactively modified fatty acid improves survival and impairs metastasis in preclinical models of acute leukemia. Clin Cancer Res. 2006;12:3525–3531. doi: 10.1158/1078-0432.CCR-05-2802. [DOI] [PubMed] [Google Scholar]
- Jensen LR, Berge K, Bathen TF, Wergedahl H, Schonberg SA, Bofin A, et al. Effect of dietary tetradecylthioacetic acid on colon cancer growth studied by dynamic contrast enhanced MRI. Cancer Biol Ther. 2007;6:1810–1816. doi: 10.4161/cbt.6.11.4887. [DOI] [PubMed] [Google Scholar]
- Jiang JG, Chen CL, Card JW, Yang S, Chen JX, Fu XN, et al. Cytochrome P450 2J2 promotes the neoplastic phenotype of carcinoma cells and is up-regulated in human tumors. Cancer Res. 2005;65:4707–4715. doi: 10.1158/0008-5472.CAN-04-4173. [DOI] [PubMed] [Google Scholar]
- Kamp F, Hamilton JA. pH gradients across phospholipid membranes caused by fast flip-flop of unionized fatty acids. Proc Natl Acad Sci USA. 1992;89:11367–11370. doi: 10.1073/pnas.89.23.11367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanazawa A, Tanaka A, Iwata S, Satoh S, Hatano E, Shinohara H, et al. The beneficial effect of phosphocreatine accumulation in the creatine kinase transgenic mouse liver in endotoxin-induced hepatic cell death. J Surg Res. 1998;80:229–235. doi: 10.1006/jsre.1998.5482. [DOI] [PubMed] [Google Scholar]
- Kharbanda S, Saxena S, Yoshida K, Pandey P, Kaneki M, Wang Q, et al. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-x(L) in response to DNA damage. J Biol Chem. 2000;275:322–327. doi: 10.1074/jbc.275.1.322. [DOI] [PubMed] [Google Scholar]
- Kim EJ, Holthuizen PE, Park HS, Ha YL, Jung KC, Park JH. Trans-10,cis-12-conjugated linoleic acid inhibits Caco-2 colon cancer cell growth. Am J Physiol Gastrointest Liver Physiol. 2002;283:G357–G367. doi: 10.1152/ajpgi.00495.2001. [DOI] [PubMed] [Google Scholar]
- Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Koehne CH, Dubois RN. COX-2 inhibition and colorectal cancer. Semin Oncol. 2004;31(2 Suppl 7):12–21. doi: 10.1053/j.seminoncol.2004.03.041. [DOI] [PubMed] [Google Scholar]
- Kondapaka SB, Singh SS, Dasmahapatra GP, Sausville EA, Roy KK. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol Cancer Ther. 2003;2:1093–1103. [PubMed] [Google Scholar]
- Korshunov SS, Korkina OV, Ruuge EK, Skulachev VP, Starkov AA. Fatty acids as natural uncouplers preventing generation of O2− and H2O2 by mitochondria in the resting state. FEBS Lett. 1998;435:215–218. doi: 10.1016/s0014-5793(98)01073-4. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, et al. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- Larsen NB, Rasmussen M, Rasmussen LJ. Nuclear and mitochondrial DNA repair: similar pathways? Mitochondrion. 2005;5:89–108. doi: 10.1016/j.mito.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Lin T, Yin X, Cai Q, Fan X, Xu K, Huang L, et al. 13-Methyltetradecanoic acid induces mitochondrial-mediated apoptosis in human bladder cancer cells. Urolog Oncol. 2012;30:339–345. doi: 10.1016/j.urolonc.2010.04.011. [DOI] [PubMed] [Google Scholar]
- Locatelli SL, Giacomini A, Guidetti A, Cleris L, Mortarini R, Anichini A, et al. Perifosine and sorafenib combination induces mitochondrial cell death and antitumor effects in NOD/SCID mice with Hodgkin lymphoma cell line xenografts. Leukemia. 2013;27:1677–1687. doi: 10.1038/leu.2013.28. [DOI] [PubMed] [Google Scholar]
- Maccarrone M, Lorenzon T, Bari M, Melino G, Finazzi-Agro A. Anandamide induces apoptosis in human cells via vanilloid receptors. Evidence for a protective role of cannabinoid receptors. J Biol Chem. 2000;275:31938–31945. doi: 10.1074/jbc.M005722200. [DOI] [PubMed] [Google Scholar]
- Maccarrone M, Melino G, Finazzi-Agro A. Lipoxygenases and their involvement in programmed cell death. Cell Death Diff. 2001;8:776–784. doi: 10.1038/sj.cdd.4400908. [DOI] [PubMed] [Google Scholar]
- Machida K, Ohta Y, Osada H. Suppression of apoptosis by cyclophilin D via stabilization of hexokinase II mitochondrial binding in cancer cells. J Biol Chem. 2006;281:14314–14320. doi: 10.1074/jbc.M513297200. [DOI] [PubMed] [Google Scholar]
- McLaughlin SGA, Dilger JP. Transport of protons across membranes by weak acids. Physiol Rev. 1980;60:825–863. doi: 10.1152/physrev.1980.60.3.825. [DOI] [PubMed] [Google Scholar]
- Marden NY, Fiala-Beer E, Xiang SH, Murray M. Role of activator protein-1 in the downregulation of the human CYP2J2 gene in hypoxia. Biochem J. 2003;373:669–680. doi: 10.1042/BJ20021903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari M, Morales A, Colell A, Garcia-Ruiz C, Fernandez-Checa JC. Mitochondrial glutathione, a key survival antioxidant. Antioxid Redox Signal. 2009;11:2685–2700. doi: 10.1089/ars.2009.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo I, Brenner C, Zamzami N, Susin SA, Beutner G, Brdiczka D, et al. The permeability transition pore complex: a target for apoptosis regulation by caspases and bcl-2-related proteins. J Exp Med. 1998a;187:1261–1271. doi: 10.1084/jem.187.8.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL, et al. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998b;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- Merad-Boudia M, Nicole A, Santiard-Baron D, Saillé C, Ceballos-Picot I. Mitochondrial impairment as an early event in the process of apoptosis induced by glutathione depletion in neuronal cells: relevance to Parkinson's disease. Biochem Pharmacol. 1998;56:645–655. doi: 10.1016/s0006-2952(97)00647-3. [DOI] [PubMed] [Google Scholar]
- Merrill AH, Jr, Jones DD. An update of the enzymology and regulation of sphingomyelin metabolism. Biochim Biophys Acta. 1990;1044:1–12. doi: 10.1016/0005-2760(90)90211-f. [DOI] [PubMed] [Google Scholar]
- Miccoli L, Beurdeley-Thomas A, De Pinieux G, Sureau F, Oudard S, Dutrillaux B, et al. Light-induced photoactivation of hypericin affects the energy metabolism of human glioma cells by inhibiting hexokinase bound to mitochondria. Cancer Res. 1998;58:5777–5786. [PubMed] [Google Scholar]
- Miller A, McGrath E, Stanton C, Devery R. Vaccenic acid (t11-18:1) is converted to c9,t11-CLA in MCF-7 and SW480 cancer cells. Lipids. 2003;38:623–632. doi: 10.1007/s11745-003-1107-8. [DOI] [PubMed] [Google Scholar]
- Milovic V, Teller IC, Turchanowa L, Caspary WF, Stein J. Effect of structural analogues of propionate and butyrate on colon cancer cell growth. Int J Colorectal Dis. 2000;15:264–270. doi: 10.1007/s003840000257. [DOI] [PubMed] [Google Scholar]
- Mollinedo F, Fernandez-Luna JL, Gajate C, Martin-Martin B, Benito A, Martinez-Dalmau R, et al. Selective induction of apoptosis in cancer cells by the ether lipid ET-18-OCH3 (Edelfosine): molecular structure requirements, cellular uptake, and protection by Bcl-2 and Bcl-X(L) Cancer Res. 1997;57:1320–1328. [PubMed] [Google Scholar]
- Mollinedo F, Fernandez M, Hornillos V, Delgado J, Amat-Guerri F, Acuna AU, et al. Involvement of lipid rafts in the localization and dysfunction effect of the antitumor ether phospholipid edelfosine in mitochondria. Cell Death Dis. 2011;2:e158. doi: 10.1038/cddis.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray M. Mechanisms and significance of inhibitory drug interactions involving cytochrome P450 enzymes. Int J Mol Med. 1999;3:227–238. [PubMed] [Google Scholar]
- Nathan C, Cunningham-Bussel A. Beyond oxidative stress: an immunologist's guide to reactive oxygen species. Nat Rev Immunol. 2013;13:349–361. doi: 10.1038/nri3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto-Miguel T, Gajate C, Mollinedo F. Differential targets and subcellular localization of antitumor alkyl-lysophospholipid in leukemic versus solid tumor cells. J Biol Chem. 2006;281:14833–14840. doi: 10.1074/jbc.M511251200. [DOI] [PubMed] [Google Scholar]
- Nyåkern M, Cappellini A, Mantovani I, Martelli AM. Synergistic induction of apoptosis in human leukemia T cells by the Akt inhibitor perifosine and etoposide through activation of intrinsic and Fas-mediated extrinsic cell death pathways. Mol Cancer Ther. 2006;5:1559–1570. doi: 10.1158/1535-7163.MCT-06-0076. [DOI] [PubMed] [Google Scholar]
- Oates JA, FitzGerald GA, Branch RA, Jackson EK, Knapp HR, Roberts LJ., II Clinical implications of prostaglandin and thromboxane A2 formation. N Engl J Med. 1988;319:689–698. doi: 10.1056/NEJM198809153191106. [DOI] [PubMed] [Google Scholar]
- Oliw EH. Oxygenation of polyunsaturated fatty acids by cytochrome P450 monooxygenases. Prog Lipid Res. 1994;33:329–354. doi: 10.1016/0163-7827(94)90029-9. [DOI] [PubMed] [Google Scholar]
- Ookhtens M, Kannan R, Lyon I, Baker N. Liver and adipose tissue contributions to newly formed fatty acids in an ascites tumor. Am J Physiol. 1984;247:R146–R153. doi: 10.1152/ajpregu.1984.247.1.R146. [DOI] [PubMed] [Google Scholar]
- Ostrander DB, Sparagna GC, Amoscato AA, McMillin JB, Dowhan W. Decreased cardiolipin synthesis corresponds with cytochrome c release in palmitate-induced cardiomyocyte apoptosis. J Biol Chem. 2001;276:38061–38067. doi: 10.1074/jbc.M107067200. [DOI] [PubMed] [Google Scholar]
- Pablo de MA, Susin SA, Jacotot E, Larochette N, Costantini P, Ravagnan L, et al. Palmitate induces apoptosis via a direct effect on mitochondria. Apoptosis. 1999;4:81–87. doi: 10.1023/a:1009694124241. [DOI] [PubMed] [Google Scholar]
- Petrosillo G, Ruggiero FM, Paradies G. Role of reactive oxygen species and cardiolipin in the release of cytochrome c from mitochondria. FASEB J. 2003;17:2202–2208. doi: 10.1096/fj.03-0012com. [DOI] [PubMed] [Google Scholar]
- Pike LS, Smift AL, Croteau NJ, Ferrick DA, Wu M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochem Biophys Acta. 2011;1807:726–734. doi: 10.1016/j.bbabio.2010.10.022. [DOI] [PubMed] [Google Scholar]
- Pizer ES, Chrest FJ, DiGiuseppe JA, Han WF. Pharmacological inhibitors of mammalian fatty acid synthase suppress DNA replication and induce apoptosis in tumor cell lines. Cancer Res. 1998;58:4611–4615. [PubMed] [Google Scholar]
- Pugazhenthi S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE, et al. Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem. 2000;275:10761–10766. doi: 10.1074/jbc.275.15.10761. [DOI] [PubMed] [Google Scholar]
- Ramirez M, Amate L, Gil A. Absorption and distribution of dietary fatty acids from different sources. Early Hum Dev. 2001;65:S95–S101. doi: 10.1016/s0378-3782(01)00211-0. [DOI] [PubMed] [Google Scholar]
- Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, Capaldi RA. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004;64:985–993. doi: 10.1158/0008-5472.can-03-1101. [DOI] [PubMed] [Google Scholar]
- Rotem R, Heyfets A, Fingrut O, Blickstein D, Shaklai M, Flescher E. Jasmonates: novel anticancer agents acting directly and selectively on human cancer cell mitochondria. Cancer Res. 2005;65:1984–1993. doi: 10.1158/0008-5472.CAN-04-3091. [DOI] [PubMed] [Google Scholar]
- Ruemmele FM, Dionne S, Qureshi I, Sarma DS, Levy E, Seidman EG. Butyrate mediates Caco-2 cell apoptosis via up-regulation of pro-apoptotic bak and inducing caspase-3 mediated cleavage of poly-(ADP-ribose) polymerase (PARP) Cell Death Diff. 1999;6:729–735. doi: 10.1038/sj.cdd.4400545. [DOI] [PubMed] [Google Scholar]
- Ruiter GA, Zerp SF, Bartelink H, Blitterswijk van WJ, Verheij M. Alkyl-lysophospholipids activate the SAPK/JNK pathway and enhance radiation-induced apoptosis. Cancer Res. 1999;59:2457–2463. [PubMed] [Google Scholar]
- Ruiter GA, Zerp SF, Bartelink H, Blitterswijk van WJ, Verheij M. Anti-cancer alkyl-lysophospholipids inhibit the phosphatidylinositol 3-kinase-Akt/PKB survival pathway. Anticancer Drugs. 2003;14:167–173. doi: 10.1097/00001813-200302000-00011. [DOI] [PubMed] [Google Scholar]
- Sabine JR, Abraham S, Chaikoff IL. Control of lipid metabolism in hepatomas: insensitivity of rate of fatty acid and cholesterol synthesis by mouse hepatoma BW7756 to fasting and to feedback control. Cancer Res. 1967;27:793–799. [PubMed] [Google Scholar]
- Samudio I, Fiegl M, Andreeff M. Mitochondrial uncoupling and the Warburg effect: molecular basis for the reprogramming of cancer cell metabolism. Cancer Res. 2009;69:2163–2166. doi: 10.1158/0008-5472.CAN-08-3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarker KP, Obara S, Nakata M, Kitajima I, Maruyama I. Anandamide induces apoptosis of PC-12 cells: involvement of superoxide and caspase-3. FEBS Lett. 2000;472:39–44. doi: 10.1016/s0014-5793(00)01425-3. [DOI] [PubMed] [Google Scholar]
- Schlame M, Rua D, Greenberg ML. The biosynthesis and functional role of cardiolipin. Prog Lipid Res. 2000;39:257–288. doi: 10.1016/s0163-7827(00)00005-9. [DOI] [PubMed] [Google Scholar]
- Schmid PC, Wold LE, Krebsbach RJ, Berdyshev EV, Schmid HH. Anandamide and other N-acylethanolamines in human tumours. Lipids. 2002;37:907–912. doi: 10.1007/s11745-002-0978-z. [DOI] [PubMed] [Google Scholar]
- Schwarz H, Blanco FJ, Lotz M. Anandamide, an endogenous cannabinoid receptor agonist inhibits lymphocyte proliferation and induces apoptosis. J Neuroimmunol. 1994;55:107–115. doi: 10.1016/0165-5728(94)90152-x. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- Shinohara N, Tsuduki T, Ito J, Honma T, Kijima R, Sugawara S, et al. Jacaric acid, a linolenic acid isomer with a conjugated triene system, has a strong antitumor effect in vitro and in vivo. Biochim Biophys Acta. 2012;1821:980–988. doi: 10.1016/j.bbalip.2012.04.001. [DOI] [PubMed] [Google Scholar]
- Skorski T, Bellacosa A, Nieborowska-Skorska M, Majewski M, Martinez R, Choi JK, et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. EMBO J. 1997;16:6151–6161. doi: 10.1093/emboj/16.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skulachev VP. Uncoupling: new approaches to an old problem of bioenergetics. Biochim Biophys Acta. 1998;1363:100–124. doi: 10.1016/s0005-2728(97)00091-1. [DOI] [PubMed] [Google Scholar]
- Spector AA, Yorek MA. Membrane lipid composition and cellular function. J Lipid Res. 1985;26:1015–1035. [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Szymczak M, Murray M, Petrovic N. Modulation of angiogenesis by ω-3 polyunsaturated fatty acids is mediated by cyclooxygenases. Blood. 2008;111:3514–3521. doi: 10.1182/blood-2007-08-109934. [DOI] [PubMed] [Google Scholar]
- Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by β-phenylethyl isothiocyanate. Cancer Cell. 2006;10:241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Tronstad KJ, Berge K, Dyroy E, Madsen L, Berge RK. Growth reduction in glioma cells after treatment with tetradecylthioacetic acid: changes in fatty acid metabolism and oxidative status. Biochem Pharmacol. 2001;61:639–649. doi: 10.1016/s0006-2952(01)00525-1. [DOI] [PubMed] [Google Scholar]
- Tronstad KJ, Gjertsen BT, Krakstad C, Berge K, Brustugun OT, Doskeland SO, et al. Mitochondrial-targeted fatty acid analog induces apoptosis with selective loss of mitochondrial glutathione in promyelocytic leukemia cells. Chem Biol. 2003;10:609–618. doi: 10.1016/s1074-5521(03)00142-x. [DOI] [PubMed] [Google Scholar]
- Tsujii M, Kawano S, DuBois RN. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci USA. 1997;94:3336–3340. doi: 10.1073/pnas.94.7.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuzuki T, Tokuyama Y, Igarashi M, Miyazawa T. Tumor growth suppression by alpha-eleostearic acid, a linolenic acid isomer with a conjugated triene system, via lipid peroxidation. Carcinogenesis. 2004;25:1417–1425. doi: 10.1093/carcin/bgh109. [DOI] [PubMed] [Google Scholar]
- Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- Venturini I, Zeneroli ML, Corsi L, Avallone R, Farina F, Alho H, et al. Up-regulation of peripheral benzodiazepine receptor system in hepatocellular carcinoma. Life Sci. 1998;63:1269–1280. doi: 10.1016/s0024-3205(98)00388-9. [DOI] [PubMed] [Google Scholar]
- Vrablic AS, Albright CD, Craciunescu CN, Salganic RI, Zeisel SH. Altered mitochondrial function and overgeneration of reactive oxygen species precede the induction of apoptosis by 1-O-octadecyl-2-O-methyl-rac-glycero-3-phosphocholine in p53-defective hepatocytes. FASEB J. 2001;15:1739–1744. doi: 10.1096/fj.00-0300com. [DOI] [PubMed] [Google Scholar]
- Wasilewski M, Wieckowski MR, Dymkowska D, Wojtczak L. Effects of N-acylethanolamines on mitochondrial energetics and permeability transition. Biochim Biophys Acta. 2004;1657:151–163. doi: 10.1016/j.bbabio.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Wieckowski MR, Wojtczak L. Fatty acid-induced uncoupling of oxidative phosphorylation is partly due to opening of the mitochondrial permeability transition pore. FEBS Lett. 1998;423:339–342. doi: 10.1016/s0014-5793(98)00118-5. [DOI] [PubMed] [Google Scholar]
- Wongtangtintharn S, Oku H, Iwasaki H, Inafuku M, Toda T, Yanagita T. Incorporation of branched-chain fatty acid into cellular lipids and caspase-independent apoptosis in human breast cancer cell line, SKBR-3. Lipids Health Dis. 2005;4:29. doi: 10.1186/1476-511X-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodfield K, Ruck A, Brdiczka D, Halestrap AP. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem J. 1998;336:287–290. doi: 10.1042/bj3360287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang QH, Church-Hajduk R, Ren J, Newton ML, Du C. Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis (IAP) irreversibly inactivates IAPs and facilitates caspase activity in apoptosis. Genes Dev. 2003;17:1487–1496. doi: 10.1101/gad.1097903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Liu S, Chen X, Chen H, Huang M, Zheng J. Induction of apoptotic cell death and in vivo growth inhibition of human cancer cells by a saturated branched-chain fatty acid, 13-methyltetradecanoic acid. Cancer Res. 2000;60:505–509. [PubMed] [Google Scholar]
- Zaugg K, Yao Y, Reilly PT, Kannan K, Kiarash R, Mason J, et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011;25:1041–1051. doi: 10.1101/gad.1987211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zborowski J, Wojtczak L. Phospholipid synthesis in rat-liver mitochondria. Biochim Biophys Acta. 1969;187:73–84. doi: 10.1016/0005-2760(69)90134-9. [DOI] [PubMed] [Google Scholar]
- Zhang G, Panigrahy D, Mahakian LM, Yang J, Liu JY, Stephen Lee KS, et al. Epoxy metabolites of docosahexaenoic acid (DHA) inhibit angiogenesis, tumor growth, and metastasis. Proc Natl Acad Sci USA. 2013;110:6530–6535. doi: 10.1073/pnas.1304321110. [DOI] [PMC free article] [PubMed] [Google Scholar]