

Abstract
Until recently, hydrogen sulfide (H2S) was exclusively viewed a toxic gas and an environmental hazard, with its toxicity primarily attributed to the inhibition of mitochondrial Complex IV, resulting in a shutdown of mitochondrial electron transport and cellular ATP generation. Work over the last decade established multiple biological regulatory roles of H2S, as an endogenous gaseous transmitter. H2S is produced by cystathionine γ-lyase (CSE), cystathionine β-synthase (CBS) and 3-mercaptopyruvate sulfurtransferase (3-MST). In striking contrast to its inhibitory effect on Complex IV, recent studies showed that at lower concentrations, H2S serves as a stimulator of electron transport in mammalian cells, by acting as a mitochondrial electron donor. Endogenous H2S, produced by mitochondrially localized 3-MST, supports basal, physiological cellular bioenergetic functions; the activity of this metabolic support declines with physiological aging. In specialized conditions (calcium overload in vascular smooth muscle, colon cancer cells), CSE and CBS can also associate with the mitochondria; H2S produced by these enzymes, serves as an endogenous stimulator of cellular bioenergetics. The current article overviews the biochemical mechanisms underlying the stimulatory and inhibitory effects of H2S on mitochondrial function and cellular bioenergetics and discusses the implication of these processes for normal cellular physiology. The relevance of H2S biology is also discussed in the context of colonic epithelial cell physiology: colonocytes are exposed to high levels of sulfide produced by enteric bacteria, and serve as a metabolic barrier to limit their entry into the mammalian host, while, at the same time, utilizing it as a metabolic ‘fuel’.
Linked Articles
This article is part of a themed issue on Mitochondrial Pharmacology: Energy, Injury & Beyond. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2014.171.issue-8
Keywords: mitochondrial electron transport, bioenergetics, cytochrome c oxidase, 3-mercaptopyruvate sulfurtransferase, gasotransmitters, blood vessels, nitric oxide, superoxide, free radicals, cysteine
Introduction: biphasic biological responses elicited by hydrogen sulfide (H2S)
H2S is a colourless, flammable, water-soluble gas with the characteristic smell of rotten eggs. Until recently, H2S was viewed exclusively as a toxic gas and environmental hazard. However, a growing body of data shows that H2S is also synthesized by mammalian tissues via two pyridoxal-5′-phosphate-dependent enzymes responsible for metabolism of L-cysteine: cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE), and a third pathway that involves the combined action of 3-mercaptopyruvate sulfurtransferase (3-MST) and cysteine aminotransferase (CAT) (overviewed in Wang, 2012; Kimura, 2013).
H2S in aqueous solution equilibrates quickly forming two different ionized forms: HS− (hydrosulfide anion) and S2− (sulfide ion). At physiological pH, H2S and HS− represent 20–30% and 70–80%, respectively; S2− is only present in negligible quantity. The terms ‘sulfide’ and H2S are used interchangeably throughout the current article and, unless stated otherwise, refer to the situation in biological fluids to collectively designate H2S and the ionized forms. These various forms remain in permanent acid-base equilibrium so that if one form of sulfide is siphoned off, the two other dissociate/protonate to restore the ratio between the different forms determined by the pH of the medium (Kabil and Banerjee, 2010; Whiteman et al., 2011; Nagy et al., 2013). It must be kept in mind that the above chemistry is the basis for the instability of sulfide solutions at physiological pH, since H2S tends to equilibrate between the liquid phase – such as cell culture or animal tissue – and the nearly infinite external atmosphere in which sulfide concentrations are very low. This phenomenon results in outgassing (complicating the experimental conditions where H2S is added to the tissue culture solution) (Whiteman et al., 2011; Olson, 2012). A related phenomenon in vivo is H2S exhalation, which represents one of the modes of the elimination of biologically produced H2S (Insko et al., 2009; Toombs et al. 2010).
The distribution and regulation of H2S producing enzymes and the wide range of biological effects of H2S are discussed in separate reviews (Fiorucci et al., 2006; Szabo, 2007; Calvert et al., 2010; Elsey et al., 2010; Gadalla and Snyder, 2010; Kimura, 2013; 2013; Predmore and Lefer, 2010; Whiteman and Winyard, 2011; Whiteman et al., 2011; Kimura et al., 2012; Wang, 2012). The biological roles of H2S span the central and peripheral nervous system, the regulation of immunological/inflammatory responses and the various aspects of cardiovascular biology. For the purposes of the current article, we will restrict our focus on the regulation of cellular bioenergetics. It is hoped that the complex biochemical mechanisms regulated by sulfide (overviewed in the current article) will help with the future design and implementation of diagnostic and therapeutic approaches based on pharmacological sulfide administration and/or therapeutic modulation of endogenous sulfide homeostasis. While the current article focused on the biochemical and physiological aspects of sulfide, the pathophysiological and therapeutic aspects of sulfide donation and sulfide biosynthesis inhibition are discussed in a separate review (Módis et al., 2013a).
Before discussing the biochemical effects of H2S on mitochondrial function, we must first familiarize the reader with a key biological feature of H2S, and this is its bell-shaped or biphasic biological dose-response curve. Strikingly, the effects of H2S are highly divergent, depending on its local concentrations. This is determined, at least in part, by its rate of production/delivery: approaches, which generate H2S at fast rates, tend to result in high local concentrations. Table 1 compares and contrasts some of the biological effects of H2S at low rates versus at high rates. An example of the context-dependent effects of H2S on cellular viability is shown in Figure 1. While the concentrations of both NaHS (to generate H2S) and of H2O2 (to induce oxidative stress) were rather high, the findings obtained in this model system illustrate that the same concentration of H2S can be either cytotoxic/inhibitory on mitochondrial respiration (in the absence of oxidative stress) or cytoprotective (in the presence of oxidative stress). Many of the effects seen at low H2S concentrations/release rates are beneficial, while many of the effects seen at high concentrations/high production rates are pathophysiological or toxicological (Table 1).
Table 1.
Representative examples illustrating the Janus-faced biological effects of H2S. At lower concentrations (or lower release rates), beneficial effects predominate, while at higher concentrations (or high release rates) H2S can exert a variety of deleterious/adverse effects. Please note that the responses shown here only serve as illustrations and generalizations and are dependent on the experimental system/species used. For detailed mechanisms and biological implications of these opposing effects of H2S, we refer to specialized review articles (e.g. Fiorucci et al., 2006; Szabo, 2007; Wagner et al., 2009; Calvert et al., 2010; Elsey et al., 2010; Gadalla and Snyder, 2010; Kimura, 2010; Predmore and Lefer, 2010; Whiteman and Winyard, 2011; Whiteman et al., 2011; Kimura et al., 2012; Wang, 2012; Kimura, 2013) and references contained therein
| Lower concentrations of H2S | Higher concentrations of H2S | |
|---|---|---|
| Bioenergetic effects | Stimulation of mitochondrial electron transport, support of cellular bioenergetics, increased cellular ATP levels | Inhibition of mitochondrial respiration, inhibition of cellular bioenergetics, decreased cellular ATP levels |
| Oxidant/antioxidant effects | Antioxidant effects, reducing agent | Pro-oxidant effects, DNA damage, genotoxicity |
| Vascular effects | Physiological vasodilatation, stimulation of angiogenesis | Toxic vasodilatation or vasoconstriction (vascular bed-dependent) |
| Modulation of inflammation | Suppression of proinflammatory gene expression, anti-inflammatory effects | Proinflammatory effects, nociceptive effects |
| Effects on cell viability | Protection from oxidative stress, improvement of cell viability | Suppression of cell viability, promotion of cell necrosis and/or apoptosis |
Figure 1.
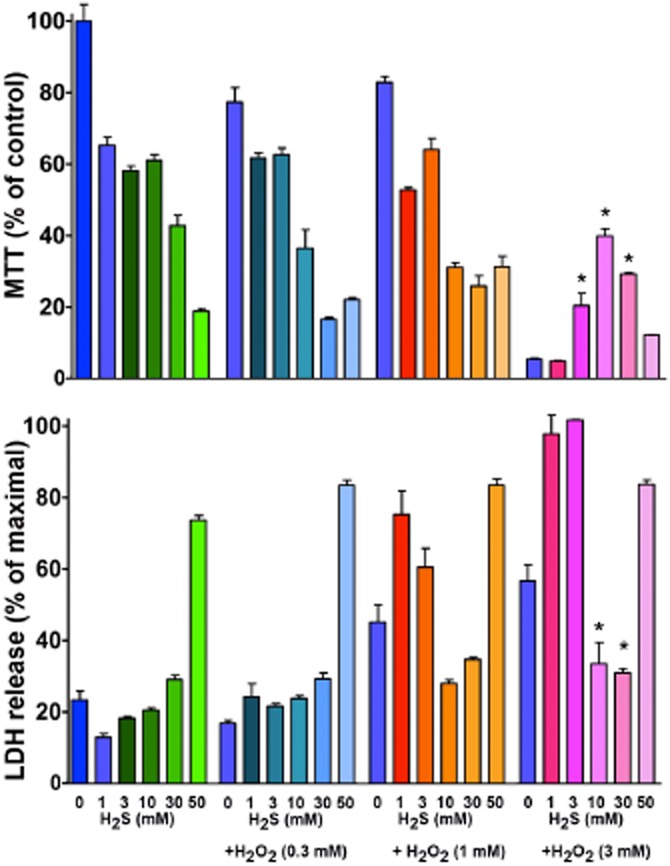
Suppression or enhancement of cellular viability by H2S, depending on the degree of oxidative stress. Murine J774.2 macrophages were grown in culture as described (Szoleczky et al., 2012) and exposed to various concentrations (0.3–3 mM) of hydrogen peroxide (H2O2) in the presence or absence of a 15-min pretreatment with various concentrations of the H2S donor NaHS (1–50 mM). At 5 h, cellular viability was measured by the lactate dehydrogenase (LDH) release method and mitochondrial activity was measured by the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium) method, as described (Szabo et al., 2011; Szoleczky et al., 2012). (This method largely measures the activity of mitochondrial succinate dehydrogenase, but it is also used as a general index of cell viability.) Note the concentration-dependent suppression of mitochondrial respiration and loss of cell viability by high concentrations of sulfide, and the protective effect of the same concentrations of sulfide in the presence of severe oxidative stress elicited by H2O2. Data are mean ± SEM of n = 6. *P < 0.05 represent significant protection by 3–30 mM H2S against 3 mM H2O2 exposure (anova, followed by Tukey's post hoc test).
Inhibitory effects of H2S on mitochondrial respiration
Prior to the last decade, essentially all biological literature on H2S focused on its environmental toxicology (Beauchamp et al., 1984; Reiffenstein et al., 1992). Indeed, H2S exposure, at high concentrations/rates is highly toxic to all mammalian species (including humans), as evidenced by multiple industrial accidents, toxic exposure to H2S in swamps, sinkholes, volcanic emissions, and, most recently, the use of H2S (produced by mixing common household reagents) as an agent of suicide in Japan (Beauchamp et al., 1984; Reiffenstein et al., 1992; Milby and Baselt, 1999; Kamijo et al., 2013). The environmental toxicity of H2S gas is both dose-dependent and dependent on the duration of exposure. The dose-response starts with local effects such as conjunctival irritation (at 50 p.p.m.) and upper airway irritation (at 100–200 p.p.m.). At higher inhalational exposures, pulmonary oedema (at 250–500 p.p.m.), and, at even higher rates of exposure (500–1000 p.p.m.) respiratory paralysis, CNS suppression and cardiovascular depression occur, which can culminate in death (Reiffenstein et al., 1992).
On the molecular level, most of the acute toxicity of H2S has been mainly attributed to the inhibition of mitochondrial Complex IV (cytochrome C oxidase) (Petersen, 1977; Hill et al., 1984). Complex IV is the final member of the mitochondrial electron transport chain in the mitochondrial inner membrane, and an essential component of aerobic cell respiration and ATP generation. It is a large integral membrane protein composed of several metal prosthetic sites and 14 protein subunits. The complex contains two hemes, a cytochrome a and cytochrome a3, and two copper centres (CuA and CuB). As the last enzyme in the respiratory electron transport chain of mitochondria, it receives an electron from each of four cytochrome c molecules, and transfers them to one oxygen molecule, converting molecular oxygen to two molecules of water. In the process, it binds four protons from the inner aqueous phase to make water, and contributes to the generation of transmembrane proton gradient (subsequently harvested by ATP synthase to synthesize ATP). Complex IV/cytochrome c oxidase catalyzes the transfer of electrons from reduced cytochrome c to molecular oxygen. It has been established that H2S, at high concentrations, reversibly and competitively binds to Complex IV, thereby inhibiting the binding of oxygen. In addition, at lower H2S concentrations a non-competitive type inhibition has also been proposed (Petersen, 1977; Cooper and Brown, 2008). Once the binding of oxygen to Complex IV is inhibited, there is a backing up of electrons in the mitochondrial electron transport chain, and, consequently, the potential of the mitochondrial inner membrane dissipates and aerobic ATP generation stops. Toxicology studies focusing on inhalational H2S exposure have established the doses (>600 p.p.m.) that induce an inhibitory effect on Complex IV in the lung and heart that is detectable ex vivo (Figure 2) and is associated with pathological alterations (Khan et al., 1990; 1991; Struve et al., 2001; Dorman et al., 2011; 2004; Wu et al., 2011). In rats, 1 h of H2S inhalation at 300 p.p.m. results in over 80% inhibition of cytochrome c oxidase activity in the heart ex vivo (Wu et al., 2011). The inhibitory effect of high concentrations (typically, 10–100 μM, but highly dependent on the experimental method used) of H2S on cytochrome c oxidase (Figure 3), and, hence, on mitochondrial respiration is well documented in isolated cytochrome c oxidase, as well as in various cell lines in vitro (e.g. Khan et al., 1991; Thompson et al., 2003; Leschelle et al., 2005; Truong et al., 2006; Buckler, 2012; Groeger et al., 2012; Sun et al., 2012). The inhibition is reversible; enzyme activity returns to near-original levels within 10–30 min after exposure of tissue homogenates to a single bolus administration of NaHS, in parallel with the decomposition of sulfide (Di Meo et al., 2011). It is interesting to note that in the studies reported earlier by Khan et al. (1990) (Figure 2), the inhibition of Complex IV persisted despite the time period between the in vivo H2S exposure and the isolation of the mitochondria and the conduction of the cytochrome c oxidase assay. It is likely that the degree of inhibition by H2S of Complex IV in vivo is substantially higher than the amount of inhibition reported in this study. It is also conceivable that H2S exerts an additional, non-reversible inhibitory effect on this enzyme, and/or in vivo exposure of whole animals may build up tissue ‘pools’ of labile sulfur in the mitochondria, which may subsequently release H2S to contribute to a more prolonged type inhibition. These possibilities must be further investigated with today's biochemical tools, as many of these earlier reports are several decades old and were conducted prior to the refinements in mitochondrial biochemistry and prior to today's understanding of the biological properties of sulfide.
Figure 2.
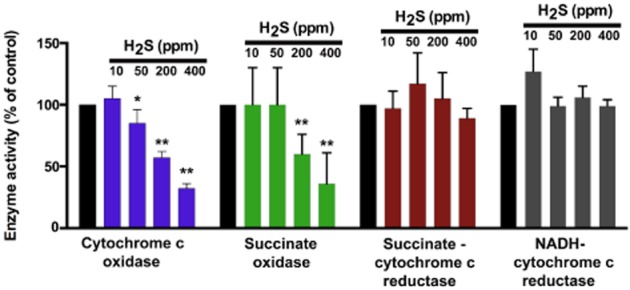
Inhalation toxicity with H2S results in a preferential inhibition of Complex IV activity in the lung ex vivo. Data show the activities of various lung mitochondrial respiratory chain enzymes following a 4-h inhalational exposure of Fischer-344 rats to various concentrations of H2S. Lungs were prepared freshly, mitochondria were prepared via differential centrifugation and mitochondrial enzyme activities were determined at saturating substrate concentrations (Khan et al., 1990). Briefly, cytochrome c oxidase activity was assayed according to the procedure of Wharton and Tzagoloff (1967) with the following modifications. The assay contained potassium phosphate buffer, pH 7.0 (10 mM), ferrocytochrome c (50 μM), distilled water and mitochondrial suspension. Except for the mitochondrial fraction, all other assay components were pre-incubated for 1 min in a spectrophotometric cuvette. Enzyme activity was measured by rapidly mixing in the mitochondrial suspension and monitoring the decrease in optical density at 550 nm. The rate of decrease during the initial 30 set was used in the calculation of enzyme activity. NADH-cytochrome c reductase and succinate-cytochrome c reductase activities and succinate oxidase activity were measured by the methods described by Mustafa et al. (1977). All assays were conducted at 25°C, except, because of low activity of succinate oxidase in lung mitochondria, this enzyme was assayed at 37°C. Data are shown as mean ± SD of n = 4–6 animals in each group. *P < 0.05 and **P < 0.01 represent significant inhibition of enzyme activity at the indicated concentration of H2S. The figure represents a recalculation and replotting of previously published data by Khan et al. (1990); the guidelines of the Canadian Council for Animal Care were followed in all animal experimentations.
Figure 3.
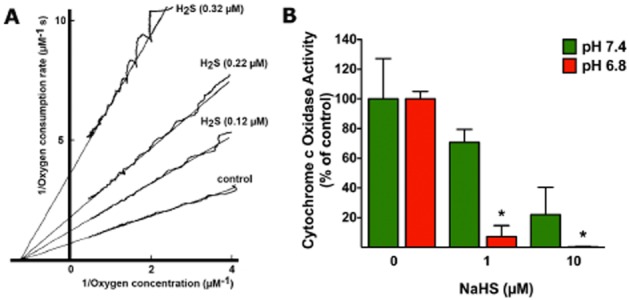
Inhibition of cytochrome c oxidase by H2S in vitro. (A) The effect on the oxygen kinetics of various concentrations of sulfide: 0.32, 0.22 and 0.12 μM is shown on bovine heart cytochrome c oxidase activity. The medium contained 23 mM ascorbate, 68 μM cytochrome c, 34 nM cytochrome aa3, 67 mM potassium phosphate, 1 mM EDTA, 0.5% Tween-80, pH 7.4, 25°C. Sulfide binding was allowed under turnover conditions as described (Petersen, 1977). The respiration rate as a function at the oxygen concentration was calculated and plotted as Lineweaver–Burk plots. The figure represents a replotting of previously published experiments by Petersen (1977). (B) Inhibition of cytochrome c oxidase by H2S is enhanced by mild acidosis in vitro. The activity of cytochrome c oxidase (Sigma, St. Louis, MO, USA) was measured in rat liver mitochondrial homogenates using the CYTOCOX1 cytochrome c oxidase kit (Sigma) according to the manufacturer's instructions in the presence of H2S generated by NaHS (1 and 20 μM) at two different pH levels, 7.4 (neutral) and 6.8 (moderate acidosis). While the basal activity of the enzyme was unaffected by pH, the inhibitory effect of sulfide was potentiated by acidosis. Mean ± SEM of n = 3 is shown; *P < 0.05 shows significant enhancement of the inhibitory effect of sulfide in acidosis (Student's t-test). The animal experimentation component of the studies was conducted with the approval of the Animal Care and Use Committee (IACUC) of the University of Texas Medical Branch and according to the applicable guidelines of the National Institute of Health.
Several series of biochemical studies, utilizing recombinant Complex IV, have characterized, in remarkable detail, the characteristics of H2S binding and the mode of the inhibitory effect. Initial work characterized the main features of the inhibition as (i) non-competitive; (ii) reversible; (iii) has a Ki (binding constant) of 0.2 μM at pH 7.4 (for comparison: Ki values for CO, azide and cyanide are 0.32, 33 and 0.2 μM respectively) (Nicholls, 1975; Petersen, 1977; Nicholls and Kim, 1981; 1982; Hill et al., 1984). In subsequent studies, certain anomalies (both kinetic and in equilibrium) were noticed in the inhibition patterns and sulfide was found to be both an inhibitor and an electron donor of Complex IV, due to complex interactions between sulfide and cytochrome aa3. According to these studies, inhibition of cytochrome aa3 requires three molecules of sulfide, with the first two molecules being ‘used up’ to form a reactive centre adapted to the binding of the third molecule. Moreover, it was suggested that the inhibition of Complex IV by sulfide involves not only the inhibition of cytochrome aa3 but also a ‘pseudosubstrate’ pathway in which a cysteine radical or copper-cysteine complex reacts directly with molecular oxygen (Nicholls and Kim, 1981; 1982). Thus, a combination of three effects has been invoked to explain the complex mode of inhibition: (i) reduction of the cytochrome a3 centre, followed by a reaction with molecular O2; (ii) reduction of other centres, for example, cytochrome a in the cytochrome aa3 complex and (iii) ligating the ferrocytochrome a3 hem group. Using a recently created functional model of cytochrome c oxidase, the mode of sulfide-mediated inhibition of the enzyme was characterized, with special attention to the biochemical mechanisms that occur at lower concentrations of sulfide (Collman et al., 2009). The model uses a synthetic model of cytochrome c oxidase that has all relevant components of the active site of the enzyme (such as heme a3, CuB and tyrosine 244) (Figure 4A). The conclusion of the study (Figure 4B) is that sulfide, at higher concentrations, binds to FeII in the reduced active site, and acts as a competitive inhibitor of the enzyme as it competes with its substrate (O2) for binding to the reduced FeIICuI active site. This inhibition is reversible: O2 easily replaces bound H2S to a reduced FeII site. At lower sulfide concentrations, however, the affinity of H2S is too low to the FeII site to produce detectable inhibition of the enzyme. However, at these concentrations, H2S quantitatively reduces the oxidized cytochrome c oxidase site. In addition, sulfide can also reduce cytochrome c, thereby providing electrons to cytochrome c and substituting for NADH during physiological O2 reduction (Figure 4B). It has been suggested (Collman et al., 2009) (but remains to be directly tested) that the latter mechanism (i.e. the one that occurs at low H2S concentrations) is the more relevant one in vivo. It must be noted, nevertheless, that if this proposed mechanism, indeed, occurs, sulfide would drive oxygen consumption by Complex IV and, unless a determination of the activity of Complexes I–III as well as the reduction state of NADH would be studied, the inhibitory effect of sulfide (on NADH re-oxidation) would not be detected during this reaction. Moreover, this model would predict that the sulfide-induced increase in oxygen consumption would not be susceptible to antimycin inhibition, but would be inhibited by cyanide. To our knowledge, this has not been demonstrated experimentally. Therefore, the relevance of this proposed reaction remains to be further investigated.
Figure 4.
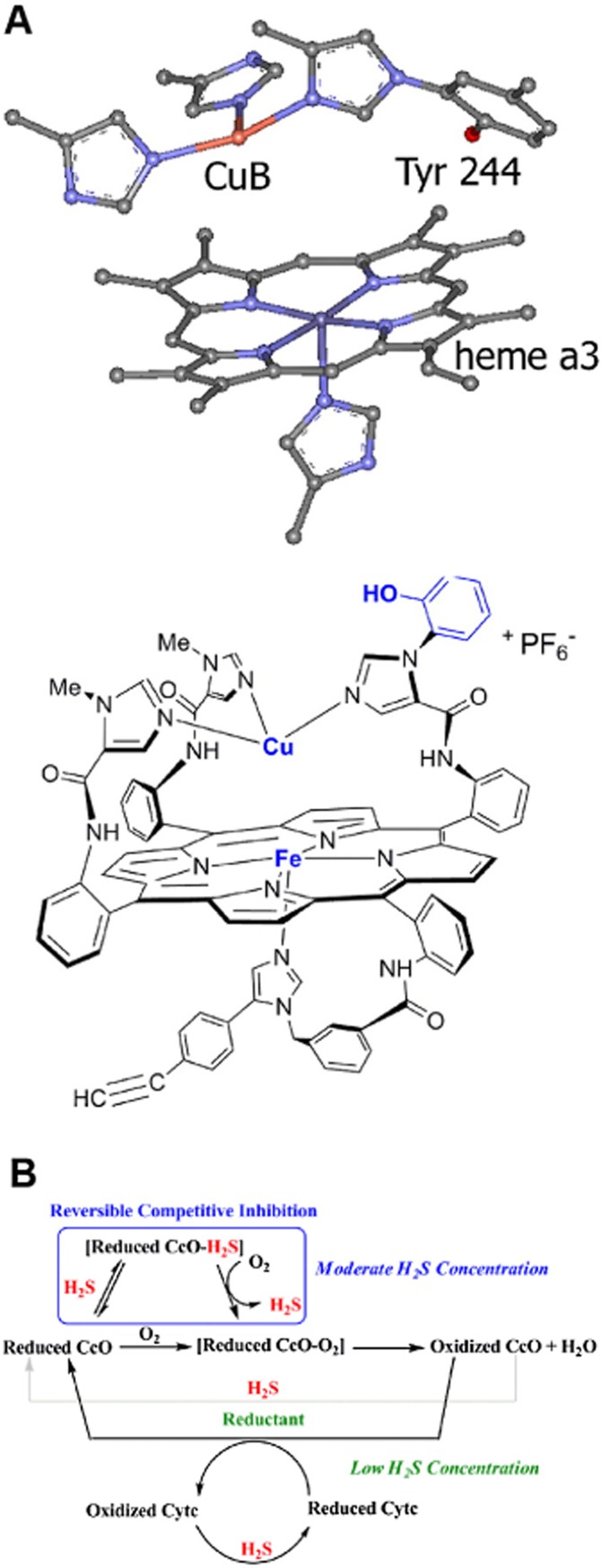
Mechanisms of inhibition of cytochrome c oxidase by H2S. (A) Active site structures of cytochrome C oxidase (CcO, top) and its synthetic model (Fe Cu and phenol analogue) used to study the mechanism of its inhibition by sulfide (Collman et al., 2009). (B) Schematic representation of the possible roles of H2S affecting CcO activity, as derived from the synthetic model shown in part A. The box indicates that H2S binds to the reduced CcO active site, but can be subsequently replaced by O2. This inhibition is hypothesized to occur at moderate H2S concentrations. At lower concentrations, H2S does not compete with the substrate O2 but can still reduce CcO's active site and/or cytochrome c (Cytc) during catalytic O2 reduction. From Collman et al. (2009), reproduced by permission.
Interestingly, the inhibition of Complex IV is pH dependent (Ki values amount to 2.6 μM, 0.55 μM and 0.07 μM at pH of 8.05, 7.48 and 6.28 respectively) (Nicholls and Kim, 1982). The enhancement of the sulfide-induced inhibition is already significant at relatively small (biologically relevant) decreases in pH (Figure 3B) (Groeger et al., 2010). While the mechanism of this enhancement has not been characterized, it is conceivable that acidosis shifts the balance of the various forms of sulfide in solution, and these various forms of sulfide may have different affinity to its enzymatic targets. However, this hypothesis remains to be tested in the future. This characteristic of the inhibitory effect may well be important in the context of conditions/sites associated with systemic acidosis (such as circulatory shock; see Módis et al., 2013a) or local acidosis (such as in physiological hypoxia sensing; see below).
Although most of the metabolic inhibitory effects of sulfide are attributed to inhibition of Complex IV, it must be mentioned that sulfide, at concentrations comparable to the ones at which it inhibits cytochrome c oxidase, also exerts an inhibitory effect on carbonic anhydrase (Nicholson et al., 1998). Although the biological consequences of this action have not been investigated in detail, it is conceivable that this effect, alone, or in concert with the inhibition of Complex IV, may contribute to the metabolic inhibitory effects of sulfide. For instance, in the CNS, sulfide may impair a number of important functions ascribed to carbonic anhydrase, including the interconversion of carbon dioxide and carbonic acid (a key element of acid/base regulation and removal of carbon dioxide from metabolically active tissues). Additional cytotoxic mechanisms described with exposure of cells to high concentrations of sulfide may include calcium mobilization, iron mobilization, mitochondrial uncoupling, mitochondrial pore opening, DNA damage, release of excitatory amino acids and intracellular acidification (Thompson et al., 2003; Roberts et al., 2006; Truong et al., 2006; Attene-Ramos et al., 2010). It is possible (but it remains to be directly tested) that many of these responses are downstream consequences of sulfide-induced metabolic inhibition and cellular energetic distress.
Studies in conscious mice demonstrated that exposure to H2S gas results in a reversible inhibition of cellular metabolism (‘on-demand’ suspended animation) (Blackstone et al., 2005; Volpato et al., 2008). These observations, as well as additional follow-up studies that have generated a diverse, and often contradictory body of studies, are discussed in a complementary review article (Módis et al., 2013a).
Stimulatory effects of H2S on mitochondrial respiration
Although sulfide is most widely recognized by its toxic biological actions (see above), sulfide is also known as the ‘sunlight of the deep ocean’, as in this dark environment the sulfide released from the deep-sea volcanic vents is a vital source of metabolic energy for the primary producers of living matter (Gaill, 1993). There are many sulfo-oxidant bacteria in this environment that derive their metabolic energy from driving electrons from sulfide to oxygen (Kato et al., 2010; Shi et al., 2012; Sylvan et al., 2012; Perner et al., 2013). Moreover, multicellular organisms (e.g. tubeworms) living in the specialized environment of these deep-sea vents are also known to utilize H2S as a primary source of energy, using a complex system where the worm takes up HS− from its environment, and, in turn, delivers sulfide to its internal bacterial symbionts that utilize it (Gaill, 1993; Goffredi et al., 1997) (Figure 5). According to one evolutionary theory, life, in fact, evolved near the deep-sea vents billions of years ago, prior the existence of oxygen on the planet. Therefore, primordial versions of sulfide-based metabolism may have preceded the current, oxygen-based life on the planet by several billions of years (MacLeod et al., 1994; Heinen and Lauwers, 1996; Kundell, 2011; Mielke et al., 2011; Mulkidjanian et al., 2012).
Figure 5.
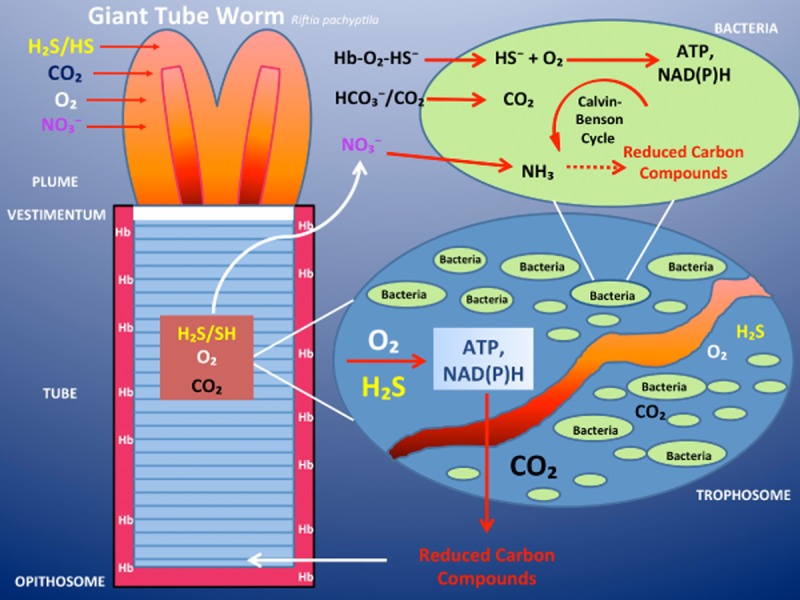
H2S is a source of energy for the tubeworm living in the oxygen-poor but sulfide-rich environment of deep-sea hydrothermal vents. Schematic illustration of the anatomical and physiological organization of the vestimentiferan tubeworm Rijiia pachypeila. The animal is anchored inside its protective tube by the vestimentum. At its anterior end is a respiratory plume. Behind the vestimentum is a region that makes up the bulk of the worm, the trunk. Inside the trunk is the trophosome, which consists primarily of symbiont containing bacteriocytes, associated cells and blood vessels. At the posterior end of the worm is a short region (opisthosome), which anchors the base of the worm to its tube. Oxygen, sulfide (most likely in the form of HS−), nitrate and carbon dioxide are absorbed through the plume and transported in the blood to the cells of the trophosome. Sulfide, such as oxygen, is bound to the sulfide-binding protein, the worm's haemoglobin, and carried to the symbionts. These tubeworm haemoglobins are capable of carrying oxygen in the presence of sulfide, without being ‘poisoned’ by it. The symbionts oxidize the sulfide and use some of the energy released to drive the Calvin-Benson cycle of net CO2 fixation. Some fraction of the reduced carbon compounds synthesized by the symbionts is translocated to the animal host. Furthermore, the chemosynthetic bacteria within the trophosome are able to convert nitrate to ammonium ions, which then become available for the production of amino acids by the bacteria. Later these organic nitrogen-containing compounds are also released to the tubeworm. Modified after Gaill (1993).
At the opposite end of the metabolic spectrum, sulfo-reductant bacteria exist, as well. These bacteria move electrons from reduced molecules towards oxidized form of sulfur (sulfate) to release H2S. It was proposed that the constitution of the eukaryotic cell stems from the association of a H2S-releasing archaeon with a sulfide-oxidizing bacterium bound for a mitochondrial fate (Searcy, 2003). While the former would be feeding the latter with sulfide, the latter would protect its archaeon mate from oxygen toxicity, opening a promising future for the ‘combined organism’ in the oxidant environment on the surface of present-day Earth. Based on these considerations, it is, in fact, predictable that the redox sulfur metabolism remains a basic metabolic feature of present-day aerobic organisms. An interesting place to observe the sulfo-oxidant/sulfo-reductant contrast is the seashore, where the surface of the sediment exposed to the oxygen is occupied by sulfide-oxidizing bacteria, while few mm or cm in depth the sediment is anoxic and releases the characteristic odour of H2S (Grieshaber and Volkel, 1998). Studies focusing on the adaptation of invertebrates to this environment demonstrate that low concentrations of sulfide stimulate oxygen consumption of animal tissues, while high concentrations of sulfide inhibit it (Powell and Somero, 1986).
Despite the above considerations, the direct demonstration that chicken mitochondria (Searcy, 2003) and mitochondria prepared from mammalian cells and tissues (Goubern et al., 2007; Lagoutte et al., 2010; Módis et al., 2013b) utilize H2S to maintain mitochondrial electron transport and to produce ATP (Figures 6–11), came as somewhat of a surprise to the biological community. We speculate that the cause of this surprise may lay in the combination of the facts that the toxicity of sulfide is contained in standard biochemistry curriculum, while the evolutionary and deep-sea vent aspects of sulfur chemistry are not contained in the standard biology curriculum.
Figure 6.
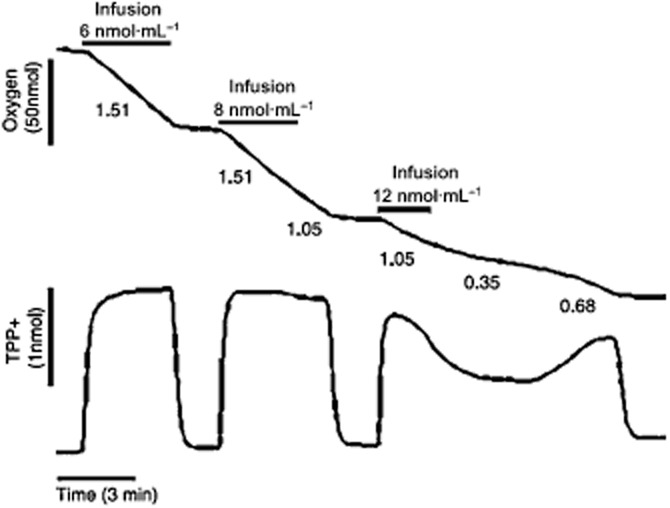
Mammalian cells consume H2S, leading to mitochondrial energization in HT-29 cells. To simultaneously study the mitochondrial bioenergetics (respiration) and membrane potential in colonocytes, permeabilized HT29 cells were used. Approximately 5 × 106 HT29 cells were resuspended in 1.5 mL of the permeabilization medium for this experiment. Top tracing: oxygen concentration; bottom tracing: TPP+ uptake (proportional to membrane potential) are plotted versus time. Sulfide infusions are indicated by horizontal bars above the oxygen tracing, with the rate of infusion shown. Respiration rates (nanomoles of O2/minute/million cells) are shown under the O2 tracing. Infusion of sulfide causes O2 consumption by the permeabilized cells. This is accompanied by an immediate increase in membrane potential as judged from the TPP+ uptake. Both responses can be blocked by antimycin and myxothiazol, which are known inhibitors of the Complex III (coenzyme Q – cytochrome c reductase) of the mitochondrial respiratory chain (not shown). Reproduced with permission from Goubern et al. (2007).
Figure 11.
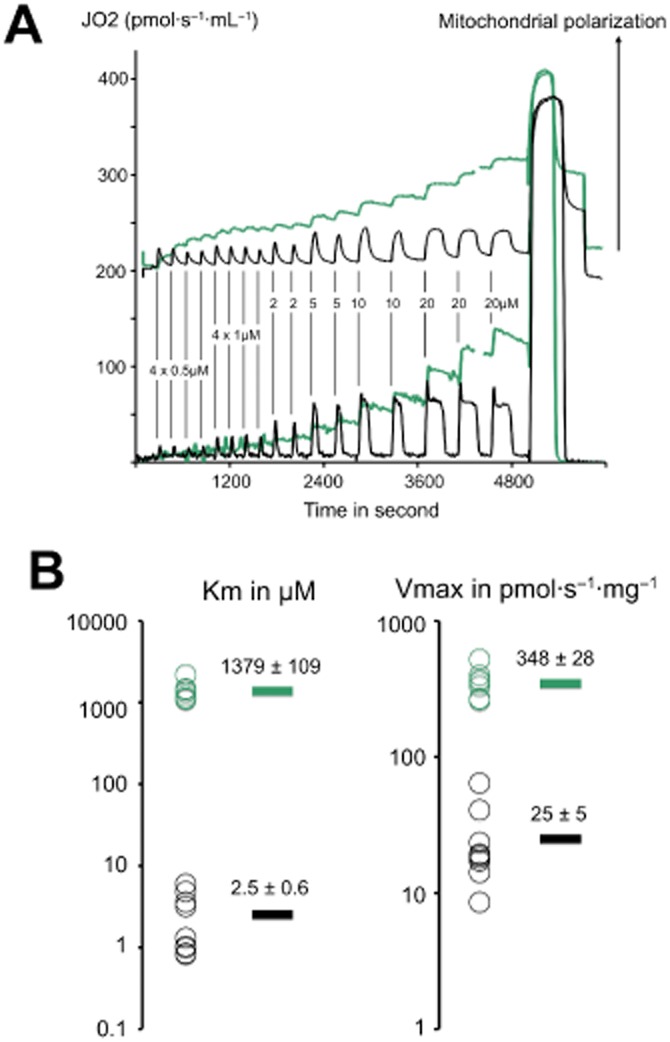
Kinetic parameters of sulfide and succinate oxidation in rat liver mitochondria. The experiments shown were made with a O2k apparatus equipped with the TiP injection pumps. A stock solution of 1 M Na2S was prepared each day and diluted at 5 mM to load in the glass syringes of the injection pump. (A) rat liver mitochondria were resuspended (1 mg·mL−1) in a KCl/sucrose medium in presence of rotenone (1 μM), ADP 1.5 mM and Rhodamine123 400 nM. This suspension was loaded in the two chambers of the O2k Oroboros apparatus equipped with the fluorescence detection module. In one chamber was injected the 5 mM solution of Na2S (black traces) and in the other the same volumes of a 50 mM solution of sodium succinate (green traces). The vertical lines indicate the injections with the resulting final concentration shown for sulfide (10 times more for succinate). The bottom traces represent the oxygen consumption rates (left Y axis). The upper trace shows the (Fmax-F)/Fmax value where Fmax is the fluorescent signal of Rhodamine123 in initial non-energized conditions, this value is proportional to the mitochondrial membrane potential. Injection of sulfide results in sharp increase in oxygen consumption that lasts the time to exhaust sulfide. A calculation based on the amount of oxygen consumed and stoichiometry confirms this (not shown). This oxygen consumption is accompanied by transient energization of mitochondria. In contrast, succinate injections caused stepwise increase in oxygen consumption and potential. The gap in the green curves corresponds to a re-oxygenation step necessary to pursue the experiment. After the end of the injections sequence a saturating concentration of succinate (+7.5 mM final) was added to evaluate the maximal rate, which led to anoxia (drop to zero value). (B) values of kinetic constants were calculated by use of the Lineweaver–Burke double reciprocal plot (1/JO2 vs. 1/[Na2S]) to represent the experimental data and the linear fitting of excel to extrapolate the ordinate at the origin (1/Vmax) and interception with the X axis (−1/Km). A logarithmic scale is used individual values (circles) are shown on the left (n = 8 with succinate and 9 with sulfide). The mean values are shown as bars on the right with their values ± SEM. The Vmax for sulfide oxidation could be used to evaluate the flux of incoming sulfide that could be neutralized by the use of the stoichiometric ratio (25/0.75 = 33). Therefore, this experiment suggests that rat liver mitochondria could neutralize a flux of incoming sulfide close to 10% of their maximal phosphorylating respiratory rate. These experiments used optimal conditions: no competition between sulfide and other substrates and high oxygen concentration. Therefore, the in vivo the efficiency of liver to remove sulfide is likely to be lower; indeed, a sulfide turnover that would represent 10% of the metabolic rate of an organ appears unrealistically high. Nevertheless, the capacity of liver mitochondria to neutralize sulfide appears to be significant. The animal experimentation component of the studies was conducted with the approval of the local Animal Care and Use Committee and according to the applicable guidelines of the European Union.
The sections below focus on the recent studies made with H2S oxidation/utilization of H2S, in support of mitochondrial function in mammalian cells. Before discussing the mechanisms, we must point out that, in addition to the concentration of sulfide used, another important parameter is the mode of its administration. In a typical pharmacological study aimed to establish a concentration-response curve, single additions of the pharmacological agent would be sufficient. However, with H2S, due to its short half-life and its opposing effects on mitochondrial electron transport components (see below), it is crucial to carefully select the infusion rates of sulfide such that the chosen rate matches the sulfide consumption of the biological material under study. With this experimental approach, it was demonstrated in 2007 that sulfide stimulated oxygen consumption and led to energization of mitochondria in permeabilized mammalian colonocytes (Goubern et al., 2007). These studies established that sulfide serves as a physiological mitochondrial substrate. Its functional effect, therefore, is comparable to Krebs cycle-derived electron donors – such as NADH or FADH2, which can be produced, experimentally, in typical mitochondrial preparations by the addition of succinate (FADH2) or glutamate/malate (NADH). The cessation of sulfide infusion leads to a drop in oxygen consumption indicating that the totality of the sulfide infused is immediately consumed (Figures 6, 7, 10). Increases above the optimal rate of sulfide infusion result in an accumulation of sulfide, which can, relatively quickly, rise to toxic levels to induce a decline in oxygen consumption (Figures 6, 7). The optimal steady state of sulfide administration (delivery rate = consumption rate) directly provides an experimental value for the stoichiometry of the oxidation process (Goubern et al., 2007). This value, in colonic epithelial cells, was found to be 0.79; a value that is different from the normal stoichiometry used for other mitochondrial substrates (0.5). This suggested that sulfide oxidation involves a supplementary oxygen consumption, which is explained by additional reactions to that of the normal mitochondrial respiratory chain (Goubern et al., 2007). Importantly, in the same series of studies it was demonstrated that sulfide oxidation was not accompanied by uncoupling of mitochondrial respiration (Goubern et al., 2007).
Figure 7.
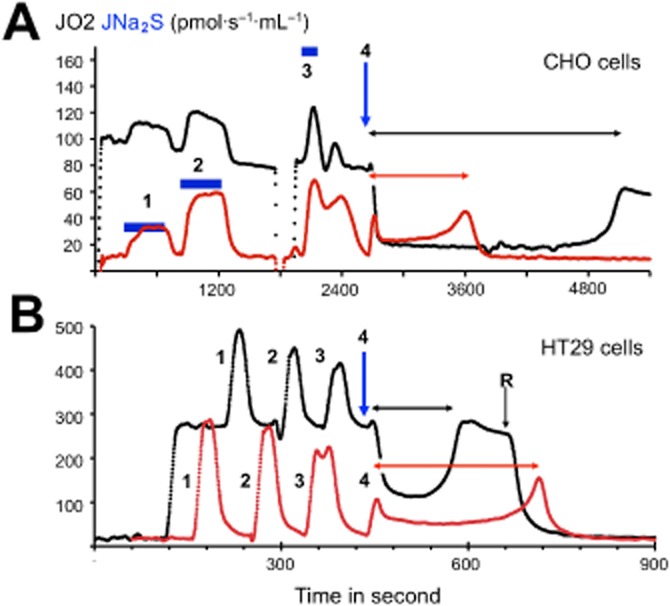
Comparison of CHO cells and colonocytes reveals reverse bioenergetics in presence of high sulfide. The experiments shown were made with a O2k apparatus equipped with the TiP injection pumps. A stock solution of 1 M Na2S was prepared each day and diluted at 5 mM to load in the glass syringes of the injection pump. CHO cells (top) or HT29 cells (immortalized human colonocytes derived from colon cancer) were subject to addition of sulfide during normal respiration (black trace) or in presence of the Complex I inhibitor rotenone (red trace) that inhibits almost completely the cellular respiration. (A) CHO cell line resuspended at 2 × 106 cells·mL−1, the additions 1, 2 and 3 were infusions of sulfide for the time indicated by the black horizontal bars at different increasing rates (same Y ordinate as oxygen consumption). During additions 2, 3 and 4, the same quantity of sulfide was administered at a rate matching with (2) or significantly higher than (3) the cellular capacity for sulfide oxidation, or in a single fast addition (4) final concentration 26 μM (thick black arrow downward). (B) HT29 cells (4–6 × 106 cells·mL−1) additions 1, 2 and 3 were repeated, leading to 25 μM final sulfide concentration. The injection 4 (thick black arrow downward) increased sulfide concentration to 50 μM. This sequence of four injections was repeated twice firstly in absence and secondly after rotenone addition indicated as ‘R’. In order to permit direct comparison, these two successive sequences of additions are shown superposed. In the absence of rotenone, the last sulfide addition (4) produced in both cell types a significant inhibition of cellular oxygen consumption (a reversible effect). In the presence of rotenone, the same addition produced a submaximal increase of oxygen consumption when compared to previous additions. This rate increased in a non-linear way, with time reaching its maximum short time before sulfide exhaustion, revealing a progressive loss of inhibition as oxidation proceeds. With the CHO cells, the time necessary to reverse the inhibition of cellular respiration (horizontal double headed black arrow) was significantly longer than the time to consume all sulfide when mitochondrial Complex I was inhibited by rotenone (horizontal double headed grey arrow). The explanation of this observation is that under conditions of inhibition the reduction state of the NADH/NAD redox couple increases dramatically and renders the Complex I a strong competitor for SOU. In contrast, with colonocytes the opposite is observed (compare horizontal arrows), as if inhibition of Complex I, in fact, lowered the rate of sulfide oxidation. This observation can be explained by a model where Complex I operates in a reverse mode (accepts electrons from quinone to reduce NAD into NADH), which, in turn, helps sulfide oxidation, rather than to antagonize it.
Figure 10.
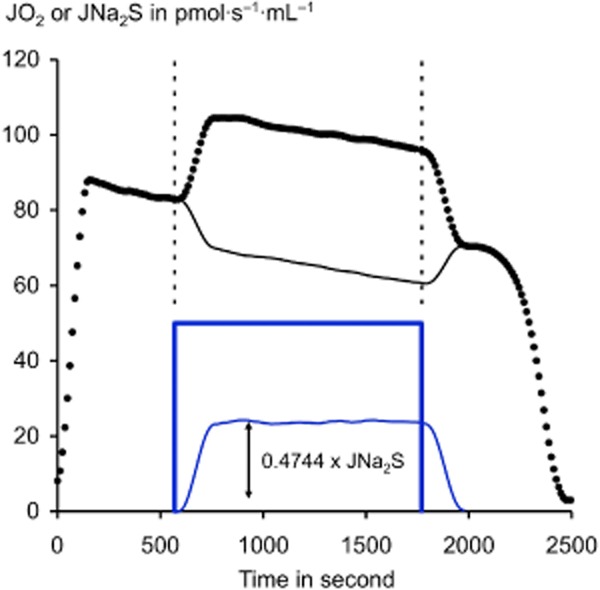
Cells can sustain intense sulfide oxidation for minutes. This experiment demonstrates that cells neutralize a continuous sulfide release over extended period of time: Cultured CHO cells (2 × 106 cells·mL−1 in the 2 mL ‘Oroboros O2k’ chamber for other experimental details, see the legend of Figure 9. The curve shows the cellular oxygen consumption rate expressed in pmol/(s·mL) (black dots). In absence of sulfide, this value declines slowly from 90 to 70 with time/decrease in oxygen concentration before the final fast drop when oxygen is exhausted. The 20-min period of sulfide infusion rate at 50 pmol/(s·mL) is indicated with the thick blue line, the dotted lines aim to emphasize on the coincidence between sulfide infusion and increase in cellular oxygen consumption. This increase is calculated and shown (thin blue line), a linear regression was used to estimate the reference value in absence of sulfide. This increase led to the calculation of a value for the stoichiometry close to 0.5, consistent with that of the dioxygenase reaction alone. Accordingly, it is concluded that the respiration based on the use of carbon containing substrates is decreased (thin black line). This experiment illustrates: (i) the importance of respiratory control: the flux of electrons through respiratory chain remains constant because the general controlling factor is ATP turnover. (ii) The fact that SOU oxidation takes precedence over carbon metabolism since the flux of electrons coming from carbon metabolism is diminished from the amount of electrons coming from sulfide oxidation the latter replacing the former. Thus these cells could accommodate for a prolonged time an incoming sulfide flux of a value representing more than 50% of their basal oxygen consumption rate preventing thus sulfide toxicity. In absence of oxidation this infusion would have raised the sulfide oxidation to 60 μM.
Inhibition of Complex III by antimycin or of Complex IV with cyanide impedes sulfide oxidation, while inhibition of Complex I with rotenone does not (Volkel and Grieshaber, 1997; Yong and Searcy, 2001; Goubern et al., 2007). This indicates that electrons from sulfide enter the mitochondrial respiratory chain at the level of coenzyme Q. Thus, a sulfide quinone reductase (SQR) activity is involved. Afterwards, electrons from sulfide follow the normal oxidative route to Complex III, cytochrome c and Complex IV to finally reduce an atom of oxygen to form water (Goubern et al., 2007). In addition to SQR, two other enzymes are involved: a dioxygenase and a sulfur transferase. This enzymatic system constitutes a sulfide oxidation unit (SOU) that, on one side, injects two electrons (and protons) to reduce the coenzyme Q in the mitochondrial respiratory chain and, on the other side, releases an oxidized form of sulfur (thiosulfate H2S2O3) (Figure 8). For this latter step, an additional molecule of dioxygen is needed (Hildebrandt and Grieshaber, 2008). The stoichiometry predicted by this reaction scheme is 0.75 (1.5 O2/2 H2S), thus, very close to the observed experimental value (Goubern et al., 2007). The human gene for a putative sulfide quinone reductase was overexpressed in CHO cells and increased their sulfide oxidation rate (Lagoutte et al., 2010). The identity of the two other members of the SOU: the dioxygenase (ETHE1 gene) and sulfur transferase (rhodanese), and the tightness of their association with SQR remain to be clarified. Large variations in the range of activity of SOU were detected among cells/tissues (see below).
Figure 8.
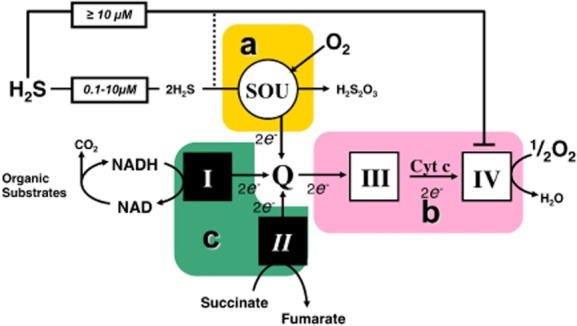
The Sulfide Oxidation Unit (SOU), and limiting steps for sulfide oxidation. Complexes of the mitochondrial respiratory chain are numbered with roman numerals. Three essential blocks of reactions important for the control of sulfide oxidation ‘a, b, c’ are boxed: ‘a’ is the SOU, which oxidizes two molecules of sulfide, uses one dioxygen molecule, releases thiosulfate (H2S2O3) and reduces quinone ‘Q’. ‘b’ accepts electrons from quinone and drives them through Complexes III and IV to oxygen. The sequential intervention of ‘a’ and ‘b’ is necessary for sulfide oxidation to proceed. 1.5 molecules of dioxygen and two molecules of H2S are used: the stoichiometry is thus 1.5/2 = 0.75. Complex IV is the target of sulfide inhibition, which becomes significant at 10 μM and above, while the SOU could operate well below this 10 μM concentration. Block ‘c’ is constituted by the other enzymatic reactions, reducing quinone and is a competitor of SOU. Mitochondrial Complex V (not shown in the scheme) imposes collectively to a, b, c, the constraint of respiratory control matching the flux through the redox reactions (a, b, c) to values imposed by the ATP turnover rate.
When compared to other substrates of the mitochondrial respiratory chain (NADH, FADH2, succinate, L-alpha-glycerophosphate), the yield of sulfide in terms of electrons to be used by the respiratory chain is relatively low: two molecules of sulfide are necessary to provide two electrons. Moreover, it is costly in terms of oxygen: for the same electron transfer in the respiratory Complexes III and IV, sulfide oxidation needs three times more oxygen. Thus, when sulfide oxidation takes place, the yield in energy per oxygen atom consumed is low in comparison with NADH or FADH2 generated by oxidation of carbon containing substrates and sulfide may appear as a poor energy substrate. For example as part of the oxidative metabolism of cysteine seen as source for one pyruvate and one sulfide, the latter would provide one electron to the respiratory chain and the former 10 through pyruvate dehydrogenase and Krebs cycle. However, this low energy yield is balanced by several unique properties of sulfide: First, H2S diffuses freely across membranes and extracellular sulfide could reach and feed mitochondria inside cells without the need of transporters. Second, sulfide does not need any ‘biochemical preparation’ which contrasts with other circulating/extracellular carbon containing substrates that needs to be activated (energy cost) and broken down by metabolic processes to generate the reduced coenzymes NADH or FADH2. Third, affinity of SOU for sulfide is high (Figures 9, 11) so that 100% of any amount released in the high nM or low μM range is oxidized. Therefore, although the exact physiological/pathophysiological role of this process remains to be clarified, sulfide may serve as an ‘emergency’ substrate, or as a substrate that balances and complements the electron-donating effect of Krebs cycle-derived electron donors, and/or play cellular roles in specialized situations (see below). Free sulfide is in equilibrium with bound forms that show no toxicity and might be seen as a reserve of this ‘emergency substrate’ although the cost of the constitution and maintenance of this sulfide reserve has to be taken into account. Finally, in the gut an abundant sulfide production occurs with no energy cost for the eukaryotic cells as sulfide is a metabolic waste of the anaerobic microbiota of the lumen (this mechanism is discussed in additional detail in a subsequent section).
Figure 9.
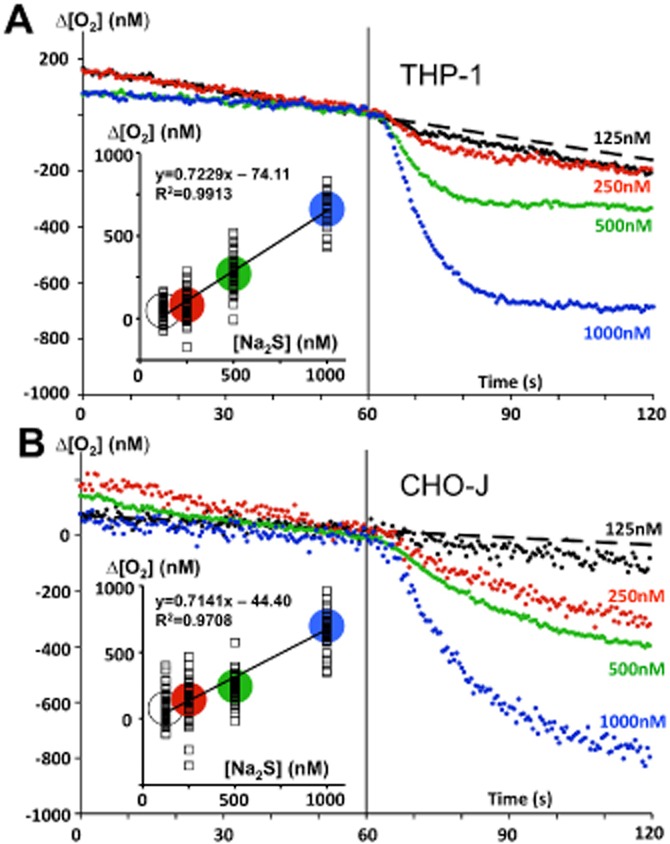
Fast cellular oxidation of sulfide in the nanomolar range. The human monocyte cell line THP1 (top) and a CHO cell line (2 × 106 cells·mL−1) expressing human SQR (bottom) were used in these experiments aiming to demonstrate that sulfide oxidation could take place when external sulfide concentration is in the nanomolar range. The experiments were made with an ‘Oroboros O2k’ apparatus equipped with the TiP injection pumps a stock solution of 1 M Na2S was prepared each day and diluted at 5 mM to load in the glass syringes of the injection pump. Single injections (indicated by the vertical line at 60 s) leading to 125, 250, 500 or 1000 nM final concentrations were made and the respiratory rate (oxygen consumption of cells) was recorded before and after the injection. To increase accuracy, the data points are calculated from the difference between two parallel experiments: one in presence of rotenone (inhibitor of Complex I allowing sulfide oxidation to proceed), and another one in presence of antimycin (inhibitor of Complex III blocking sulfide oxidation). The data points are the mean values of up to 40–50 measurements (10 successive injections of Na2S 125 nM during four to five independent experiments). The dotted line prolongs the slope before sulfide injection for the 125 nM injections. The drop in oxygen concentration caused by sulfide oxidation was determined 1 min after the injection, and was used for the linear regression analysis (insets). The slope relates the stoichiometry (O2/sulfide) for the oxidation process (>95% of the theoretical value of 0.75). There was no correction for Na2S purity/content. While experimental error/difficulties could explain the ordinate at the origin different from zero, this may also suggest that a small part of the sulfide injected escaped to SOU.
The net mitochondrial functional effects of sulfide are complex, because its two radically opposing effects (electron donation at SQR, inducing a stimulation of electron flow and inhibition of mitochondrial oxygen binding at Complex IV, thereby stopping electron flow) counteract each other (Figure 8). At nanomolar to low micromolar concentrations, sulfide is a substrate for most mitochondria (Lagoutte et al., 2010). At higher concentration, sulfide is a poison that blocks mitochondrial respiration in all cell types (Lagoutte et al., 2010). This latter situation results in positive feedback effects; on the one hand, if sulfide delivery rate exceeds the oxidation rate, sulfide accumulates and depresses oxidation rate, which accelerates accumulation, leading to further suppression of the mitochondrial function. On the other hand, when the rate of sulfide oxidation outperforms the rate of sulfide supply, the elimination proceeds faster and faster. This latter situation is particularly relevant to experiments in which a single bolus of sulfide is administered to a biological system (animals or cells). The stimulating and inhibiting concentrations of H2S overlap (Yong and Searcy, 2001 Lagoutte et al., 2010) at the low micromolar range, while a significant inhibition takes place above 10 μM sulfide.
The cell's free sulfide concentration is likely to be permanently in dynamic equilibrium with a much larger pool of ‘bound forms of sulfur’, including thiol groups of proteins (e.g. Wintner et al., 2010). In this respect, sulfide can be viewed as a ‘redox shuttle’ linking mitochondrial bioenergetics with a sulfur-based redox world remnant of an archaic metabolism. Although, in terms of quantitative bioenergetics, it is clearly inferior to carbon metabolism (Krebs cycle; re-oxidation of glycolytic NADH; FADH2-NADH derived from beta oxidation etc.), the data demonstrating that under physiological conditions or under stress conditions, H2S production by endogenous H2S-producing enzymes can contribute to cellular bioenergetics (Fu et al., 2012; Módis et al., 2013b; Szabo et al., 2013) suggest that this mechanism is active even in mammals living in present-day, oxygen-rich environment. In addition, we speculate that its conservation through the ages may be explained by several additional (and not mutually exclusive) biological necessities. For instance, in some cases an active neutralization of excessive fluxes of endogenous sulfide may be necessary, because passive diffusion of this gas would not be sufficient to decrease its concentration to nontoxic values. In addition, the sulfide redox shuttle may serve the purposes of redox control and signal transduction, with mitochondrial bioenergetics as one of its targets. This latter hypothesis makes even the disappearance of SOU in certain biological sites (see below) a relevant event, possibly serving to increase the range of sulfide signalling and/or to withdraw mitochondria from its control (Bouillaud and Blachier, 2011).
In a recent set of experiments (Figure 9), the determination of the cellular affinity for sulfide showed that a short time (seconds to minutes) is sufficient to reduce μM concentrations of sulfide to near-zero levels in a large volume of external medium. Extrapolation to tissues in animals (where cells are much more concentrated) is not straightforward because SOU activity depends on its relative expression level and on multiple interactions with other respiratory enzymes with which SOU is in competition to reduce quinone. Thus, to be consistent with sulfide physiology in mammals, sulfide oxidation was considered in mitochondria isolated from animal tissues in presence of substrates plus ADP to simulate normal mitochondrial activity or with suspension of cultured cells in presence of their endogenous respiratory rate. The dioxygenase activity of SOU has the consequence that sulfide oxidation results in a significant increase in the oxygen consumption rate of respiring cells/mitochondria (Figure 10). This increase is linearly proportional to the sulfide infusion rate, as defined by a stoichiometric relationship.
Although sulfide oxidation and sulfide-related bioenergetic effects have now been demonstrated in a wide variety of cell types (including colonocytes, macrophages, hepatocytes, neurons etc.) in vitro (Goubern et al., 2007; Lagoutte et al., 2010; Groeger et al., 2012; Mimoun et al., 2012; Módis et al., 2013b), there are significant variations between various cell types with respect to their ability to oxidize sulfide as well as with respect to their ability to utilize it for the generation of energy. The first conclusion that can be drawn from this observation is that the importance of the SOU ranges from conditions in which all the electrons travelling to Complex IV and oxygen come from sulfide (e.g. in colonocytes) to conditions in which no sulfide oxidation is detectable (e.g. neuroblastoma derived cell lines and mouse brain mitochondria). Most of the cells in culture show intermediate characteristics in which the oxidation rate of sulfide could account for a significant percentage of the maximal respiratory rate (Lagoutte et al., 2010) (Figure 7) and thus appears disproportionately higher than the expected endogenous sulfide releasing rates. However, with mitochondria isolated from animal organs, the relative activity of the SOU is, when present, of much lower amplitude. The current examples in rodents converge towards values between zero and no more than 10–20% of the maximal respiratory rate (Lagoutte et al., 2010). This is shown here with rat liver mitochondria (Figure 11): the high affinity of the SOU contrasts with the low affinity of succinate dehydrogenase since there is a more than two orders of magnitude gap between their respective affinity constants. At the opposite, the maximal velocity of the succinate dehydrogenase is more than 10 times that of the SOU. However, less than a few percent of the basal metabolic rate would be sufficient to annihilate endogenous sulfide release rates. The explanation of the difference in the SOU activity between cultured cells and isolated mitochondria is still unclear: one possibility could be that culture conditions stimulate the expression of the components of the SOU. It should be also noted that the activity and the role of SOU may be dependent on species, gender, age and other factors. While we have now data in CHO cells and in several rodent species, additional data (especially additional data involving human mitochondria) would be necessary to test the overall validity and applications of this concept. One should also remember that cultured cells are lacking in respiratory enzymes and rely heavily on the glycolytic metabolism in comparison to the differentiated cells present in the organs used to prepare mitochondria. If a constant expression of SQR is maintained, the decreased expression of respiratory enzymes in cultured cells would raise the ratio between the SOU and other complexes of respiratory chain.
A further conclusion from cell-based studies focusing on the function of the SOU is that the SOU has a strong tendency to take over other electrons donors so that there is priority for electrons coming from sulfide (Lagoutte et al., 2010). This, in addition to the high affinity of SQR (Figures 9, 11), guarantees that sulfide oxidation takes place and maintains low (i.e. non-toxic) concentrations of free sulfide (Figure 10), while other mitochondrial substrates would be present at concentrations several orders of magnitude higher (Figure 11 estimates the succinate concentrations for a significant Krebs cycle activity). However, when the carbon metabolism is driven to a more ‘reduced state’ it could severely slow down the activity of the SOU. This more reduced state could be obtained by inhibition of the respiratory chain, including by sulfide (Figure 7). The specific adaptation of colonocytes aims to reduce this competition process (see below), and thus indicates its relevance when exposure to sulfide is high.
The biphasic mitochondrial effects of sulfide have now been demonstrated in various cell types (Goubern et al., 2007; Lagoutte et al., 2010; Groeger et al., 2012; Mimoun et al., 2012; Módis et al., 2013b) using a variety of methods – primarily, the Oroboros system, as in Figures 6–11; as well as the Seahorse system (extracellular flux analysis) (Figures 12, 13). The bell-shaped concentration responses have also been confirmed, with low (nanomolar/low micromolar) concentrations of sulfide being stimulatory, while higher (mid-to high micromolar) concentrations of sulfide being inhibitory. Because of the short half-life of sulfide, direct sulfide exposure studies using the Oroboros system provides a more direct method of analysis than the Seahorse system, which measures with time delay (‘Seahorse reading cycles’). On the other hand, the same method is highly useful when investigating endogenous sulfide producing systems, when sulfide is produced by endogenous, biological enzyme systems at relatively constant rates; see below. The same Seahorse method, furthermore, does not permit differential ‘rates of infusion’, which is highly instructive in experiments involving sulfide (see above and in Figure 7). The stoichiometry of sulfide oxidation predicts that the consumption of 1 μM sulfide via the SOU would consume 0.75 μM of oxygen while its concentration in the medium is around 200 μM at 30°C so that the drop in oxygen concentration would be of less than 0.5%. Therefore, the sulfide-mediated alterations in oxygen consumption/mitochondrial electron transport observed may be amplified by additional pharmacological effects of sulfide beyond direct electron donation to SQR. In support of this hypothesis, recent data indicate that the sulfide-induced increase in oxygen consumption/electron transport in isolated rat liver mitochondria observed in the Seahorse system at 1–3 μM sulfide was attenuated by inhibition of PKA (the downstream effector of cAMP signalling) (Módis et al., 2013c). Our interpretation of these findings is that sulfide, which is a known inhibitor of cAMP/cGMP phosphodiesterases (Bucci et al., 2010; Coletta et al., 2012), enhances mitochondrial cAMP levels, which, in turn, contribute to the stimulation of mitochondrial electron transport. Indeed, sulfide is a direct inhibitor of the activity of the mitochondrial phosphodiesterase isoform (PDEA2), as evidenced by in vitro experiments using recombinant PDEA2 enzyme (Módis et al., 2013c). It is conceivable that additional effects of sulfide on mitochondrial function may involve activation/opening of ATP-sensitive potassium channels, redox/antioxidant responses, post-transcriptional modification of mitochondrial proteins (e.g. via sulfhydration), effects on the PDE5/cGMP/PKG system, mitochondrial calcium handling and other factors. Even though the stimulation of electron transport by sulfide may involve multiple mechanisms, it is important to stress that in the Seahorse experimental system (similar to the Oroboros system) the stimulation of electron transport occurs at the level of Complex II, and the positive bioenergetic responses seen at lower sulfide concentrations are diminished, at higher concentrations of sulfide (10–100 μM) by a direct inhibition of Complex IV (i.e. cytochrome c oxidase) (Figures 12, 13). Another feature of sulfide in this system is its stimulatory bioenergetic effect, which is most prominent at intermediate degree of fluxes of Krebs cycle-derived electron donors (Módis et al., 2013b). This is consistent with the hypothesis that sulfide acts as a secondary/back-up or perhaps ‘emergency’ source of electron donors compared to ‘classical’ (i.e. Krebs cycle-derived) electron donors.
Figure 12.
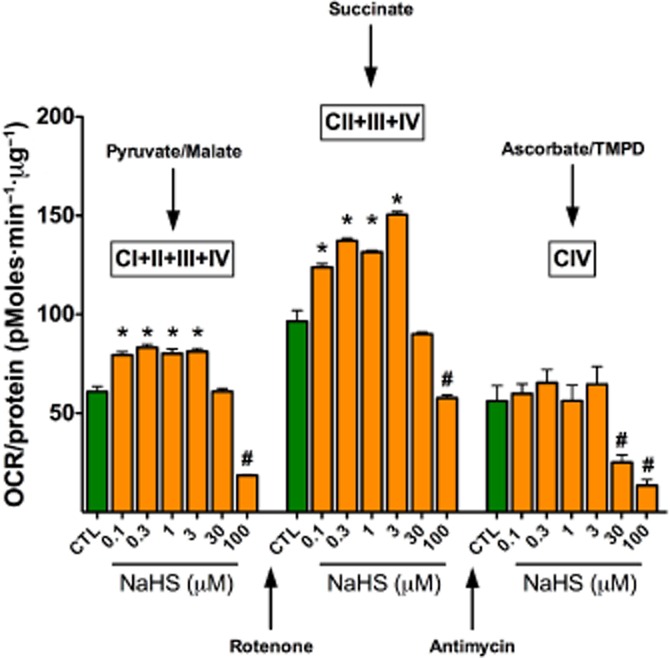
Bell-shaped dose responses to the sulfide donor NaHS in isolated rat liver mitochondria. Rat liver mitochondria were isolated and subjected to bioenergetic analysis using the Extracellular Flux Analysis method as described (Módis et al., 2013b). In separate sets of experiments, mitochondrial electron transport was stimulated by the addition of pyruvate/malate (10 mM/2 mM, respectively, in order to enable the activity of all Complexes; left panel); with succinate (10 mM, in the presence of the Complex I inhibitor rotenone, 2 μM, in order to direct the electron flow exclusively through Complexes II, III and IV only; middle panel) or with the artificial substrates ascorbate/TMPD (10 mM/100 μM, respectively, in the presence of the Complex III inhibitor antimycin at 4 μM, in order to selectively activate Complex IV only; right panel) in the presence of various concentrations of NaHS. Please note that sulfide exerted a biphasic effect on mitochondrial O2 consumption when Complexes I–IV or II–IV were active (stimulation at 0.1–3 μM, followed by inhibition at 30–100 μM), while it only exerted inhibitory effect when Complex IV was active only (at 30–100 μM). Data are shown as mean ± SEM of n = 6 experiments; *P < 0.05 indicates significant enhancement; #P < 0.05 indicates significant inhibition (anova, followed by Tukey's test). The animal experimentation component of the studies was conducted with the approval of the Animal Care and Use Committee (IACUC) of the University of Texas Medical Branch and according to the applicable guidelines of the National Institute of Health.
Figure 13.
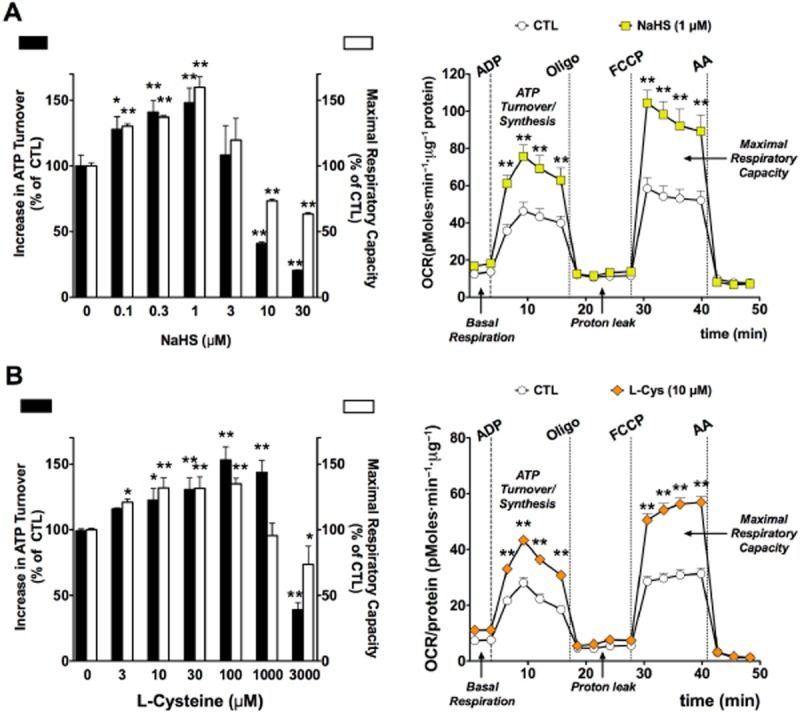
Biphasic effect of (A): hydrogen sulfide (NaHS) or (B): L-cysteine (L-Cys) on mitochondrial oxygen consumption rate (OCR) in isolated rat liver mitochondria. Rat liver mitochondria were isolated and subjected to bioenergetic analysis using the Extracellular Flux Analysis method as described (Módis et al., 2013b). At lower concentrations, H2S (0.1–1 μM) or L-cysteine (10–1000 μM) elicit a significant increase in mitochondrial activity, as evidenced by the measurement of two parameters, calculated ATP Turnover (state 3) and FCCP-stimulated Maximal Respiratory Capacity (state 3u). Higher concentrations of H2S (3–30 μM) or L-cysteine (3000 μM) suppress mitochondrial activity. The right panels show representative experiments, depicting the effect of H2S (1 μM, top panel) or L-cysteine (L-Cys, bottom panel). All experiments were conducted in the presence of 10 mM succinate. Data represent mean ± SEM of six experiments. *P < 0.05 and **P < 0.01 shows the effect of NaHS or L-cysteine, as compared to vehicle control (anova, followed by Tukey's test). Part A is reproduced with permission from Módis et al. (2013b). The animal experimentation component of the studies was conducted with the approval of the Animal Care and Use Committee (IACUC) of the University of Texas Medical Branch and according to the applicable guidelines of the National Institute of Health.
Regulation of cellular bioenergetics by endogenously produced H2S
Similar to administration of low concentrations/low release of exogenous H2S, which acts as an electron donor and stimulatory bioenergetic factor (Goubern et al., 2007; Módis et al., 2013b), endogenously produced H2S also exerts stimulatory bioenergetic effects in various cell types in vitro. The enzyme that is responsible for the majority of mitochondrial H2S production is 3-MST, which shows partial mitochondrial localization. It must be noted that, although many articles consider this enzyme as strictly mitochondrial, several studies show that the enzyme is present in the cytosol, as well (Shibuya et al., 2009a,b2009b; Módis et al., 2013b). The 3-MST pathway of mitochondrial production of H2S starts out with L-cysteine and involves several enzymes. The first one is CAT (cysteine/aspartate aminotransferase), which produces 3-mercaptopyruvate (3-MP) from L-cysteine. 3-MP is then converted by 3-MST to produce pyruvate and an enzyme-bound persulfide. This enzyme-bound persulfide acts as the precursor of H2S: the release of H2S requires reducing agents such as thioredoxin and glutathione (Mikami et al., 2011; Tanizawa, 2011; Kimura, 2013; Yadav et al., 2013). The biochemistry of human recombinant 3-MST has been recently characterized in substantial detail (Yadav et al., 2013). There is evidence that 3-MST is regulated by intra-and intermolecular cysteines in an oxidant-sensitive fashion. Using recombinant rat 3-MST, it has been demonstrated that an intermolecular switch consisting of Cys 154 and Cys 263 can be oxidized to form a dimer between Cys 154 and Cys 154, Cys 154 and Cys 263, and/ or Cys 263 and Cys 263, thereby rendering the enzyme inactive (Nagahara et al., 2012). In addition, more severe oxidative stress can affect an intramolecular switch, whereby a catalytic site Cys 247 is oxidized to form a cysteine sulfenate, which also results in an inhibition of the enzyme (Nagahara and Katayama, 2005; Nagahara et al., 2012; Nagahara, 2013). This interaction has been recently implicated in the oxidative inactivation of 3-MST and a subsequent inhibition of the bioenergetic role of endogenously produced H2S (see below).
The role of 3-MST in the regulation of cellular bioenergetics is supported by several sets of data. First, stable silencing of 3-MST reduces basal bioenergetic parameters in cultured hepatoma cells (Módis et al., 2013b). Second, low concentrations of 3-MP produce H2S and exert a stimulatory effect on bioenergetic parameters both in isolated mitochondria and in cultured cells, and this response is suppressed by the silencing of 3-MST (Módis et al., 2013b). Third, (and similar to the biphasic nature of exogenously administered H2S) higher concentrations of 3-MST suppress the cellular bioenergetic response; this suppression, too, is attenuated by 3-MST silencing (Módis et al., 2013b). Fourth, SQR silencing suppresses both basal and 3-MP mediated stimulation of bioenergetic function (Módis et al., 2013b), as well as the L-cysteine-mediated stimulation of mitochondrial oxygen consumption (Figure 14). Fifth, alpha-ketoglutarate and cysteine both stimulate mitochondrial electron transport, and these effects are attenuated by the CAT inhibitor aspartate (Figure 15). The simplest interpretation of these findings is that 3-MP-derived, 3-MST-mediated production of H2S feeds electrons into the mitochondrial electron transport chain via SQR at the level of Complex II, although additional indirect effects of sulfide may also contribute. Interestingly, oxidative stress impairs the bioenergetic role of 3-MST: incubation of recombinant 3-MST with hydrogen peroxide suppresses the ability of the enzyme to produce H2S (Módis et al., 2013d). Similarly, incubation of isolated mitochondria with H2O2 suppresses the production of H2S from 3-MP (Módis et al., 2013d). Finally, challenge of hepatoma cells with hydrogen peroxide markedly attenuates the stimulatory bioenergetic effect of the 3-MST substrate 3-MP (Módis et al., 2013d). Interestingly, in mitochondria prepared from aged mice (which show a marked baseline suppression of mitochondrial function), the 3-MP-mediated stimulation of electron transport is absent (Figure 16), indicative that the importance of this bioenergetic pathway is diminished during physiological aging.
Figure 14.
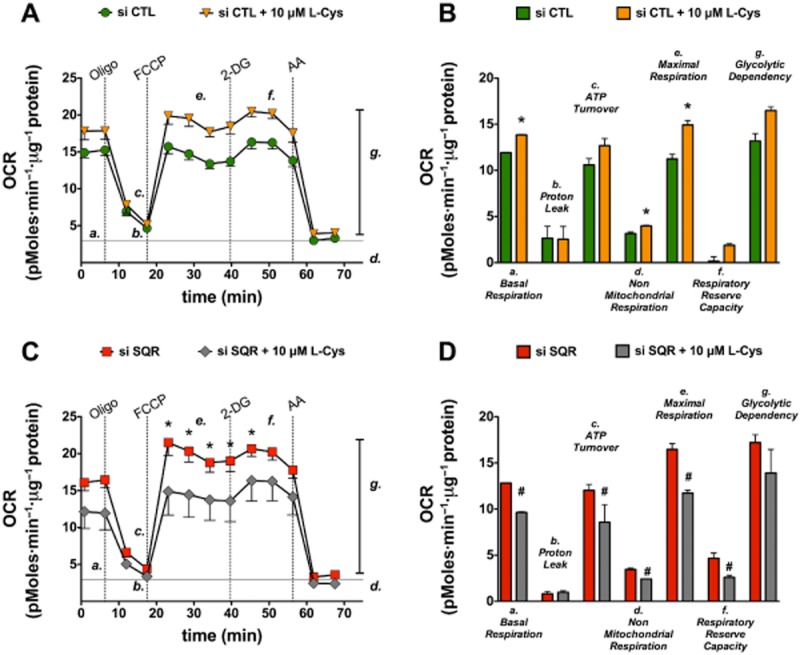
SQR attenuates the stimulatory effect of cysteine on mitochondrial function. Mitochondria were isolated from cultured Hepa1c1c7 cells and subjected to bioenergetic analysis using the Extracellular Flux Analysis method as described (Módis et al., 2013b). (A, B): Mitochondrial function under basal conditions and in the presence of L-cysteine (10 μM) in cultured murine hepatoma cells during the sequential administration of oligomycin (1 μg·mL−1, to inhibit ATP synthase), FCCP (0.3 μM, to induce mitochondrial uncoupling), 2-deoxyglucose (50 mM, to inhibit glycolysis) and antimycin A (2 μg·mL−1, to inhibit Complex III, for determination of non-mitochondrial cellular respiration). (C, D): Mitochondrial function under basal conditions and in the presence of L-cysteine (10 μM) in cultured murine hepatoma cells after SQR silencing (conducted as described in Módis et al., 2013b). Please note that L-cysteine stimulates mitochondrial function in wild-type cells, while it inhibits mitochondrial function in cells with SQR silencing. Data represent mean ± SEM of n = 15 collected from three experiments performed on three different experimental days. Statistical analysis was performed by anova followed by Bonferroni's post hoc test. *P < 0.05 shows significant difference between groups as indicated.
Figure 15.
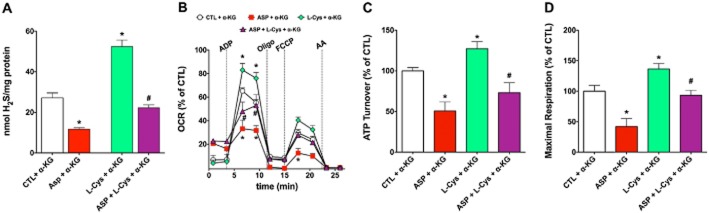
Effect of the cysteine aminotransferase (CAT) inhibitor aspartate on H2S production and function of isolated rat liver mitochondria. Rat liver mitochondria were isolated and studied as described (Módis et al., 2013b). In part (A) mitochondrial homogenates were subjected to H2S measurements using the methylene blue method in the presence or absence of α-ketoglutarate (0.5 mM), aspartate (2 mM) and L-cysteine (1 mM) and their combinations; in parts (B-D), bioenergetic analysis was performed using the Extracellular Flux Analyzer (Seahorse) in the presence or absence of α-ketoglutarate (0.5 mM), aspartate (2 mM) and L-cysteine (10 μM) and their combinations. Please note that L-cysteine stimulated H2S production and mitochondrial function, while the CAT inhibitor aspartate inhibited these responses. Data represent mean ± SEM of three experiments; *P < 0.05. The animal experimentation component of the studies was conducted with the approval of the Animal Care and Use Committee (IACUC) of the University of Texas Medical Branch and according to the applicable guidelines of the National Institute of Health.
Figure 16.
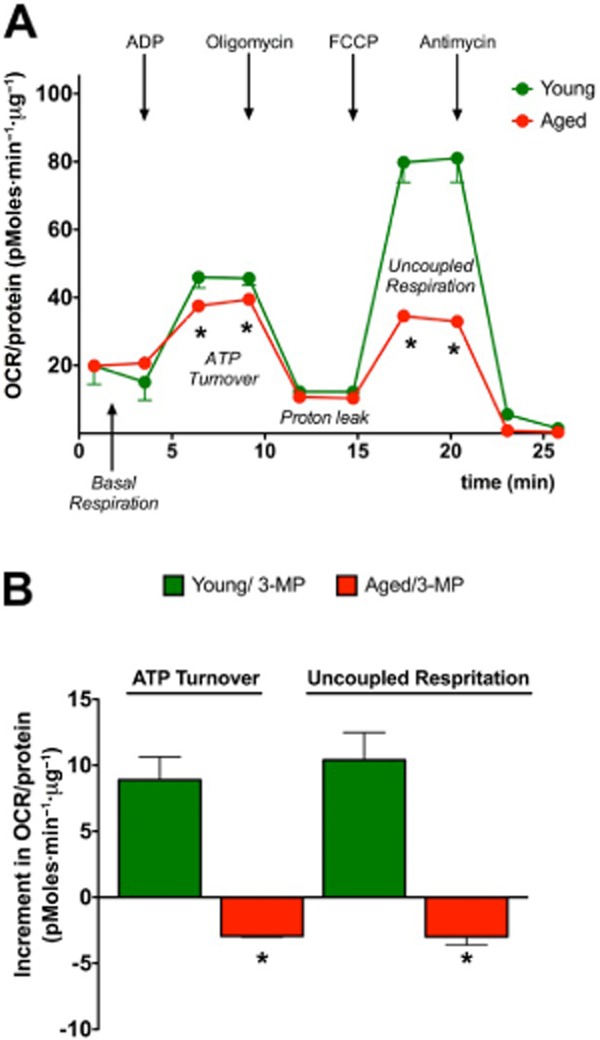
Basal and 3-mercaptopyruvate-induced bioenergetic responses are suppressed in mitochondria prepared from aged mice. Liver mitochondria were isolated from young (2 months old) and aged (18 months old) C57BL/6 mice and subjected to bioenergetic analysis using the Extracellular Flux Analysis method as described (Módis et al., 2013b) in basal conditions or in the presence of 3-mercaptopyruvate (10 μM). Panel A shows the marked suppression of mitochondrial function in aged mice; Panel B shows that the stimulatory effect of 3-MP on mitochondrial function is absent in aged mice. Data represent mean ± SEM of three experiments; *P < 0.05 shows significant difference between the responses of the young and aged groups. The animal experimentation component of the studies was conducted with the approval of the Animal Care and Use Committee (IACUC) of the University of Texas Medical Branch and according to the applicable guidelines of the National Institute of Health.
Currently, there are only limited data to answer the question as to whether the other two enzymes, CSE and CBS also contribute to the physiological regulation of bioenergetic function. Although both CSE and CBS are predominantly cytosolic, the H2S produced by them is highly diffusible. Although there are intracellular sulfide gradients and intracellular enzymes that react with sulfide and impair its free diffusion, it is likely that some of the cytosolic sulfide will reach the mitochondrial inner membrane to modulate electron transport. Moreover, in isolated rat liver mitochondria, partial digestion studies show that CBS is associated with the outer mitochondrial membrane and mitochondrial matrix (Figure 17). Since the distribution of both CSE and CBS is cell type and tissue dependent, one would expect that the bioenergetic modulatory effects of CBS/CSE are cell type and organ dependent. It was demonstrated that CSE plays a role as a supporter of cellular bioenergetics that occurs under conditions of cell dysfunction, and is dependent on the cytosolic-to-mitochondrial translocation of this enzyme (Fu et al., 2012), while in colon cancer cells, endogenous H2S production by CBS supports cellular bioenergetics and cell proliferation (Szabo et al., 2013). Moreover, in the liver tissue and in cultured hepatocytes, ischaemia leads to mitochondrial translocation of CBS, resulting in mitochondrial protective effects (Teng et al., 2013).
Figure 17.
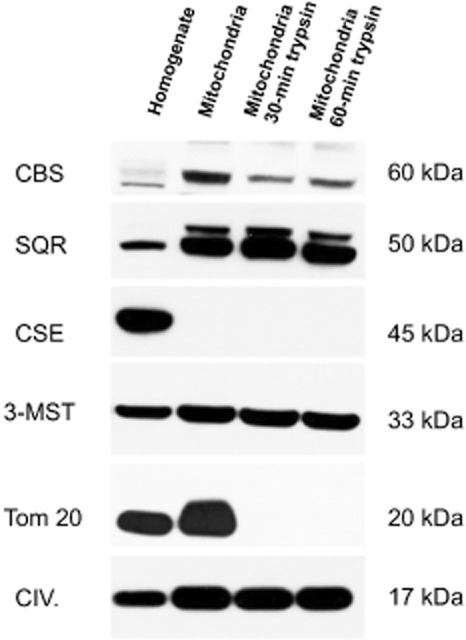
Mitochondrial localization of H2S-producing enzymes. Rat liver mitochondria were isolated as described (Módis et al., 2013b) and subjected to in vitro partial trypsin digestion by treatment with trypsin for 30 or 60 min at room temperature, followed by addition of an equivalent amount of bovine trypsin inhibitor in order to stop the proteolysis, as described (Szabo et al., 2013). Using this method, mitoplasts (mitochondrial preparation without the outer mitochondrial membrane) can be produced. For Western blotting, rat liver homogenate and rat liver isolated mitochondria were lysed in denaturing loading buffer (20 mM Tris, 2% SDS, 10% glycerol, 6 M urea, 100 mg·mL−1 bromophenol blue, 200 mM β-mercaptoethanol), sonicated and boiled. Lysates (25 μg·well−1) were resolved on 4–12% NuPage Bis–Tris acrylamide gels (Invitrogen, Grand Island, NY, USA) and transferred to PVDF membranes. Membranes were blocked in starting BlockTM T20 (TBS), a commercially available blocking buffer solution (Fischer Scientific, Hampton, NH, USA). Membranes were probed overnight with anti-sulfide quinone reductase-like (SQRDL) antibody (anti-SQR; 1:1500; ProteinTech, Chicago, IL, USA), anti 3-mercaptopyruvate sulfurtransferase antibody (anti-3-MST, 1:1000; Sigma-Aldrich, St. Louis, MO, USA), anti-cystathionine β-synthase (anti-CBS, 1:1000) and anti-cystathionine γ-lyase (anti-CSE, 1:1000) primary antibodies. Moreover, the anti-Tom20 antibody (anti-Tom20, Santa Cruz Biotechnology, Dallas, TX, USA) and anti-Complex IV antibody (anti-CIV, 1:1000, Abcam, Cambridge, MA, USA) were used as different mitochondrial markers, Tom20 localized into the outer mitochondrial membrane, while Complex IV being present exclusively in the inner mitochondrial membrane. On the following day, anti-rabbit-HRP conjugate secondary antibody (1:3000) was applied and enhanced chemiluminescent substrate (ECL, Pierce) was used for detecting the signals. SQR was detected at 50 kDa, 3-MST at 33 kDa, CBS at 60 kDa, CSE at 45 kDa, Complex IV at 17 kDa, and Tom20 was detected at 20 kDa. The Western blot shows that CBS is localized to the mitochondrial outer membrane and largely disappears in the samples that were subjected to limited proteolysis, indicating its primary association to the outer membrane and its low expression in mitoplasts (inner mitochondrial membrane). CSE was not mitochondrially associated. 3-MST and SQR were localized to the mitochondrial inner membrane. As expected from their known respective localizations, trypsin digestion abolished Tom 20 immunoreactivity, but maintained Complex IV levels. A representative Western blot of n = 3 independent experiments is shown. The animal experimentation component of the studies was conducted with the approval of the Animal Care and Use Committee (IACUC) of the University of Texas Medical Branch and according to the applicable guidelines of the National Institute of Health.
Taken together, the above results support the conclusion that not only the ability to utilize sulfide as a bioenergetic ‘fuel’ is maintained in mammalian mitochondria, but mammalian cells also produce H2S, to support bioenergetic function, under basal conditions or during cellular emergencies (Figure 18). Silencing of either 3-MST of SQR impairs basal cellular bioenergetics (as evidenced by reduced basal oxygen consumption rate) (Figures 14, 15) (Módis et al., 2013b) and so does a mild degree of oxidative stress (Módis et al., 2013d).
Figure 18.
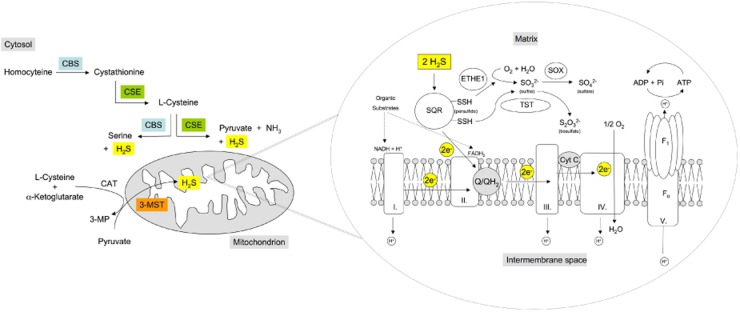
Constitutive and inducible sources of H2S production and associated bioenergetic effects in mitochondria. Left panel: H2S in the mitochondria is typically produced by the constitutively expressed 3-mercaptopyruvate sulfurtransferase (3-MST). In vascular smooth muscle cells, cystathionine γ-lyase (CSE) translocates to the mitochondria in response to cellular calcium overload, while in hepatocytes, hypoxia induces the mitochondrial stabilization and accumulation of cystathionine-β-synthase enzyme (CBS). Moreover, in some cells/tissues (e.g. rat liver; HCT116 human colon carcinoma cell line) CBS is associated to the mitochondrial outer membrane and mitochondrial matrix and produces H2S. CBS and CSE, as well as 3-MST produce H2S from their respective substrates; L-cystathionine/L-cysteine for CSE; L-homocysteine/L-cysteine for CBS and 3-mercaptopyruvate for 3-MST (produced from L-cysteine/α-ketoglutarate by the enzyme CAT). Right panel: mitochondrial electron transport is primarily fuelled by the oxidation of carbon-based substrates, which leads to the reduction of the NAD or FAD coenzymes. The reduced forms of these coenzymes (NADH + H, FADH2) deliver electrons to coenzyme Q of the mitochondrial respiratory chain. NADH + H is oxidized by mitochondrial Complex I. FADH2 coenzymes yield electrons through the function of Complex II (succinate dehydrogenase). Both Complexes (I and II) donate electrons via coenzyme Q of the mitochondrial electron transport chain. H2S, produced in the vicinity of the mitochondria, working in close cooperation with the sulfide-oxidizing unit (SOU), stimulates and balances mitochondrial electron transport. The SOU is constituted of the mitochondrial membrane-bound sulfide quinone reductase (SQR) and of two other enzymes the sulfur dioxygenase (ETHE1, also called dioxygenase ethylmalonic encephalopathy) and the thiosulfate sulfur transferase (TST, also known as one isoenzyme of the rhodanese), ensuring the final oxidation of the two disulfides (-SSH) bound to SQR into oxidized cysteine linked by a disulfide bond. The sulfur dioxygenase in the mitochondrial matrix oxidizes persulfides to sulfite (SO32−), consuming molecular oxygen and water. Sulfite, then, is further oxidized to sulfate (SO42−) by sulfite oxidase (SOX). The thiosulfate sulfur transferase (TST) produces thiosulfate (S2O32−) by transferring the second persulfide from the SQR to sulfite. SQR is responsible for the oxidation of H2S in the mitochondria. While from two H2S molecules, two disulfides (-SSH) bounds are created on the SQR, two electrons derived from two H2S molecules also enter the mitochondrial electron transport chain, promoting mitochondrial ATP generation. Higher concentrations of H2S can also inhibit Complex IV, thereby inhibiting mitochondrial respiration.
All of the above data supporting the bioenergetic role of endogenously produced H2S were obtained in in vitro studies in cultured cells, or in isolated mitochondria. The concept that endogenously produced H2S can contribute to basal energetic function in vivo is supported in recent data in the model organism Caenorhabditis elegans, in which silencing of its CBS homologue (the main H2S-producing enzyme in this animal) (Vozdek et al., 2012; Módis et al., 2013e) attenuates both basal oxygen consumption and the maximal oxygen consumption elicited by the uncoupling agent trifluorocarbonylcyanide phenylhydrazone (FCCP) (Figure 19). Future studies are necessary to probe the role of these pathways in mammalian systems in vivo.
Figure 19.
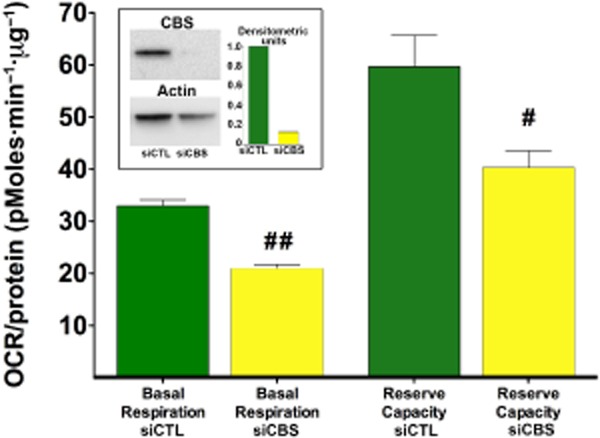
CBS silencing attenuates the bioenergetics in the model organism Caenorhabditis elegans. The C. elegans wild-type N2 Bristol strain was obtained from the C. elegans Genetics Center, University of Minnesota, Minneapolis, MN, USA. Nematode culturing, synchronization and RNAi feeding were performed according to standard techniques (Vozdek et al., 2012). Egg isolation was done using the bleaching method. The resulting embryos were placed in S-complete medium without OP50 bacteria, and allowed to hatch overnight at 20°C with constant rotation. The L1 larvae were counted and supplemented with strain OP50 bacteria with pL4440-CBS or pL4440-scrambled RNAi feeding vector containing Ampicillin resistance marker to resume the life cycle. RNAi induction of bacteria was induced by IPTG. L1 larvae were transferred to a flask in a concentration of 80–100 worms·mL−1 in S-complete. To sterilize the animals, FUDR (5-fluoro-2'-deoxyuridine, Sigma-Aldrich) stock solution was added to each well 50 h after hatching. Five days after reaching adult stage, worms were washed with water and collected for subsequent Western blot analysis. For Western blot analysis followed by immuno-detection of CBS-1 a rabbit polyclonal antibody against recombinant CBS-1 was used. The general technique of the functional analysis of the metabolic activity of the worms was conducted using a 24-well Seahorse Extracellular Flux Analyzer as described (Houtkooper et al., 2013) with 50 worms per well under basal conditions and upon the addition of the uncoupling agent FCCP (10 μM). CBS silencing attenuated CBS-1 protein expression, and suppressed basal and FCCP-stimulated oxygen consumption. Data are shown as mean ± SEM of n = 20 determinations collected from three experimental days. Oxygen consumption was normalized to protein content. Statistical analysis was performed by anova followed by Bonferroni's post hoc test. #P < 0.05 and ##P < 0.01 show lower oxygen consumption in the CBS-1 silenced group, when compared to the respective sham-silenced controls.
Role of mitochondria in the physiological and adaptive responses of intestinal epithelial cells to H2S
The bacteria present in the colonic lumen constitute a source of sulfide. This sulfide derives from bacterial amino acid metabolism and from anaerobic sulfo-reducing bacteria using oxidized ions, such as sulfate, as a final electron acceptor (reviewed in Blachier et al., 2010; Carbonero et al., 2012a,b2012b) (Figure 20). The sources of sulfur are amino acids and peptides that escaped assimilation in the upper part of the digestive tract, proteins of cells detached from the epithelium, and mucins with sulfated sugars and sulfur-containing ions such as sulfate or sulfite (used as conservative agents). The final result is that the sulfide content of the colon is high. Values as high as one millimolar have been reported for the total content (Blachier et al., 2010; Medani et al., 2011; Carbonero et al., 2012a,2012b). A much lower value of 60 μM is reported for free H2S. Antibiotic treatment (ciprofloxacin) has been shown to markedly reduce sulfide production of the intestinal flora (Ohge et al., 2003).
Figure 20.
H2S production by the intestinal microbiota: its clearance and utilization by the gastrointestinal tract. Intestinal bacteria produce H2S from various substrates of alimentary origin (for the overview of the exact biochemical pathways involved, see Carbonero et al., 2012a,b2012b). Colonic epithelial cells take up and degrade, as well as utilize sulfide. A small portion of sulfide enters the circulation, and is further neutralized by the liver. The potential contribution of intestinal microbiota as a source of systemic/tissue sulfide remains to be defined by further studies.
What then could be the relevance of the content in sulfide in the colon lumen with regard to the intestinal epithelial cell? The HT29 cell line derived from human colon cancer was used to study the effect of sulfide on the metabolism of colonocytes. In initial studies, these HT29 cells were exposed to high (1 mM) concentrations of sulfide (Leschelle et al., 2005) and studied 24 h later, a time at which no more sulfide was present as no precaution was taken to avoid/compensate its evaporation. With regard to mitochondria, two parameters changed: (i) the activity of cytochrome oxidase (Complex IV) was decreased and (ii) the proton leak of the inner membrane increased. Both indicated a general suppression of mitochondrial bioenergetics. Cell proliferation was inhibited and lactate release was increased. Therefore, both an increase of anaerobic glycolysis rate and spare of energy expenses took place in response to this sulfide challenge, which were orders of magnitude higher than the Ki of cytochrome oxidase, which was found unchanged in these cells (0.3 μM). However, even if the sulfide challenge was intense, it became apparent that the cells themselves could neutralize the exogenous sulfide (Leschelle et al., 2005).
Subsequent studies explained how cellular respiration participates in this detoxification process: HT29 cells, as well as freshly isolated rat colonocytes, were the first mammalian cells in which mitochondrial oxidation of sulfide were demonstrated (Goubern et al., 2007). The maximal rate of sulfide oxidation increases with differentiation (Mimoun et al., 2012). HT29 cells with the highest differentiation state (day 10–11 and 2 mM butyrate) show a maximal rate of sulfide oxidation, that is very close to the theoretical limit, according to which all the electrons travelling in the respiratory Complexes III–IV come from SQR (Lagoutte et al., 2010). There is no increase in the expression of SOU components with differentiation (Mimoun et al., 2012), most likely due to the fact that even resting cells contain sufficient levels of SQR to oxidize sulfide. In contrast, differentiation increased the maximal possible respiratory rate observed in absence of sulfide but in presence of an uncoupler (Mimoun et al., 2012). This second observation is consistent with an increase in the content of respiratory complexes. However, after inhibition of the activity of the respiratory chain by a high sulfide concentration (Leschelle et al., 2005), the restoration of the respiratory rate after sulfide elimination seems to take place with a reduced intensity whether sulfide exposure was short term (minutes) or long term (hours) (Leschelle et al., 2005; Mimoun et al., 2012), possibly producing irreversible damage.
As mentioned before, the competition with the other pathways creates a problem when sulfide exposure is maximal and, therefore, colonocytes show adaptation (Blachier et al., 2010). Mitochondrial Complex IV catalyzes the reduction of oxygen to form water (an irreversible reaction). In contrast, the other respiratory complexes could work in opposite directions according to the redox conditions. The observation of the sulfide oxidation rates led to the conclusion that Complex I could work in reverse mode when sulfide is oxidized. This phenomenon appears to be specific to colonocytes (Lagoutte et al., 2010). In a previous study, indication of the reversion of Complex II was obtained (Goubern et al., 2007). When Complexes I or II operate in reverse mode, they are no longer competitors of the SOU. Instead, they help the oxidation of sulfide as they accept electrons from the coenzyme Q reduced by the SQR. This could increase the rate of sulfide oxidation but more importantly this oxidation could continue even if Complex IV is severely impaired by high sulfide concentrations. The impact of these reverse reactions on ATP production should be considered as null or negative as, for example, if Complex III or IV are still operating, the reversion of Complex I would cause uncoupling (Lagoutte et al., 2010). If complete inhibition of Complex IV is taking place, in fact, no energy is ‘lost’, but the cell must use anaerobic glycolysis to produce energy instead. Then, in the presence of high sulfide, both the anaerobic glycolysis and mitochondrial reverse reactions in colonocytes would be expected to build up a ‘redox debt’ that might be exported to other unexposed tissues, or could provide colonocytes with a reductive power unmatched in the normal physiology of any other cell.
Looking beyond the intestinal epithelial cell, what could be the relevance of the content in sulfide in the colon lumen with regard to the rest of the organism? Simple calculations produce the following estimations. A plausible range of the colonic internal volume is around 1 L (length 140 cm; diameter 3–4 cm). The total volume is not occupied by sulfide releasing material and the concentration of sulfide mentioned above can be estimated to occupy between 0.1 and 1% of the body volume. Global dilution of colonic free sulfide would increase the sulfide concentration (to 60–600 nM), consistent with SQR activity. Dilution of total sulfide (3–30 μM) may yield potentially harmful concentrations (Blachier et al., 2010). Dilution in the entire body seems unrealistic, but this calculation demonstrates that the colonic content could potentially generate biologically relevant systemic sulfide levels.
While the colonocytes seem to use every possible strategy to increase their rate of sulfide oxidation (see above) to limit the transfer of sulfide to the host (while also using sulfide as source of bioenergetic ‘fuel’), it is likely that a second line of defence exists with the SQR present in other organs and notably the liver, which receives venous blood from the digestive tract via the portal vein (Figure 20). Indeed, the capacity of liver mitochondria to neutralize sulfide appears to be significant, a finding also corroborated by perfused liver studies with sulfide infusion (Norris et al., 2011). Recent studies have directly investigated whether the colon (and the liver) is able to remove all sulfide produced by the bacterial flora, or, alternatively, whether sulfide-producing bacteria are producers of sulfide for the mammalian host organism (conceptually similarly to many vitamins and metabolites that the bacterial flora generates for the host). These studies demonstrated that germ-free mice have significantly reduced free H2S levels in the plasma, as well as they have lower levels of bound sulfane sulfur levels, compared to conventional mice (Shen et al., 2013). There were also marked differences in free H2S levels in some organs (e.g. cecum and colon free H2S levels were lower in the germ-free mice than in the conventional mice), but no differences were noted in the other organs studied. Interestingly, in many organs, bound sulfane sulfur levels tended to be lower in the germ-free mice than in the conventional mice (Shen et al., 2013). Surprisingly, tissue CSE activity was also suppressed in the germ-free mice, for reasons that are currently unclear. Based on these data, it appears that colonic, bacterial sulfide production contributes to the circulating H2S levels, as well as to the H2S levels of the colonic tissue, but probably does not serve as the principal remote source of H2S to most other parenchymal tissues, presumably due to the fact that these tissues have significant autonomous sources of H2S, as well.
Role of mitochondria in other physiological responses to H2S
Another area where H2S-associated metabolic effects may play a physiological regulatory role is the carotid body. In organs specialized for physiological hypoxia sensing (e.g. the carotid body or the kidney medulla), lowered O2 tension may lead to a physiological inhibition of Complex IV by endogenous H2S (produced by CSE). During hypoxia, H2S production increased in glomus cells due to an increased activity of CSE and/or by reduced oxidative inactivation of sulfide, resulting in increased accumulation of sulfide. Increased H2S may directly activate voltage-dependent Ca2+ channels (VDCC) or indirectly activate VDCC by causing membrane depolarization via the decrease in the activity of large-conductance Ca2+-activated K+ channels (Beltowski, 2010; Olson and Whitfield, 2010; Peng et al., 2010; Makarenko et al., 2012; Smith and Yuan, 2012; Olson, 2013). Activation of VDCC raises cytosolic Ca2+ concentration in the glomus cells and increases the sensory activity. In addition, hypoxia-mediated suppression of mitochondrial electron transport via inhibition of Complex IV activity (an effect, which may be enhanced by intracellular acidification during hypoxia) results in the production of reactive oxygen species, which, in turn, may induce additional signalling responses, in part via mobilization of Ca2+ from the endoplasmic reticulum and, in part by activation of the Ca2+-activated Cl− channels (Smith and Yuan, 2012). The hypoxia-mediated increases in intracellular H2S levels and intracellular ROS levels may work synergistically to induce membrane depolarization, resulting in increased sensory activity.
Although the biological roles of H2S span the central and peripheral nervous system, the regulation of immunological/inflammatory responses and various aspects of cardiovascular biology (Fiorucci et al., 2006; Szabo, 2007; Calvert et al., 2010; Elsey et al., 2010; Gadalla and Snyder, 2010; Kimura, 2010; 2013; Predmore and Lefer, 2010; Whiteman and Winyard, 2011; Whiteman et al., 2011; Kimura et al., 2012; Wang, 2012), at present time, there is insufficient information available to implicate H2S-associated mitochondrial or bioenergetic responses in these physiological regulatory mechanisms. It has been demonstrated (Kiss et al., 2008) that the H2S-induced vascular relaxant effect in isolated rat aortic rings resembles in its kinetics and in its bioenergetic aspects (reduction in ATP levels in the smooth muscle) to the vascular relaxation induced by sodium cyanide, and hence may be, at least in part, of metabolic nature. However, the potential physiological relevance of such a regulatory mechanism has not yet been evaluated in vivo. As discussed elsewhere (Módis et al., 2013a), exogenously administered H2S can induce a hypometabolic state. Whether endogenous H2S plays a role in the physiological process of hibernation is a logical question, and one that has not yet been explored. In this context, it is interesting to note, nevertheless, that torpor in hamsters is associated with an increased expression of CBS and an increased H2S tissue content (Talaei et al., 2012). We anticipate that additional work in the field of H2S will unveil many other aspects of H2S-regulated mitochondrial and metabolic function in various physiological contexts. With respect to pathophysiological conditions where the mitochondrial effects of H2S are relevant, the reader is referred to a complementary review article (Módis et al., 2013a).
Acknowledgments
The work of C. S. is supported by grants from the National Institutes of Health (P50GM060338), the Shriners Burns Hospitals (#8661), the American Diabetes Association (7-12-BS-184) and the John Sealy Memorial Endowment Fund for Biomedical Research. C. S. and G. O. thank for the advice of Dr. H. Epstein (Department of Neuroscience and Cell Biology, University of Texas Medical Branch) with setting up the C. elegans experiments. Sadly, Dr. Epstein has passed away since then, but his guidance and his positive spirit will remain with us. The editorial assistance of Ms. Lili Szabo is appreciated.
Glossary
- 3-MP
3-mercaptopyruvate
- 3-MST
3-mercaptopyruvate sulfurtransferase
- CAT
cysteine aminotransferase
- CBS
cystathionine β-synthase
- CSE
cystathionine γ-lyase
- H2S
hydrogen sulfide
- SOU
sulfide oxidizing unit
- VDCC
voltage-dependent Ca2+ channel
Conflict of interest
C. S. is a stockholder and corporate officer of CBS Therapeutics Inc., a for-profit organization involved in the therapeutic exploitation of sulfide biosynthesis inhibitors for cancer therapy.
References
- Attene-Ramos MS, Nava GM, Muellner MG, Wagner ED, Plewa MJ, Gaskins HR. DNA damage and toxicogenomic analyses of hydrogen sulfide in human intestinal epithelial FHs 74 Int cells. Environ Mol Mutagen. 2010;51:304–314. doi: 10.1002/em.20546. [DOI] [PubMed] [Google Scholar]
- Beauchamp RO, Jr, Bus JS, Popp JA, Boreiko CJ, Andjelkovich DA. A critical review of the literature on hydrogen sulfide toxicity. Crit Rev Toxicol. 1984;13:25–97. doi: 10.3109/10408448409029321. [DOI] [PubMed] [Google Scholar]
- Beltowski J. Hypoxia in the renal medulla: implications for hydrogen sulfide signaling. J Pharmacol Exp Ther. 2010;334:358–363. doi: 10.1124/jpet.110.166637. [DOI] [PubMed] [Google Scholar]
- Blachier F, Davila AM, Mimoun S, Benetti PH, Atanasiu C, Andriamihaja M, et al. Luminal sulfide and large intestine mucosa: friend or foe? Amino Acids. 2010;39:335–347. doi: 10.1007/s00726-009-0445-2. [DOI] [PubMed] [Google Scholar]
- Blackstone E, Morrison M, Roth MB. H2S induces a suspended animation-like state in mice. Science. 2005;308:518. doi: 10.1126/science.1108581. [DOI] [PubMed] [Google Scholar]
- Bouillaud F, Blachier F. Mitochondria and sulfide: a very old story of poisoning, feeding, and signaling? Antioxid Redox Signal. 2011;15:379–391. doi: 10.1089/ars.2010.3678. [DOI] [PubMed] [Google Scholar]
- Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Pyriochou A, Roussos C, et al. Hydrogen sulfide is an endogenous inhibitor of phosphodiesterase activity. Arterioscler Thromb Vasc Biol. 2010;30:1998–2004. doi: 10.1161/ATVBAHA.110.209783. [DOI] [PubMed] [Google Scholar]
- Buckler KJ. Effects of exogenous hydrogen sulphide on calcium signalling, background (TASK) K channel activity and mitochondrial function in chemoreceptor cells. Pflugers Arch. 2012;463:743–754. doi: 10.1007/s00424-012-1089-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvert JW, Coetzee WA, Lefer DJ. Novel insights into hydrogen sulfide-mediated cytoprotection. Antioxid Redox Signal. 2010;12:1203–1217. doi: 10.1089/ars.2009.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonero F, Benefiel AC, Alizadeh-Ghamsari AH, Gaskins HR. Microbial pathways in colonic sulfur metabolism and links with health and disease. Front Physiol. 2012a;3:448. doi: 10.3389/fphys.2012.00448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonero F, Benefiel AC, Gaskins HR. Contributions of the microbial hydrogen economy to colonic homeostasis. Nat Rev Gastroenterol Hepatol. 2012b;9:504–518. doi: 10.1038/nrgastro.2012.85. [DOI] [PubMed] [Google Scholar]
- Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Módis K, Panopoulos P, et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc Natl Acad Sci U S A. 2012;109:9161–9166. doi: 10.1073/pnas.1202916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collman JP, Ghosh S, Dey A, Decréau RA. Using a functional enzyme model to understand the chemistry behind hydrogen sulfide induced hibernation. Proc Natl Acad Sci U S A. 2009;106:22090–22095. doi: 10.1073/pnas.0904082106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper CE, Brown GC. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J Bioenerg Biomembr. 2008;40:533–539. doi: 10.1007/s10863-008-9166-6. [DOI] [PubMed] [Google Scholar]
- Di Meo I, Fagiolari G, Prelle A, Viscomi C, Zeviani M, Tiranti V. Chronic exposure to sulfide causes accelerated degradation of cytochrome c oxidase in ethylmalonic encephalopathy. Antioxid Redox Signal. 2011;15:353–362. doi: 10.1089/ars.2010.3520. [DOI] [PubMed] [Google Scholar]
- Dorman DC, Moulin FJ, McManus BE, Mahle KC, James RA, Struve MF. Cytochrome oxidase inhibition induced by acute hydrogen sulfide inhalation: correlation with tissue sulfide concentrations in the rat brain, liver, lung, and nasal epithelium. Toxicol Sci. 2002;65:18–25. doi: 10.1093/toxsci/65.1.18. [DOI] [PubMed] [Google Scholar]
- Dorman DC, Struve MF, Gross EA, Brenneman KA. Respiratory tract toxicity of inhaled hydrogen sulfide in Fischer-344 rats, Sprague-Dawley rats, and B6C3F1 mice following subchronic (90-day) exposure. Toxicol Appl Pharmacol. 2004;198:29–39. doi: 10.1016/j.taap.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Elsey DJ, Fowkes RC, Baxter GF. Regulation of cardiovascular cell function by hydrogen sulfide (H2S) Cell Biochem Funct. 2010;28:95–106. doi: 10.1002/cbf.1618. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Distrutti E, Cirino G, Wallace JL. The emerging roles of hydrogen sulfide in the gastrointestinal tract and liver. Gastroenterology. 2006;131:259–271. doi: 10.1053/j.gastro.2006.02.033. [DOI] [PubMed] [Google Scholar]
- Fu M, Zhang W, Wu L, Yang G, Li H, Wang R. Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proc Natl Acad Sci U S A. 2012;109:2943–2948. doi: 10.1073/pnas.1115634109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadalla MM, Snyder SH. Hydrogen sulfide as a gasotransmitter. J Neurochem. 2010;113:14–26. doi: 10.1111/j.1471-4159.2010.06580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaill F. Aspects of life development at deep sea hydrothermal vents. FASEB J. 1993;7:558–565. doi: 10.1096/fasebj.7.6.8472894. [DOI] [PubMed] [Google Scholar]
- Goffredi SK, Childress JJ, Desaulniers NT, Lallier FJ. Sulfide acquisition by the vent worm Riftia pachyptila appears to be via uptake of HS-, rather than H2S. J Exp Biol. 1997;200:2609–2616. doi: 10.1242/jeb.200.20.2609. [DOI] [PubMed] [Google Scholar]
- Goubern M, Andriamihaja M, Nubel T, Blachier F, Bouillaud F. Sulfide, the first inorganic substrate for human cells. FASEB J. 2007;21:1699–1706. doi: 10.1096/fj.06-7407com. [DOI] [PubMed] [Google Scholar]
- Grieshaber MK, Volkel S. Animal adaptations for tolerance and exploitation of poisonous sulfide. Annu Rev Physiol. 1998;60:33–53. doi: 10.1146/annurev.physiol.60.1.33. [DOI] [PubMed] [Google Scholar]
- Groeger M, Wagner F, Baumgart K, Huber-Lang M, Knoeferl M, Georgieff M, et al. Mitochondrial respiration and cytochrome c inhibition by sulfide in peritoneal macrophages in vitro: effects of temperature and pH. Crit Care. 2010;14:P6. [Google Scholar]
- Groeger M, Matallo J, McCook O, Wagner F, Wachter U, Bastian O, et al. Temperature and cell-type dependency of sulfide effects on mitochondrial respiration. Shock. 2012;38:367–374. doi: 10.1097/SHK.0b013e3182651fe6. [DOI] [PubMed] [Google Scholar]
- Heinen W, Lauwers AM. Organic sulfur compounds resulting from the interaction of iron sulfide, hydrogen sulfide and carbon dioxide in an anaerobic aqueous environment. Orig Life Evol Biosph. 1996;26:131–150. doi: 10.1007/BF01809852. [DOI] [PubMed] [Google Scholar]
- Hildebrandt TM, Grieshaber MK. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 2008;275:3352–3361. doi: 10.1111/j.1742-4658.2008.06482.x. [DOI] [PubMed] [Google Scholar]
- Hill BC, Woon TC, Nicholls P, Peterson J, Greenwood C, Thomson AJ. Interactions of sulphide and other ligands with cytochrome c oxidase. An electron-paramagnetic-resonance study. Biochem J. 1984;224:591–600. doi: 10.1042/bj2240591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature. 2013;497:451–457. doi: 10.1038/nature12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insko MA, Deckwerth TL, Hill P, Toombs CF, Szabo C. Detection of exhaled hydrogen sulphide gas in rats exposed to intravenous sodium sulphide. Br J Pharmacol. 2009;157:944–951. doi: 10.1111/j.1476-5381.2009.00248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabil O, Banerjee R. Redox biochemistry of hydrogen sulfide. J Biol Chem. 2010;285:21903–21907. doi: 10.1074/jbc.R110.128363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamijo Y, Takai M, Fujita Y, Hirose Y, Iwasaki Y, Ishihara S. A multicenter retrospective survey on a suicide trend using hydrogen sulfide in Japan. Clin Toxicol (Phila) 2013;51:425–428. doi: 10.3109/15563650.2013.799676. [DOI] [PubMed] [Google Scholar]
- Kato S, Takano Y, Kakegawa T, Oba H, Inoue K, Kobayashi C, et al. Biogeography and biodiversity in sulfide structures of active and inactive vents at deep-sea hydrothermal fields of the Southern Mariana Trough. Appl Environ Microbiol. 2010;76:2968–2979. doi: 10.1128/AEM.00478-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AA, Schuler MM, Prior MG, Yong S, Coppock RW, Florence LZ, et al. Effects of hydrogen sulfide exposure on lung mitochondrial respiratory chain enzymes in rats. Toxicol Appl Pharmacol. 1990;103:482–490. doi: 10.1016/0041-008x(90)90321-k. [DOI] [PubMed] [Google Scholar]
- Khan AA, Yong S, Prior MG, Lillie LE. Cytotoxic effects of hydrogen sulfide on pulmonary alveolar macrophages in rats. J Toxicol Environ Health. 1991;33:57–64. doi: 10.1080/15287399109531505. [DOI] [PubMed] [Google Scholar]
- Kimura H. Hydrogen sulfide: from brain to gut. Antioxid Redox Signal. 2010;12:1111–1123. doi: 10.1089/ars.2009.2919. [DOI] [PubMed] [Google Scholar]
- Kimura H. Production and physiological effects of hydrogen sulfide. Antioxid Redox Signal. 2013 doi: 10.1089/ars.2013.5309. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Shibuya N, Kimura Y. Hydrogen sulfide is a signaling molecule and a cytoprotectant. Antioxid Redox Signal. 2012;17:45–57. doi: 10.1089/ars.2011.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss L, Deitch EA, Szabo C. Hydrogen sulfide decreases adenosine triphosphate levels in aortic rings and leads to vasorelaxation via metabolic inhibition. Life Sci. 2008;83:589–594. doi: 10.1016/j.lfs.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundell FA. A suggested pioneer organism for the Wächtershäuser origin of life hypothesis. Orig Life Evol Biosph. 2011;41:175–198. doi: 10.1007/s11084-010-9217-y. [DOI] [PubMed] [Google Scholar]
- Lagoutte E, Mimoun S, Andriamihaja M, Chaumontet C, Blachier F, Bouillaud F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim Biophys Acta. 2010;1797:1500–1511. doi: 10.1016/j.bbabio.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Leschelle X, Goubern M, Andriamihaja M, Blottiere HM, Couplan E, Gonzalez-Barroso MD, et al. Adaptative metabolic response of human colonic epithelial cells to the adverse effects of the luminal compound sulfide. Biochim Biophys Acta. 2005;1725:201–212. doi: 10.1016/j.bbagen.2005.06.002. [DOI] [PubMed] [Google Scholar]
- MacLeod G, McKeown C, Hall AJ, Russell MJ. Hydrothermal and oceanic pH conditions of possible relevance to the origin of life. Orig Life Evol Biosph. 1994;24:19–41. doi: 10.1007/BF01582037. [DOI] [PubMed] [Google Scholar]
- Makarenko VV, Nanduri J, Raghuraman G, Fox AP, Gadalla MM, Kumar GK, et al. Endogenous H2S is required for hypoxic sensing by carotid body glomus cells. Am J Physiol Cell Physiol. 2012;303:C916–C923. doi: 10.1152/ajpcell.00100.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medani M, Collins D, Docherty NG, Baird AW, O'Connell PR, Winter DC. Emerging role of hydrogen sulfide in colonic physiology and pathophysiology. Inflamm Bowel Dis. 2011;17:1620–1625. doi: 10.1002/ibd.21528. [DOI] [PubMed] [Google Scholar]
- Mielke RE, Robinson KJ, White LM, McGlynn SE, McEachern K, Bhartia R, et al. Iron-sulfide-bearing chimneys as potential catalytic energy traps at life's emergence. Astrobiology. 2011;11:933–950. doi: 10.1089/ast.2011.0667. [DOI] [PubMed] [Google Scholar]
- Mikami Y, Shibuya N, Kimura Y, Nagahara N, Ogasawara Y, Kimura H. Thioredoxin and dihydrolipoic acid are required for 3-mercaptopyruvate sulfurtransferase to produce hydrogen sulfide. Biochem J. 2011;439:479–485. doi: 10.1042/BJ20110841. [DOI] [PubMed] [Google Scholar]
- Milby TH, Baselt RC. Hydrogen sulfide poisoning: clarification of some controversial issues. Am J Ind Med. 1999;35:192–195. doi: 10.1002/(sici)1097-0274(199902)35:2<192::aid-ajim11>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Mimoun S, Andriamihaja M, Chaumontet C, Atanasiu C, Benamouzig R, Blouin JM, et al. Detoxification of H2S by differentiated colonic epithelial cells: implication of the sulfide oxidizing unit and of the cell respiratory capacity. Antioxid Redox Signal. 2012;17:1–10. doi: 10.1089/ars.2011.4186. [DOI] [PubMed] [Google Scholar]
- Módis K, Coletta C, Papapetropoulos A, Bos EM, Calzia E, van Goor H, et al. Regulation of mitochondrial function by hydrogen sulfide. Part II. Pathophysiological and therapeutic implications. Br J Pharmacol. 2013a doi: 10.1111/bph.12368. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Módis K, Coletta C, Erdélyi K, Papapetropoulos A, Szabo C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013b;27:601–611. doi: 10.1096/fj.12-216507. [DOI] [PubMed] [Google Scholar]
- Módis K, Panopoluos P, Coletta C, Papapetropoulos A, Szabo C. Hydrogen sulfide-mediated stimulation of mitochondrial electron transport involves inhibition of the mitochondrial phosphodiesterase 2A, elevation of cAMP and activation of protein kinase A. Biochem Pharmacol. 2013c doi: 10.1016/j.bcp.2013.08.064. (in press) [DOI] [PubMed] [Google Scholar]
- Módis K, Asimakopoulou A, Coletta C, Papapetropoulos A, Szabo C. Oxidative stress suppresses the cellular bioenergetic effect of the 3-mercaptopyruvate sulfurtransferase/hydrogen sulfide pathway. Biochem Biophys Res Commun. 2013d;433:401–407. doi: 10.1016/j.bbrc.2013.02.131. [DOI] [PubMed] [Google Scholar]
- Módis K, Wolanska K, Wozdek R. Hydrogen sulfide in cell signaling, signal transduction, cellular bioenergetics and physiology in C. elegans. Gen Physiol Biophys. 2013e;32:1–22. doi: 10.4149/gpb_2013001. [DOI] [PubMed] [Google Scholar]
- Mulkidjanian AY, Bychkov AY, Dibrova DV, Galperin MY, Koonin EV. Open questions on the origin of life at anoxic geothermal fields. Orig Life Evol Biosph. 2012;42:507–516. doi: 10.1007/s11084-012-9315-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa MG, Hacker AD, Ospital JJ, Hussain MZ, Lee SD. Biochemical effects of environmental pollutants in animal lungs. In: Lee SD, editor. Biochemical Effects of Environmental Pollutants. Ann Arbor: Ann Arbor Science; 1977. pp. 59–96. [Google Scholar]
- Nagahara N. Regulation of mercaptopyruvate sulfurtransferase activity via intrasubunit and intersubunit redox-sensing switches. Antioxid Redox Signal. 2013 doi: 10.1089/ars.2012.5031. (in press) [DOI] [PubMed] [Google Scholar]
- Nagahara N, Katayama A. Post-translational regulation of mercaptopyruvate sulfurtransferase via a low redox potential cysteine-sulfenate in the maintenance of redox homeostasis. J Biol Chem. 2005;280:34569–34576. doi: 10.1074/jbc.M505643200. [DOI] [PubMed] [Google Scholar]
- Nagahara N, Nirasawa T, Yoshii T, Niimura Y. Is novel signal transducer sulfur oxide involved in the redox cycle of persulfide at the catalytic site cysteine in a stable reaction intermediate of mercaptopyruvate sulfurtransferase? Antioxid Redox Signal. 2012;16:747–753. doi: 10.1089/ars.2011.4468. [DOI] [PubMed] [Google Scholar]
- Nagy P, Pálinkás Z, Nagy A, Budai B, Tóth I, Vasas A. Chemical aspects of hydrogen sulfide measurements in physiological samples. Biochim Biophys Acta. 2013 doi: 10.1016/j.bbagen.2013.05.037. (in press) [DOI] [PubMed] [Google Scholar]
- Nicholls P. The effect of sulphide on cytochrome aa3. Isosteric and allosteric shifts of the reduced alpha-peak. Biochim Biophys Acta. 1975;396:24–35. doi: 10.1016/0005-2728(75)90186-3. [DOI] [PubMed] [Google Scholar]
- Nicholls P, Kim JK. Oxidation of sulphide by cytochrome aa3. Biochim Biophys Acta. 1981;637:312–320. doi: 10.1016/0005-2728(81)90170-5. [DOI] [PubMed] [Google Scholar]
- Nicholls P, Kim JK. Sulphide as an inhibitor and electron donor for the cytochrome c oxidase system. Can J Biochem. 1982;60:613–623. doi: 10.1139/o82-076. [DOI] [PubMed] [Google Scholar]
- Nicholson RA, Roth SH, Zhang A, Zheng J, Brookes J, Skrajny B, et al. Inhibition of respiratory and bioenergetic mechanisms by hydrogen sulfide in mammalian brain. J Toxicol Environ Health A. 1998;54:491–507. doi: 10.1080/009841098158773. [DOI] [PubMed] [Google Scholar]
- Norris EJ, Culberson CR, Narasimhan S, Clemens MG. The liver as a central regulator of hydrogen sulfide. Shock. 2011;36:242–250. doi: 10.1097/SHK.0b013e3182252ee7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohge H, Furne JK, Springfield J, Sueda T, Madoff RD, Levitt MD. The effect of antibiotics and bismuth on fecal hydrogen sulfide and sulfate-reducing bacteria in the rat. FEMS Microbiol Lett. 2003;228:137–142. doi: 10.1016/S0378-1097(03)00748-1. [DOI] [PubMed] [Google Scholar]
- Olson KR. A practical look at the chemistry and biology of hydrogen sulfide. Antioxid Redox Signal. 2012;17:32–44. doi: 10.1089/ars.2011.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson KR. Hydrogen sulfide as an oxygen sensor. Clin Chem Lab Med. 2013;51:623–632. doi: 10.1515/cclm-2012-0551. [DOI] [PubMed] [Google Scholar]
- Olson KR, Whitfield NL. Hydrogen sulfide and oxygen sensing in the cardiovascular system. Antioxid Redox Signal. 2010;12:1219–1234. doi: 10.1089/ars.2009.2921. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Nanduri J, Raghuraman G, Souvannakitti D, Gadalla MM, Kumar GK, et al. H2S mediates O2 sensing in the carotid body. Proc Natl Acad Sci U S A. 2010;107:10719–10724. doi: 10.1073/pnas.1005866107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perner M, Hansen M, Seifert R, Strauss H, Koschinsky A, Petersen S. Linking geology, fluid chemistry, and microbial activity of basalt-and ultramafic-hosted deep-sea hydrothermal vent environments. Geobiology. 2013;11:340–355. doi: 10.1111/gbi.12039. [DOI] [PubMed] [Google Scholar]
- Petersen LC. The effect of inhibitors on the oxygen kinetics of cytochrome c oxidase. Biochim Biophys Acta. 1977;460:299–307. doi: 10.1016/0005-2728(77)90216-x. [DOI] [PubMed] [Google Scholar]
- Powell MA, Somero GN. Hydrogen sulfide oxidation is coupled to oxidative phosphorylation in mitochondria of Solemya reidi. Science. 1986;233:563–566. doi: 10.1126/science.233.4763.563. [DOI] [PubMed] [Google Scholar]
- Predmore BL, Lefer DJ. Development of hydrogen sulfide-based therapeutics for cardiovascular disease. J Cardiovasc Transl Res. 2010;3:487–498. doi: 10.1007/s12265-010-9201-y. [DOI] [PubMed] [Google Scholar]
- Reiffenstein RJ, Hulbert WC, Roth SH. Toxicology of hydrogen sulfide. Annu Rev Pharmacol Toxicol. 1992;32:109–134. doi: 10.1146/annurev.pa.32.040192.000545. [DOI] [PubMed] [Google Scholar]
- Roberts ES, Wong VA, McManus BE, Marshall MW, Lancianese S, Dorman DC. Changes in intracellular pH play a secondary role in hydrogen sulfide-induced nasal cytotoxicity. Inhal Toxicol. 2006;18:159–167. doi: 10.1080/08958370500434156. [DOI] [PubMed] [Google Scholar]
- Searcy DG. Metabolic integration during the evolutionary origin of mitochondria. Cell Res. 2003;13:229–238. doi: 10.1038/sj.cr.7290168. [DOI] [PubMed] [Google Scholar]
- Shen X, Carlström M, Borniquel S, Jädert C, Kevil CG, Lundberg JO. Microbial regulation of host hydrogen sulfide bioavailability and metabolism. Free Radic Biol Med. 2013;60:195–200. doi: 10.1016/j.freeradbiomed.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Shao M, Zhang L, Ma Y, Zhang Z. Screening of genes related to sulfide metabolism in Urechis unicinctus (Echiura, Urechidae) using suppression subtractive hybridization and cDNA microarray analysis. Comp Biochem Physiol Part D Genomics Proteomics. 2012;7:254–259. doi: 10.1016/j.cbd.2012.04.001. [DOI] [PubMed] [Google Scholar]
- Shibuya N, Mikami Y, Kimura Y, Nagahara N, Kimura H. Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. J Biochem. 2009a;146:623–626. doi: 10.1093/jb/mvp111. [DOI] [PubMed] [Google Scholar]
- Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K, et al. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid Redox Signal. 2009b;11:703–714. doi: 10.1089/ars.2008.2253. [DOI] [PubMed] [Google Scholar]
- Smith KA, Yuan JX. H2S, a gasotransmitter for oxygen sensing in carotid body. Focus on ‘Endogenous H2S is required for hypoxic sensing by carotid body glomus cells’. Am J Physiol Cell Physiol. 2012;303:C911–C912. doi: 10.1152/ajpcell.00307.2012. [DOI] [PubMed] [Google Scholar]
- Struve MF, Brisbois JN, James RA, Marshall MW, Dorman DC. Neurotoxicological effects associated with short-term exposure of Sprague-Dawley rats to hydrogen sulfide. Neurotoxicology. 2001;22:375–385. doi: 10.1016/s0161-813x(01)00021-3. [DOI] [PubMed] [Google Scholar]
- Sun WH, Liu F, Chen Y, Zhu YC. Hydrogen sulfide decreases the levels of ROS by inhibiting mitochondrial Complex IV and increasing SOD activities in cardiomyocytes under ischemia/reperfusion. Biochem Biophys Res Commun. 2012;421:164–169. doi: 10.1016/j.bbrc.2012.03.121. [DOI] [PubMed] [Google Scholar]
- Sylvan JB, Toner BM, Edwards KJ. Life and death of deep-sea vents: bacterial diversity and ecosystem succession on inactive hydrothermal sulfides. Mbio. 2012;3:e00279–e00211. doi: 10.1128/mBio.00279-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- Szabo C, Coletta C, Chao C, Módis K, Szczeszny B, Papapetropoulos A, et al. Tumor-derived H2S, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation and angiogenesis in colon cancer. Proc Natl Acad Sci U S A. 2013;110:12974–12979. doi: 10.1073/pnas.1306241110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo G, Veres G, Radovits T, Gero D, Módis K, Miesel-Gröschel C, et al. Cardioprotective effects of hydrogen sulfide. Nitric Oxide. 2011;25:201–210. doi: 10.1016/j.niox.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szoleczky P, Módis K, Nagy N, Dóri Tóth Z, DeWitt D, Szabo C, et al. Identification of agents that reduce renal hypoxia-reoxygenation injury using cell-based screening: purine nucleosides are alternative energy sources in LLC-PK1 cells during hypoxia. Arch Biochem Biophys. 2012;517:53–70. doi: 10.1016/j.abb.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talaei F, Bouma HR, Hylkema MN, Strijkstra AM, Boerema AS, Schmidt M, et al. The role of endogenous H2S formation in reversible remodeling of lung tissue during hibernation in the Syrian hamster. J Exp Biol. 2012;215:2912–2919. doi: 10.1242/jeb.067363. [DOI] [PubMed] [Google Scholar]
- Tanizawa K. Production of H2S by 3-mercaptopyruvate sulphurtransferase. J Biochem. 2011;149:357–359. doi: 10.1093/jb/mvr018. [DOI] [PubMed] [Google Scholar]
- Teng H, Wu B, Zhao K, Yang G, Wu L, Wang R. Oxygen-sensitive mitochondrial accumulation of cystathione β-synthase mediated by Lon protease. Proc Natl Acad Sci U S A. 2013;110:12679–12684. doi: 10.1073/pnas.1308487110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RW, Valentine HL, Valentine WM. Cytotoxic mechanisms of hydrosulfide anion and cyanide anion in primary rat hepatocyte cultures. Toxicology. 2003;188:149–159. doi: 10.1016/s0300-483x(03)00079-9. [DOI] [PubMed] [Google Scholar]
- Toombs CF, Insko MA, Wintner EA, Deckwerth TL, Usansky H, Jamil K, et al. Detection of exhaled hydrogen sulphide gas in healthy human volunteers during intravenous administration of sodium sulphide. Br J Clin Pharmacol. 2010;69:626–636. doi: 10.1111/j.1365-2125.2010.03636.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong DH, Eghbal MA, Hindmarsh W, Roth SH, O'Brien PJ. Molecular mechanisms of hydrogen sulfide toxicity. Drug Metab Rev. 2006;38:733–744. doi: 10.1080/03602530600959607. [DOI] [PubMed] [Google Scholar]
- Volkel S, Grieshaber M. Mitochondrial sulfide oxidation in Arenicola marina. Eur J Biochem. 1997;235:231–237. doi: 10.1111/j.1432-1033.1996.00231.x. [DOI] [PubMed] [Google Scholar]
- Volpato GP, Searles R, Yu B, Scherrer-Crosbie M, Bloch KD, Ichinose F, et al. Inhaled hydrogen sulfide: a rapidly reversible inhibitor of cardiac and metabolic function in the mouse. Anesthesiology. 2008;108:659–668. doi: 10.1097/ALN.0b013e318167af0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vozdek R, Hnízda A, Krijt J, Kostrouchová M, Kožich V. Novel structural arrangement of nematode cystathionine β-synthases: characterization of Caenorhabditis elegans CBS-1. Biochem J. 2012;443:535–547. doi: 10.1042/BJ20111478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner F, Asfar P, Calzia E, Radermacher P, Szabo C. Bench-to-bedside review: hydrogen sulfide – the third gaseous transmitter: applications for critical care. Crit Care. 2009;13:213. doi: 10.1186/cc7700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- Wharton DC, Tzagoloff A. Cytochrome oxidase from beef heart mitochondria. In: Estabrook RW, Pullman ME, editors. Methods in Enzymology. Vol. 10. New York: Academic Press; 1967. pp. 245–250. Vol. [Google Scholar]
- Whiteman M, Winyard PG. Hydrogen sulfide and inflammation: the good, the bad, the ugly and the promising. Expert Rev Clin Pharmacol. 2011;4:13–32. doi: 10.1586/ecp.10.134. [DOI] [PubMed] [Google Scholar]
- Whiteman M, Le Trionnaire S, Chopra M, Fox B, Whatmore J. Emerging role of hydrogen sulfide in health and disease: critical appraisal of biomarkers and pharmacological tools. Clin Sci (Lond) 2011;121:459–488. doi: 10.1042/CS20110267. [DOI] [PubMed] [Google Scholar]
- Wintner EA, Deckwerth TL, Langston W, Bengtsson A, Leviten D, Hill P, et al. A monobromobimane-based assay to measure the pharmacokinetic profile of reactive sulphide species in blood. Br J Pharmacol. 2010;160:941–957. doi: 10.1111/j.1476-5381.2010.00704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, Du X, Wang D, Hao F. Myocardial and lung injuries induced by hydrogen sulfide and the effectiveness of oxygen therapy in rats. Clin Toxicol (Phila) 2011;49:161–166. doi: 10.3109/15563650.2011.565419. [DOI] [PubMed] [Google Scholar]
- Yadav PK, Yamada K, Chiku T, Koutmos M, Banerjee R. Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J Biol Chem. 2013;288:20002–20013. doi: 10.1074/jbc.M113.466177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong R, Searcy DG. Sulfide oxidation coupled to ATP synthesis in chicken liver mitochondria. Comp Biochem Physiol B Biochem Mol Biol. 2001;129:129–137. doi: 10.1016/s1096-4959(01)00309-8. [DOI] [PubMed] [Google Scholar]