

Abstract
Mitochondrially bound hexokinase II (mtHKII) has long been known to confer cancer cells with their resilience against cell death. More recently, mtHKII has emerged as a powerful protector against cardiac cell death. mtHKII protects against ischaemia-reperfusion (IR) injury in skeletal muscle and heart, attenuates cardiac hypertrophy and remodelling, and is one of the major end-effectors through which ischaemic preconditioning protects against myocardial IR injury. Mechanisms of mtHKII cardioprotection against reperfusion injury entail the maintenance of regulated outer mitochondrial membrane (OMM) permeability during ischaemia and reperfusion resulting in stabilization of mitochondrial membrane potential, the prevention of OMM breakage and cytochrome C release, and reduced reactive oxygen species production. Increasing mtHK may also have important metabolic consequences, such as improvement of glucose-induced insulin release, prevention of acidosis through enhanced coupling of glycolysis and glucose oxidation, and inhibition of fatty acid oxidation. Deficiencies in expression and distorted cellular signalling of HKII may contribute to the altered sensitivity of diabetes to cardiac ischaemic diseases. The interaction of HKII with the mitochondrion constitutes a powerful endogenous molecular mechanism to protect against cell death in almost all cell types examined (neurons, tumours, kidney, lung, skeletal muscle, heart). The challenge now is to harness mtHKII in the treatment of infarction, stroke, elective surgery and transplantation. Remote ischaemic preconditioning, metformin administration and miR-155/miR-144 manipulations are potential means of doing just that.
LINKED ARTICLES
This article is part of a themed issue on Mitochondrial Pharmacology: Energy, Injury & Beyond. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2014.171.issue-8
Keywords: ischaemia-reperfusion injury, mPTP, cell death, diabetes, glycolysis, mitochondria
Introduction
The two leading causes of human mortality are cancer and cardiovascular disease. Curiously, the directed goal of therapy against these diseases is diametrically opposed: in cancer, we strive to kill the tumour cells, whereas in heart disease, we strive to protect cardiac cells from being killed. The question arose whether the processes that promote cell survival in cancer cells could potentially be used in promoting survival in the heart, and whether our understanding of the death processes in the heart could be harnessed in the treatment of cancer. The cancer literature has suggested that the resilience of cancer cells against cell death is, at least partly, due to highly elevated levels of hexokinase II (HKII) bound to mitochondria (Pedersen, 2007). Knowing that mitochondrial dysfunction has emerged as a major mediator of cell death in ischaemia-reperfusion (IR) injury of the heart, we considered the possibility that HKII is one of the critical regulators of mitochondrial dysfunction in cardiac IR injury. This consideration was primarily based on the pioneering work by others, demonstrating the pivotal role of HK in mitochondrial function and cell death in non-cardiovascular research fields. Instrumental in these HK pioneering studies were the discoveries of hexokinase (HK) binding to and regulation of mitochondrial voltage-dependent anion channel (VDAC; Fiek et al., 1982; Forte et al., 1987; Brdiczka, 1990; Rostovtseva et al., 2005; Rostovtseva and Bezrukov, 2008) and the regulatory role of HK in mitochondrial pore formation (Nakashima et al., 1986; Marzo et al., 1998).
Various work by us and others have now clearly demonstrated that mitochondrially bound hexokinase II (mtHKII) is indeed a major determinant of infarct size and may constitute one of the end-effectors of ischaemic preconditioning. Mitochondrial hexokinase II (and I) affects mitochondrial membrane potential and reactive oxygen species (ROS) production, regulates mitochondrial permeability transition pore (mPTP) and may also determine the direction of cardiac metabolic flux. This review summarizes current knowledge about the role of mitochondrially bound HK in cardioprotection and cardiac IR injury, adding mtHKII as a new target that may prove useful in the quest to reduce mortality due to cardiovascular disease.
The HK family
The first step in the metabolism of glucose is its phosphorylation upon entrance into the cell. Sufficient and rapid phosphorylation is important to maintain the steep gradient of glucose concentration over the plasma membrane to drive continuous glucose uptake through the GLUT transporters, as well as to render glucose polar, and therefore incapable of exiting the cell. Glucose phosphorylation is catalysed by the enzyme HK, of which four isozymes are present within mammalian tissue. HKI, II and III are isozymes of 100 kDa, displaying high affinity for glucose (Km ≤ 0.3 mM) and product inhibition for glucose-6-phosphate (G6P) (Ki ≤ 0.1 mM), whereas HKIV (glucokinase) is 50 kDa in size, has low affinity for glucose (half-saturation at about 8 mM glucose) and does not show product inhibition at physiological levels of G6P (Wilson, 2003). Importantly, HKI and II contain a hydrophobic amino terminal mitochondrial binding motif, which is not present in the HKIII and IV isoforms.
HKI is ubiquitously expressed in almost all mammalian tissues, is largely unresponsive to hormonal and prevailing metabolic conditions, and can be considered more of a housekeeping protein. Surprisingly, despite being a glycolytic enzyme, and with glycolysis mainly being thought of as a cytosolic process, HKI is predominantly associated with mitochondria (Crane and Sols, 1953; Johnson, 1960; Abraham et al., 1964). It is suggested (Wilson, 1995; John et al., 2011) that HKI principally performs a catabolic function, channelling glucose into glycolysis for ATP production. In contrast, HKII is more variably located in either the cytosol or at the mitochondrial outer membrane and is mostly expressed in insulin-sensitive tissue, such as heart, skeletal muscle and adipose tissue. When situated in the cytosol, it directs glucose into glycogen synthesis, while when bound to mitochondria, it primarily directs glucose into glycolysis (John et al., 2011). HKII expression levels and localization are highly regulated by (patho)physiology, hormones and metabolic state (Heikkinen et al., 2000; Wilson, 2003). In contrast to the abundance of HKI and HKII, HKIII shows low expression in most mammalian tissues, being most highly represented in lung, liver and spleen (Heumann et al., 1974; Furuta et al., 1996). Finally, HKIV is traditionally regarded as a glucose-sensing enzyme (although other HK isoforms also display glucose-sensing properties), associated with regulating insulin release by pancreatic beta cell. This glucokinase is mainly expressed in the liver and pancreas, but can also be found in certain parts of the brain and gut (Postic et al., 2001).
Mitochondrial HK in cancer
As early as 1924, the pioneering work of the Nobel prize winner Otto Warburg (Warburg et al., 1924) associated malignant, aggressive tumour growth with increased rates of aerobic glycolysis and increased lactate production, a signature now known as the Warburg effect (Pedersen, 2007). It took another 50 years to demonstrate mitochondrial HKII as a key molecular governor of this increased glycolysis (Bustamante and Pedersen, 1977; Bustamante et al., 1981), with the expression of HKII (sometimes HKI) being increased often more than 100-fold. Activation of the PI3K/Akt pathway, one of the most frequently mutated pathways in cancer (Shaw and Cantley, 2006), and activation of PKA and PKC pathways, also commonly seen in cancers, may explain this increased HKII expression. DNA demethylation and HKII gene amplification has also been suggested to play a role (Mathupala et al., 2009), as has the increased expression of hypoxia-inducible factor HIF1α (Semenza, 2003; Keith et al., 2012). Although Warburg originally hypothesized that the increased reliance of tumours on glycolysis was due to impairments in mitochondrial function, it is now known that this is not the case. Mitochondria from tumours can still have normal oxidative phosphorylation with intact ATP synthetic capacity; however, they are often reprogrammed towards biosynthetic pathways supporting tumour proliferation, such that glucose and glutamine become important substrates feeding rewired anabolic pathways (Ward and Thompson, 2012). Thus, the increased HKII expression and its binding to mitochondria facilitates not only increased aerobic glycolysis and lactate production (John et al., 2011) but also the channelling of glycolytic substrates into biosynthetic pathways for which mitochondria play a crucial role. The increase in the proportion of HKII that is bound to mitochondria also provides the cancer cell with resilience to cell death. The exact mechanism of this protection has not yet been elucidated, but it is known that glucose is necessary in order for mtHKII to inhibit apoptosis, indicating that this is an active process requiring glucose phosphorylation (Gottlob et al., 2001). Targeting the binding between HKII and mitochondria is currently actively pursued as a possible treatment against aggressive proliferative tumours. Such targeting may be achieved with 3-bromopyruvate (Mathupala et al., 2009), methyl jasmonate (Goldin et al., 2008), dichloroacetate (Michelakis et al., 2010), the antifungal compounds clotrimazole and bifonazole (Penso and Beitner, 1998), and some traditional Chinese medicinal plants (Wei et al., 2013). However, such treatment comes with a price. Our recent data (Smeele et al., 2011a), showing the high sensitivity of heart towards disruption of mtHKII binding and the immediate development of cardiac cell death, are a direct warning against any such treatment being a global, whole-body treatment. A nice example of this phenomenon is the use of anthracyclines in cancer chemotherapy, which, while very effective in cancer treatment, have to be very carefully titrated because of their severe cardiotoxicity. It has been suggested that this cardiotoxicity may be mediated in part by mito-HK dissociation via inhibition of Akt signalling (Pastorino et al., 2005). Thus, it is of paramount importance that these drugs target the cancer cell through localized delivery to the tumour (Ko et al., 2012) or using compounds that are only taken up by the cancer cell through selective cancer-expressed transporters (Birsoy et al., 2013).
Mitochondrial HK and protection in non-cardiac tissues
Although the primary focus of this review is on the heart, mitochondrial HK has been shown to protect against stressors in several other organs and tissues. We demonstrated that decreased (mitochondrial) HKII increased IR injury in skeletal muscle (Smeele et al., 2010; 2012,). In fact, skeletal muscle IR injury was very sensitive to reductions in HKII (50% HKII reduction increased IR-induced cell necrosis from 36 to 76%; Smeele et al., 2012), probably because HKII is the major HK isoform in skeletal muscle. Bryson et al. (2002) and Gall et al. (2011) showed that increased HK activity protects kidney epithelial cells against oxidant injury, whereas Ahmad et al. (2002) demonstrated that HKII protected human lung epithelial cells against hyperoxia and oxidative stress. Finally, mtHKII protects against neurodegeneration in models of Parkinson's disease (Gimenez-Cassina et al., 2009; Corona et al., 2010). Therefore, it seems that the interaction of HK with mitochondria constitute an endogenous cellular protective mechanism against cell death that is operative in many tissues and organs.
Mitochondrial HK and metabolism
Early observations (Bessmann and Geiger, 1980; Wilson, 1995), later supported by various other researchers, demonstrated that mitochondrially bound HK preferentially uses mitochondrially produced ATP (in contrast to cytosolic ATP), with direct channelling of the produced ADP back into the mitochondria. Thus, one major metabolic consequence of HK translocation to the mitochondria is that the ATP sensitivity of glucose phosphorylation is shifted from a cytosolic to a mitochondrial ATP source. Surprisingly, however, it remains unknown what the metabolic consequences of this translocation per se are in terms of oxygen consumption and energy substrate selection in intact organs/tissues. The role of mtHK in cell death however is better understood. Several seminal studies have now clearly shown the association between mtHK disruption and cell death, induced by stimuli such as H2O2 and/or UV irradiation in cellular studies (Gottlob et al., 2001; Ahmad et al., 2002; Bryson et al., 2002; Pastorino et al., 2002) or IR in intact organs and tissues (Smeele et al., 2011a, 2012; Pasdois et al., 2013). There are indications that enhancing mtHKII may increase (glucose-mediated) oxidative phosphorylation and therefore overall energy production in permeabilized human fibres from dilated atria (Roosimaa et al., 2013). These findings are in line with older literature showing that HK can display high control strength over respiration in isolated mitochondria (Groen et al., 1982). Interestingly, it has also been suggested that an increased translocation of HKI to mitochondria increases the glucose sensitivity of the pancreatic beta cell to release insulin (Rabuazzo et al., 1997). Whether mtHK may affect cardiac (and skeletal muscle) insulin sensitivity is unknown. Clearly, more work needs to be done to answer these important questions as to how mtHKII affects metabolism of the intact heart.
Older literature indicates that mtHK not only affects glucose metabolism but may also regulate cardiac fatty acid oxidation. Mitochondrial HK inhibits palmitoyl-CoA synthetase through competition for ATP and thereby inhibiting palmitate activation at the outer mitochondrial membrane (OMM) in isolated mitochondria (De Jong and Hülsmann, 1970). The reported decrease in mtHK associated with diabetes (Katzen et al., 1970) may therefore be a contributing factor to the often increased fatty acid metabolism in diabetic hearts. The reciprocal relationship was demonstrated by Southworth et al. who showed that perfusion of isolated hearts with fatty acids dislodged HKI and HKII from mitochondria (Southworth et al., 2007). Thus, mtHK seems to be an ideal localization hub for the well-known competition between glucose and fatty acid metabolism and the regulation thereof.
Mitochondrial HK in cardiovascular diseases (Figure 1)
Figure 1.
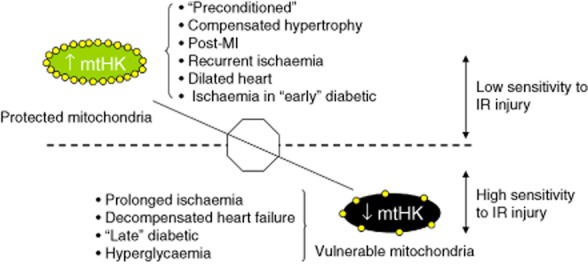
Schematic representation of cardiac disease states with low or high sensitivity to IR injury, which is associated with protected mitochondria (high mtHK) or vulnerable mitochondria (low mtHK) respectively.
HKI and HKII are both present in the heart. In adult mouse heart, HKI and HKII contribute approximately equally to total cardiac HK activity (Smeele et al., 2011b), depending on the age and the nutritional/pathophysiological condition of the animal. Few data are available for human heart, except for a recent report by Roosimaa et al. (2013), which indicates that in non-dilated human atrial tissue, HKI is the most abundant isoform. In mouse and human skeletal muscle (Mandarino et al., 1995; Smeele et al., 2011b), however, HKII accounts for >80% of total HK activity.
Ischaemic preconditioning (IPC) protection against I/R injury through increased mtHKII
The discovery that short, non-lethal periods of ischaemia activate an endogenous cardiac protection programme against long, lethal, periods of ischaemia led to a highly intensified research effort to elucidate the cellular mechanisms. This phenomenon was called IPC (Murry et al., 1986). Subsequent research demonstrated that IPC cellular signalling converged on the mitochondrion and was associated with alterations in glycolysis (Murry et al., 1986; Murphy and Steenbergen, 2008). Work from our laboratory at that time also noticed this interaction between glycolysis and mitochondrial function following non-lethal periods of ischaemia. We demonstrated that the activation of mitochondrial oxygen consumption, due to an instantaneously increased cardiac workload (Zuurbier and van Beek, 1997; Van Beek et al., 1998), was slowed following non-lethal periods of ischaemia. However, no such slowing was observed when glycolysis was bypassed using high concentrations of pyruvate or lactate (Zuurbier and Ince, 2002). In other words, the changes observed in mitochondrial function following non-lethal ischaemia were precipitated through changes evoked in glycolysis. After having established that the alterations in glycolysis were not due to alterations in the pentose phosphate pathway (Zuurbier et al., 2004), translocation of HK with non-lethal periods of ischaemia became a likely candidate (Zuurbier et al., 2009). Finally, we demonstrated that reversible ischaemia such as IPC induced a translocation of the glycolytic enzyme HK to the mitochondria (Zuurbier et al., 2005; Gürel et al., 2009). This leads us to the hypothesis that IPC could, at least partly, be attributed to increased HKII trafficking to the mitochondria (Zuurbier et al., 2009). This hypothesis was confirmed by subsequent studies showing loss of IPC protective effects with a peptide blocking mito-HKII binding (Smeele et al., 2011a) and increased IR injury with partial deletion of the HKII gene in both heart and skeletal muscle (Smeele et al., 2010; 2012,; Wu et al., 2011). This work was recently confirmed by findings in Halestrap's laboratory, showing a close correlation between cardiac infarct size and the extent of mitochondrial HKII dissociation (Pasdois et al., 2013).
IR, post-myocardial infarction, regeneration
HK expression and cellular localization changes dramatically during and after periods of ischaemia. During prolonged cardiac ischaemia, there is an increase in cytosolic HK activity (Correa et al., 2008) that can be explained by solubilization of HKII (but not HKI) from the mitochondria (Gürel et al., 2009; Pasdois et al., 2011). The mechanism of HK detachment from the mitochondria during ischaemia is, at least partly, related to acidification and increases in G6P (Pasdois et al., 2013). The endogenous adaptation following ischaemia in cardiac tissue is associated with increases (∼30–70%) in total and mitochondrial HK (McFalls et al., 2002; Miyamoto et al., 2010; Wu et al., 2011; Yeih et al., 2011). Large increases (200–300%) in HK activity were also observed in regenerated skeletal muscle 2 weeks after the IR insult (Smeele et al., 2012). It thus seems that following an ischaemic episode, there is increased HK expression, presumably enhancing biosynthetic pathways for regeneration and growth and to offer protection against recurrent episodes of ischaemia.
Hypertrophy and heart failure
During pressure overload-induced cardiac hypertrophy, mtHKII and total HKII protein content have been shown to increase (Riehle et al., 2011; Wu et al., 2012). Increased HKII expression was also recently reported in dilated human atria (Roosimaa et al., 2013). Genetic reductions in HKII resulted in exaggerated cardiac hypertrophy in a pressure-overload model (Wu et al., 2012). These data suggest that the increased HKII expression in pressure-overloaded hearts is an adaptive response, possibly attenuating hypertrophy through diminishing oxidative stress (Wu et al., 2012). Therapeutic enhancement of tissue HKII during this hypertrophic stage may therefore be a potentially beneficial approach. Interestingly, while mild pressure-overload results in a relatively compensated hypertrophy associated with increases in HKII, the same procedure in animals deficient for PPAR gamma co-activator 1 beta (PGC-1β) resulted in decreased HKII levels, decompensated hypertrophy and evidence of increased oxidative stress (Riehle et al., 2011). These data suggest that PGC-1β, a regulator of mitochondrial biogenesis and genes encoding for mitochondrial metabolism, is needed to maintain cardiac function following pressure overload by possibly preserving HKII expression and preventing oxidative stress. Cardiac HKII has also been shown to be severely reduced in a pacing-induced heart failure model in pigs (Lionetti et al., 2009). While these data suggest that the development of heart failure is associated with decreases in HKII, further studies are warranted to examine whether the reduced HKII protein levels are cause or consequence of cardiac failure.
Hyperglycaemia and diabetes
Hyperglycaemia in the clinical condition is currently viewed as an important risk factor for poor clinical outcome. In addition, the presence of hyperglycaemia in the pre-diabetic state is a strong predictor of developing diabetic disease. Acute hyperglycaemia has been shown to induce the detachment of HK from mitochondria (Da-Silva et al., 2004; Pasdois et al., 2013), which may underlie its pathology. During the development of diabetes, there is a shift in cardiac metabolism away from glucose metabolism towards fatty acid metabolism. This shift is generally associated with significant decreases in cardiac and muscle HKII protein content without alterations in HKI protein content (Katzen et al., 1970; Vestergaard et al., 1995). The diabetic heart displays an altered response to IR and IPC, with short-term diabetes frequently offering protection against IR and attenuated IPC potential, and long-term diabetes resulting in worsened outcome after IR and loss of IPC protective effects (Miki et al., 2012). It is possible that changes in cardiac HK contribute to such an altered response of diabetic heart to IR and IPC. Indeed, we have demonstrated that the increased protection against IR and the attenuation of IPC in the short-term type I diabetic heart are associated with altered mitochondrial HK binding characteristics (Gürel et al., 2013). These data suggest that the known association between diabetes mellitus and ischaemic cardiovascular disease may partly stem from alterations at the level of cardiac HKII expression. More work is needed to fully explore the role of HKII in diabetic cardiomyopathy.
Mechanisms of mtHKII-induced protection against IR injury (Figure 2)
Figure 2.
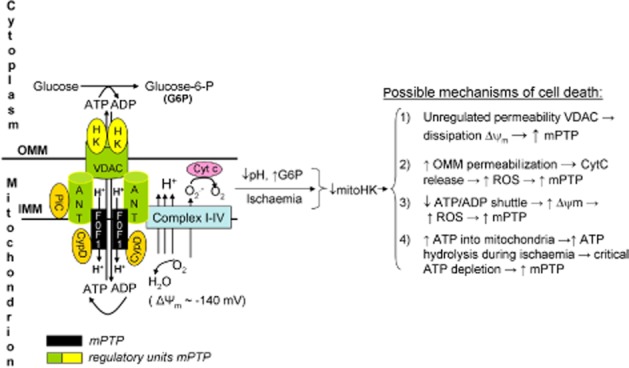
Schematic drawing of the mitochondrial permeability transition pore complex dictating cell death through decreases in mtHK induced by ischaemia-reperfusion. The most likely mechanisms (1–4) through which disruption of hexokinase II-mitochondrial binding may cause cell death are displayed (see text for further discussion).
There is currently no consensus concerning the exact mechanism of mtHK protection against IR injury. Below, we restrict our discussion to those four mechanisms for which experimental evidence can be found. The overarching action of mtHK is its regulatory role in the homeostatic crosstalk between the mitochondria and the cell. This crosstalk is set by the degree of permeability of the OMM and the IMM to extramitochondrial metabolites.
The permeability of the OMM is determined by VDAC, the mitochondrial binding partner of HK. HK, at least partly, regulates the permeability of VDAC for many important metabolites, such as ATP, ADP and NADH (Rostovtseva et al., 2005). The permeability barrier of the IMM to protons and ions is essential to ATP synthesis by mitochondria; loss of this permeability barrier prevents the generation of an electrochemical gradient (mitochondrial potential) that drives ATP synthesis through the F0F1ATPase. This loss of the IMM permeability barrier, as reflected by the opening of a non-specific pore in the inner mitochondrial membrane (IMM), the so-called mPTP, is currently considered the final event causing IR injury and irreversible cell death (Halestrap, 2009). The molecular identity of the mPTP was recently suggested to consist of dimers of the F0F1ATPase localized on the IMM (Giorgio et al., 2013), with HK, VDAC, adenine nucleotide transporter (ANT) and cyclophilin D (CypD) as important regulators of the mPTP (see also Figure 2). It is through this mPTP complex that HK mediates its decisive role in IR injury and cardioprotection. It should be noted that there may also be a role for the mitochondrial phosphate carrier (PiC) as important regulator of the mPTP (Leung et al., 2008). The reader is directed to literature that discusses this component in detail (Leung and Halestrap, 2008; Halestrap, 2009). For this review, we focus primarily on how HK may affect the mPTP.
Prevention of conformational change in molecular mPTP regulation complex to stabilize mitochondrial membrane potential
It has been shown that mitochondrial depolarization occurs during cardiac ischaemia and may be an early sign of irreversible injury (Lyon et al., 2010). Mitochondrial depolarization during ischaemia and early reperfusion may cause direct mPTP opening (Bernardi, 1992), and there is evidence that mtHKII may prevent such depolarization. We have recently demonstrated that acute HKII detachment from mitochondria in the beating heart per se (without any concomitant stress signal), using medium-to-high dosage (≥2.5 μM) of an HKII dislodging peptide, acutely depolarized mitochondria and induced cell death, an effect that could not be explained by vascular obstruction or ensuing ischaemia in the intact organ (Smeele et al., 2011a; Nederlof et al., 2013). In these conditions of normoxic perfusion with a pH > 7 buffer, this depolarization can cause immediate mPTP opening and cell death (Bernardi, 1992). Previously, Chiara et al. (2008) demonstrated that the sensitivity of isolated cardiomyocytes to ROS-induced mPTP opening was much increased with low concentrations (<1 μM) of the HKII dislodging peptide. They hypothesized that detachment of HKII induced a conformational change in the molecular complex connecting the OMM with the IMM and the mPTP (Chiara et al., 2008). We would suggest that the HKII dislodging peptide fully opens VDAC, either due to loss of HK or other VDAC-regulating proteins, such as tubulin (Sheldon et al., 2013). VDAC is the most abundant protein of the OMM and is responsible for the transport of ADP/ATP and other metabolites (e.g. NADH, Ca2+) across the OMM (Colombini, 2004; Rostovtseva et al., 2005; Rostovtseva and Bezrukov, 2008). VDAC can change between an open and ‘closed’ state, which is almost impermeable to ADP and ATP, and may thereby regulate mitochondrial respiration. The binding of several cytosolic proteins (tubulin, tBid, Bcl-xL, HK) with VDAC, in combination with OMM lipid composition and transmembrane potential, regulate VDAC conductance. It was recently demonstrated that dimeric tubulin, known to interact with mitochondria in vivo, induced voltage-sensitive closure of VDAC, reducing ADP availability and thereby mitochondrial respiration (Rostovtseva et al., 2008). Preliminary data (Sheldon et al., 2013) now show that the HKII dislodging peptide disrupts HKII and tubulin binding to VDAC in planar lipid bilayers, resulting in unregulated permeability of VDAC that may ultimately results in large ADP influx, decreasing the mitochondrial membrane potential to a critical level of spontaneous mPTP opening.
Stabilization of mitochondrial contact sites during ischaemia preventing cytochrome C (CytC) release
mtHKII may alternatively prevent OMM rupture and/or permeabilization by stabilization of mitochondrial contact sites. HK is preferentially bound to mitochondria at locations where the IMM comes closest to the OMM (Brdiczka et al., 2006). These contact sites contain large protein complexes consisting of VDAC, ANT, CytC, benzodiazepine receptor, CypD, cardiolipin, HK and creatine kinase. In reconstituted vesicles, disruption of HK from these protein complexes increases their permeability, which could be interpreted as analogous to increased permeability of the OMM and/or mPTP (Beutner et al., 1998). It has been suggested that during ischaemia, progressive acidosis and G6P accumulation dislodge HK from mitochondria (Pasdois et al., 2013). Such decrease in mtHK, together with increased Ca2+, disrupts the contact sides resulting in an increased permeability of the OMM for CytC release. Knowing that oxidized CytC is an important antioxidant, the loss of CytC will result in increased ROS production at early reperfusion (Pasdois et al., 2011; 2013,), finally resulting in mPTP opening and infarction.
Maintaining mitochondrial ADP to inhibit mPTP and reduce ROS
The activity of HK at the mitochondrial surface rapidly returns ADP back to the inner mitochondrial compartment via VDAC and ANT, thereby ensuring high levels of ADP in the vicinity of F0F1 ATPase dimers at early reperfusion. This ‘ATP/ADP’ shuttle has been shown to reduce the mitochondrial membrane potential and limit ROS production (Da-Silva et al., 2004; Santiago et al., 2008; Wu et al., 2012). The reduction in ROS production offered by active mtHK can range from >90% (going from zero to normal mtHK levels in isolated brain mitochondria; Da-Silva et al., 2004) to 70% (with activation of mtHK in isolated rat heart mitochondria; Santiago et al., 2008) and to 20% (with 40% increase in mtHKII in neonate rat cardiomyocytes; Wu et al., 2012).
Inhibition of ATP hydrolysis during ischaemia
Maintaining HKII at the mitochondria during ischaemia may impair cytosolic ATP entrance into the mitochondria (Perevoshchikova et al., 2010) during ischaemia, thereby attenuating mitochondrial hydrolysis of cytosolic ATP (through reversed mode of mitochondrial F1F0 ATPsynthase). Prevention of a critical depletion of ATP by the mitochondria during ischaemia can significantly reduce anaerobic glycolysis and lactate accumulation, cardiac contracture and cell death induced by IR interventions (Steenbergen et al., 1990; Jennings, 2013).
Structural obstruction of pro-apoptotic protein binding to mitochondria
It has been proposed, in cellular studies employing oxidant agents, that mtHKII may be cytoprotective through structural opposition to the mitochondrial binding of pro-apoptotic proteins such as Bax or Bad (Pastorino et al., 2002). However, we and others have been unable to show a role for Bax in mtHKII-mediated protection against IR injury in isolated hearts (Smeele et al., 2011a; Pasdois et al., 2013) or skeletal muscle (Smeele et al., 2012). The purely structural basis of mtHKII cytoprotection by steric hindrance is also challenged by several studies which demonstrate that glucose must be present in order for HK to offer protection against cell death (Gottlob et al., 2001; Mergenthaler et al., 2012). Furthermore, Majewski et al. (2004) demonstrated that the protection offered through HK-mitochondria interaction does not necessitate the presence of Bax and Bak. Thus, it seems that Bax and Bad do not play a significant role in mtHKII protection against IR injury.
Targeting HK to mitochondria (Figure 3)
Figure 3.
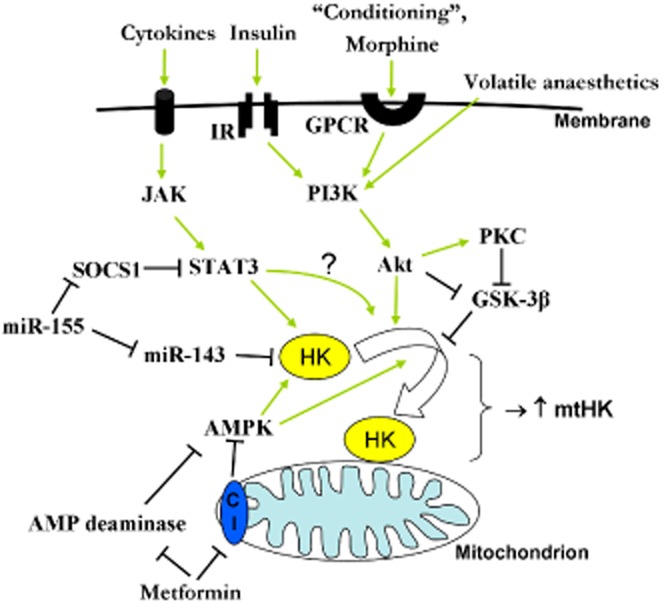
Intracellular signalling pathways showing how different interventions (‘conditioning’) and compounds such as cytokines, insulin, metformin, miRNA's and anaesthetics may increase HKII expression and/or HKII translocation to mitochondria. IR, insulin receptor; SOCS1, suppressor of cytokine signalling 1; C1, complex I of the electron transport chain.
It is clearly established in numerous cardiovascular disease models and in skeletal muscle IR interventions that the amount of HKII bound to mitochondria is a major determinant of infarct size and/or disease progression (Smeele et al., 2010; 2011a; 2012,,; Wu et al., 2011; 2012,). In fact, the Halestrap laboratory recently reported a strong inverse correlation between the amount of end-ischaemic mtHKII and infarct size in the isolated rat heart subjected to different perfusion condition: tripling of mtHKII reduced infarct size from 65 to 5% (see figure 9, Pasdois et al., 2013). Development of new therapies directed at increasing and/or keeping HKII at the mitochondria therefore seems like an attractive cardioprotective approach. Below we summarize the most promising interventions that have been shown to raise total HK or specifically mtHKII, for exploration of their (clinical) potential as adjunct therapy in settings of ischaemia-reperfusion and possible chronic diseases such as diabetes, cardiac hypertrophy and heart failure.
‘Conditioning’ of the heart
In 2005, we demonstrated that an IPC stimulus immediately translocates HK to the mitochondria in isolated rat hearts (Zuurbier et al., 2005). Subsequently, we have also shown that IPC also prevents HKII detachment from mitochondria during the irreversible period of ischaemia (Gürel et al., 2009). These studies were later partly confirmed by Pasdois et al. (2011; 2013,). Such IPC interventions can be performed clinically for leg or arm surgeries, but applicability for the heart is not directly feasible. However, the discovery that the heart can also be protected through remote ischaemic preconditioning (RIPC) or post-conditioning opened an avenue to cardiac application of the ‘conditioning’ phenomenon. Since the intracellular signalling pathways providing protection with IPC overlap to a large extent with those of RIPC (e.g. Li et al., 2011), it is anticipated that RIPC may also translocate HKII to mitochondria. This information is however lacking in the literature. Our currently active clinical mtHK-RIPC trial (NTR2915, Nederlands Trial Register) of RIPC in coronary artery bypass grafting (CABG) patients examines the relationship between RIPC protection and mtHKII in human atrial tissue and will hopefully provide this much needed information.
Insulin/PI3/Akt/glycogen synthase kinase (GSK)-3β axis
Several studies have demonstrated that activation of this pathway will acutely (<30 min) increase HKII trafficking to the mitochondria (Russell et al., 1992; Pastorino et al., 2005; Zuurbier et al., 2005; Southworth et al., 2007; Miyamoto et al., 2008) and later on increases HKII expression. Juhaszova et al. (2004) demonstrated that many cardioprotective agents confer protection through phosphorylation of GSK-3β and consequently inhibition of mPTP. Although many other studies can subsequently be found that have demonstrated cardioprotection with activation of this pathway, none of them have directly examined whether this was due to increased mtHKII (e.g. Hausenloy et al., 2005; Terashima et al., 2010; Ng et al., 2012). A disadvantage of using this pathway to advance mtHKII is of course its pleiotropic character, with unwanted side effects increasing with every step upstream above HKII. For example, insulin may increase HKII translocation to mitochondria, but at the same time may not only result in hypoglycaemia, but also in hypolipidaemia (Zuurbier et al., 2008a2002). It therefore seems advisable to use more specific treatment for increasing mtHKII.
Anaesthesia
The use of specific anaesthetics for providing protection against IR injury is an attractive scenario in surgeries that necessitate anaesthesia. Pre-clinical studies in healthy, young animals almost all report protective effects of certain volatile anaesthetics (sevoflurane, isoflurane) and opiates (morphine). These protective anaesthetics also commonly activate the PI3/Akt/GSK-3β axis. However, these protective effects were not always observed in clinical studies employing, for example, CABG procedures (De Hert, 2011). The ambiguity in the clinical scenario potentially results from co-morbidities (diabetes, ageing and hypertrophy) and co-medications (e.g. statins, dexamethasones, opioids, nitroglycerine, β-blockers) that disturb the signalling pathway. Using several different anaesthetic regimens in healthy rats, we were able to demonstrate that the cardioprotective volatile anaesthetics sevoflurane and isoflurane did indeed maintain HK at cardiac mitochondria at a level similar to the non-anaesthetized animal (Zuurbier et al., 2008b). An anaesthetic regimen of propofol–sufentanil–morphine, also often used in the clinical arena, resulted in solubilization of HK from the mitochondria. It can be speculated that the divergent effects of these two clinically most used anaesthetic regimens (volatile anaesthesia vs. fentanyl–propofol anaesthesia) on mtHKII may also explain the dissipating effect of propofol anaesthesia on RIPC protection (Kottenberg et al., 2012). Further studies will be needed to test this hypothesis directly. In conclusion, although certain anaesthetics may indeed offer protection through increases in mtHKII, the many cellular signalling steps that exist between the start of protection induced by an anaesthetic agent to the final subcellular HKII translocation is prone to be disturbed by many clinical and disease factors, thereby decreasing its likeliness as an ideal option offering IR protection under clinical conditions.
Metformin
The glucose-lowering anti-diabetic drug metformin has been shown to be cardioprotective beyond its anti-hyperglycaemic properties. It reduces infarct size in both diabetic and non-diabetic animals (Bhamra et al., 2008; Calvert et al., 2008; Solskov et al., 2008; Whittington et al., 2013). In addition, multiple clinical studies showed reduced cardiovascular mortality in diabetic patients treated with metformin (El Messaoudi et al., 2013). These cardioprotective effects may be caused by increased mtHK activity. Metformin treatment reverses the down-regulation in total HK activity and has been shown to increase HK translocation to the mitochondria in diabetic hearts without adversely affecting normal hearts (Da Silva et al., 2012). Whether metformin will increase (mt)HK in hearts during IR to afford protection remains to be examined, but 2 weeks of metformin treatment has previously been demonstrated to increase HK activity in rat white gastrocnemius muscle (Suwa et al., 2006). This increase in HK might be explained by an increase in Akt phosphorylation after metformin treatment during reperfusion (Bhamra et al., 2008). However, Akt phosphorylation was not observed when metformin was given before ischaemia (Bhamra et al., 2008; Calvert et al., 2008). Alternatively, metformin may increase HK activity through activation of AMP-activated PK (AMPK). Increases in AMPK activity have been associated with increased HK activity in skeletal muscle (Holmes et al., 1999; Dieni and Storey, 2011). Metformin may activate AMPK through increases in AMP resulting from inhibition of complex I (Owen et al., 2000) and/or AMP deaminase (Ouyang et al., 2011; Vytla and Ochs, 2013). Metformin is one of the most long-standing prescribed drugs in diabetes, showing relatively high efficacy. It will be interesting to elucidate whether a common mechanism of this drug entails the translocation of HK to the mitochondria, to identify a cellular mechanisms for its cardioprotective effects.
miR-155/miR-143 therapy
MicroRNAs (miR) have recently been discovered as yet another level of regulation in biological function. miRs are non-protein-coding RNAs of 20–30 nucleotides, which silence gene expression at the post-transcriptional level by targeting the 3′-untranslated region of messenger RNA. The use of miR therapeutics in cardiovascular medicine is only starting to develop, although several studies have already demonstrated the feasibility of such an approach (Thum, 2011). HKII has been shown to be regulated by miR-155 and miR-143 in cancer cells (Jiang et al., 2012; Peschiaroli et al., 2012). Increases in miR-155 induced HKII expression through activation of a HKII transcriptional activator (STAT3) and repression of a negative regulator of HKII, miR-143. Matkovich et al. (2013) recently demonstrated that cardiac overexpression of miR-143 was also able to suppress HKII mRNA in the heart with no indirect target regulation. Therapies may therefore be developed which employ synthetic complementary oligonucleotides against miR-143 and/or adeno-associated virus containing miR-155 to specifically increase cardiac HKII and offer protection during IR interventions.
Heat shock proteins
HKII has recently shown to be released from mitochondria in tumour cells during inhibition of mitochondrial heat shock protein 90 (HSP90; Chae et al., 2012). HSP90 has previously been demonstrated to have cardioprotective capacity (Latchman, 2001; Xiang et al., 2010) and the mitochondrial heat shock protein, tumour necrosis factor receptor-associated protein 1 (TRAP1), is known to be present in the heart (Xiang et al., 2010). A therapy directed at increasing mitochondrial HSPs, either through heat treatment or gene therapy, may therefore also have cardioprotective potential by increasing mtHKII.
Detrimental effects of persistent elevated mtHKII in the heart?
The previous sections illustrate the many pro-survival effects of augmenting HKII-mitochondria binding within the heart. This raises the question that if mtHKII is such a powerful protector of the myocardium, why is HKII not permanently bound to mitochondria in the healthy heart? We hypothesize that irreversibly bound mtHK would hamper the natural occurrence of mitochondrial depolarization needed to maintain mitochondria healthy. Since mitochondrial depolarization is a trigger signal for mitochondrial recycling through mitophagy, prevention of depolarization may result in accumulation of dysfunctional mitochondria and ultimately a dysfunctional heart. This is supported by the apparent benefit afforded by dichloroacetate (DCA) in the treatment of pulmonary hypertension, right heart failure (McMurtry et al., 2004), hyperthyroid hypertrophy (Atherton et al., 2011) and cancer (Bonnet et al., 2007; Michelakis et al., 2010). DCA induces mitochondrial depolarization, which may at least partly be explained through disruption of HK-mitochondrial binding (Michelakis et al., 2010). The often observed hyperpolarized mitochondria in cancer cells (Chen, 1988; Bonnet et al., 2007) is consistent with their increased mtHKII as characterized by the Warburg effect. Permanently increasing mtHKII binding would also increase baseline aerobic glycolysis, and subsequently retard cellular signalling and mitochondrial activation, required for rapid responsive change in cardiac workload (Zuurbier and Ince, 2002; Harrison et al., 2003). Such retardation of mitochondrial activation may temporarily impair the free energy of ATP hydrolysis through elevation of cytosolic ADP (Van Beek et al., 1998). Finally, the up-regulated glycolysis caused by permanent association of HK to cardiac mitochondria would presumably switch off fatty acid oxidation through the Randle effect, resulting in a less efficient heart when oxygen is not limiting, and a redirection of fatty acids towards lipid accumulation in the heart. The chronic elevated glucose uptake may also result in an increased accumulation of G6P,, resulting in chronic mammalian target of rapamycin (mTOR) activation concomitant with an activated endoplasmic reticulum (ER) stress response (Sen et al., 2013). Interestingly, lipid accumulation, chronic mTOR activation and ER stress are all biochemical markers of cardiac failure (Shioi et al., 2003; Sharma et al., 2004), a known condition with elevated cardiac HK. It should be realized that the schemes discussed above are purely hypothetical; no evidence currently exists that has demonstrated detrimental effects of excessive mtHKII levels for the heart. However, future work may be devised to provide answers to these important questions of possible detrimental effects of too much mtHKII for the heart.
In conclusion, the association of HKII with mitochondria is a critical determinant of cardiomyocyte death, making it a potential drug target in the treatment of cardiovascular ischaemic diseases. It seems that in HK-mitochondrial binding, the two human diseases with the largest impact on human mortality and morbidity, cancer and cardiovascular disease, share a similar, but directionally opposing, cellular mechanism: mtHK binding is increased in malignant cancer providing it with resilience against cell death, whereas it is decreased during cardiac infarction and thereby contributes to cardiac cell death. The challenge is now to target this mechanism that governs whether cells live or die in the treatment of cardiovascular diseases, with a minimum of collateral damage.
Glossary
- AMPK
AMP-activated PK
- ANT
adenine nucleotide transporter
- CABG
coronary artery bypass grafting
- CypD
cyclophilin D
- CytC
cytochrome C
- ER
endoplasmic reticulum
- G6P
glucose-6-phosphate
- HK
hexokinase
- HSP90
heat shock protein 90
- IMM
inner mitochondrial membrane
- IPC
ischaemic preconditioning
- IR
ischaemia-reperfusion
- miR
micro RNA
- mPTP
mitochondrial permeability transition pore
- mtHK
mitochondrially bound hexokinase
- OMM
outer mitochondrial membrane
- PGC-1β
PPAR gamma co-activator 1 beta
- RIPC
remote ischaemic preconditioning
- ROS
reactive oxygen species
- VDAC
voltage-dependent anion channel
Conflict of interests
None.
References
- Abraham S, Borrebaeck B, Chaikoff IL. Effect of dietary carbohydrates on glucokinase and mannokinase activities of various rat tissues. J Nutr. 1964;83:273–288. doi: 10.1093/jn/83.3.273. [DOI] [PubMed] [Google Scholar]
- Ahmad A, Ahmad S, Schneider BK, Allen CB, Chang L-Y, White CW. Elevated expression of hexokinase II protects human lung epithelial-like A549 cells against oxidative injury. Am J Physiol Lung Cell Mol Physiol. 2002;283:L573–L584. doi: 10.1152/ajplung.00410.2001. [DOI] [PubMed] [Google Scholar]
- Atherton HJ, Dodd MS, Heather LC, Schroeder MA, Griffin JL, Radda JL, et al. Role of pyruvate dehydrogenase inhibition in the development of hypertrophy in the hyperthyroid rat heart: a combined magnetic resonance imaging and hyperpolorized magnetic resonance spectroscopy study. Circulation. 2011;123:2552–2561. doi: 10.1161/CIRCULATIONAHA.110.011387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by the proton electrochemical gradient. J Biol Chem. 1992;267:8834–8839. [PubMed] [Google Scholar]
- Bessmann SP, Geiger PJ. Compartmentation of hexokinase and creatine kinase phosphokinase, cellular regulation, and insulin action. In: Horecker BL, Stadtman ER, editors. Current Topics in Cellular Regulation. Vol. 16. New York: Academic; 1980. pp. 55–86. [DOI] [PubMed] [Google Scholar]
- Beutner G, Ruck A, Riede B, Brdiczka D. Complexes between porin, hexokinase, mitochondrial creatine kinase and adenylate translocator display properties of the permeability transition pore. Implications for regulation of permeability transition by the kinases. Biochim Biophys Acta. 1998;1368:7–18. doi: 10.1016/s0005-2736(97)00175-2. [DOI] [PubMed] [Google Scholar]
- Bhamra GS, Hausenloy DJ, Davidson SM, Carr RD, Paiva M, Wynne AM, et al. Metformin protects the ischemic heart by the Akt-mediated inhibition of mitochondrial permeability transition pore opening. Basic Res Cardiol. 2008;103:274–284. doi: 10.1007/s00395-007-0691-y. [DOI] [PubMed] [Google Scholar]
- Birsoy K, Wang T, Possemato R, Yilmaz OH, Koch CE, Chen WW, et al. MCT1-mediated transport of a toxic molecule is an effective strategy for targeting glycolytic tumors. Nat Gen. 2013;45:104–108. doi: 10.1038/ng.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- Brdiczka D. Interaction of mitochondrial porin with cytosolic proteins. Experientia. 1990;46:161–167. doi: 10.1007/BF02027312. [DOI] [PubMed] [Google Scholar]
- Brdiczka DG, Zorov DB, Sheu SS. Mitochondrial contact sites: their role in energy metabolism and apoptosis. Biochim Biophys Acta. 2006;1762:148–163. doi: 10.1016/j.bbadis.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Bryson JM, Coy PE, Gottlob K, Hay N, Robey RB. Increased hexokinase expression of either ectopic or endogenous origin, protects renal epithelial cells against acute oxidant-induced cell death. J Biol Chem. 2002;277:11392–11400. doi: 10.1074/jbc.M110927200. [DOI] [PubMed] [Google Scholar]
- Bustamante E, Pedersen PL. High aerobic glycolysis of rat hepatoma cells in culture: role of mitochondrial hexokinase. Proc Natl Acad Sci U S A. 1977;74:3735–3739. doi: 10.1073/pnas.74.9.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante E, Morris HP, Pedersen PL. Energy metabolism of tumor cells. Requirements for a form of hexokinase with a propensity for mitochondrial binding. J Biol Chem. 1981;256:8699–8704. [PubMed] [Google Scholar]
- Calvert JW, Gundewar S, Jha S, Greer JJ, Bestermann WH, Tian R, et al. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes. 2008;57:696–705. doi: 10.2337/db07-1098. [DOI] [PubMed] [Google Scholar]
- Chae YC, Caino MC, Lisanti S, Ghosh JC, Dohi T, Danial NN, et al. Control of tumor bioenergetics and survival stress signalling by mitochondrial HSP90s. Cancer Cell. 2012;22:331–344. doi: 10.1016/j.ccr.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LB. Mitochondrial membrane potential in living cells. Annu Rev Cell Biol. 1988;4:155–181. doi: 10.1146/annurev.cb.04.110188.001103. [DOI] [PubMed] [Google Scholar]
- Chiara F, Castellaro D, Marin O, Petronilli V, Brusilow WS, Juhaszova M, et al. Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS ONE. 2008;3:e1852. doi: 10.1371/journal.pone.0001852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombini M. VDAC: the channel at the interface between mitochondria and the cytosol. Mol Cell Biochem. 2004;256–257:107–115. doi: 10.1023/b:mcbi.0000009862.17396.8d. [DOI] [PubMed] [Google Scholar]
- Corona JC, Gimenez-Cassina A, Lim F, Diaz-Nido J. Hexokinase II gene transfer protects against neurodegeneration in the rotenone and MPTP mouse models of Parkinson's disease. J Neurosci Res. 2010;88:1943–1950. doi: 10.1002/jnr.22357. [DOI] [PubMed] [Google Scholar]
- Correa F, Garcia N, Gallardo-Perez JC, Carreno-Fuentes L, Rodrigues-Enriques S, Marin-Hernandez A, et al. Post-conditioning preserves glycolytic ATP during early reperfusion: a survival mechanism for the reperfused heart. Cell Physiol Biochem. 2008;22:635–644. doi: 10.1159/000185547. [DOI] [PubMed] [Google Scholar]
- Crane RK, Sols A. The association of hexokinase with particulate fractions of brain and other tissue hoogenates. J Biol Chem. 1953;203:273–292. [PubMed] [Google Scholar]
- Da Silva D, Ausina P, Alencar EM, Coelho WS, Zancan P, Sola-Penna M. Metformin reverses hexokinase and phosphofructokinase downregulation and intracellular distribution in the heart of diabetic mice. IUBMB Life. 2012;64:766–774. doi: 10.1002/iub.1063. [DOI] [PubMed] [Google Scholar]
- Da-Silva WS, Gomez-Puyou A, Gomez-Puyou de MT, Moreno-Sanchez R, Felice de FG, Meis de L, et al. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: steady-state ADP formation as a regulatory mechanism of membrane potential and reactive species generation in mitochondria. J Biol Chem. 2004;279:39846–39855. doi: 10.1074/jbc.M403835200. [DOI] [PubMed] [Google Scholar]
- De Hert SG. Cardioprotection by volatile anesthetics: what about noncardiac surgery? J Cardiothorac Vasc Anesth. 2011;25:899–901. doi: 10.1053/j.jvca.2011.08.004. [DOI] [PubMed] [Google Scholar]
- De Jong JW, Hülsmann WC. Effects of nagarse, adenosine and hexokinase on palmitate activation and oxidation. Biochim Biophys Acta. 1970;210:499–501. doi: 10.1016/0005-2760(70)90050-0. [DOI] [PubMed] [Google Scholar]
- Dieni CA, Storey KB. Regulation of hexokinase by reversible phosphorylation in skeletal muscle of a freeze-tolerant frog. Comp Biochem Physiol B Biochem Mol Biol. 2011;159:236–243. doi: 10.1016/j.cbpb.2011.05.003. [DOI] [PubMed] [Google Scholar]
- El Messaoudi S, Rongen GA, Riksen NP. Metformin therapy in diabetes: the role of cardioprotection. Curr Atheroscler Rep. 2013;15:314. doi: 10.1007/s11883-013-0314-z. [DOI] [PubMed] [Google Scholar]
- Fiek C, Benz R, Roos N, Brdiczka D. Evidence for identity between the hexokinase-binding protein and the mitochondrial porin in the outer membrane of rat liver mitochondria. Biochim Biophys Acta. 1982;688:429–440. doi: 10.1016/0005-2736(82)90354-6. [DOI] [PubMed] [Google Scholar]
- Forte M, Adelsberger-Mangan D, Colombini M. Purification and characterization of the voltage-dependent anion channel from the other mitochondrial membrane of yeast. J Membr Biol. 1987;99:65–72. doi: 10.1007/BF01870622. [DOI] [PubMed] [Google Scholar]
- Furuta H, Nishi S, Le Beau MML, Fernald AA, Yano H, Bell GI. Sequence of human hexokinase III cDNA and assignment of the human hexokinase III gene (HK3) to chromosome band 5q35.2 by fluorescence in situ hybridization. Genomics. 1996;36:206–209. doi: 10.1006/geno.1996.0448. [DOI] [PubMed] [Google Scholar]
- Gall JM, Wong V, Pimental DR, Havasi A, Wang Z, Pastorino JG, et al. Hexokinase regulates Bax-mediated mitochondrial membrane injury following ischemic stress. Kidney Int. 2011;79:1207–1216. doi: 10.1038/ki.2010.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez-Cassina A, Lim F, Palomo GM, Diaz-Nido J. Mitochondrial hexokinase II promotes neuronal survival and acts downstream of glycogen synthase kinase-3. J Biol Chem. 2009;284:3001–3011. doi: 10.1074/jbc.M808698200. [DOI] [PubMed] [Google Scholar]
- Giorgio V, Stockum von S, Antoniel M, Fabbro A, Fogolari F, Forte M, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin N, Arzoine L, Heyfets A, Israelson A, Zaslavsky Z, Bravman T, et al. Methyl jasmonate binds to and detaches mitochondria-bound hexokinase. Oncogene. 2008;27:4636–4643. doi: 10.1038/onc.2008.108. [DOI] [PubMed] [Google Scholar]
- Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptosis events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406–1418. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groen AK, Wanders RJA, Westerhoff HV, Meer van der R, Tager JM. Quantification of the contribution of various steps to the control of mitochondrial respiration. J Biol Chem. 1982;257:2754–2757. [PubMed] [Google Scholar]
- Gürel E, Smeele KM, Eerbeek O, Koeman A, Demirci C, Hollmann MW, et al. Ischemic preconditioning affects hexokinase activity and HKII in different subcellular compartments throughout cardiac ischemia-reperfusion. J Appl Physiol. 2009;106:1909–1916. doi: 10.1152/japplphysiol.90537.2008. [DOI] [PubMed] [Google Scholar]
- Gürel E, Ustunova S, Kapucu A, Yilmazer N, Eerbeek O, Nederlof R, et al. Hexokinase cellular trafficking in ischemia-reperfusion and ischemic preconditioning is altered in type I diabetic heart. Mol Biol Rep. 2013;40:4153–4160. doi: 10.1007/s11033-013-2495-5. [DOI] [PubMed] [Google Scholar]
- Halestrap A. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009;46:821–831. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- Harrison GJ, Van Wijhe MH, Groot de B, Dijk FJ, Gustafson LA, Beek van JH. Glycolytic buffering affects cardiac bioenergetic signaling and contractile reserve similar to creatine kinase. Am J Physiol Heart Circ Physiol. 2003;285:H883–H890. doi: 10.1152/ajpheart.00725.2002. [DOI] [PubMed] [Google Scholar]
- Hausenloy D, Tseng A, Mocanu MM, Yellon DM. Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am J Physiol Heart Circ Physiol. 2005;288:H971–H976. doi: 10.1152/ajpheart.00374.2004. [DOI] [PubMed] [Google Scholar]
- Heikkinen S, Suppola S, Malkki M, Deeb SS, Jänne J, Laakso M. Mouse hexokinase II gene: structure, cDNA, promotor analysis, and expression pattern. Mamm Genome. 2000;11:91–96. doi: 10.1007/s003350010019. [DOI] [PubMed] [Google Scholar]
- Heumann S, Falkenberg F, Pfleiderer G. Purification and immunological characterization of the human hexokinase isozymes I and III (ATP-D-hexose 6-phosphotransferase EC 2.7.1.1) Biochim Biophys Acta. 1974;334:328–342. [Google Scholar]
- Holmes BF, Kurth-Kraczek EJ, Winder WW. Chronic activation of 5′-AMP-activated protein kinase increases GLUT-4, hexokinase, and glycogen in muscle. J Appl Physiol. 1999;87:1990–1995. doi: 10.1152/jappl.1999.87.5.1990. [DOI] [PubMed] [Google Scholar]
- Jennings RB. Historical perspective on the pathology of myocardial ischemia/reperfusion injury. Circ Res. 2013;113:428–438. doi: 10.1161/CIRCRESAHA.113.300987. [DOI] [PubMed] [Google Scholar]
- Jiang S, Zhang LF, Zhang HW, Hu S, Lu MH, Liang S, et al. A novel miR-155/miR-143 cascade controls glycolysis by regulating hexokinase 2 in breast cancer cells. EMBO J. 2012;31:1985–1998. doi: 10.1038/emboj.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S, Weiss JN, Ribalet B. Subcellular localization of hexokinase I and II directs the metabolic fate of glucose. PLoS ONE. 2011;6:e17674. doi: 10.1371/journal.pone.0017674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MK. The intracellular distribution of glycolytic and other enzymes in rat brain homogenates and mitochondrial preparations. Biochem J. 1960;77:610–618. doi: 10.1042/bj0770610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzen HM, Soderman DD, Wiley CE. Multiple forms of hexokinase. Activities associated with subcellular particulate and soluble fractions of normal and streptozotocin diabetic rat tissues. J Biol Chem. 1970;245:4081–4096. [PubMed] [Google Scholar]
- Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2012;12:9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko YH, Verhoeven HA, Lee MJ, Corbin DJ, Vogl TJ, Pedersen PL. A translational study ‘case report’ on the small molecule ‘energy blocker’ 3-bromopyruvate (3BP) as a potent anticancer agent: from bench side to bedside. J Bioenerg Biomembr. 2012;44:163–170. doi: 10.1007/s10863-012-9417-4. [DOI] [PubMed] [Google Scholar]
- Kottenberg E, Thielmann M, Bergmann L, Heine T, Jakob H, Heusch G, et al. Protection by remote ischemic preconditioning during coronary artery bypass graft surgery with isoflurane but not propofol – a clinical trial. Acta Anaesthesiol Scand. 2012;56:30–38. doi: 10.1111/j.1399-6576.2011.02585.x. [DOI] [PubMed] [Google Scholar]
- Latchman DS. Heat shock proteins and cardiac protection. Cardiovasc Res. 2001;51:637–646. doi: 10.1016/s0008-6363(01)00354-6. [DOI] [PubMed] [Google Scholar]
- Leung AW, Halestrap A. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim Biophys Acta. 2008;1777:946–952. doi: 10.1016/j.bbabio.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Leung AW, Varanyuwatana P, Halestrap AP. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J Biol Chem. 2008;283:26312–26323. doi: 10.1074/jbc.M805235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Xuan W, Yan R, Tropak MB, Jean-St-Michel E, Liang W, et al. Remote preconditioning provides potent cardioprotection via PI3K/Akt activation and is associated with nuclear accumulation of β-catenin. Clin Sci. 2011;120:451–462. doi: 10.1042/CS20100466. [DOI] [PubMed] [Google Scholar]
- Lionetti V, Aquaro GD, Simioniuc A, Cristofano di C, Forini F, Cecchetti F, et al. Severe mechanical dyssynchrony causes regional hibernation-like changes in pigs with nonischemic heart failure. J Cardiac Fail. 2009;15:920–928. doi: 10.1016/j.cardfail.2009.06.436. [DOI] [PubMed] [Google Scholar]
- Lyon AR, Joudrey PJ, Jin D, Nass RD, Aon MA, O'Rourke B, et al. Optical imaging of mitochondrial function uncovers actively propagating waves of mitochondrial membrane potential collapse across intact heart. J Mol Cell Cardiol. 2010;49:565–575. doi: 10.1016/j.yjmcc.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFalls EO, Murad B, Liow JS, Gannon MC, Haspel HC, Lange A, et al. Glucose uptake and glycogen levels are increased in pig heart after repetitive ischemia. Am J Physiol Heart Circ Physiol. 2002;282:H205–H211. doi: 10.1152/ajpheart.2002.282.1.H205. [DOI] [PubMed] [Google Scholar]
- McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, et al. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res. 2004;95:830–840. doi: 10.1161/01.RES.0000145360.16770.9f. [DOI] [PubMed] [Google Scholar]
- Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, et al. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16:819–830. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Mandarino LJ, Printz RL, Cusi KA, Kinchington P, O'Doherty R, Osawa H, et al. Regulation of hexokinase II and glycogen synthase mRNA, protein, and activity in human muscle. Am J Physiol. 1995;269:E701–E708. doi: 10.1152/ajpendo.1995.269.4.E701. [DOI] [PubMed] [Google Scholar]
- Marzo I, Brenner C, Zamzami N, Susin SA, Beutner G, Brdiczka D, et al. The permeability transition pore complex: a target for apoptosis regulation by caspases and Bcl-2-related proteins. J Exp Med. 1998;187:1261–1271. doi: 10.1084/jem.187.8.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathupala SP, Ko YH, Pedersen PL. Hexokinase-2 bound to mitochondria: cancer's stygian link to the ‘Warburg effect’ and a pivotal target for effective therapy. Semin Cancer Biol. 2009;19:17–24. doi: 10.1016/j.semcancer.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matkovich SJ, Hu Y, Dorn GW., II Regulation of cardiac microRNAs by cardiac microRNAs. Circ Res. 2013;113:62–71. doi: 10.1161/CIRCRESAHA.113.300975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergenthaler P, Kahl A, Kamitz A, Laak van V, Stohlmann K, Thomsen S, et al. Mitochondrial hexokinase II (HKII) and phosphoprotein enriched in astrocytes (PEA15) form a molecular switch governing cellular fate depending on the metabolic state. Proc Natl Acad Sci U S A. 2012;109:1518–1523. doi: 10.1073/pnas.1108225109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelakis ED, Sutendra G, Dromparis P, Webster L, Harmony A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2:31ra34. doi: 10.1126/scitranslmed.3000677. [DOI] [PubMed] [Google Scholar]
- Miki T, Itoh T, Sunaga D, Miura T. Effects of diabetes on myocardial infarct size and cardioprotection by preconditioning and postconditioning. Cardiovasc Diabetol. 2012;11:67. doi: 10.1186/1475-2840-11-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S, Murphy AN, Brown JH. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008;15:521–529. doi: 10.1038/sj.cdd.4402285. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Purcell NH, Smith JM, Gao T, Whittaker R, Huang K, et al. PHLPP-1 negatively regulates Akt activity and survival in the heart. Circ Res. 2010;107:476–484. doi: 10.1161/CIRCRESAHA.109.215020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–590. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- Nakashima RA, Mangan PS, Colombini M, Pedersen PL. Hexokinase receptor complex in hepatoma mitochondria: evidence from N,N'-dicyclohexylcarbodiimide-labeling studies for the involvement of the pore-forming protein VDAC. Biochemistry. 1986;25:1015–1021. doi: 10.1021/bi00353a010. [DOI] [PubMed] [Google Scholar]
- Nederlof R, Xie C, Eerbeek O, Koeman A, Milstein DMJ, Hollmann MW, et al. Pathophysiological consequences of TAT-HKII peptide administration are independent of impaired vascular function and ensuing ischemia. Circ Res. 2013;112:e8–e13. doi: 10.1161/CIRCRESAHA.112.274308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng KW, Allen ML, Desai A, Macrae D, Pathan N. Cardioprotective effects of insulin: how intensive insulin therapy may benefit cardiac surgery patients. Circulation. 2012;125:721–728. doi: 10.1161/CIRCULATIONAHA.111.063784. [DOI] [PubMed] [Google Scholar]
- Ouyang J, Parakhia RA, Ochs RS. Metformin activates AMP kinase through inhibition of AMP deaminase. J Biol Chem. 2011;286:1–11. doi: 10.1074/jbc.M110.121806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348:607–614. [PMC free article] [PubMed] [Google Scholar]
- Pasdois P, Parker JC, Griffiths EJ, Halestrap AP. The role of oxidized cytochrome c in regulating mitochondrial reactive oxygen species production and its perturbation in ischaemia. Biochem J. 2011;436:493–505. doi: 10.1042/BJ20101957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasdois P, Parker JE, Halestrap AP. Extent of mitochondrial hexokinase II dissociation during ischemia correlates with mitochondrial cytochrome c release, reactive oxygen species production, and infarct size on reperfusion. J Am Heart Assoc. 2013;2:e005645. doi: 10.1161/JAHA.112.005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits bax-induced cytochrome c release and apoptosis. J Biol Chem. 2002;277:7610–7618. doi: 10.1074/jbc.M109950200. [DOI] [PubMed] [Google Scholar]
- Pastorino JG, Hoek JB, Shulga N. Activation of glycogen synthase kinase 3{beta} disrupts the binding of hexokinase iI to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005;65:10545–10554. doi: 10.1158/0008-5472.CAN-05-1925. [DOI] [PubMed] [Google Scholar]
- Pedersen PL. Warburg, me and hexokinase 2: multiple discoveries of key molecular events underlying one of cancers' most common phenotypes, the ‘Warburg effect’, i.e. elevated glycolysis in the presence of oxygen. J Bioenerg Biomembr. 2007;39:211–222. doi: 10.1007/s10863-007-9094-x. [DOI] [PubMed] [Google Scholar]
- Penso J, Beitner R. Clotrimazole and bifonazole detach hexokinase from mitochondria of melanoma cells. Eur J Pharmacol. 1998;342:113–117. doi: 10.1016/s0014-2999(97)01507-0. [DOI] [PubMed] [Google Scholar]
- Perevoshchikova IV, Zorov SD, Kotova EA, Zorov DB, Antonenko YN. Hexokinase inhibits flux of fluorescently labeled ATP through mitochondrial outer membrane porin. FEBS Lett. 2010;584:2397–2402. doi: 10.1016/j.febslet.2010.04.033. [DOI] [PubMed] [Google Scholar]
- Peschiaroli A, Giacobbe A, Formosa A, Markert EK, Bongiorno-Borbone L, Levine AJ, et al. miR-143 regulates hexokinase 2 expression in cancer cells. Oncogene. 2012;32:797–802. doi: 10.1038/onc.2012.100. [DOI] [PubMed] [Google Scholar]
- Postic C, Shiota M, Magnuson MA. Cell-specific roles of glucokinase in glucose metabolism. Recent Prog Horm Res. 2001;56:195–217. doi: 10.1210/rp.56.1.195. [DOI] [PubMed] [Google Scholar]
- Rabuazzo AM, Patane G, Anello M, Piro S, Vigneri R, Purrello F. Hexokinase shift to mitochondria is associated with an increased sensitivity to glucose in rat pancreatic islets. Diabetes. 1997;46:1148–1152. doi: 10.2337/diab.46.7.1148. [DOI] [PubMed] [Google Scholar]
- Riehle C, Wende AR, Zaha VG, Pires KM, Wayment B, Olsen C, et al. PCG-1β deficiency accelerates the transition to heart failure in pressure overload hypertrophy. Circ Res. 2011;109:783–793. doi: 10.1161/CIRCRESAHA.111.243964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roosimaa M, Põdramägi T, Kadaja L, Ruusalepp A, Paju K, Puhke R, et al. Dilation of human atria: increase diffusion restrictions for ADP, overexpression of hexokinase 2 and its coupling to oxidative phosphorylation in cardiomyocytes. Mitochondrion. 2013;13:399–409. doi: 10.1016/j.mito.2012.12.005. [DOI] [PubMed] [Google Scholar]
- Rostovtseva TK, Bezrukov SM. VDAC regulation: role of cytosolic proteins and mitochondrial lipids. J Bioenerg Biomembr. 2008;40:163–170. doi: 10.1007/s10863-008-9145-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostovtseva TK, Tan W, Colombini M. On the role of VDAC in apoptosis: fact and fiction. J Bioenerg Biomembr. 2005;37:129–142. doi: 10.1007/s10863-005-6566-8. [DOI] [PubMed] [Google Scholar]
- Rostovtseva TK, Sheldon KL, Hassanzadeh E, Monge C, Saks V, Bezrukov SM, et al. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc Natl Acad Sci U S A. 2008;105:18746–18751. doi: 10.1073/pnas.0806303105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RR, 3rd, Mrus JM, Mommessin JI, Taegtmeyer H. Compartmentation of hexokinase in rat heart. A critical factor for tracer kinetic analysis of myocardial glucose metabolism. J Clin Invest. 1992;90:1972–1977. doi: 10.1172/JCI116076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago AP, Chaves EA, Oliveira MF, Galina A. Reactive oxygen species generation is modulated by mitochondrial kinases: correlation with mitochondrial antioxidant peroxidises in rat tissues. Biochimie. 2008;90:1566–1577. doi: 10.1016/j.biochi.2008.06.013. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- Sen S, Kundu BK, Wu HC, Hashmi SS, Guthrie P, Locke LW, et al. Glucose regulation of load-induced mTOR signalling and ER stress in mammalian heart. J Am Heart Assoc. 2013;2:e004796. doi: 10.1161/JAHA.113.004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, et al. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004;18:1692–1700. doi: 10.1096/fj.04-2263com. [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- Sheldon KL, Zuurbier CJ, Bezrukov SM, Rostovtseva TK. Novel mechanism of mitochondrial respiration control through competition between hexokinase-2 and tubulin for VDAC binding. Biophys J. 2013;104(Suppl. 1):p655a. [Google Scholar]
- Shioi T, McMullen JR, Tarnavski O, Concerso K, Sherwood MC, Manning WJ, et al. Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation. 2003;107:1664–1670. doi: 10.1161/01.CIR.0000057979.36322.88. [DOI] [PubMed] [Google Scholar]
- Smeele KM, Eerbeek O, Koeman A, Bezemer R, Ince C, Heikkinen S, et al. Partial hexokinase II knockout results in acute ischemia-reperfusion damage in skeletal muscle of male, but not female, mice. Pflüg Arch. 2010;459:705–712. doi: 10.1007/s00424-010-0787-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeele KM, Horst ter LH, Koeman A, Heikkinen S, Laakso M, Weber NC, et al. The effect of standard chow and reduced hexokinase II on growth, cardiac and skeletal muscle hexokinase and low-flow cardiac ischaemia-reperfusion injury. Lab Anim. 2011b;45:160–166. doi: 10.1258/la.2011.010096. [DOI] [PubMed] [Google Scholar]
- Smeele KM, Eerbeek O, Schaart G, Koeman A, Bezemer R, Nelson JK, et al. Reduced hexokinase II impairs muscle function 2 wk after ischemia-reperfusion through increased cell necrosis and fibrosis. J Appl Physiol. 2012;113:608–618. doi: 10.1152/japplphysiol.01494.2011. [DOI] [PubMed] [Google Scholar]
- Smeele KMA, Southworth R, Wu R, Xie C, Nederlof R, Warley A, et al. Disruption of hexokinase II-mitochondrial binding blocks ischemic preconditioning and causes rapid cardiac necrosis. Circ Res. 2011a;108:1165–1169. doi: 10.1161/CIRCRESAHA.111.244962. [DOI] [PubMed] [Google Scholar]
- Solskov L, Lofgren B, Kristiansen SB, Jessen N, Pold R, Nielsen TT, et al. Metformin induces cardioprotection against ischaemia/reperfusion injury in the rat heart 24 hours after administration. Basic Clin Pharmacol Toxicol. 2008;103:82–87. doi: 10.1111/j.1742-7843.2008.00234.x. [DOI] [PubMed] [Google Scholar]
- Southworth R, Davey KA, Warley A, Garlick PB. A reevaluation of the roles of hexokinase I and II in the heart. Am J Physiol Heart Circ Physiol. 2007;292:H378–H386. doi: 10.1152/ajpheart.00664.2006. [DOI] [PubMed] [Google Scholar]
- Steenbergen C, Murphy E, Watts JA, London RE. Correlation between cytosolic free calcium, contracture, ATP, and irreversible ischemic injury in perfused rat heart. Circ Res. 1990;66:135–146. doi: 10.1161/01.res.66.1.135. [DOI] [PubMed] [Google Scholar]
- Suwa M, Egashira T, Nakano H, Sasaki H, Kumagai S. Metformin increases the PGC-1alpha protein and oxidative enzyme activities possibly via AMPK phosphorylation in skeletal muscle in vivo. J Appl Physiol. 2006;101:1685–1692. doi: 10.1152/japplphysiol.00255.2006. [DOI] [PubMed] [Google Scholar]
- Terashima Y, Sato T, Yano T, Maas O, Itoh T, Miki T, et al. Role of phospho-GSK-3β in myocardial protection afforded by activation of the mitochondrial K ATP channel. J Mol Cell Cardiol. 2010;49:762–770. doi: 10.1016/j.yjmcc.2010.08.001. [DOI] [PubMed] [Google Scholar]
- Thum T. MicroRNA therapeutics in cardiovascular medicine. EMBO Mol Med. 2011;4:3–14. doi: 10.1002/emmm.201100191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Beek JHGM, Tian X, Zuurbier CJ, Groot de B, Van Echteld CJA, Eijgelshoven MHJ, et al. The dynamic regulation of myocardial oxidative phosphorylation: analysis of the response time of oxygen consumption. Mol Cell Biochem. 1998;184:321–344. [PubMed] [Google Scholar]
- Vestergaard H, Bjørbaek C, Hansen T, Larsen FS, Granner DK, Pedersen O. Impaired activity and gene expression of hexokinase II in muscle from non-insulin-dependent diabetes mellitus patients. J Clin Invest. 1995;96:2639–2645. doi: 10.1172/JCI118329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vytla VS, Ochs RS. Metformin increases mitochondrial energy formation in L6 muscle cell cultures. J Biol Chem. 2013;288:20369–20377. doi: 10.1074/jbc.M113.482646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O, Posener K, Negelein E. Ueber den Stoffwechsel der Tumoren. Biochem Zeitschrift. 1924;152:319–344. [Google Scholar]
- Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Zhou Y, Dai Q, Qiao C, Zhao L, Lu N, et al. Oroxylin A induces dissociation of hexokinase II from the mitochondria and inhibits glycolysis by SIRT3-mediated deacetylation of cyclophilin D in breast carcinoma. Cell Death Dis. 2013;4:e601. doi: 10.1038/cddis.2013.131. doi: 10.1038/cddis.2013.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington HJ, Hall AR, McLaughlin CP, Hausenloy DJ, Yellon DM, Mocanu MM. Chronic metformin associated cardioprotection against infarction: not just a glucose lowering phenomenon. Cardiovasc Drugs Ther. 2013;27:5–16. doi: 10.1007/s10557-012-6425-x. [DOI] [PubMed] [Google Scholar]
- Wilson JE. Hexokinases. Rev Physiol Biochem Pharmacol. 1995;126:65–198. doi: 10.1007/BFb0049776. [DOI] [PubMed] [Google Scholar]
- Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003;206:2049–2057. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
- Wu R, Smeele KM, Wyatt E, Ichikawa Y, Eerbeek O, Sun L, et al. Reduction in hexokinase II levels results in decreased cardiac function and altered remodelling after ischemia-reperfusion injury. Circ Res. 2011;108:60–69. doi: 10.1161/CIRCRESAHA.110.223115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R, Wyatt E, Chawla K, Tran M, Ghanefar M, Laakso M, et al. Hexokinase II knockdown results in exaggerated cardiac hypertrophy via increased ROS production. EMBO Mol Med. 2012;4:633–646. doi: 10.1002/emmm.201200240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang F, Huang YS, Shi XH, Zhang Q. Mitochondrial chaperone tumour necrosis factor receptor-associated protein 1 protects cardiomyocytes from hypoxic injury by regulating mitochondrial permeability transition pore opening. FEBS J. 2010;277:1929–1938. doi: 10.1111/j.1742-4658.2010.07615.x. [DOI] [PubMed] [Google Scholar]
- Yeih DF, Yeh HI, Lin LY, Tsay YG, Chiang FT, Tseng CD, et al. Enhanced activity and subcellular redistribution of myocardial hexokinase after acute myocardial infarction. Int J Cardiol. 2011;149:74–79. doi: 10.1016/j.ijcard.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Zuurbier CJ, Beek van JHGM. Mitochondrial response to heart rate steps in isolated rabbit heart is slowed after myocardial stunning. Circ Res. 1997;81:69–75. doi: 10.1161/01.res.81.1.69. [DOI] [PubMed] [Google Scholar]
- Zuurbier CJ, Ince C. Post-ischaemic changes in the response time of oxygen consumption to demand in the isolated rat heart are mediated partly by calcium and glycolysis. Pflügers Arch. 2002;443:908–916. doi: 10.1007/s00424-001-0744-2. [DOI] [PubMed] [Google Scholar]
- Zuurbier CJ, Eerbeek O, Goedhart PT, Struys EA, Verhoeven NM, Jakobs C, et al. Inhibition of the pentose phosphate pathway decreases ischemia-reperfusion-induced creatine kinase release in the heart. Cardiovasc Res. 2004;62:145–153. doi: 10.1016/j.cardiores.2004.01.010. [DOI] [PubMed] [Google Scholar]
- Zuurbier CJ, Eerbeek O, Meijer AJ. Ischemic preconditioning, insulin, and morphine all cause hexokinase redistribution. Am J Physiol Heart Circ Physiol. 2005;289:H496–H499. doi: 10.1152/ajpheart.01182.2004. [DOI] [PubMed] [Google Scholar]
- Zuurbier CJ, Hoek FJ, Dijk van J, Abeling NG, Meijers JC, Levels JH, et al. Perioperative hyperinsulinaemic normoglycaemic clamp causes hypolipidaemia after coronary artery surgery. Br J Anaesth. 2008a;100:442–450. doi: 10.1093/bja/aen018. [DOI] [PubMed] [Google Scholar]
- Zuurbier CJ, Keijzers PJM, Koeman A, Wezel van HB, Hollmann MW. Anesthesia's effects on plasma glucose and insulin and cardiac hexokinase at similar hemodynamics and without major surgical stress in fed rats. Anesth Analg. 2008b;106:135–142. doi: 10.1213/01.ane.0000297299.91527.74. [DOI] [PubMed] [Google Scholar]
- Zuurbier CJ, Smeele KMA, Eerbeek O. Mitochondrial hexokinase and cardioprotection of the intact heart. J Bioenerg Biomembr. 2009;41:181–185. doi: 10.1007/s10863-009-9209-7. [DOI] [PubMed] [Google Scholar]