

Abstract
Background and Purpose
Cardiac ischaemia–reperfusion (IR) injury remains a significant clinical problem with limited treatment options available. We previously showed that cardioprotection against IR injury by nitro-fatty acids, such as nitro-linoleate (LNO2), involves covalent modification of mitochondrial adenine nucleotide translocase 1 (ANT1). Thus, it was hypothesized that conjugation of LNO2 to the mitochondriotropic triphenylphosphonium (TPP+) moiety would enhance its protective properties.
Experimental Approach
TPP+-LNO2 was synthesized from aminopropyl-TPP+ and LNO2, and characterized by direct infusion MS/MS. Its effects were assayed in primary cultures of cardiomyocytes from adult C57BL/6 mice and in mitochondria from these cells, exposed to simulated IR (SIR) conditions (oxygen and metabolite deprivation for 1h followed by normal conditions for 1h) by measuring viability by LDH release and exclusion of Trypan blue. Nitro-alkylated mitochondrial proteins were also measured by Western blots, using antibodies to TPP+.
Key Results
TPP+-LNO2 protected cardiomyocytes from SIR injury more potently than the parent compound LNO2. In addition, TPP+-LNO2 modified mitochondrial proteins, including ANT1, in a manner sensitive to the mitochondrial uncoupler carbonylcyanide-p-trifluoromethoxyphenylhydrazone (FCCP) and the ANT1 inhibitor carboxyatractyloside. Similar protein nitro-alkylation was obtained in cells and in isolated mitochondria, indicating the cell membrane was not a significant barrier to TPP+-LNO2.
Conclusions and Implications
Together, these results emphasize the importance of ANT1 as a target for the protective effects of LNO2, and suggest that TPP+-conjugated electrophilic lipid compounds may yield novel tools for the investigation of cardioprotection.
Linked Articles
This article is part of a themed issue on Mitochondrial Pharmacology: Energy, Injury & Beyond. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2014.171.issue-8
Keywords: TPP+-LNO2, cardiomyocyte, ischaemia, mitochondria, nitroalkene
Introduction
Cardiac mitochondria sit at a critical axis in determining the fate of myocardium during ischaemia–reperfusion (IR) injury (Murphy and Steenbergen, 2008). Some of the key events that determine cardiomyocyte fate during IR and are governed by mitochondria include dys-regulation of Ca2+ homeostasis (Shintani-Ishida et al., 2012), generation of reactive oxygen species (Zorov et al., 2006), disruption of membrane integrity (Sparagna and Lesnefsky, 2009) and bioenergetic crisis (Grossman et al., 2013). Numerous studies have demonstrated that pharmacological interventions that preserve mitochondrial function can be beneficial in cardiac IR injury (see Walters et al., 2012), although few such therapies have achieved clinical usage. As such, there is a need for new small molecule therapeutic agents to limit cardiac IR injury.
The nitroalkenes are a novel series of NO-derived anti-inflammatory lipid-signalling molecules of potential therapeutic benefit in the setting of cardiac IR. These molecules, for example nitro-linoleate (LNO2), are electrophilic nitration products of polyunsaturated fatty acids (Lim et al., 2002), and as such can covalently modify nucleophilic cysteine and lysine residues in proteins (nitro-alkylation) (Koenitzer and Freeman, 2010). In support of a cardioprotective role for nitroalkenes, we recently reported that LNO2 can be generated inside cardiac mitochondria during the innate cardioprotective paradigm of ischaemic preconditioning. In addition, acute administration of synthetic LNO2 elicited protection against IR injury in an ex vivo perfused heart model (Nadtochiy et al., 2009). Furthermore, we identified adenine nucleotide translocase 1 (ANT) as a potential molecular target of LNO2, mediating its cardioprotective effects via activation of mild mitochondrial uncoupling (Nadtochiy et al., 2009; 2012).
A possible limiting factor in the therapeutic utility of LNO2 (and other electrophilic lipids of interest) is its interaction with non-mitochondrial proteins (Batthyany et al., 2006). As such, limiting these side reactions by targeting LNO2 to mitochondria might enhance its ability to engage specific mitochondrial targets such as ANT1. A popular approach to enhance drug accumulation in mitochondria is conjugation to the triphenylphosphonium cation (TPP+) (Murphy, 2008). Several studies have demonstrated that mitochondrially targeted drugs can confer pharmacological effects at lower concentrations than their parent non-targeted compounds (Prime et al., 2009; Diers et al., 2010). Furthermore, numerous mitochondrially targeted antioxidants and other drugs have been shown to elicit cardioprotection against IR injury, at in vivo doses of as low as 100 ng·kg−1, including mitoQ (Adlam et al., 2005), mitoTEMPO (Luo et al., 2013), mitoSkQ1 (Lakomkin and Kapel'ko, 2009) and mitoSNO (Chouchani et al., 2010; 2013).
In this study, we hypothesized that mitochondrial targeting of LNO2 by conjugation to TPP+ would enhance its protective efficacy. Furthermore, because mitochondrial targeting of LNO2 would be expected to decrease its reaction with potential cytosolic targets, protection by TPP+-LNO2 would provide important supporting evidence for the concept that the bona fide protective target of LNO2 is indeed mitochondrial. Finally, the TPP+ moiety provides a convenient antigen for detection of LNO2 covalently linked to mitochondrial proteins, by Western blot.
Methods
Animals
All animal care and experimental procedures complied with the recommendations of the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Rochester Committee on Animal Resources. Male 2-month-old mice on the C57BL/6 background were maintained in a pathogen-free vivarium, under a 12 h light–dark cycle and food and water available ad libitum. A total of 31 mice were used in the experiments described here.
Synthesis, purification and quantitation of TPP+-LNO2
LNO2 was synthesized by nitroselynation (Lim et al., 2002) and purified by TLC (Cui et al., 2006), as previously described (Nadtochiy et al., 2009). Amino-propyl-TPP+ (NH2-TPP+) was synthesized according to Wagner et al., (1991). LNO2 and NH2-TPP+ (Supporting Information Figure S1) were coupled by combining in a 1:1:1 molar ratio with 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (Thermo Scientific Inc., Waltham, MA, USA) in acetonitrile/ethanol (8:2, v/v). The mixture was gassed under argon and stirred at room temperature in the dark for 18 h. Products were separated by TLC, and TPP+-LNO2 (Rf = 0.56) was then recovered and subjected to MS/MS analysis. The single-charged species with m/z = 627 was analysed by a Thermo Finnigan LCQ Deca Max XP (Thermo Fisher Scientific Inc.) using collision-induced dissociation (normalized collision energy 40 V, fragmentation scan range m/z = 170–800, positive ion mode). Figure 1 shows MS/MS fragmentation of TPP+-LNO2.
Figure 1.
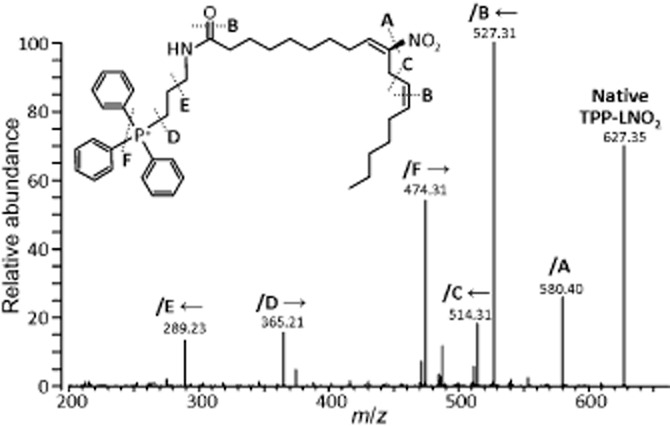
Fragmentation of TPP+-LNO2 by MS/MS. TPP+-LNO2 was analysed by collision-induced dissociation as described in the Methods section. Cleavage at sites A–F results in ions to the left or right of the cleavage site (indicated by arrows), as labelled on the spectrum.
TPP+-LNO2 was quantified using tri-iodide chemiluminescence analysis (Sievers NOA-280) (Samouilov and Zweier, 1998; Burwell et al., 2006). TPP+-LNO2 and synthetic LNO2 were decomposed in phosphate buffer in sealed clear vials at 37°C for 1 h. Aliquots were injected into the purge vessel containing a mixture of acidified potassium iodide (1% w/v) and iodine (0.65% w/v), causing reduction of NO2− to NO•. Inside the NOA, NO• reacts with ozone, emitting light proportional to [NO•] generated in the purge vessel. Concentrations were calibrated to a standard curve constructed using NaNO2. Aliquots of TPP+-LNO2 were stored under argon in amber vials at −80°C.
Seahorse XF-24 measurements in adult mouse cardiomyocytes
Cardiomyocytes were isolated from adult mice as follows. Mice were anesthetized using freshly prepared tribromoethanol (100 μl of a 2.5 mg mL−1 solution in sterile saline, given i.p.) and the heart rapidly excised and perfused for 3 min. (flow rate 3 mL min−1) with perfusion buffer (PB) comprising (in mM): NaCl (120), KCl (15), Na2HPO4 (0.6), KH2PO4 (0.6), MgSO4 (1.2), HEPES (10), NaHCO3 (4.6), taurine (30), butadione monoxime (10), glucose (5.5), pH 7.4. PB was then replaced by 10 min. perfusion with digestion buffer (DB): i.e., PB plus 2.5% (w/v) trypsin + 6.525U collagenase A + 15.375U collagenase D (Roche, Indianapolis, IN). Ventricular tissue was then chopped and filtered through 200 μm mesh. Cells were settled by gravity for 10 min. then resuspended in 2.5 ml of stop buffer (SB): i.e., PB plus 10% (v/v) FBS, 12.5 μM CaCl2. A series of settling and wash steps were then used to raise [Ca2+] to 1.8 mM and the final pellet was resuspended in MEM with penicillin/streptomycin and 2.5% (v/v) heat inactivated FBS. The protocol yielded ∼4.5x105 cells per heart, ∼85% rod-shaped and excluding Trypan blue (Figure S2).
Seahorse XF V7-PET plates (Seahorse Bioscience, Billerica, MA, USA) were coated with laminin (No. 354232, BD Bioscience, San Jose, CA, USA), 1.5 μg per well in PBS, incubated at 37°C for 1 h (Readnower et al., 2012). Excess laminin was aspirated and cardiomyocytes plated at 10 000 cells per well by centrifugation (44× g, 2 min) and then incubated at 37°C in a 5% CO2 atmosphere for 1 h.
Cell media were replaced with a bicarbonate-free DMEM containing (in mM) L-glutamine (4.0), pyruvate (0.1), glucose (5.0) and palmitate (0.1, conjugated to fat-free BSA), pH 7.4 at 37°C. The 20 measurement wells on each plate were divided into five groups: (i) simulated IR (SIR) alone; (ii) linoleic acid (LA) + SIR; (iii) LNO2 + SIR; (iv) TPP+-LNO2 + SIR; and (v) bromopropyl-TPP+ (TPP+) + SIR. All agents were administered 60 min before the onset of SIR. The Seahorse XF-24 analyser was adapted for infusion of gases in the cartridge headspace to provide an in vitro model of IR injury as previously described (Guo et al., 2012). Following SIR, LDH release into the supernatant was measured using a kit (Roche, Indianapolis, IN, USA) according to the manufacturer's protocol, and expressed as a percentage of the total LDH content in both the supernatant and the Triton X-100 (Sigma, St. Louis, MO, USA) lysed cell pellet. Cell death was confirmed using Trypan blue (see below).
Cell viability assay with Trypan blue
At the end of SIR, 20 μl of a 0.4% (w/v) solution of Trypan blue dye was added to each well of the XF24 plate (1 mL. total media volume), and cardiomyocytes were incubated for 15 min. at 37°C. Dead cardiomyocytes were stained blue. Images were collected by digital photography using a Canon Powershot SD990-IS and analyzed by ImageJ 1.47t software (NIH). Representative images are shown in Figure S3.
Mitochondrial incubations
Cardiac mitochondria were isolated as follows. Hearts were removed and immersed in 2 mL ice-cold isolation medium (IM) comprising (in mM) sucrose (300), Tris-HCl (20), EGTA (2), pH 7.35 @ 4°C. Tissue was chopped, washed and homogenized in 2 mL of IM using a ‘Tissumizer’ device (IKA Instruments). The homogenate was centrifuged at 1000 x g for 5 min., the pellet discarded, and the supernatant centrifuged at 7,000 x g for 10 min. The pellet was resuspended in 1.5 mL IM and centrifuged at 10,000 x g for 5 min. The top layer of the resulting pellet (broken mitochondria and microsomes) was discarded, and the remaining pellet resuspended in 50μL IM. Protein was determined by the Folin-Phenol method (Lowry et al. 1951) against a standard curve constructed using bovine serum albumin. A single mouse heart typically yielded 3-4 mg mitochondrial protein. Mitochondria were suspended at a concentration of 0.5 mg protein·mL−1 in buffer comprising (in mM) KCl (120), KH2PO4 (3), Tris (50), succinate (5), glutamate (5) and malate (2.5), pH 7.35 at 37°C. Optionally, the following reagents were added: TPP+-LNO2 (1 μM), carbonylcyanide-p-trifluoromethoxyphenylhydrazone (FCCP; 1 μM), TPP+ (1 μM), carboxyatractyloside (CATr, 5 μM; Calbiochem EMD Millipore, Billerica, MA, USA). In some experiments, TPP+-LNO2 was pre-incubated with NaOH (5 mM) to destroy its electrophilic moiety via the Nef reaction (Schopfer et al., 2005), and pH was corrected before the product was added to mitochondria. All reactions were incubated for 20 min, followed by centrifugation at 14 000× g to pellet mitochondria.
Mitochondrial membrane potential (ΔΨm) and mitochondrial permeability transition (PT) pore opening
Methods for measuring these variables are presented in the Supporting Information.
For ΔΨm measurements, cells (5x104 mL−1) were incubated with 20 nM TMRE (tetramethylrhodamine ethyl ester) in non-quench mode, for 20 min. in bicarbonate free DMEM containing (in mM): L-glutamine (4), pyruvate (0.1), glucose (5), and palmitate (0.1, conjugated to fat-free BSA), pH 7.4, 37°C. TMRE fluorescence was measured (555 nm λEX, 577 nm λEM) in a Cary Eclipse spectrofluorimeter (Varian, Australia). The mitochondrial uncoupler carbonylcyanide-p-trifluoromethoxyphenylhydrazone (FCCP, 5 μM) was added to dissipate ΔΨm, and the ΔTMRE fluorescence recorded. Two groups were established: (i) Ctrl (no treatments) and (ii) TPP+ (25nM, added before TMRE).
Mitochondrial swelling
Mitochondrial permeability transition (PT) pore opening was measured as the decrease in light scattering (absorbance at 520 nm) caused by swelling, in a Beckman DU800 spectrophotometer. Mitochondria were suspended at 0.5mg mL−1 in buffer comprising KCl (120 mM), KH2PO4 (3 mM), Tris (50 mM), succinate (5 mM), rotenone (5 μM), pH 7.35 at 37°C. The following groups were established: (i) Ctrl (no treatment); (ii) Ca2+ (100 μM); (iii) Ca2+ + CsA (2 μM, added prior to Ca2+); (iv) TPP+-LNO2 (25 nM) and (v) TPP+ (25 nM). Tested agents were added as indicated by the arrow in Figure S6.
Immunoprecipitation and Western blotting
After treatments, mitochondria or cell pellets were incubated for 5 min at 37°C in a high-SDS sample loading buffer. In some experiments, ANT1 was immunoprecipitated from cell lysate (10 000 cells in RIPA buffer) using protein G agarose (EMD Millipore Corporation, Billerica, MA, USA) and monoclonal anti-ANT1 antibodies (Abcam-MitoScience, Cambridge, MA, USA) (Nadtochiy et al., 2012). Samples were resolved on SDS-PAGE gels, transferred to nitrocellulose and probed with antibodies against TPP+ (a kind gift of Mike Murphy, Cambridge, UK) or ANT1. HRP-linked secondary antibodies (GE Biosciences, Pittsburgh, PA, USA) were used with ECL detection (GE Biosciences, Pittsburgh, PA, USA). Blots were quantified by densitometry using open-source Scion Image software (National Institutes of Health, Bethesda, MD, USA).
Statistics
Results are presented as means ± SEM. Significance between two groups was determined using Student's t-test. For comparisons between multiple groups, normality of data was calculated using Prism (v6.02, GraphPad, La Jolla, CA, USA). Normally distributed data were analysed by anova with Tukey's multiple comparison tests.
Materials
All chemicals were the highest grade obtainable from Sigma, unless specified otherwise.
Results
Previously, we have shown that LNO2 can protect cardiac-derived H9c2 cells from SIR injury in a manner sensitive to ANT1 knock-down (Nadtochiy et al., 2012). Thus, we sought to test the cytoprotective efficacy of TPP+-LNO2 in a similar SIR injury model using primary adult mouse cardiomyocytes. The model, based on the Seahorse XF-24 plate reader platform, was essentially as previously described (Guo et al., 2012), wherein the instrument was adapted for internal gas flow, permitting control of pO2 and real-time measurement of pO2 and pH in the media throughout the experiment. Figure 2 shows averaged pO2 and pH readings for a typical SIR injury experiment.
Figure 2.
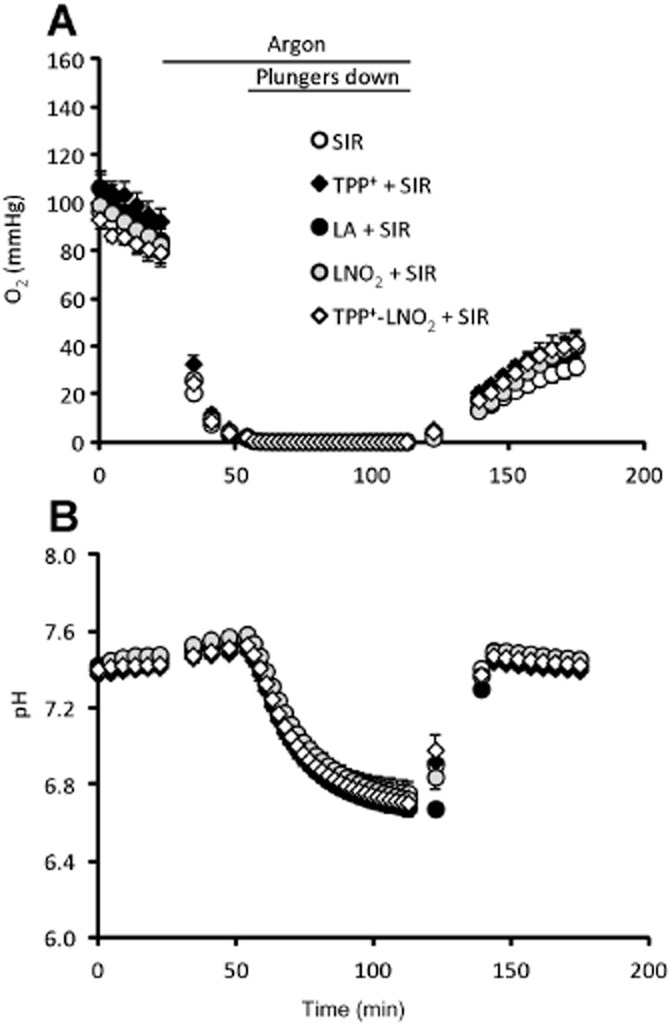
Representative pO2 and pH traces during simulated IR. (SIR). Values of pO2 (A) and of pH (B) were measured by fluorescent probes mounted on each plunger of the XF-24 cartridge. Times of argon flow and lowered plungers are noted above the graph in panel A. pO2 and pH values were averaged from n = 8 runs, and data are shown as means ± SEM.
A concentration-response study (Figure 3A) revealed the optimal protective effect (lower cell death after IR injury) of TPP+-LNO2 occurred at 25 nM. In contrast, the most protective dose of LNO2 was 100 nM, that is, four times higher than that of TPP+-LNO2. A direct comparison of 25 nM LNO2 versus TPP+-LNO2 showed that only the latter exhibited significant protection (Figure 3B), while at this same dose LA and TPP+ were without effect. The morphological appearance of cells following IR injury (i.e. live cells rod shaped, dead cells rounded) was consistent with LDH release data (Figure 3B, micrographs). Furthermore, assessment of cell death by Trypan blue exclusion assay yielded similar results (Supporting Information Figure S3).
Figure 3.
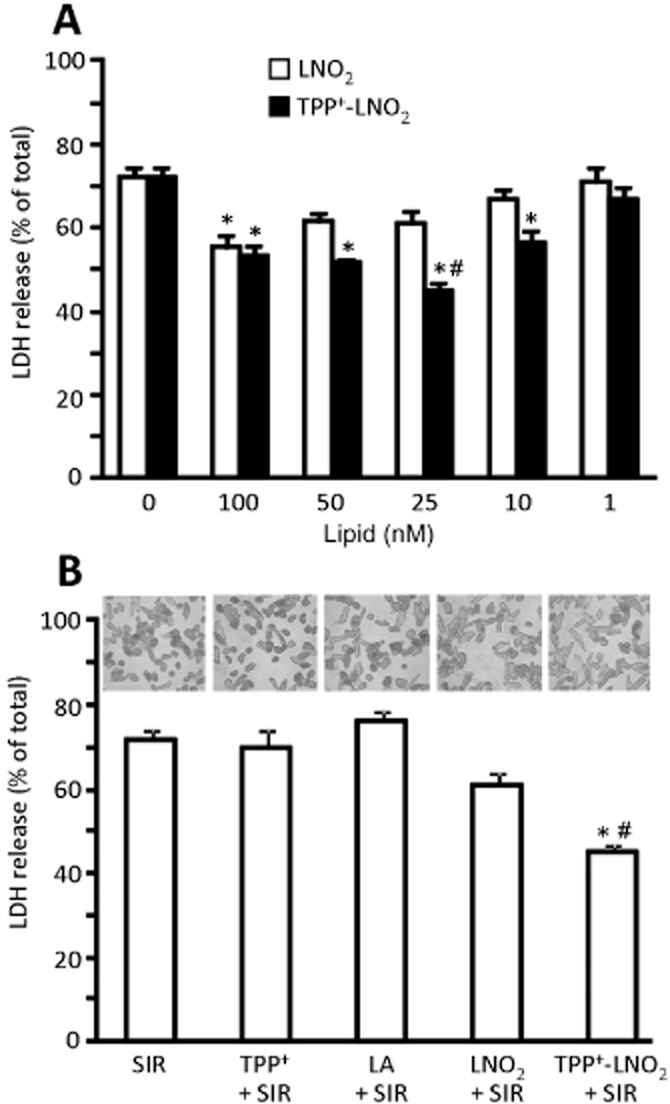
TPP+-LNO2 protects cardiomyocytes during SIR injury. Myocytes were isolated, plated on V7-PET plates and subjected to SIR. LDH release was measured in supernatants and expressed as percentage of total LDH (obtained after lysing cells with Triton X-100). (A) Dose response for LNO2 and TPP+-LNO2. Nitroalkene concentrations are shown below the graph. Data are means ± SEM, *P < 0.01 versus control (no added lipid), #P < 0.01 versus LNO2. (B) Comparison of cytoprotection from SIR injury by LA, TPP+, LNO2 and TPP+-LNO2, each at 25 nM, using LDH assay. Above the graph are representative photomicrographs, showing typical changes in cardiomyocyte morphology after SIR. Data are means ± SEM, *P < 0.01 versus control (SIR alone), #P < 0.01 versus LNO2.
The TPP+ moiety, in addition to being mitochondriotropic, provides a convenient antigen for detection by immunological techniques. Such technology has been used previously to detect thiol modifications with the TPP+-tagged alkylator iodobutyl-TPP+ (Lin et al., 2002; Tompkins et al., 2006). Here, we have used anti-TPP+ antibodies to detect covalent modification (i.e. nitro-alkenylation) of mitochondrial proteins by TPP+-LNO2.
Figure 4 demonstrates that upon incubation of mitochondria with TPP+-LNO2, a number of mitochondrial proteins were labelled (lane 2). These modifications were due to covalent interactions as decomposition of the electrophilic moiety of LNO2 via the Nef reaction (Schopfer et al., 2005) significantly reduced protein nitro-alkylation (lane 3). Furthermore, TPP+-LNO2 labelled mitochondrial proteins in an FCCP-sensitive manner (lane 4), indicating that mitochondrial membrane potential (ΔΨm) was the driving force for TPP+-LNO2 accumulation. Importantly, TPP+ (i.e. the TPP+ moiety alone) did not label proteins (lane 5), indicating that the protein modification was due to the LNO2 part of the molecule and not simply hydrophobic interaction. Densitometry and statistics are shown below the blots. Furthermore, we observed no effect of TPP+ alone on cellular oxygen consumption measured by the Seahorse method, or on mitochondrial membrane potential measured by TMRE fluorescence in cells (Supporting Information Figure S4), suggesting the TPP+ moiety did not affect mitochondrial function at the concentrations used here.
Figure 4.
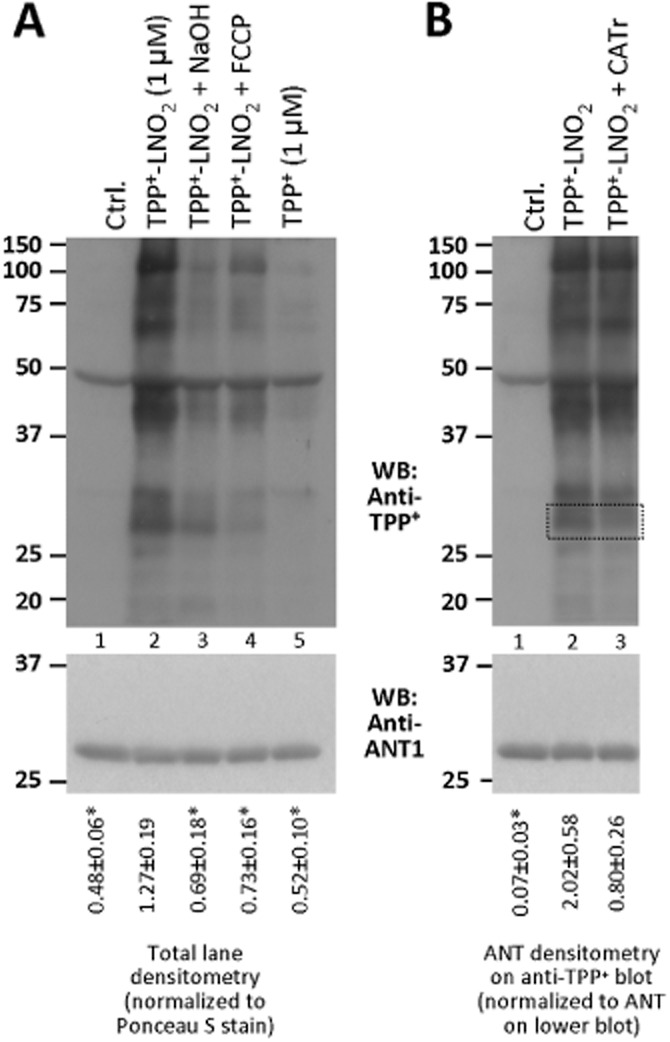
Modification of mitochondrial proteins by TPP+-LNO2. Cardiac mitochondria were incubated as described. (A) Upper blot shows TPP+–LNO2-mediated protein modifications detected by Western blot with anti-TPP+ antibodies. Experimental conditions are indicated above each lane (labels 1–5 below are to simplify explanations in the text). The membrane was stripped and re-probed with anti-ANT1 antibodies for loading control (lower blot). Densitometry was performed in the range 20–150 kDa and normalized to protein across the same molecular mass range from Ponceau S-stained membranes (Supporting Information Figure S5). Values are shown below the blot. *P < 0.05 significantly different from TPP+-LNO2. (B) Upper blot shows protein modifications by TPP+-LNO2, with or without CATr (5 μM). The membrane was stripped and re-probed with anti-ANT1 antibodies for loading control (lower blot). Regions corresponding to ANT1 are shown by dotted lines. Densitometry of ANT on the anti-TPP+ blot was performed and normalized to ANT1 densitometry on the lower blot. Values are shown below the blot. * P < 0.05, significantly different from TPP+-LNO2. All images are representative of, and densitometry values are means ± SEM of, at least five independent experiments. Corresponding Ponceau S-stained membranes are shown in Supporting Information Figure S5.
Previously, we demonstrated that the cytoprotective effect of LNO2 was blocked in cells with ANT1 knocked-down (Nadtochiy et al., 2012). We also identified Cys57 of ANT1 as a mitochondrial target for LNO2, and showed that this modification could be blocked by a conformational shift in ANT1 induced by the inhibitor CATr (Nadtochiy et al., 2009; 2012). Figure 4B shows that TPP-LNO2 also modified a protein of about 32 kDa, a modification blocked by CATr, consistent with ANT1 being a target. Importantly, these changes were not due to altered levels of ANT1 on the gel (lower panels).
To test whether TPP+-LNO2 could modify ANT in intact cells, cardiomyocytes were incubated with TPP+-LNO2 followed by anti-TPP+ Western blotting. Figure 5A shows that TPP+-LNO2 modified several proteins in cells. Furthermore, immunoprecipitation of ANT1 from cells, followed by anti-TPP+ Western blot revealed that ANT1 was indeed modified by TPP+-LNO2 (Figure 5B). Ponceau S stained membranes for blots in Figures 4 and 5, indicating equal protein loading, are shown in Supporting Information Figure S5.
Figure 5.
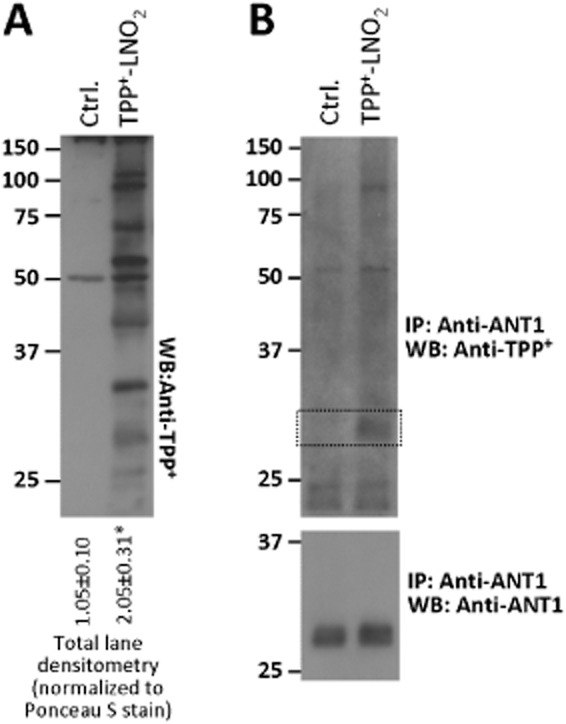
Modification of cardiomyocyte proteins by TPP+-LNO2. Cardiomyocytes were treated with TPP+-LNO2 as described. (A) Blot shows TPP+–LNO2-mediated protein modifications detected by Western blot of whole cell lysates with anti-TPP+ antibodies. Densitometry was performed in the range 20–150 kDa and normalized to protein across the same molecular mass range from Ponceau S-stained membranes (Supporting Information Figure S5). Values shown below the blot are means ± SEM, from 4 independent blots. *P < 0.05, significantly different from Ctrl (Student's t-test). (B) ANT1 was immunoprecipitated from Ctrl (non-treated) and TPP+–LNO2-treated samples, and Western blotted with anti-TPP+ antibodies (upper blot). Regions corresponding to ANT1 are shown by dotted lines. Lower panel shows anti-ANT1 blot, demonstrating equal amounts of ANT1 were pulled down in each sample. Images are representative of 3 independent experiments.
Finally, although we had previously shown that LNO2 was only capable of inducing mitochondrial permeability transition (PT) pore opening at very high concentrations (>10 μM), it might be possible that intra-mitochondrial concentration of TPP+-LNO2 could elicit PT pore opening at lower doses. However, as shown in Supporting Information Figure S6, the protective concentration of TPP+-LNO2 (25 nM) was without effect in the standard isolated mitochondrial PT pore-swelling assay.
Discussion and conclusions
The key advance of this study is the development of a novel mitochondrially targeted nitroalkene (TPP+-LNO2) for use as both an investigational tool and potentially as a therapeutic agent. The use of this reagent revealed that TPP+ conjugation of LNO2 enhanced its cytoprotective efficacy approximately fourfold in a model of IR injury in primary cardiomyocytes (i.e. peak protection by TPP+-LNO2 was seen at a fourfold lower dose than for LNO2; Figure 3). Notably, however, the protective index of the reagent was not altered significantly, that is, the reduction in cell death was approximately the same for the peak doses of LNO2 or TPP+-LNO2.
It is somewhat surprising that TPP+ conjugation of LNO2 only moderately enhanced its protective efficacy. Previous studies on TPP+-conjugated drugs have shown that this strategy can achieve increases in the intra-mitochondrial concentration of the targeted agent, on the order of 100-to 1000-fold (Lin et al., 2002; Prime et al., 2009). However, the present results could be due to the fact that LNO2 is already a very potent cardioprotective agent (Nadtochiy et al., 2009) and is known to enter mitochondria, as shown by its modification of a thiol on the matrix side of ANT1 (Nadtochiy et al., 2012). Alternatively, this result may represent a limitation of the cardiomyocyte model of SIR injury used here, such that a ceiling of cytoprotection may have been reached, and further increases in efficacy may not be achievable. In addition, assuming that the protective effect of LNO2 requires its reaction with proteins, the drop in pH observed during SIR (Figure 2B) may compromise the efficacy of protein nitroalkenylation, which is known to be a pH-sensitive process (Baker et al., 2007). A third possibility is that addition of the TPP+ moiety to LNO2 may indeed enhance its targeting to specific proteins within the mitochondrial membrane such as ANT, but may simultaneously prevent LNO2 from engaging an alternative target required for its cardioprotective benefit (e.g. a cytosolic protein or a non-membrane mitochondrial protein). Although a direct comparison of the binding targets of TPP+-LNO2 versus LNO2 would be desirable, unfortunately, the latter has so far only been visualized using biotin-tagged LNO2 (Nadtochiy et al., 2009; 2012). Because a number of mitochondrial proteins contain biotin, this makes comparison between biotin-based and antibody-based detection difficult.
Nevertheless, the data presented here are consistent with our previous findings using biotin-tagged LNO2. Specifically, conjugation of the –COOH moiety of the nitro-fatty acid [either with biotin (Nadtochiy et al., 2009; 2012) or with TPP+ (as here) does not affect its biological activity. This suggests that the biological effect of LNO2, in terms of a protective phenotype, is not mediated by the –COOH end of the molecule. This has important implications for ANT1 as a target of LNO2, as it has been suggested that one mechanism of mitochondrial uncoupling involves flip-flop of protonated/de-protonated fatty acids across the mitochondrial membrane, catalysed by carrier proteins such as ANT or the uncoupling proteins (Skulachev, 1991; Jezek et al., 1997). Because biotin-LNO2 and TPP+-LNO2 both have their –COOH moiety blocked, they cannot be protonated/de-protonated, so this mechanism is not possible. Furthermore, it is known that TPP+ accumulates on the matrix side of the mitochondrial membrane, and this is consistent with our previous finding that Cys57 of ANT, also on the matrix side of the membrane, is a target for LNO2 (Nadtochiy et al., 2012).
TPP+ conjugation has been used to target several different classes of therapeutic drugs to mitochondria, including antioxidants (Adlam et al., 2005; Lyamzaev et al., 2011), S-nitrosothiols (Prime et al., 2009), lipids (Diers et al., 2010), spin traps (Quin et al., 2009) and fluorescent reporter molecules (Mavri-Damelin et al., 2009). This is the first report of a mitochondrial-targeted nitroalkene, and it may be interesting for TPP+ to conjugate to other nitroalkenes that elicit cardioprotective effects, such as nitro-oleate (Rudolph et al., 2010).
Finally, it is important to emphasize that TPP+-LNO2 should currently be considered only as a tool. As with all potentially cardioprotective molecules, significant obstacles remain in translating these findings into actual therapeutic usage in humans (see Kloner, 2013; Ovize et al., 2013; Vander Heide and Steenbergen, 2013). Nevertheless, TPP+-conjugated nitroalkenes may still be useful in determining the modification of mitochondrial proteins by electrophilic lipids. As such, the identification of additional mitochondrial targets of TPP+-LNO2 seen in Figures 4 and 5 is an ongoing project.
Acknowledgments
This work was funded by a grant from the US National Institutes of Health (RO1 HL-071158) to P. S. B. We thank Mike Murphy (Cambridge, UK) for the gift of anti-iodobutyl-TPP+ antiserum, William Urciuoli (Rochester, NY, USA) for technical assistance and George Porter Jr. (Rochester, NY, USA) for help with statistical analysis.
Glossary
- ANT1
adenine nucleotide translocase 1
- CATr
carboxyatractyloside
- FCCP
carbonylcyanide-p-trifluoromethoxyphenylhydrazone
- LNO2
nitro-linoleate
- SIR
simulated ischaemia-reperfusion
- TMRE
tetramethylrhodamine ethyl ester
- TPP+
triphenylphosphonium cation
Conflict of interest
The authors declare no conflicts of interest with regard to the content of this paper.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12405
Figure S1 Detection of amino-propyl-triphenylphosphonium (NH2-TPP+) by MS. NH2-TPP+ (m/z = 320) was detected using Thermo Fisher Finnigan LCQ Deca Max XP in positive ion mode. Inset shows a structure of NH2-TPP+.
Figure S2 Freshly isolated adult mouse cardiomyocytes. Approximately 85% of cells were rod shaped, as shown. For scale, the single grid lines on the haemocytometer are spaced every 200 microns.
Figure S3 Measurement, by Trypan blue exclusion, of cell death in simulated IR (SIR) injury. Myocytes were isolated, plated on V7-PET plates and subjected to SIR as in Figure 3 of the main manuscript. (A–E) Representative photomicrographs of cardiomyocytes (10× objective). Cells were subjected to the following conditions: (A) SIR, (B) TPP+ + SIR, (C) LA + SIR, (D) LNO2 + SIR and (E) TPP+-LNO2 + SIR, and incubated with Trypan blue dye after SIR. All agents were at 25 nM final dose. (F) Quantitation of cell death (i.e. number of Trypan blue positive cells as a percentage of total number of cells). Data are means ± SEM, *P < 0.01 versus SIR alone, n = 5.
Figure S4 Effect of TPP+ on mitochondrial function. Cardiomyocyte isolation and incubations were performed as detailed in the methods. (A) Oxygen consumption rate (OCR) in Ctrl-and TPP+-treated cardiomyocytes. OCR was measured using a Seahorse XF-24 analyser. (B) Membrane potential in Ctrl-and TPP+-treated cardiomyocytes. ΔTMRE fluorescence represents the difference before and after FCCP (5 μM) addition, (a.u., arbitrary units). All data are means ± SEM, n > 5 independent experiments.
Figure S5 Ponceau S staining ofWestern blot membranes for Figures 4 and 5 in the main manuscript. (A) corresponds to Figure 4A. (B) corresponds to Figure 4B. (C) corresponds to Figure 5A.
Figure S6 Effect of TPP+-LNO2 and TPP+ on mitochondrial PT pore opening/swelling. Upper panel shows typical PT poreswelling traces, and lower panel shows magnitude of swelling measured at 15 min, expressed as percentage decrease relative to initial optical density (OD). Legend indicates traces/bars corresponding to addition of Ca2+ (100 μM), TPP+-LNO2 (25 nM) or TPP+ (25 nM), at the arrow. PT pore inhibitor cyclosporin A (CsA, 2 μM) was added 5 min before Ca2+. All data are means ± SEM, n = 5, **P < 0.05 versus Ctrl.
References
- Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, et al. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- Baker LM, Baker PR, Golin-Bisello F, Schopfer FJ, Fink M, Woodcock SR, et al. Nitro-fatty acid reaction with glutathione and cysteine. Kinetic analysis of thiol alkylation by a Michael addition reaction. J Biol Chem. 2007;282:31085–31093. doi: 10.1074/jbc.M704085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batthyany C, Schopfer FJ, Baker PR, Duran R, Baker LM, Huang Y, et al. Reversible post-translational modification of proteins by nitrated fatty acids in vivo. J Biol Chem. 2006;281:20450–20463. doi: 10.1074/jbc.M602814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burwell LS, Nadtochiy SM, Tompkins AJ, Young S, Brookes PS. Direct evidence for S-nitrosation of mitochondrial complex I. Biochem J. 2006;394:627–634. doi: 10.1042/BJ20051435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Hurd TR, Nadtochiy SM, Brookes PS, Fearnley IM, Lilley KS, et al. Identification of S-nitrosated mitochondrial proteins by S-nitrosothiol difference in gel electrophoresis (SNO-DIGE): implications for the regulation of mitochondrial function by reversible S-nitrosation. Biochem J. 2010;430:49–59. doi: 10.1042/BJ20100633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S, et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat Med. 2013;19:753–759. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui T, Schopfer FJ, Zhang J, Chen K, Ichikawa T, Baker PR, et al. Nitrated fatty acids: endogenous anti-inflammatory signaling mediators. J Biol Chem. 2006;281:35686–35698. doi: 10.1074/jbc.M603357200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diers AR, Higdon AN, Ricart KC, Johnson MS, Agarwal A, Kalyanaraman B, et al. Mitochondrial targeting of the electrophilic lipid 15-deoxy-Delta12,14-prostaglandin J2 increases apoptotic efficacy via redox cell signalling mechanisms. Biochem J. 2010;426:31–41. doi: 10.1042/BJ20091293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman AN, Opie LH, Beshansky JR, Ingwall JS, Rackley CE, Selker HP. Glucose-insulin-potassium revived: current status in acute coronary syndromes and the energy-depleted heart. Circulation. 2013;127:1040–1048. doi: 10.1161/CIRCULATIONAHA.112.130625. [DOI] [PubMed] [Google Scholar]
- Guo S, Olm-Shipman A, Walters A, Urciuoli WR, Devito S, Nadtochiy SM, et al. A cell-based phenotypic assay to identify cardioprotective agents. Circ Res. 2012;110:948–957. doi: 10.1161/CIRCRESAHA.111.263715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jezek P, Modriansky M, Garlid KD. A structure-activity study of fatty acid interaction with mitochondrial uncoupling protein. FEBS Lett. 1997;408:166–170. doi: 10.1016/s0014-5793(97)00335-9. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloner RA. Current state of clinical translation of cardioprotective agents for acute myocardial infarction. Circ Res. 2013;113:451–463. doi: 10.1161/CIRCRESAHA.112.300627. [DOI] [PubMed] [Google Scholar]
- Koenitzer JR, Freeman BA. Redox signaling in inflammation: interactions of endogenous electrophiles and mitochondria in cardiovascular disease. Ann N Y Acad Sci. 2010;1203:45–52. doi: 10.1111/j.1749-6632.2010.05559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakomkin VL, Kapel'ko VI. [Protective effect of mitochondrial antioxidant SkQl at cardiac ischemia and reperfusion] Kardiologiia. 2009;49:55–60. [PubMed] [Google Scholar]
- Lim DG, Sweeney S, Bloodsworth A, White CR, Chumley PH, Krishna NR, et al. Nitrolinoleate, a nitric oxide-derived mediator of cell function: synthesis, characterization, and vasomotor activity. Proc Natl Acad Sci U S A. 2002;99:15941–15946. doi: 10.1073/pnas.232409599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TK, Hughes G, Muratovska A, Blaikie FH, Brookes PS, rley-Usmar V, et al. Specific modification of mitochondrial protein thiols in response to oxidative stress: a proteomics approach. J Biol Chem. 2002;277:17048–17056. doi: 10.1074/jbc.M110797200. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Luo M, Guan X, Luczak ED, Lang D, Kutschke W, Gao Z, et al. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J Clin Invest. 2013;123:1262–1274. doi: 10.1172/JCI65268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyamzaev KG, Pustovidko AV, Simonyan RA, Rokitskaya TI, Domnina LV, Ivanova OY, et al. Novel mitochondria-targeted antioxidants: plastoquinone conjugated with cationic plant alkaloids berberine and palmatine. Pharm Res. 2011;28:2883–2895. doi: 10.1007/s11095-011-0504-8. [DOI] [PubMed] [Google Scholar]
- Mavri-Damelin D, Wilden J, Mani AR, Selden C, Hodgson HJ, Damelin LH. The use of 3-aminophthalimide as a pro-chemiluminescent label in chemiluminescence and fluorescence-based cellular assays. Bioconjug Chem. 2009;20:266–273. doi: 10.1021/bc800361r. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia–reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP. Targeting lipophilic cations to mitochondria. Biochim Biophys Acta. 2008;1777:1028–1031. doi: 10.1016/j.bbabio.2008.03.029. [DOI] [PubMed] [Google Scholar]
- Nadtochiy SM, Baker PR, Freeman BA, Brookes PS. Mitochondrial nitroalkene formation and mild uncoupling in ischaemic preconditioning: implications for cardioprotection. Cardiovasc Res. 2009;82:333–340. doi: 10.1093/cvr/cvn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadtochiy SM, Zhu Q, Urciuoli W, Rafikov R, Black SM, Brookes PS. Nitroalkenes confer acute cardioprotection via adenine nucleotide translocase 1. J Biol Chem. 2012;287:3573–3580. doi: 10.1074/jbc.M111.298406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovize M, Thibault H, Przyklenk K. Myocardial conditioning: opportunities for clinical translation. Circ Res. 2013;113:439–450. doi: 10.1161/CIRCRESAHA.113.300764. [DOI] [PubMed] [Google Scholar]
- Prime TA, Blaikie FH, Evans C, Nadtochiy SM, James AM, Dahm CC, et al. A mitochondria-targeted S-nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia–reperfusion injury. Proc Natl Acad Sci U S A. 2009;106:10764–10769. doi: 10.1073/pnas.0903250106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quin C, Trnka J, Hay A, Murphy MP, Hartley RC. Synthesis of a mitochondria-targeted spin trap using a novel Parham-type cyclization. Tetrahedron. 2009;65:8154–8160. doi: 10.1016/j.tet.2009.07.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Readnower RD, Brainard RE, Hill BG, Jones SP. Standardized bioenergetic profiling of adult mouse cardiomyocytes. Physiol Genomics. 2012;44:1208–1213. doi: 10.1152/physiolgenomics.00129.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph V, Rudolph TK, Schopfer FJ, Bonacci G, Woodcock SR, Cole MP, et al. Endogenous generation and protective effects of nitro-fatty acids in a murine model of focal cardiac ischaemia and reperfusion. Cardiovasc Res. 2010;85:155–166. doi: 10.1093/cvr/cvp275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samouilov A, Zweier JL. Development of chemiluminescence-based methods for specific quantitation of nitrosylated thiols. Anal Biochem. 1998;258:322–330. doi: 10.1006/abio.1998.2609. [DOI] [PubMed] [Google Scholar]
- Schopfer FJ, Baker PR, Giles G, Chumley P, Batthyany C, Crawford J, et al. Fatty acid transduction of nitric oxide signaling. Nitrolinoleic acid is a hydrophobically stabilized nitric oxide donor. J Biol Chem. 2005;280:19289–19297. doi: 10.1074/jbc.M414689200. [DOI] [PubMed] [Google Scholar]
- Shintani-Ishida K, Inui M, Yoshida K. Ischemia–reperfusion induces myocardial infarction through mitochondrial Ca2+ overload. J Mol Cell Cardiol. 2012;53:233–239. doi: 10.1016/j.yjmcc.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Skulachev VP. Fatty acid circuit as a physiological mechanism of uncoupling of oxidative phosphorylation. FEBS Lett. 1991;294:158–162. doi: 10.1016/0014-5793(91)80658-p. [DOI] [PubMed] [Google Scholar]
- Sparagna GC, Lesnefsky EJ. Cardiolipin remodeling in the heart. J Cardiovasc Pharmacol. 2009;53:290–301. doi: 10.1097/FJC.0b013e31819b5461. [DOI] [PubMed] [Google Scholar]
- Tompkins AJ, Burwell LS, Digerness SB, Zaragoza C, Holman WL, Brookes PS. Mitochondrial dysfunction in cardiac ischemia–reperfusion injury: ROS from complex I, without inhibition. Biochim Biophys Acta. 2006;1762:223–231. doi: 10.1016/j.bbadis.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Vander Heide RS, Steenbergen C. Cardioprotection and myocardial reperfusion: pitfalls to clinical application. Circ Res. 2013;113:464–477. doi: 10.1161/CIRCRESAHA.113.300765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner DS, Salari A, Gage DA, Leykam J, Fetter J, Hollingsworth R, et al. Derivatization of peptides to enhance ionization efficiency and control fragmentation during analysis by fast atom bombardment tandem mass spectrometry. Biol Mass Spectrom. 1991;20:419–425. doi: 10.1002/bms.1200200705. [DOI] [PubMed] [Google Scholar]
- Walters AM, Porter GA, Jr, Brookes PS. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circ Res. 2012;111:1222–1236. doi: 10.1161/CIRCRESAHA.112.265660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 2006;1757:509–517. doi: 10.1016/j.bbabio.2006.04.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Detection of amino-propyl-triphenylphosphonium (NH2-TPP+) by MS. NH2-TPP+ (m/z = 320) was detected using Thermo Fisher Finnigan LCQ Deca Max XP in positive ion mode. Inset shows a structure of NH2-TPP+.
Figure S2 Freshly isolated adult mouse cardiomyocytes. Approximately 85% of cells were rod shaped, as shown. For scale, the single grid lines on the haemocytometer are spaced every 200 microns.
Figure S3 Measurement, by Trypan blue exclusion, of cell death in simulated IR (SIR) injury. Myocytes were isolated, plated on V7-PET plates and subjected to SIR as in Figure 3 of the main manuscript. (A–E) Representative photomicrographs of cardiomyocytes (10× objective). Cells were subjected to the following conditions: (A) SIR, (B) TPP+ + SIR, (C) LA + SIR, (D) LNO2 + SIR and (E) TPP+-LNO2 + SIR, and incubated with Trypan blue dye after SIR. All agents were at 25 nM final dose. (F) Quantitation of cell death (i.e. number of Trypan blue positive cells as a percentage of total number of cells). Data are means ± SEM, *P < 0.01 versus SIR alone, n = 5.
Figure S4 Effect of TPP+ on mitochondrial function. Cardiomyocyte isolation and incubations were performed as detailed in the methods. (A) Oxygen consumption rate (OCR) in Ctrl-and TPP+-treated cardiomyocytes. OCR was measured using a Seahorse XF-24 analyser. (B) Membrane potential in Ctrl-and TPP+-treated cardiomyocytes. ΔTMRE fluorescence represents the difference before and after FCCP (5 μM) addition, (a.u., arbitrary units). All data are means ± SEM, n > 5 independent experiments.
Figure S5 Ponceau S staining ofWestern blot membranes for Figures 4 and 5 in the main manuscript. (A) corresponds to Figure 4A. (B) corresponds to Figure 4B. (C) corresponds to Figure 5A.
Figure S6 Effect of TPP+-LNO2 and TPP+ on mitochondrial PT pore opening/swelling. Upper panel shows typical PT poreswelling traces, and lower panel shows magnitude of swelling measured at 15 min, expressed as percentage decrease relative to initial optical density (OD). Legend indicates traces/bars corresponding to addition of Ca2+ (100 μM), TPP+-LNO2 (25 nM) or TPP+ (25 nM), at the arrow. PT pore inhibitor cyclosporin A (CsA, 2 μM) was added 5 min before Ca2+. All data are means ± SEM, n = 5, **P < 0.05 versus Ctrl.