

Abstract
Emerging work demonstrates the dual regulation of mitochondrial function by hydrogen sulfide (H2S), including, at lower concentrations, a stimulatory effect as an electron donor, and, at higher concentrations, an inhibitory effect on cytochrome C oxidase. In the current article, we overview the pathophysiological and therapeutic aspects of these processes. During cellular hypoxia/acidosis, the inhibitory effect of H2S on complex IV is enhanced, which may shift the balance of H2S from protective to deleterious. Several pathophysiological conditions are associated with an overproduction of H2S (e.g. sepsis), while in other disease states H2S levels and H2S bioavailability are reduced and its therapeutic replacement is warranted (e.g. diabetic vascular complications). Moreover, recent studies demonstrate that colorectal cancer cells up-regulate the H2S-producing enzyme cystathionine β-synthase (CBS), and utilize its product, H2S, as a metabolic fuel and tumour-cell survival factor; pharmacological CBS inhibition or genetic CBS silencing suppresses cancer cell bioenergetics and suppresses cell proliferation and cell chemotaxis. In the last chapter of the current article, we overview the field of H2S-induced therapeutic ‘suspended animation’, a concept in which a temporary pharmacological reduction in cell metabolism is achieved, producing a decreased oxygen demand for the experimental therapy of critical illness and/or organ transplantation.
Linked Articles
This article is part of a themed issue on Mitochondrial Pharmacology: Energy, Injury & Beyond. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2014.171.issue-8
Keywords: mitochondrial electron transport, bioenergetics, 3-mercaptopyruvate sulfurtransferase, gasotransmitters, blood vessels, nitric oxide, superoxide, free radicals, cysteine, ischaemia, shock, suspended animation
Introduction
H2S, a colourless, flammable, water-soluble gas, is gaining increased attention as an endogenous biological mediator. The distribution and regulation of the three H2S-producing enzymes [cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE) and 3-mercaptopyruvate sulfurtransferase (3-MST)], and the wide range of biological effects of H2S are discussed in separate reviews (Fiorucci et al., 2006; Szabo, 2007; Calvert et al., 2010; Elsey et al., 2010; Gadalla and Snyder, 2010; Kimura, 2010; 2013; Predmore and Lefer, 2010; Whiteman and Winyard, 2011; Whiteman et al., 2011; Kimura et al., 2012; Wang, 2012). In a recent article, we have overviewed the dual mitochondrial effects of H2S, which range from stimulatory effects, occurring at lower concentrations, to the suppression of mitochondrial function, which occurs at higher concentrations (Szabo et al., 2013a). The purpose of the current article is to outline the physiological, pathophysiological and therapeutic aspects of this regulation. Similar to our approach in the companion article (Szabo et al., 2013a), in the current article we use the terms ‘sulfide’ and H2S interchangeably to collectively refer to H2S gas as well as its two ionized forms in solution: HS- and S2-.
H2S as a potential inducer of cytopathic hypoxia in circulatory shock
Various forms of circulatory shock lead to an intrinsic impairment of mitochondrial function, a phenomenon often called ‘cytopathic hypoxia’ (Fink, 2001; Levy, 2007; LaRosa and Opal, 2008). This phenomenon entails the intrinsic impairment of the mitochondria to utilize oxygen, and manifests itself in a narrow arteriovenous oxygen difference due to the inability of the ‘poisoned’ tissue to ‘extract’ oxygen during circulatory shock.
There are several lines of published studies using shock models induced by bacterial LPS, and by cecal ligation and puncture (CLP, a polymicrobial model of sepsis), showing overproduction of H2S and/or the beneficial effect of inhibitors of H2S production such as propargylglycine (PAG) (reviewed in Wagner et al., 2009; Coletta and Szabo, 2013). There are also several clinical studies demonstrating the overproduction of sulfide in patients with various forms of circulatory shock (Goslar et al., 2011). It is a logical assumption, therefore, that endogenously produced sulfide may contribute to the pathogenesis of cytopathic hypoxia in circulatory shock, especially in light of the findings showing that acidosis (a common feature of shock) enhances the inhibitory effect of sulfide on complex IV (Groeger et al., 2010; Szabo et al., 2013a). Nevertheless, the question whether endogenously produced sulfide contributes to cytopathic hypoxia has not yet been directly or comprehensively addressed; nor has it been explored how much the specific pattern of perfusion and oxygenation occurring in the tissue during the course of a shock state may modulate this response. For example, in a porcine model of haemorrhagic shock (Bracht et al., 2012b), it was found that a pronounced reduction of mitochondrial respiratory activity occurs in terms of oxygen flux (JO2) per amount of tissue in permeabilized skeletal muscle biopsies after 3 h of shock, when compared with the control before the haemorrhage (control, JO2 = 44 ± 12 [pmol/(s*mg tissue)] vs. 3 h shock, JO2 = 25 ± 7 [pmol/(s*mg tissue)], P < 0.001). However, 12 h of reperfusion allowed mitochondrial respiration to almost completely recover (JO2 = 40 ± 6 [pmol/(s*mg tissue)], P < 0.001), suggesting that mitochondria highly dynamically adapt to the actual circulatory and oxygenation conditions in this experimental model. Continuous infusion of the sulfide donor Na2S did not affect the response to shock and reperfusion (control, JO2 = 35 ± 13 [pmol/(s*mg tissue)] vs. 3 h shock, JO2 = 22 ± 8 [pmol/(s*mg tissue)], P < 0.001, and 12 h recovery, JO2 = 39 ± 15 [pmol/(s*mg tissue)], P < 0.001), suggesting that exogenously administered sulfide does not induce or exacerbate cytopathic hypoxia in this model. It is important to note that the direct answer as to whether endogenous sulfide contributes to the cytopathic hypoxia in circulatory shock would require studies in which H2S overproduction in shock is inhibited (by pharmacological tools or by genetically modified animals), followed by metabolic analysis of tissues ex vivo. Such studies have not yet been conducted.
Indirect lines of data, nevertheless, indicate that sulfide may have an impact on mitochondrial respiration during circulatory shock. For example, as shown in Figure 1, CLP-induced septic shock reduces the maximum mitochondrial respiratory capacity (ETS capacity) in the liver of mice, this effect being more pronounced under hypothermia than under normothermia (diagram A). Comparison of the respiratory activity compensating for the proton leak at the inner mitochondrial membrane (LEAK respiration) with the OxPhos capacity to the maximum respiration in the uncoupled (ETS) state (L/E and P/E ratios, respectively, diagram C) reveals that the reduction of maximum mitochondrial respiratory capacity is pronounced even under normothermic conditions. Interestingly, treating the animals with sulfide fully compensated for these CLP-related effects, thus maintaining normal mitochondrial activity regardless of body temperature in septic shock (diagrams B and D). These findings would be consistent with a therapeutic effect of exogenous sulfide administration, rather than a potential causative role of sulfide in inducing cytopathic hypoxia in shock. The different effects of sulfide under normo-and hypothermia are clearly explained by the strong temperature-dependent inhibition of mitochondrial respiration by sulfide, which is much weaker at low body temperature (Baumgart et al., 2010).
Figure 1.

Effect of H2S and hypothermia on mitochondrial respiration in mechanically permeabilized small murine liver biopsies. Eight groups of anaesthetized and mechanically ventilated mice were studied in order to combine all the treatments of interest, namely sham operation versus CLP (closed white and black bars vs. light and grey bars), normothermia (38°C, white and light grey bars) versus hypothermia (27°C, black and dark grey bars) and i.v. application of the sulfide donor Na2S (diagrams B and D) or placebo (diagrams A and C). The study design as well as the methods used to measure mitochondrial respiratory activity in the tissue samples is described in Baumgart et al. (2010). Briefly, mitochondrial respiration was measured in terms of oxygen flux (JO2) per mg wet tissue under maximum stimulation by complex I + II substrates and ADP in the coupled (OxPhos) and FCCP-induced uncoupled (ETS) conditions. The Leak state (Leak) was obtained in the OxPhos state by inhibiting the ATP synthesis by oligomycin, and represents the respiratory activity necessary to compensate for the proton leakage, slipping and cations exchange along the inner mitochondrial membrane. Relating the Leak state to the OxPhos state and to the ETS state yields the L/P and L/E ratios, respectively, the P/E ratio is the ratio OxPhos to ETS state (see diagrams C and D). Data are shown as mean ± SEM of n = 6 determinations, *P < 0.05.
The study of combined hypothermia and sulfide is interesting because both of these interventions have been previously linked to metabolic suppression, and the combination of them may offer therapeutic possibilities in the context of ‘on-demand suspended animation’ (see below). Hypothermia not only assumes the importance for H2S-induced inhibition of mitochondrial respiration, but also markedly influences substrate utilization and thereby may even improve the activity of the mitochondrial respiratory chain. In anaesthetized and ventilated mice, inhaling 100 p.p.m. H2S did not affect endogenous glucose production (as calculated from the rate of appearance of 1,2,3,4,5,6-13C6-glucose during continuous i.v. isotope infusion), whole body CO2 production, or direct, aerobic glucose oxidation rate (as derived from VCO2 and the expiratory 13CO2/12CO2 ratio) during normothermia. However, combining inhaled H2S with hypothermia (core temperature 27°C) increased glucose oxidation, suggesting a shift towards preferential carbohydrate utilization (Figure 2) (Baumgart et al., 2010). Such a switch in fuel utilization is associated with an improved yield of oxidative phosphorylation (Korvald et al., 2000): the ATP synthesis/oxygen consumption ratio is higher for glycolysis than for oxidation because NADH (as an electron donor) provides three coupling sites rather than the two that FADH2 provides (Leverve, 2007). This effect on substrate metabolism coincides with a significantly attenuated responsiveness of hepatic mitochondrial respiration to stimulation with exogenous cytochrome C oxidase, that is, an improved maintenance of the outer mitochondrial membrane (Kuznetsov et al., 2004). It should be noted, however, that this protective effect of inhaled H2S partially disappeared during CLP-induced septic shock: not only did inhaled H2S not affect the sepsis-induced metabolic acidosis (Wagner et al., 2011). Finally, during sepsis, inhaled H2S did not improve the responsiveness to stimulation with exogenous cytochrome-c-oxidase either. It remains open whether the lacking effects of H2S on substrate utilization were due to the sepsis per se and/or the ongoing noradrenaline infusion needed to maintain target haemodynamics: noradrenaline causes oxidative stress and may inhibit mitochondrial respiration (Bracht et al., 2012a).
Figure 2.
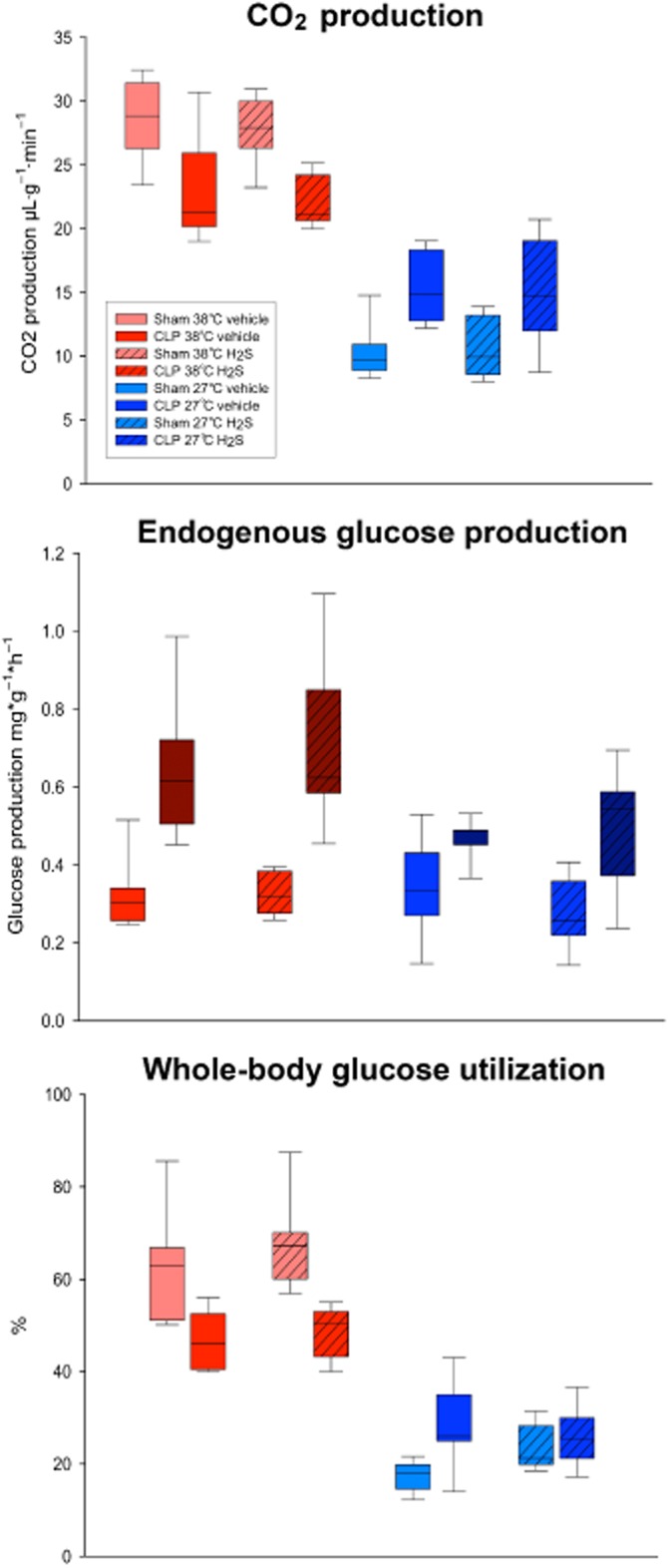
Glucose utilization in anaesthetized and ventilated mice and inhaling 100 p.p.m. gaseous H2S or the carrier gas N2 (vehicle) during deliberate normothermia (38°C) or hypothermia (27°C) with CLP-induced septic shock or after sham operation. All data are median (quartiles), n = 8–11 per group. #P < 0.05 shows significant difference versus respective normothermic vehicle-treated animals, §P < 0.05 shows significant difference between vehicle and respective H2S-treated mice, $P < 0.05 shows significant difference versus respective sham-operated group. Data in sham-operated mice were adapted from a prior set of studies by Baumgart et al. (2010).
Taken together, the above data, as well as multiple prior studies with sulfide donation or sulfide inhibition (reviewed in Wagner et al., 2009; Coletta and Szabo, 2013), reveal a complex, and only partially understood, regulation of sulfide in circulatory shock. While in some experimental models sulfide donation is clearly beneficial, in other models, sulfide donation actually exacerbates the outcome. In yet other models, however, pharmacological inhibitors of sulfide biosynthesis are therapeutically effective. These findings may reflect the diversity of the roles of sulfide in various models, forms and stages of shock (ranging from sulfide deficiency to sulfide overproduction), and may also relate to tissue/organ-specific differences or species-specific differences, as well as may be related to the complex, biphasic or bell-shaped biological effects of sulfide. Clearly, additional work needs to be conducted in this area, and it needs to be directly tested whether endogenously produced sulfide contributes to cytopathic hypoxia in certain models. In addition, further work needs to be conducted to test whether metabolic effects or mitochondrial protective mechanisms contribute to the therapeutic effects of sulfide donation in those models where sulfide administration was found beneficial.
H2S as a mitochondrial protectant in diabetes
Chronic or intermittent elevations in circulating glucose concentration damage the blood vessels, especially the innermost cell layer, the endothelium. Diabetic endothelial dysfunction is now recognized as a central pathophysiological event in the development of many, if not all, complications of diabetes, ranging from vascular events (peripheral vascular disease, coronary artery disease, diabetic retinopathy, diabetic neuropathy) to other diabetic complications such as impaired wound healing, cardiomyopathy and male erectile dysfunction (Szabo, 2005; 2009; Pacher and Szabo, 2006; Horváth et al., 2009; Rosenson et al., 2011; Shin et al., 2011; Stitt et al., 2013).
The mechanisms and experimental therapy of diabetic/hyperglycaemic endothelial dysfunction have been characterized in significant detail. Several lines of studies show that elevated extracellular glucose induces mitochondrial dysfunction in endothelial cells (Nishikawa et al., 2000; Du et al., 2003; Giacco and Brownlee, 2010; Gerö et al., 2013). On one hand, this entails the suppression of cellular bioenergetics through the impairment of the mitochondrial electron transport/ATP generation (Sivitz and Yorek, 2010; Pangare and Makino, 2012). On the other hand, intracellular production of reactive oxygen and nitrogen species from the dysfunctional mitochondria initiates multiple pathways of pro-inflammatory signalling (Nishikawa et al., 2000; Du et al., 2003; Giacco and Brownlee, 2010), as well as deleterious, self-amplifying cycles of cell injury such as the activation of the nuclear enzyme PARP (Garcia Soriano et al., 2001; Horváth et al., 2009).
Several lines of studies demonstrate that hydrogen sulfide exerts protective effects against the development of diabetic complications (reviewed in Szabo, 2012), at least in part by protecting the diabetic mitochondria. Circulating sulfide levels are decreased in diabetes, in part due to an increased consumption of sulfide by reactive oxygen species (ROS) production and in part due to the down-regulation of CSE, the principal H2S-producing enzyme in endothelial cells (Brancaleone et al., 2008; Jain et al., 2010; Whiteman et al., 2010; Suzuki et al., 2011; Kundu et al., 2013; Yamamoto et al., 2013). These events are paralleled with impaired cellular bioenergetics as well as reduced endothelium-dependent relaxant responses and increased pro-inflammatory signalling events, ultimately leading to cell death. Silencing of CSE further exacerbates the hyperglycaemic ROS production, while supplementation of CBS (by pharmacological tools or by overexpression of H2S-producing enzymes) suppresses it (Suzuki et al., 2011). Pharmacological supplementation or genetic expression of H2S-producing enzymes in cells subjected to hyperglycaemia reduces mitochondrial ROS formation (Suzuki et al., 2011; Guan et al., 2012; Zhong et al., 2012; Zhou and Lu, 2013) and exerts cytoprotective effects (Suzuki et al., 2011), including a partial normalization of mitochondrial bioenergetics (restoration of oxidative phosphorylation and inhibition of glycolysis, which is elevated in hyperglycaemic endothelial cells as a compensatory mechanism) (Suzuki et al., 2011) (Figure 3). Sulfide also protects against the activation of pro-inflammatory signalling pathways in hyperglycaemic endothelial cells (e.g. NF-κB activation, inflammatory cytokine production) (Guan et al., 2013; Manna and Jain, 2013; Si et al., 2013), reduction in matrix protein synthesis/protection against remodelling (Yuan et al., 2011; Lee et al., 2012; Kundu et al., 2013), as well as against the activation of PARP (Suzuki et al., 2011). The beneficial effects of sulfide supplementation have also been demonstrated in multiple models of diabetes in vivo, resulting in improved cardiac contractility, improved endothelium-dependent relaxations, and improved retinal and renal function and histopathology (Gao et al., 2011; Suzuki et al., 2011; Yang et al., 2011; Ahmad et al., 2012; Zhong et al., 2012; Si et al., 2013; Yamamoto et al., 2013). Given the fact that the direct reaction of H2S with various oxidant species is relatively slow (Carballal et al., 2011) and is outcompeted by many other reactions in the mitochondrion, it is likely that indirect antioxidant effects (e.g. via the regulation of signalling processes and/or via up-regulation of various antioxidant pathways) are also involved in the observed mitochondrial protective effects of sulfide. The beneficial effects on bioenergetic/mitochondrial processes are likely to play a role in the therapeutic effect of H2S in diabetic complication models. However, it is likely that additional effects of H2S (on signal transduction, redox balance and, perhaps, vasodilatory/haemodynamic effects) also contribute.
Figure 3.
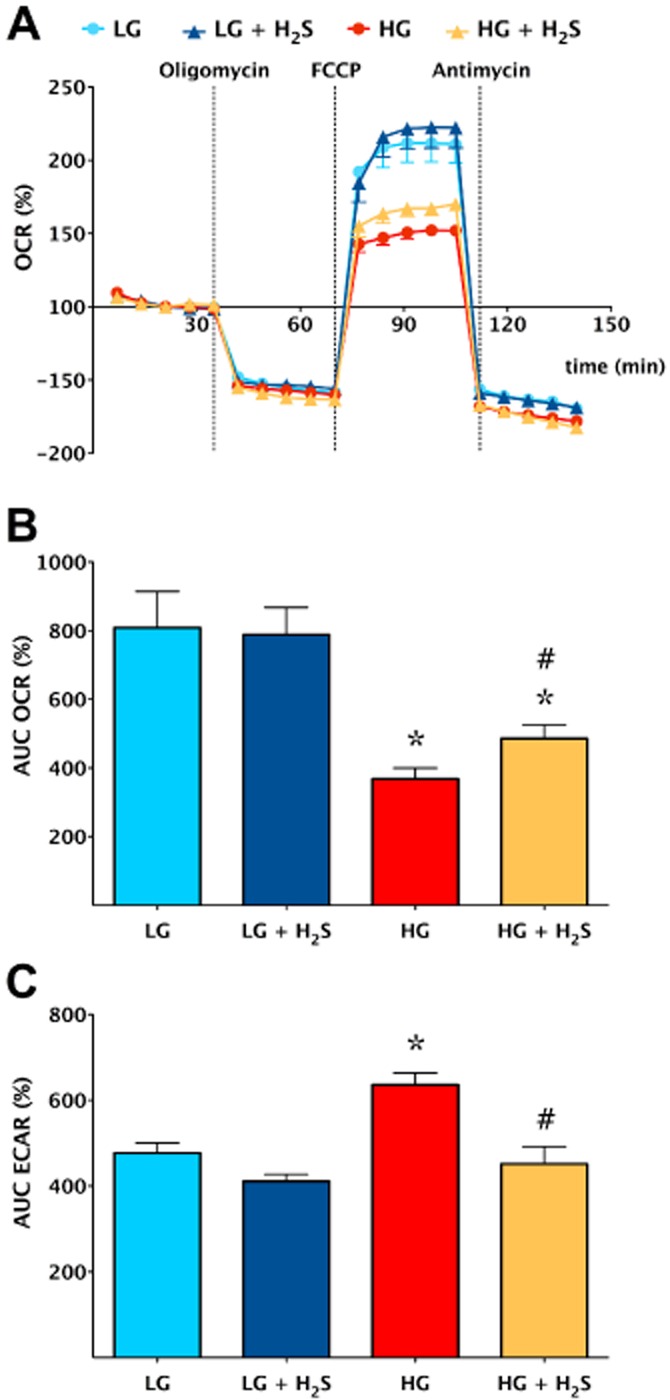
H2S reduces the degree of the bioenergetic derangements in endothelial cells placed in high extracellular glucose. Bioenergetic analysis of bEnd3 murine microvascular endothelial placed in low (5.5 mM, LG) or high (40 mM, HG) glucose conditions for 7 days in the absence or presence of H2S treatment (300 μM every 8 h) was performed using the Seahorse XF24 Analyzer as described (Suzuki et al., 2011). (A) A time course for measurement of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) under basal conditions, followed by the sequential addition of oligomycin (1 μg·mL−1), FCCP (0.3 μM) and antimycin A (2 μg·mL−1). (B) OCR and (C) ECAR values, representing indices of mitochondrial oxidative phosphorylation and glycolysis respectively. Increased extracellular glucose induced a suppression of oxidative phosphorylation and an increase in glycolysis (presumably a compensatory response), as compared with normal glucose (*P < 0.05). Treatment with H2S partially restored these changes (#P < 0.05). Data represent mean ± SEM of n = 15 wells from three experiments performed on different experimental days. Reproduced with permission from Suzuki et al. (2011).
H2S as endogenously produced metabolic ‘fuel’ in cancer cells
Cellular bioenergetics is essential for invasive tumour growth and metastasis and constitutes a key point in the control of cancer progression (Pathania et al., 2009; VanderHeiden et al., 2009; Ramsay et al., 2011; Bailey et al., 2012). Tumour cells rely both on oxidative phosphorylation (mitochondrial activity) and on increased glycolytic activity (‘aerobic glycolysis’ to produce ATP), which is used to support tumour cell growth, division cell movement. Tumour cell bioenergetics is intricately intertwined with pro-inflammatory and pro-growth tumour cell signalling processes (Cairns et al., 2011; Schulze and Harris, 2012). Therapeutic targeting of tumour cell energetics – especially using approaches that confer selectivity to the tumour cell – is an emerging concept in contemporary cancer therapy (Ramsay et al., 2011; Bailey et al., 2012).
Recent data from a colorectal cancer models indicate that cancer cells can utilize H2S as an endogenous bioenergetic ‘fuel’ (Szabo et al., 2013b). A comparison of human colon cancer specimens with patient-matched normal mucosa tissue revealed the selective up-regulation of CBS in the cancers. In contrast, the expression of the other two H2S-producing enzymes (CSE and 3-MST) remained unchanged. Similar to colon tumours, colon adenocarcinoma-derived cell lines (HCT116, HT-29, LoVo) exhibit selective up-regulation of CBS when compared with NCM356 cells (a non-tumorigenic control human epithelial cell line) (Figure 4A,B). Homogenates of the patient-derived colon tumour specimens, as well as homogenates of the colon cancer-derived cell lines, exhibit an increased rate of H2S production, which is blocked by the CBS inhibitor aminooxyacetic acid (AOAA), a widely used CBS inhibitor (Abe and Kimura, 1996; Julian et al., 2002; Austgen et al., 2011; Han et al., 2011; Asimakopoulou et al., 2013; Rashid et al., 2013; Wang et al., 2013). However, H2S production was not significantly inhibited by the CSE inhibitor PAG. (For additional discussion on the limitations of pharmacological inhibitors of CBS and CSE, see the final section of the current article.)
Figure 4.
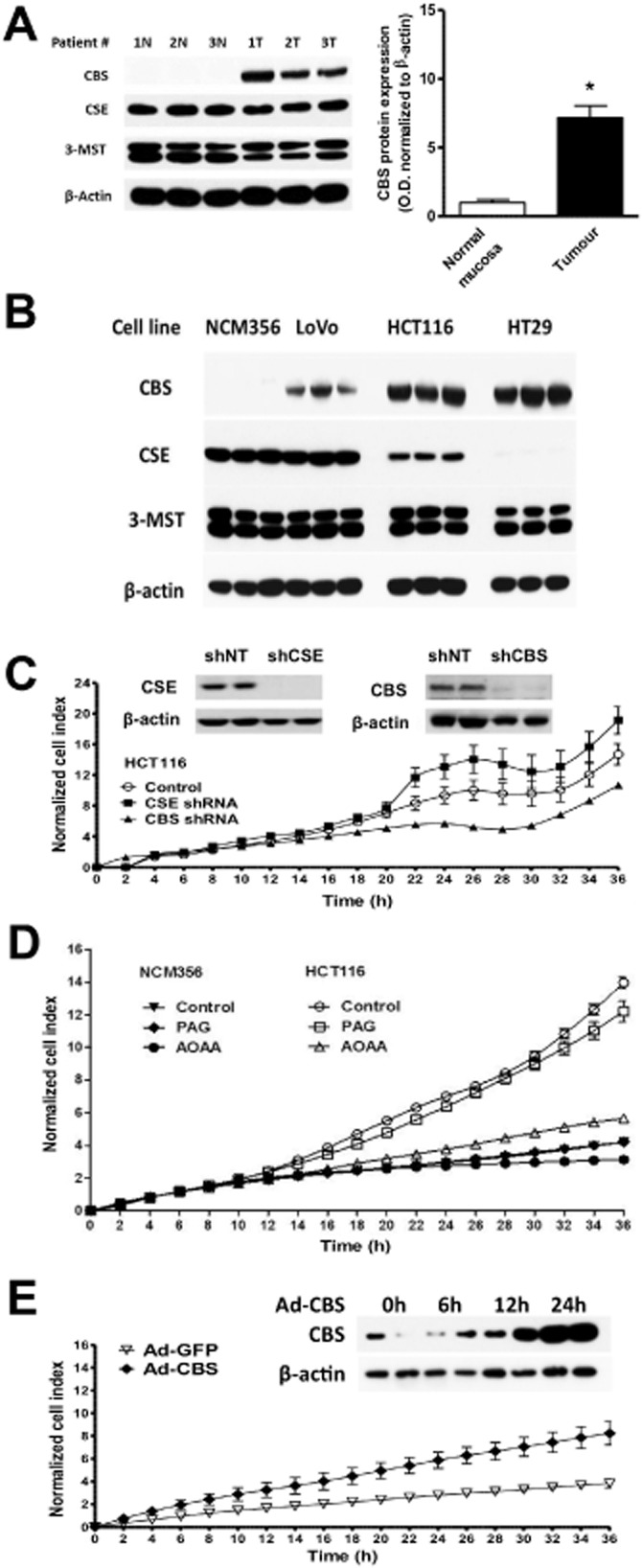
CBS is up-regulated in colon cancer cells and supports cancer cell proliferation. (A) High levels of CBS expression in human colon cancer tissues as compared with non-cancerous margin tissue. (B) High levels of CBS expression in various human colon cancer lines. Please note that the non-transformed colonic epithelial cell NCM356 did not show high CBS expression. Furthermore, please note that 3-MST expression was uniform among all tissues, while CSE expression was variable. (C) Lentiviral stable silencing of CBS, but not of CSE attenuates the growth of HCT116 colon cancer cells. Inset shows the efficiency of (D) pharmacological inhibition of CBS with AOAA (1 mM) but not of CSE with PAG (3 mM) attenuates the growth of HCT116 colon cancer cells. (E) Forced adenoviral expression of CBS in NCM356 cells increases their rate of proliferation. Inset shows the adenoviral up-regulation of CBS over time. Data represent mean ± SEM; reproduced with permission from Szabo et al. (2013b).
To investigate the functional consequence of CBS up-regulation, gene-specific shRNA sequences in lentiviral vectors were used to suppress the expression of either CBS or CSE in HCT116 cells. Densitometric analyses of Western blots revealed an approximately 50% decrease in CBS expression with comparable reductions in both cell proliferation and H2S production (Figure 4C). In contrast to CBS, silencing of CSE or its inhibition by PAG did not significantly affect either HCT116 cell proliferation or H2S production (Szabo et al., 2013b). To further define the function of CBS in colon cancer cells, both NCM356 cells (which express CSE, but only low levels of CBS) were treated and HCT116 cells (which abundantly express CBS) with the pharmacological CBS inhibitor, AOAA. Consistent with the CBS knock-down experiments, AOAA has also been utilized. AOAA inhibited the growth of HCT116 colon cancer cells, but did not affect the proliferation of the slower growing non-malignant NCM356 cell line (Szabo et al., 2013b) (Figure 4D). Conversely, forced overexpression of CBS in NCM356 cells significantly increased their basal rate of proliferation (Figure 4E). AOAA treatment also suppressed the migration and invasion of HCT116 cells. Finally, inhibition of CBS with AOAA reduced endothelial cell migration in colon cancer/endothelial cell co-cultures (Szabo et al., 2013b).
The above data demonstrated that CBS and CBS-derived H2S play an important role in promoting tumour cell proliferation. In the context of the current article, the most relevant question is the following: What role, if any, do H2S-associated energetic alterations play in these actions? In order to answer this question, HCT116 colonic cancer cells, and NCM356 cells as non-transformed controls, were subjected to bioenergetic measurements using the Extracellular Flux Analysis (Szabo et al., 2013b). In agreement with the physiological role of endogenous H2S in promoting cellular bioenergetics in various cell types including colonocytes (reviewed in Szabo et al., 2013a), low concentrations of H2S caused a stimulation of mitochondrial function in mitochondria isolated from HCT116 cells. Moreover, shRNA-mediated silencing of CBS, or CBS inhibition with AOAA, reduced basal cellular respiration, suppressed the calculated ATP synthesis and attenuated the spare respiratory capacity (Figure 5A–C). CBS silencing or CBS inhibition also reduced glycolytic functions, while CSE silencing was without significant effect (Figure 5D). This latter effect can be attributed, at least in part, to inhibition of GAPDH activity, as H2S is known to be an activator of GAPDH via sulfhydration (Mustafa et al., 2009; Paul and Snyder, 2012). In mitochondria prepared from HCT116 cells, L-cysteine stimulated mitochondrial electron transport; this effect was markedly attenuated in mitochondria prepared from shCBS cell expression (Szabo et al., 2013b). In AOAA-treated mitochondria (which showed suppressed electron transport), sulfide addition improved and restored mitochondrial function (Figure 6). This mitochondrion-autonomous response was consistent with the results of cell fractionation studies, showing that a significant portion of the total amount of cellular CBS was associated with the mitochondria in HCT116 cells. CBS was primary associated with the outer mitochondrial membrane (Szabo et al., 2013b).
Figure 5.
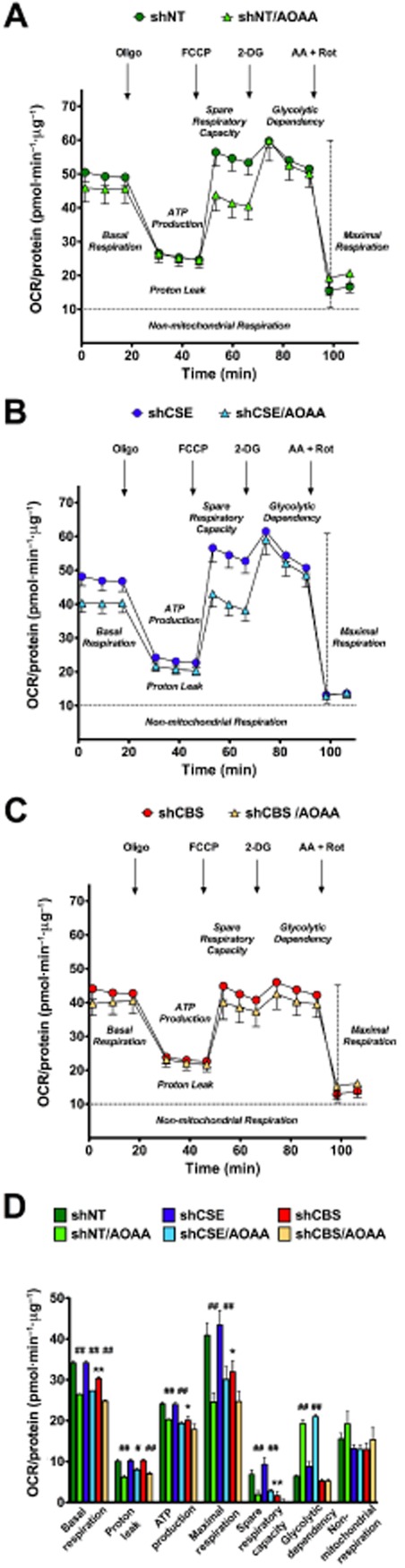
CBS supports colon cancer bioenergetics. Effect of the CBS inhibitor AOAA on cellular bioenergetics is shown in (A) HTC116 cells transfected with non-targeting vector (shNT), in (B) cells transfected with a CSE-silencing vector (shCSE) and (C) in cells transfected with a CBS-silencing vector (shCBS). Part (D) shows a comparison of the various bioenergetic responses. Please note that HCT116 cells with CBS silencing show a suppression basal bioenergetic function. In addition, the FCCP-induced increase in oxygen consumption is markedly attenuated in CBS-silenced cells. AOAA causes a comparable suppression of the bioenergetic responses in the shNT and the shCSE cells, while in the shCBS cells (in which the bioenergetic response is already suppressed), AOAA only exerts a slight degree of additional effect. (*P < 0.05 shows significantly lower bioenergetic parameters in shCBS cells as compared with shNT cells; #P < 0.05 shows significant effect of AOAA in either the shNT, shCBS or shCSE cells, when compared with the respective response in the absence of AOAA.) Data represent mean ± SEM of n = 3 determinations. Reproduced with permission from Szabo et al. (2013b).
Figure 6.
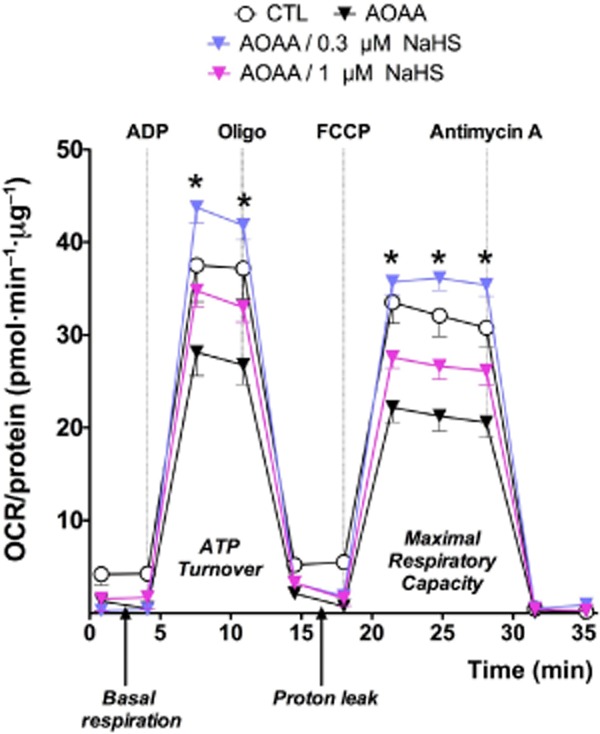
Restoration of mitochondrial function by sulfide in colon cancer cells treated with AOAA. Mitochondria were prepared from vehicle-treated HCT116 cells or from cells treated with AOAA (1 mM, 90 min) and subjected to bioenergetic analysis in the presence or absence of sulfide administration (applied as NaHS) at 0.3 and 1 μM, as described (Szabo et al., 2013b). Please note that AOAA suppressed mitochondrial OCR, while sulfide restored the functionality of the mitochondria. Data represent mean ± SEM of n = 3 determinations; *P < 0.05 shows the significant stimulatory effect of sulfide.
These findings indicate that endogenous sulfide, produced from CBS, stimulates tumour cell bioenergetics (both oxidative phosphorylation and glycolysis). The effect CBS silencing or CBS inhibition has on tumour progression in vivo has also been investigated by utilizing nude mice subjected to heterotopic transplantation of either HCT116 cells or patient-derived colon cancer xenografts (Szabo et al., 2013b). Consistent with the in vitro findings showing that CBS inhibition suppresses tumour cell proliferation, shRNA-mediated knock-down of CBS expression (shCBS) or treatment of the mice with AOAA significantly reduced the growth rate and size (i.e. volume) of HCT116 tumour xenografts (Szabo et al., 2013b). In contrast, silencing of CSE (shCSE) or pharmacological inhibition of CSE with PAG did not affect tumour growth. CBS suppression caused a significant reduction in the density of CD31-positive blood vessels within the tumour tissue as well as the prevalence of larger blood vessels and the extent of vessel branching, consistent with the hypothesis that CBS-derived H2S acts locally in a paracrine manner to stimulate tumour angiogenesis (Szabo et al., 2013b); the pro-angiogenic role of H2S is supported by multiple lines of in vitro and in vivo studies (Cai et al., 2007; Papapetropoulos et al., 2009; Szabo and Papapetropoulos, 2011; Coletta et al., 2012). Finally, in a series of translational studies, AOAA treatment showed a reduced rate of the growth of patient-derived tumour transplants (Szabo et al., 2013b).
Taken together, these data support the hypothesis that CBS-derived endogenously produced H2S plays a key role in cancer cell bioenergetics and cell proliferation via autocrine mechanisms. Moreover, via paracrine mechanisms, H2S also supports tumour angiogenesis and acts as a local vasodilator, thereby stimulating the supply of the tumour with nutrients via the bloodstream (Figure 7). Several independent lines of studies show that H2S can produce an activation of kinase pathways and inhibition of phosphatase pathways (Cai et al., 2007; 2010; Hu et al., 2008; 2011; Krishnan et al., 2011; Manna and Jain, 2011), leading to the regulation of the cell cycle (Deplancke and Gaskins, 2003; Cai et al., 2010). Because these pathways are crucial contributors to tumour cell proliferation, it is conceivable that such effects may also contribute to the proliferative effects of the CBS/H2S axis in colorectal tumours (and possibly other tumours as well).
Figure 7.
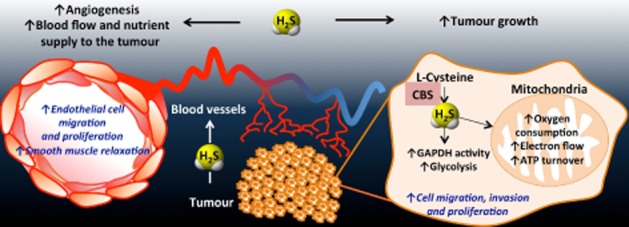
Multiple mechanisms of the cancer-cell-survival function of the CBS/H2S axis in vivo. As a result of the CBS overexpression, H2S is overproduced in colon cancer cells. H2S serves as an inorganic electron donor, stimulating mitochondrial electron transport, increasing ATP turnover. In addition, it increases the glycolytic activity of the tumour cell by activating GAPDH. Via these autocrine bioenergetic effects, H2S stimulates cancer cell proliferation, migration and invasion. In addition, H2S diffuses into the surrounding cells and tissues, stimulating angiogenesis, as well as acting as a vascular relaxant. Via these paracrine effects, CBS-derived H2S promotes the supply of blood and nutrients to the tumour. Reproduced with permission from Szabo et al. (2013b).
To put the above findings in the context of prior findings in the literature, it is interesting to note that a number of papers have previously reported that CBS expression is increased in various tumours (Goss, 1986; Zhang et al., 2005; Guo et al., 2012a). Moreover, in one prior study, CBS expression has been associated with increased cell proliferation (MacLean et al., 2002) (even though none of these prior investigations raised the potential involvement of H2S). In addition, several studies demonstrated increased H2S production in the exhaled air and flatus of cancer patients (Kumar et al., 2012; Yamagishi et al., 2012; Altomare et al., 2013) and increased urinary excretion of thiosulfate, the stable breakdown product of H2S (Chwatko et al., 2013) (even though these investigations have not identified the enzymatic source). Finally, several recent studies noted the inhibitory effect of AOAA on cancer cell proliferation in vitro and/or in vivo (Thornburg et al., 2008; Weinberg et al., 2010; Anso et al., 2013; Son et al., 2013) (even though these investigators attributed the effects of AOAA to pharmacological actions other than CBS inhibition).
The pharmacology of AOAA is, indeed, complex. This compound is generally known as a non-specific inhibitor of transaminases. It inhibits the activity of these enzymes because it irreversibly binds to the prosthetic group pyridoxal phosphate, which is commonly found in the active site of transaminases. In the context of recent studies, its inhibitory effect on GOT1 [an essential component of the malate/aspartate shuttle, a mechanism that links glycolysis to the transfer of electron donors into the mitochondria of cancer cells (Greenhouse and Lehninger, 1976; 1977; Dang et al., 2011)] has been linked to antitumor actions (Thornburg et al., 2008). In addition, the function of GOT1 is linked to glutaminolysis, a metabolic pathway that is at least partially selective to cancer cells (and, hence, may be therapeutically targetable) (Le et al., 2012; Son et al., 2013); AOAA has been shown to inhibit the metabolic activity of cancer cells by interfering with glutaminolysis (Son et al., 2013). The studies discussed above show that CBS/H2S production can also be considered as a partially cancer-selective metabolic pathway. It can be argued that with AOAA, one can simultaneously block the energetic ‘fuelling’ of the cancer cell via the malate/aspartate shuttle and the energetic ‘fuelling’ of the cancer cell via the CBS/sulfide route (Figure 8), thereby inducing ‘synthetic lethality’ with two independent pharmacological effects of a single compound. In order to compare the effects of AOAA with the effects of glycolysis inhibition and inhibition of ATP synthase, we have evaluated the effect of the glycolysis inhibitor 2-deoxyglucose, the ATP synthase inhibitor oligomycin, and AOAA (alone and in various combinations) on ATP levels in HCT116 tumour cells. AOAA reduced ATP levels to 80% of control, whereas oligomycin reduced them to 71%. Addition of AOAA to oligomycin did not exert a substantial further inhibitory effect. Likewise, addition of AOAA to 2-deoxyglucose (which, on its own, reduced ATP levels to 45%) did not further inhibit cellular ATP levels (Figure 9). These findings point to a coordinated interaction between sulfide-dependent modulation of both oxidative phosphorylation and glycolysis; in both cases more likely in a role as an enhancer/potentiator, rather than an independent activator. It must also be emphasized that the effects of sulfide in cancer cells are likely to be more complex and are likely to include additional effects beyond mitochondrial function (such as effects on glycolysis) as well as pharmacological effects beyond bioenergetics (such as effects on kinase pathways and cell cycle). While it is clear that much further work needs to be conducted in this area, the above series of studies may open a previously unexplored, and potentially clinically translatable aspect of tumour biology.
Figure 8.
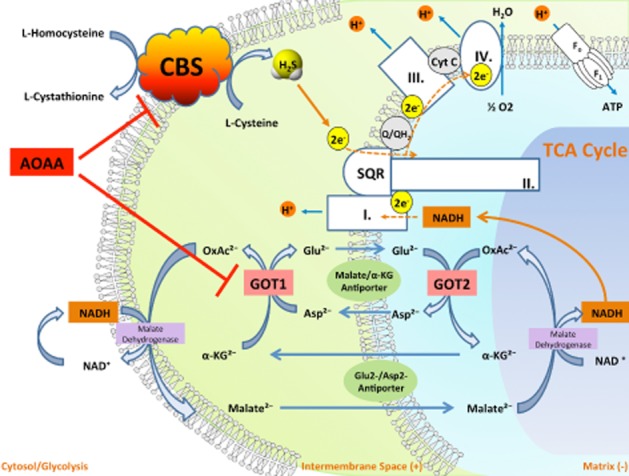
By simultaneously inhibiting CBS activity and inhibiting GOT1, a key enzyme of the malate/aspartate shuttle, AOAA acts as an inducer of ‘synthetic lethality’ in cancer cells. CBS-derived H2S supports mitochondrial electron transport and cancer cell bioenergetics by donating electrons at complex II. By inhibiting CBS, AOAA suppresses this bioenergetic pathway. The malate-aspartate shuttle translocates electrons that are produced in glycolysis across the semipermeable inner membrane of the mitochondrion in order to support oxidative phosphorylation. These electrons enter the electron transport chain at complex I. The shuttle system is required because the mitochondrial inner membrane is impermeable to NADH (a primary reducing equivalent of the electron transport chain). In humans, the cytoplasmic enzyme (GOT1) is one of the key enzymes in the malate shuttle: it functions to catalyse the interconversion of aspartate and α-ketoglutarate to oxaloacetate and glutamate using pyridoxal phosphate as a cofactor. By inhibiting GOT1, AOAA reduces the transfer of electron donors to the mitochondria, thereby suppressing cancer cell bioenergetics. By the simultaneous inhibition of CBS and GOT1, AOAA interferes with two key pathways of cancer cell mitochondrial function.
Figure 9.
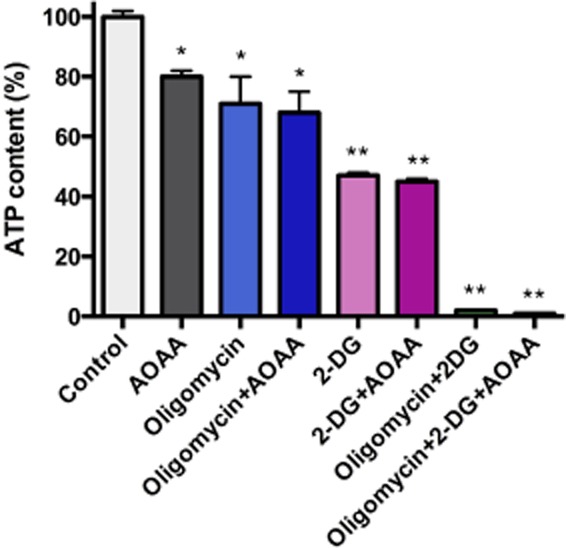
Relative importance of oxidative phosphorylation and glycolysis in the maintenance of ATP levels in HCT-116 cells. HCT-116 cells were cultured for 8 h in the presence of the ATP synthase inhibitor oligomycin (1 μg·mL−1), the glycolysis inhibitor 2-deoxyglucose (100 mM), the CBS/GOT1 inhibitor AOAA (1 mM) and their various combinations, followed by the measurement of cellular ATP levels, using a bioluminescent kit Sigma (St. Louis, MO, USA) as described (Módis et al., 2013a). Data represent mean ± SEM of n = 3 determinations; *P < 0.05 and **P < 0.01 show significant inhibitory effect of the various pharmacological inhibitors compared with the vehicle control group.
The mechanism of the up-regulation of CBS in colon cancer cells is not known. Possibilities include a transcriptional up-regulation, a blockade of its degradation, or targeted delivery to the mitochondria via not yet understood mechanisms. A recent study by Teng and colleagues demonstrating in ischaemic liver and in hypoxic hepatocytes that CBS is targeted to the mitochondria via the hypoxia-sensitive regulation of the mitochondrial matrix protease L on protease (Teng et al., 2013) offers a plausible mechanism (especially in light of the well-known phenomenon of tumour hypoxia), which, nevertheless, remains to be directly tested in future studies.
We are well aware of reports in the literature, demonstrating that it is H2S donation (rather than H2S biosynthesis inhibition) that can kill cancer cells (e.g. Shin et al., 2010; Lee et al., 2011; Chattopadhyay et al., 2012a,2012b; Lim et al., 2012). These studies tend to use relatively high concentrations of various H2S donors, often linked with molecular scaffolds that have their own sulfide-independent pharmacological actions (e.g. NOSH-aspirin, which has a non-steroidal anti-inflammatory component). These findings are in no way in conflict with the findings related to the bioenergetic and pro-angiogenic effects of endogenously produced H2S in cancer cells, but, rather, illustrate the complex pharmacology of H2S, where low concentrations/fluxes of the mediator and higher concentrations/release rates of the same mediator can have radically different biological effects (as discussed in Szabo et al., 2013a). The interpretation of the findings related to the anticancer effect of H2S donors may therefore be related to the high concentrations of H2S used, at which concentrations H2S has cytostatic and cytotoxic properties (possibly, at least in part related to the inhibition of mitochondrial electron transport chain system at complex IV). We predict that one of the technical hurdles of exploiting therapeutic H2S donation for anticancer therapy will be the delivery: one needs to generate high local levels of H2S, for reasonably prolonged time, in order to produce anticancer effects. It is hoped that further studies will solve this issue; a similar problem has previously arisen in the context of NO donation and anticancer therapy (NO, too, has bell-shaped dose responses and vital, physiological effects at low concentrations and cytostatic effects at higher concentrations). In this context, photodynamic therapy, with NO-releasing photolabile molecules, has been put forward as one possible technical solution (Bhowmick and Girotti, 2013; Rapozzi et al., 2013).
Therapeutic induction of ‘on-demand suspended animation’ with H2S
One of the most fascinating biological features of H2S is the ‘suspended animation-like state’ that it can induce. The landmark paper on that subject (Blackstone et al., 2005), which utilized gaseous H2S to induce a hibernation-like state, has sparked interest in the use of sulfide as an inducer of hypometabolism to protect organs and cells against temporary insults. By exposing mice to subtoxic concentrations of H2S (20–80 p.p.m.), a concentration-dependent state of deeply reduced metabolism was induced, as evidenced by a fast reduction of total-body CO2 production and O2 consumption (60–70% within 5–10 min), followed by a slow decline of metabolism to around 5% of basal levels. This slower decline parallels the reduction in body temperature that follows the exposure. Core body temperature steadily declines to around 2°C above ambient temperature over the course of hours. After cessation of H2S exposure, metabolism recovers to basal levels with no apparent toxic effects (Blackstone et al., 2005). The sulfide-mediated reduction of metabolic demand has been shown to protect mice against the mortality associated with otherwise lethal hypoxia (Blackstone and Roth, 2007).
Although the above findings of metabolic suppression have been repeatedly confirmed in mice (Blackstone and Roth, 2007; Volpato et al., 2008; Bos et al., 2009) ( Figure 10), it has been challenging to translate these findings to larger mammals. Different studies have been performed with varying results. Some studies showed no effect on metabolic parameters in rats (Haouzi et al., 2009), sheep (Haouzi et al., 2008) or swine (Li et al., 2008; Drabek et al., 2011), while others showed metabolic suppression in rats (Florian et al., 2008; Aslami et al., 2010; 2013a; Seitz et al., 2012) and swine (Simon et al., 2008). One human toxicity study shows that inhalation of 10 p.p.m. H2S during exercise reduces V·O2, possibly by reducing aerobic capacity (Bhambhani et al., 1997). A comparison of the effects of sulfide to the effects of anaesthetics shows that H2S does not have the hypnotic or immobilizing effects of anaesthetic compounds (Li et al., 2012). The last word has not been written on the interspecies variability, but it seems that the translation to larger mammals is more difficult than initially presumed. A possible explanation lies in the different types of metabolism between smaller and larger animals. Mice have a relatively high metabolic rate in relation to their body weight, and a larger proportion of their metabolism is geared towards maintaining core body temperature. It is well established that in response to hypoxia, small rodents can reduce their energy expenditure by decreasing ‘non-shivering thermogenesis’, irrespective of any pharmacological intervention. Hypoxia-induced hypometabolism is present in numerous mammals (Mortola, 2004) and humans (van Marken Lichtenbelt and Schrauwen, 2011), but due to the high area/volume ratio and, consequently, the higher heat dissipation, it is inversely related to body size (Mortola, 2004), that is, to the ratio of O2 consumption and body weight (VO2/kg). This is substantiated by the fact that (i) no matter the species, newborns present with more pronounced hypoxia-induced hypometabolism than adults, and (ii) in adults with low VO2/kg, hypoxia-induced hypometabolism becomes manifest when normoxic VO2 is increased, for example, during exercise (Mortola, 2004). Because non-shivering thermogenesis does not result in ATP production due to the activity of uncoupling proteins (van Marken Lichtenbelt and Schrauwen, 2011), reduced VO2 and the consecutive hypothermia represent a unique adaptation to hypoxia only present in small animals but not in larger species or humans. Consequently, with respect to the putative induction of a ‘suspended animation'-like hypometabolism, it has been questioned whether any therapeutic potential of H2S observed in mice and rats can be transferred to the clinical setting (Leslie, 2008, Derwall et al., 2010, Drabek et al., 2011, Haouzi, 2011; 2012). Clearly, data in sheep and swine subjected to shock, ischaemia/reperfusion (Sodha et al., 2008; Osipov et al., 2009; Sodha et al., 2009; Osipov et al., 2010; Simon et al., 2011; Hunter et al., 2012), haemorrhage (Bracht et al., 2012b) or burn injury (Esechie et al., 2009) suggest that the beneficial effects of infusing Na2S are independent of core temperature: any induction of moderate hypothermia is due to an attenuation of systemic inflammation rather than an effect on energy expenditure (Simon et al., 2011; Bracht et al., 2012b). Further studies need to be conducted to study dose responses and to compare different methods of administration (i.e. gaseous, i.v./i.a. soluble salt injection, slow release donor injection).
Figure 10.
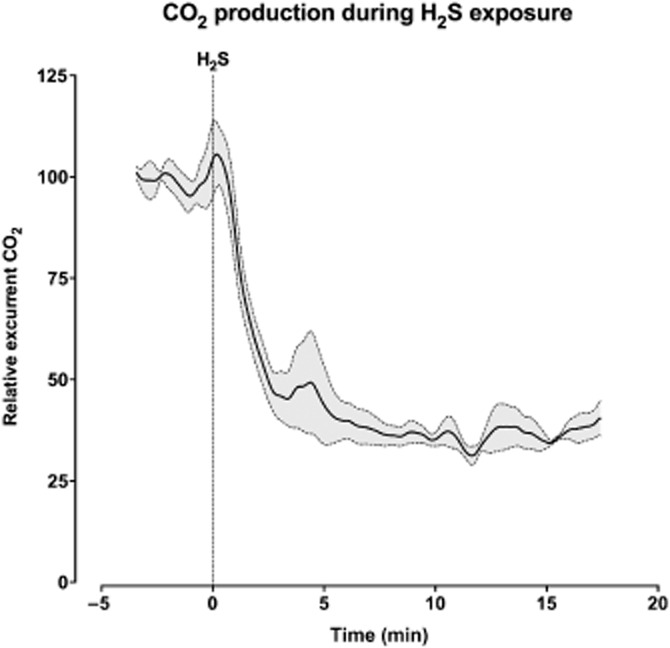
‘On-demand suspended animation’ induced by H2S: reduced CO2 production of mice exposed to H2S. Mean CO2 production (solid line) and standard error of the mean (dashed lines) of three mice, shown before and after exposure to 100 p.p.m. H2S. Reproduced with permission from Bos et al. (2012).
Sulfide-induced hypometabolism is highly protective in different models of hypoxia or ischaemia. During H2S-hypometabolism, mice can survive hypoxia (5% O2) for up to 6 h, while all the animals in the control group died within 17 min (Blackstone and Roth, 2007). Similarly, hydrogen sulfide-induced hypometabolism has been shown to improve survival in lethal haemorrhagic shock in the rat; the mechanism has been attributed to suppression of tissue oxygen demand by sulfide (Morrison et al., 2008). Tokuda and colleagues have reported that inhaled H2S gas suppresses inflammatory response and prolongs survival in endotoxin shock, effects that have been attributed to suppression of metabolism (Tokuda et al., 2012). Inducing a state of hypometabolism using 100 p.p.m. H2S before clamping the renal artery can prevent the onset of kidney damage in mice (Figure 11 and Figure 12) (Bos et al., 2009). Induction of hypometabolism during the reperfusion phase has some protective potency, but much less than pretreatment does (Bos et al., 2009). Similar results were obtained for hepatic ischaemia, in which hypometabolic pretreatment protected livers from ischaemic damage (Bos et al., 2012). In both of these studies, hypometabolism was induced while core body temperature was kept at 37°C (thus preventing hypothermia). This shows that hypothermia itself is not needed for hypometabolic protection (but, in theory, it could offer additional protection). H2S-induced hypometabolism, when established before the onset of ischaemia, was also protective in cardiac ischaemia (Snijder et al., 2013). Sub-hypometabolic concentrations of H2S are mainly associated with anti-inflammatory and anti-fibrotic action, while higher concentrations of H2S (those that induce hypometabolism) offer additional protection against early myocardial necrosis.
Figure 11.
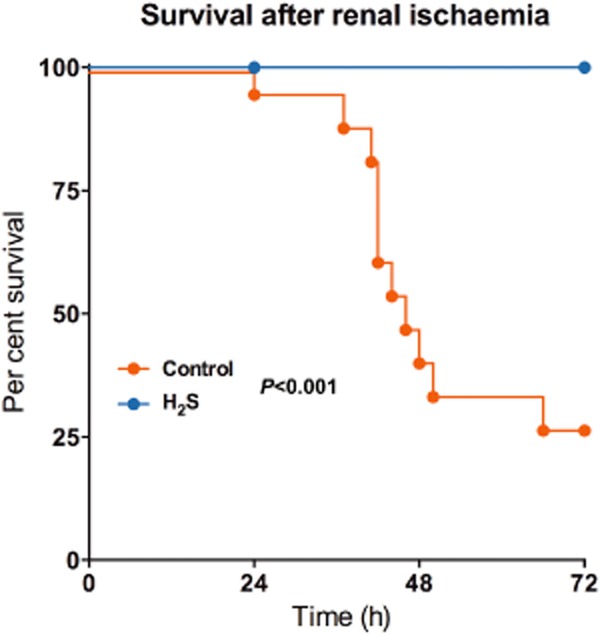
H2S improves survival after bilateral renal ischaemia. Control mice or mice that were in a hypometabolic state (caused by exposure of 100 p.p.m. H2S for 30 min) were subjected to bilateral renal ischaemia at 37°C. While in the control group 73% mortality was observed, the H2S-pretreated mice all survived the insult. Adapted from Bos et al. (2009).
Figure 12.
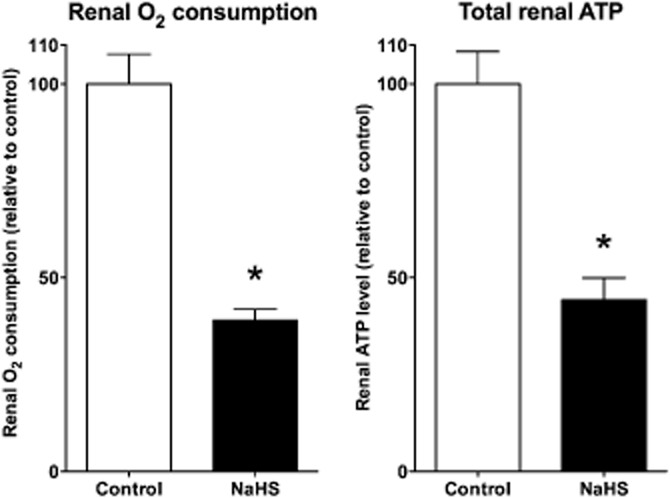
H2S induces organ hypometabolism ex vivo. Isolated perfused rat kidneys treated with NaHS show reduced oxygen consumption and reduced total renal ATP levels. Reproduced with permission from Bos et al. (2009).
It is likely that the protective effects of H2S can be attributed to several factors, hypometabolism being just one of them. As noted in the previous sections, H2S has many pharmacological effects in addition to metabolic suppression, as it can act as an anti-oxidant, anti-fibrotic, anti-inflammatory and anti-apoptotic substance (Zanardo et al., 2006; Yao et al., 2010; Bos et al., 2013; Snijder et al., 2013). In addition, treatment with H2S during hypoxia protects the integrity and function of mitochondria (Elrod et al., 2007; Bos et al., 2009).
Although it is of a mixed pharmacological action, sulfide remains to be an intriguing molecule as an on-demand inducer of hypometabolism, with the capability to protect organs from ischaemia or hypoxia. Several sets of studies show the ability of sulfide to hibernate isolated organs which could be promising in the setting of organ transplantation, where warm and cold ischaemia times affect transplant survival and function (Table 1, Figure 12) (Hu et al., 2007; Henderson et al., 2010; Hosgood and Nicholson, 2010; Balaban et al., 2011; Bos et al., 2012; George et al., 2012; Hunter et al., 2012; Lobb et al., 2012; Siriussawakul et al., 2012; Xie et al., 2012; Zhu et al., 2012; Wu et al., 2013). In addition, hypometabolic effects (if they prove to be translatable to large animals and humans) could be of potential use in other clinical situations as well, such as vascular surgery, infarction and stroke.
Table 1.
Protective effects of hydrogen sulfide during organ storage and transplantation
| Organ | Experimental conditions | Findings | Reference |
|---|---|---|---|
| Heart | Isolated rat hearts; Langendorff perfusion system; Krebs–Henseleit solution. Cold heart storage for 6 h, followed by warm reperfusion for 30 min with or without sulfide (1 μM). | Sulfide accelerated the recovery of contractility during reperfusion, improved myocardial contractility, increased ATP content and decreased myocardial apoptosis index. | Hu et al., 2007. |
| Lung | Rabbit heart/lung blocs stored in cold low-potassium dextrane sulfate solution for 18 h, followed by a 2 h reperfusion with donor blood in an in vitro perfusion system. Lungs from donor animals ventilated with room were compared with lungs of donor animals ventilated for 1 h with 150 p.p.m. H2S prior to the start of the excision/storage of the heart/lung blocs. | Perfusion pressures were improved in the sulfide group. In addition, H2S-treated lungs had better oxygenation and ventilation indices. Finally, sulfide treatment reduced ROS formation during reperfusion and resulted in a better maintenance of mitochondrial cytochrome c content. | George et al., 2012. |
| Rabbit heart/lung blocs stored in cold Perfadex solution for 18 h, followed by a 2 h reperfusion with donor blood in an in vitro perfusion system in the presence or absence of a bolus dose of NaHS (100 μg·kg−1 bolus + 1 mg·kg−1·h−1 infusion) starting at reperfusion. | Sulfide treatment reduced reactive oxygen species formation during reperfusion. Perfusion pressures were similar in all groups. | George et al., 2012. | |
| Single left lung transplantation in rats, with 3 h of cold storage/ischaemia. NaHS was administered to the recipient animal at 14 μmol·kg−1 15 min before the start of the transplantation. | Sulfide improved pulmonary function (e.g. PaO2/FiO2 ratio) and pulmonary histology, reduced lung oedema formation and reduced the accumulation of neutrophils in the lung. Sulfide resulted in a reduction in IL-1β and an increase in IL-10 levels. | Wu et al., 2013. | |
| Kidney | Porcine kidneys subjected to 25 min of warm ischaemia followed by 18 h of cold storage on ice after perfusion with a hyperosmolar citrate solution. Sulfide (or vehicle treatment) was applied as 0.5 mM NaHS infused 10 min prior and during reperfusion in an in vitro perfusion system. | Renal blood flow and renal function (measured as creatinine clearance, fractional excretion of sodium and urine output) was improved in the sulfide group. Sulfide treatment also resulted in reduced isoprostane and NO levels. No significant differences were observed in histological parameters between groups. | Hosgood and Nicholson, 2010. |
| Liver | Cold storage of rat livers in Wisconsin solution for 48 h, in the presence or absence of the sulfide donor diallyl disulfide (3.4 mM), followed by perfusion in an isolated constant-pressure perfusion system. | Hepatic clearance (bromosulfophthalein depuration) was enhanced by sulfide treatment; most other parameters (vascular resistance, oxygen consumption, LDH release as an index of cell injury) were unaffected by sulfide. | Balaban et al., 2011. |
| Cutaneous tissue | In vitro model of cutaneous tissue transplantation: endothelial cells and fibroblasts exposed to hypoxia or anoxia for 24 h, followed by 6 h of normoxia, in the absence or presence of NaHS (10 μM-1 mM). | Sulfide reduced apoptotic index (percentage of TUNEL-positive cells). | Henderson et al., 2010. |
| Stem cells | In vitro model of stem cell transplantation: rat mesenchymal stem cells exposed to hypoxia or anoxia for 6 h, in the absence or presence of NaHS (200 μM), followed by intramyocardial injection of stem cells and measurement of cardiac function in an ischaemia/reperfusion model in the rat. | Sulfide reduced hypoxia-induced stem cell apoptosis and enhanced donor cell survival in the heart after intramyocardial injection, resulting in improved cardiac contractility. Sulfide treatment was associated with increased Akt, Erk and GSK-3β activation in the stem cells, which has been suggested to contribute to the functional changes observed in vivo. | Xie et al., 2012. |
Therapeutic sulfide administration: pharmacological and biochemical considerations
The unique characteristics of the bioenergetic consequences of sulfide exposure must be carefully considered when designing experimental therapeutic approaches related to its administration. The same characteristics of sulfide must also be considered in formulating a critical analysis of the published findings showing the biological effects of sulfide administration in vitro or in vivo. With regard to in vitro models versus in vivo models, first of all it must be recalled that – while mammalian cells in vivo cannot survive for more than a few minutes when oxidative bioenergetics (i.e. mitochondrial function) is shut off (e.g. complete tissue ischaemia or severe cyanide poisoning) – the vast majority of mammalian cells in culture can survive to a prolonged eclipse or even a permanent inactivation of mitochondrial bioenergetics, if glucose concentrations are sufficient to sustain an intense anaerobic glycolysis. [Part of this may be explained by the fact that most cells grown in culture are transformed cells, with an up-regulation of glycolysis (Warburg effect).] It must therefore be kept in mind that cultured cells can, in fact, tolerate a severe transient or even permanent shutdown of mitochondrial function by high doses/steady rates of sulfide exposure, without impairing cell viability. (In fact, some cultured cells can even tolerate combined oxygen/glucose deprivation fairly well by mobilizing emergency pathways of ATP generation, e.g. by metabolizing purine nucleotides via adenosine kinase; Módis et al., 2009; Szoleczky et al., 2012). At the same time, such gross alterations in mitochondrial function may, in fact, initiate mitochondrion-mediated stress signalling. It has been suggested (Whitfield et al., 2008; Olson and Whitfield, 2010; Olson, 2012) that transient inhibitory effects of sulfide on mitochondrial electron transport may, in fact, be at least in part responsible for some of the observed cytoprotective effects of sulfide in vitro (and possibly in some of the in vivo studies as well). In other words, experiments aiming to promote or explain a sulfide-based pharmacological treatment have thus to be considered with regard to the mitochondrial bioenergetics. The first issue is whether or not a partially toxic concentration (tenth of micromolar) has been reached. If so, the cells concerned would experience something similar to an ischaemic period (sulfide toxicity period) followed by a reperfusion-like episode in which sulfide concentration would be brought back to non-toxic levels. In this case, complex cellular responses such as those induced by a drop in the ATP/ADP ratio would be triggered and while mobilization of such defences might be of interest (as they induce subsequent protection via preconditioning) the physiological responses would not necessarily represent specific ‘sulfide-signalling’ events. An indirect support of this hypothesis is that, so far, all of the pathways identified as downstream effectors of sulfide-mediated preconditioning (e.g. Nrf2, KATP channels, etc.) are, in fact, largely identical to the ones involved in the protective effect of all other forms of preconditioning (e.g. ischaemic, free radical/oxidant mediated, etc.) (e.g. Broadhead et al., 2004; Halestrap et al., 2007; Sadat, 2009; Yang et al., 2010; Bell et al., 2011). An alternative interpretation of the above-listed mechanisms may be, however, that sulfide (and possibly other gaseous transmitters) maintain a ‘controlled, adverse bioenergetic effect’ that may control cell signalling, cell viability and/or cardiovascular function.
With respect to the implications of the above mechanisms for in vivo experiments, similar considerations can be made. With a bolus administration of sulfide (or a short-acting sulfide donor), the fact that the animal tolerated the sulfide exposure means that a critical toxic concentration of sulfide was not reached/maintained for a significant amount of time in the vicinity of cells that control vital biological functions (such as a set of neurons controlling basic physiological functions). After entering the circulation, due to its diffusive properties, free sulfide will quickly distribute into the entire organism, where the cells equipped with the sulfide-oxidizing unit (SOU) will tend to metabolize it to low levels. [Part of the circulating sulfide may also exit in the gas phase via the lungs (Insko et al., 2009; Toombs et al., 2010).] We estimate that with fast-acting sulfide donors (sulfide salts), the rise in sulfide concentration compatible with survival probably does not exceed a few tenths of a micromolar for a few minutes. The resulting transient inhibition of mitochondrial function, and the subsequent preconditioning-type responses would then (similar to the in vitro considerations detailed in the previous paragraph) be expected to be at least in part responsible for subsequent protective/beneficial biological effects. When administering long-acting sulfide donors, or when applying a continuous infusion of short-acting hydrogen sulfide donors (e.g. infusion of H2S gas dissolved in physiological solutions), the above-mentioned ‘peak effects’ would not occur. In the latter case, steady-state levels of circulating sulfide would be maintained when equilibrium exists between sulfide administration (or the release rate of the sulfide donor) and the rate of sulfide elimination (i.e. overall sulfide-oxidizing capacities of the animal plus sulfide exhalation). An extreme example illustrates the biological consequences of impaired sulfide elimination: genetic deletion of ETHE1 (encoding for a β-lactamase-like, iron-coordinating metalloprotein, with sulfur dioxygenase activity) in mice results in a severe, toxic accumulation of sulfide, producing a severe inhibition of cytochrome c oxidase activity as well as a severe form of encephalopathy (Tiranti et al., 2009; Tiranti and Zeviani, 2013).
Even if the physiological sulfide degradation pathways are functioning normally, a steady-state plasma level of sulfide will produce heterogeneous cell/tissue sulfide ‘exposure levels’ for several reasons. First of all, a ‘Krogh cylinder’ type phenomenon (similar to that of oxygen) is likely to exist for sulfide, in which sulfide concentrations diminish as one moves away from the blood vessel and as one moves from the arterial to the venous side of the circulation. In addition, different cells and tissues have different levels of ‘anti-sulfide defences’ (e.g. sulfide binding and SOU in tissues). Moreover, within each cell, there may be subcellular organelles in which the degradation of sulfide is higher than in other areas of the cell. As discussed elsewhere (Bouillaud and Blachier, 2011), mitochondria are expected to produce steep decreases in the intracellular or transcellular sulfide gradients. Sulfide gradients and ‘anti-sulfide defences’ are also important in the context of the microflora resident in the colonic lumen and the colonic wall (Bouillaud and Blachier, 2011).
Notwithstanding the complex theoretical issues related to therapeutic sulfide administration, many pathophysiological conditions are associated with a relative deficiency of H2S (due to impaired production and/or enhanced degradation), and, in these circumstances, therapeutic H2S donation is be warranted, either to serve as a pharmacological replacement (as in diabetes, see above) or to induce transient metabolic suppression (as in the context of suspended animation, see above). It is hoped that future therapeutic H2S donation approaches will benefit from recent studies enabling the direct comparison of the doses and associated pharmacological actions of gaseous (inhalational) sulfide versus circulating, blood-borne sulfide. These studies yielded an approximate biological equivalency between 200 p.p.m. inhaled H2S and 10 mg·kg−1·h−1 i.v. infusion of a H2S solution (Wintner et al., 2010), and concluded that the therapeutic doses of sulfide in vivo are below those inhalational exposure levels that are associated with systemic toxicity.
Pharmacological exploitation of the mitochondrial effects of H2S: future directions
The above sections only represent selected examples of the pathophysiological and therapeutic aspects related to the complex mitochondrial modulatory effects of H2S. Based on several lines of experimental data (Hu et al., 2009; 2010; Pun et al., 2010; Caro et al., 2012; Guo et al., 2012b; Tang et al., 2012; Aslami et al., 2013b; Lu et al., 2013; Wen et al., 2013), it is likely that H2S as a mitochondrial protectant – similar to the example of diabetic endothelial dysfunction discussed above – also protects the mitochondria of neurons, stem cells, cardiac myocytes and various other cell types. It is also likely that bioenergetic effects play important roles in H2S-mediated angiogenesis (Cai et al., 2007; Papapetropoulos et al., 2009; Coletta et al., 2012), where it may serve to support the increased energy demand of the growing, migrating and dividing endothelial cells. At the opposite end of the spectrum is cancer therapy, in which high concentrations of H2S donors can cause inhibition of cell division and promotion of cell death (Lee et al., 2011; Tsubura et al., 2011; Wu et al., 2012; Kashfi and Olson, 2013), effects that are, at least in part, likely to be related to the inhibition of mitochondrial electron transport by high local concentrations of H2S. This latter concept does not conflict with the role of endogenously produced H2S in cancer, as the exogenously administered H2S donors produce much higher concentrations than endogenous sulfide levels, yielding a different biological response.
The interaction between the effects of H2S on mitochondrial function and the effects of H2S on cell signalling needs to be characterized in future studies as well. There are convincing data in the field of carbon monoxide (CO, another gaseous transmitter) showing that partial inhibition of mitochondrial electron transport, followed by a ‘leakage’ of ROS from the mitochondrial electron transport chain, plays a key role in the initiation of CO-dependent cellular signalling (Zuckerbraun et al., 2007). Whether similar processes are also at play in the case of H2S remains to be elucidated. In fact, the complex relationship between the three gasotransmitters (NO, CO and H2S) in the regulation of cellular signalling and cellular bioenergetics (Lane, 2006; Pun et al., 2010; Szabo, 2010) needs to be studied in further detail, in light of the effect of each of these transmitters on mitochondrial electron transport and glycolysis (Cooper and Brown, 2008; Pun et al., 2010). Furthermore, their emerging interdependence and mutual regulation in the context of many biological processes (e.g. Whiteman et al., 2006; Minamishima et al., 2009; Tomaskova et al., 2009; Yusof et al., 2009; Coletta et al., 2012; Carballal et al., 2013; Kondo et al., 2013; Polhemus et al., 2013) need to be further studied as well. We must also point out that in the current review, very little discussion was devoted to the ‘third’ H2S-producing enzyme, 3-MST (while its physiological roles have been detailed in the complementary article; Szabo et al., 2013a). At this point, there is essentially no information on the potential pathophysiological role of the 3-MST system; future work may identify pathophysiological conditions where the 3-MST system plays a role. The recent generation of the 3-MST deficient mice (Nagahara et al., 2013) may catalyse further work in this direction. Finally, the potential modulation of the H2S-dependent direct mitochondrial effects by other downstream effector pathways of H2S [such as cysteine modification (sulfhydration)] (Gadalla and Snyder, 2010), modulation of potassium channels (Zhao et al., 2001; Cheng et al., 2004; Cheang et al., 2010) and effects on calcium signalling (Bauer et al., 2010) remain to be explored in future studies.
There are a number of contentious, intensively debated issues in the field of H2S (for further reading, please see: Whiteman et al., 2011; Olson, 2013b,2013c), one of them being the circulating/plasma levels of H2S, and the methods used to measure it (especially the most commonly used method, the ‘methylene blue method’, which yields low-to-mid-micromolar plasma concentrations). As discussed elsewhere (Whitfield et al., 2008; Wintner et al., 2010; Whiteman et al., 2011; Olson, 2013b; Szabo et al., 2013a), there are various ‘pools’ of sulfide (or reactive sulfur species) in the circulation (free H2S gas being only a minor component of it, if any), and the methylene blue method is almost certainly measuring a net sum of these various pools, and it does not represent ‘free’ H2S levels. There may also be methodological issues that can result in a significant overestimation of the absorbance values detected by the methylene blue method (e.g. Van de Louw and Haouzi, 2012). Due to the fact that there are various biological ‘sinks’ of sulfide (e.g. in red blood cells), it is also difficult to estimate ‘steady-state’ levels of sulfide, or to be certain that such ‘steady-state’ levels even exist in biological systems (Olson, 2013c). Clearly, additional work is needed to clarify these issues and to improve the methods to detect H2S in biological systems. While this refinement is ongoing, we do not have a better option than to keep these limitations in mind, and to interpret the reported differences in the sulfide levels between two experimental groups (when reported in the same publication) as probably directionally correct, while remaining critical about the absolute values reported. Another issue that represents a severe limitation of the field is the lack of selective pharmacological tools. As discussed elsewhere (Whiteman et al., 2011; Asimakopoulou et al., 2013), the available compounds (PAG, AOAA, etc.) are neither particularly potent nor are they sufficiently selective or specific; they show cross-inhibition between the two pyridoxal 5′-phosphate (PLP)-dependent enzymes CSE and CBS, and they also inhibit a variety of other enzymes. One way to minimize this limitation is by using genetically modified animals or transient of permanent silencing of each enzyme (with the caveat that these tools, too, have certain limitations). While these issues remain to be refined, we must remain careful with the interpretation of most of the published studies utilizing these inhibitors (especially when they are not accompanied by corroborating studies using genetic tools).
Finally, we would like to emphasize that a substantial portion of the data reviewed in the current article, as well as its companion paper (Szabo et al., 2013a), stems from in vitro studies, and the number of studies investigating the stimulatory or inhibitory effect of exogenously administered or endogenously produced sulfide in vivo is limited (Table 2). As discussed elsewhere (Budde and Roth, 2011; Vozdek et al., 2012; Módis et al., 2013d), the model organism Caenorhabditis elegans provides a convenient system to study H2S-mediated biological regulatory mechanisms. In this organism, a CBS homologue enzyme is primarily responsible for endogenous H2S production (Vozdek et al., 2012). SiRNA-mediated silencing of this enzyme attenuates the bioenergetic function of C. elegans, consistent with the hypothesis that endogenous H2S production plays a stimulatory bioenergetic role (Szabo et al., 2013a). One known example of endogenously produced H2S suppressing mitochondrial function is the phenomenon of physiological hypoxia sensing (e.g. in the arterial chemoreceptors or the kidney) where, at biological sites associated with low O2 tension (e.g. the carotid body or the kidney medulla), the inhibition of complex IV by endogenous H2S (produced by CSE) plays a physiological regulatory role (Beltowski, 2010; Olson and Whitfield, 2010; Peng et al., 2010; Makarenko et al., 2012; Olson, 2013a). (Although it must be noted that the role of H2S in physiological hypoxia sensing remains a field of intensive research and debate, and other lines of data suggest that it may be a decreased oxidation of H2S during hypoxia, and the subsequent accumulation of H2S, and a subsequent signalling process that may be involved; see Olson, 2013c). Pharmacological inhibition of H2S production in the carotid body may be a potential future therapeutic approach for the therapy of chemoreceptor hypersensitivity and the associated cardiovascular imbalance, such as the one associated with chronic heart failure (Del Rio et al., 2013). It is also interesting to note that in a recent study, the CSE inhibitor PAG has been shown to block the hypoxia-induced decrease in oxygen uptake (VO2) in rats (Del Rio et al., 2013), perhaps indicative of a role endogenous H2S production in hypoxia-induced hypometabolism. One must keep in mind, nevertheless, that the selectivity of PAG is limited. Thus, it is hoped that the results of the above study will be confirmed and extended in future studies using CSE–/– mice. Additional studies in mammalian systems are necessary to clarify the complex bioenergetic roles of sulfide in various diseases in vivo and the interrelationship between bioenergetic regulation and the various other facets of sulfide biology (such as its diverse roles in the cardiovascular, central nervous and immune systems), in order to allow the design and testing of future therapeutic approaches.
Table 2.
Metabolic/bioenergetic effects of exogenously administered or endogenously produced hydrogen sulfide in cultured cells in vitro and in various species in vivo
 |
Acknowledgments
The work of C. S. is supported by grants from the National Institutes of Health (P50GM060338), the Shriners Burns Hospitals (#8661), the American Diabetes Association (7-12-BS-184) and the John Sealy Memorial Endowment Fund for Biomedical Research. The work of P. R. is supported by the Deutsche Forschungsgemeinschaft (Klinische Forschergruppe 200) and the Land Baden-Württemberg (Innovationsfond Medizin). H. v. G. is supported by the Dutch Kidney Foundation (C08-2254). F. B and A. P. are supported by COST Action BM1005 (European Network of Gasotransmitters). The editorial assistance of Ms. Lili Szabo is appreciated.
Glossary
- 3-MST
3-mercaptopyruvate sulfurtransferase
- AOAA
aminooxyacetic acid
- CBS
cystathionine β-synthase
- CSE
cystathionine γ-lyase
- H2S
hydrogen sulfide
- KATP
ATP-sensitive potassium channel
- PAG
propargylglycine
- SOU
sulfide-oxidizing unit
Conflict of interest
C.S and M.H. are stockholders and corporate officers and A.P. is a stockholder and consultant to CBS Therapeutics Inc., a for-profit organization involved in the therapeutic exploitation of sulfide biosynthesis inhibitors for cancer therapy.
References
- Abe K, Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci. 1996;16:1066–1071. doi: 10.1523/JNEUROSCI.16-03-01066.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad FU, Sattar MA, Rathore HA, Abdullah MH, Tan S, Abdullah NA, et al. Exogenous hydrogen sulfide (H2S) reduces blood pressure and prevents the progression of diabetic nephropathy in spontaneously hypertensive rats. Ren Fail. 2012;34:203–210. doi: 10.3109/0886022X.2011.643365. [DOI] [PubMed] [Google Scholar]
- Altomare DF, Di Lena M, Porcelli F, Trizio L, Travaglio E, Tutino M, et al. Exhaled volatile organic compounds identify patients with colorectal cancer. Br J Surg. 2013;100:144–150. doi: 10.1002/bjs.8942. [DOI] [PubMed] [Google Scholar]
- Anso E, Mullen AR, Felsher DW, Matés JM, DeBerardinis RJ, Chandel NS. Metabolic changes in cancer cells upon suppression of MYC. Cancer Metab. 2013;1:7. doi: 10.1186/2049-3002-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asimakopoulou A, Panopoulos P, Chasapis CT, Coletta C, Zhou Z, Cirino G, et al. Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE) Br J Pharmacol. 2013;169:922–932. doi: 10.1111/bph.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslami H, Heinen A, Roelofs JJTH, Zuurbier CJ, Schultz MJ, Juffermans NP. Suspended animation inducer hydrogen sulfide is protective in an in vivo model of ventilator-induced lung injury. Intensive Care Med. 2010;36:1946–1952. doi: 10.1007/s00134-010-2022-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslami H, Beurskens CJP, de Beer FM, Kuipers MT, Roelofs JJTH, Hegeman MA, et al. A short course of infusion of a hydrogen sulfide-donor attenuates endotoxemia induced organ injury via stimulation of anti-inflammatory pathways, with no additional protection from prolonged infusion. Cytokine. 2013a;61:614–621. doi: 10.1016/j.cyto.2012.11.018. [DOI] [PubMed] [Google Scholar]
- Aslami H, Pulskens WP, Kuipers MT, Bos AP, van Kuilenburg AB, Wanders RJ, et al. Hydrogen sulfide donor NaHS reduces organ injury in a rat model of pneumococcal pneumosepsis, associated with improved bio-energetic status. PLoS ONE. 2013b;8:e63497. doi: 10.1371/journal.pone.0063497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austgen JR, Hermann GE, Dantzler HA, Rogers RC, Kline DD. Hydrogen sulfide augments synaptic neurotransmission in the nucleus of the solitary tract. J Neurophysiol. 2011;106:1822–1832. doi: 10.1152/jn.00463.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey KM, Wojtkowiak JW, Hashim AI, Gillies RJ. Targeting the metabolic microenvironment of tumors. Adv Pharmacol. 2012;65:63–107. doi: 10.1016/B978-0-12-397927-8.00004-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban CL, Rodriguez JV, Guibert EE. Delivery of the bioactive gas hydrogen sulfide during cold preservation of rat liver: effects on hepatic function in an ex vivo model. Artif Organs. 2011;35:508–515. doi: 10.1111/j.1525-1594.2011.01256.x. [DOI] [PubMed] [Google Scholar]
- Bauer CC, Boyle JP, Porter KE, Peers C. Modulation of Ca2+ signalling in human vascular endothelial cells by hydrogen sulfide. Atherosclerosis. 2010;209:374–380. doi: 10.1016/j.atherosclerosis.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Baumgart K, Wagner F, Gröger M, Weber S, Barth E, Vogt JA, et al. Cardiac and metabolic effects of hypothermia and inhaled hydrogen sulfide in anesthetized and ventilated mice. Crit Care Med. 2010;38:588–595. doi: 10.1097/ccm.0b013e3181b9ed2e. [DOI] [PubMed] [Google Scholar]
- Bell KF, Fowler JH, Al-Mubarak B, Horsburgh K, Hardingham GE. Activation of Nrf2-regulated glutathione pathway genes by ischemic preconditioning. Oxid Med Cell Longev. 2011;2011:689524. doi: 10.1155/2011/689524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltowski J. Hypoxia in the renal medulla: implications for hydrogen sulfide signaling. J Pharmacol Exp Ther. 2010;334:358–363. doi: 10.1124/jpet.110.166637. [DOI] [PubMed] [Google Scholar]
- Bhambhani Y, Burnham R, Snydmiller G, MacLean I. Effects of 10-ppm hydrogen sulfide inhalation in exercising men and women. Cardiovascular, metabolic, and biochemical responses. J Occup Environ Med. 1997;39:122–129. doi: 10.1097/00043764-199702000-00009. [DOI] [PubMed] [Google Scholar]
- Bhowmick R, Girotti AW. Cytoprotective signaling associated with nitric oxide upregulation in tumor cells subjected to photodynamic therapy-like oxidative stress. Free Radic Biol Med. 2013;57:39–48. doi: 10.1016/j.freeradbiomed.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackstone E, Roth MB. Suspended animation-like state protects mice from lethal hypoxia. Shock. 2007;27:370–372. doi: 10.1097/SHK.0b013e31802e27a0. [DOI] [PubMed] [Google Scholar]
- Blackstone E, Morrison M, Roth MB. H2S induces a suspended animation-like state in mice. Science. 2005;308:518. doi: 10.1126/science.1108581. [DOI] [PubMed] [Google Scholar]
- Bos EM, Leuvenink HG, Snijder PM, Kloosterhuis NJ, Hillebrands JL, Leemans JC, et al. Hydrogen sulfide-induced hypometabolism prevents renal ischemia/reperfusion injury. J Am Soc Nephrol. 2009;20:1901–1905. doi: 10.1681/ASN.2008121269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos EM, Snijder PM, Jekel H, Weij M, Leemans JC, van Dijk MC, et al. Beneficial effects of gaseous hydrogen sulfide in hepatic ischemia/reperfusion injury. Transpl Int. 2012;25:897–908. doi: 10.1111/j.1432-2277.2012.01514.x. [DOI] [PubMed] [Google Scholar]
- Bos EM, Wang R, Snijder PM, Boersema M, Damman J, Fu M, et al. Cystathionine γ-lyase protects against renal ischemia/reperfusion by modulating oxidative stress. J Am Soc Nephrol. 2013;24:759–770. doi: 10.1681/ASN.2012030268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillaud F, Blachier F. Mitochondria and sulfide: a very old story of poisoning, feeding, and signaling? Antioxid Redox Signal. 2011;15:379–391. doi: 10.1089/ars.2010.3678. [DOI] [PubMed] [Google Scholar]
- Bracht H, Calzia E, Georgieff M, Singer J, Radermacher P, Russell JA. Inotropes and vasopressors: more than hemodynamics! Br J Pharmacol. 2012a;165:2009–2011. doi: 10.1111/j.1476-5381.2011.01776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracht H, Scheuerle A, Gröger M, Hauser B, Matallo J, McCook O, et al. Effects of intravenous sulfide during resuscitated porcine hemorrhagic shock. Crit Care Med. 2012b;40:2157–2167. doi: 10.1097/CCM.0b013e31824e6b30. [DOI] [PubMed] [Google Scholar]
- Brancaleone V, Roviezzo F, Vellecco V, De Gruttola L, Bucci M, Cirino G. Biosynthesis of H2S is impaired in non-obese diabetic (NOD) mice. Br J Pharmacol. 2008;155:673–680. doi: 10.1038/bjp.2008.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadhead MW, Kharbanda RK, Peters MJ, MacAllister RJ. KATP channel activation induces ischemic preconditioning of the endothelium in humans in vivo. Circulation. 2004;110:2077–2082. doi: 10.1161/01.CIR.0000144304.91010.F0. [DOI] [PubMed] [Google Scholar]
- Buckler KJ. Effects of exogenous hydrogen sulphide on calcium signalling, background (TASK) K channel activity and mitochondrial function in chemoreceptor cells. Pflugers Arch. 2012;463:743–754. doi: 10.1007/s00424-012-1089-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde MW, Roth MB. The response of Caenorhabditis elegans to hydrogen sulfide and hydrogen cyanide. Genetics. 2011;189:521–532. doi: 10.1534/genetics.111.129841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai WJ, Wang MJ, Moore PK, Jin HM, Yao T, Zhu YC. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc Res. 2007;76:29–40. doi: 10.1016/j.cardiores.2007.05.026. [DOI] [PubMed] [Google Scholar]
- Cai WJ, Wang MJ, Ju LH, Wang C, Zhu YC. Hydrogen sulfide induces human colon cancer cell proliferation: role of Akt, ERK and p21. Cell Biol Int. 2010;34:565–572. doi: 10.1042/CBI20090368. [DOI] [PubMed] [Google Scholar]
- Cairns RA, Harris I, McCracken S, Mak TW. Cancer cell metabolism. Cold Spring Harb Symp Quant Biol. 2011;76:299–311. doi: 10.1101/sqb.2011.76.012856. [DOI] [PubMed] [Google Scholar]
- Calvert JW, Coetzee WA, Lefer DJ. Novel insights into hydrogen sulfide-mediated cytoprotection. Antioxid Redox Signal. 2010;12:1203–1217. doi: 10.1089/ars.2009.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carballal S, Trujillo M, Cuevasanta E, Bartesaghi S, Möller MN, Folkes LK, et al. Reactivity of hydrogen sulfide with peroxynitrite and other oxidants of biological interest. Free Radic Biol Med. 2011;50:196–205. doi: 10.1016/j.freeradbiomed.2010.10.705. [DOI] [PubMed] [Google Scholar]
- Carballal S, Cuevasanta E, Marmisolle I, Kabil O, Gherasim C, Ballou DP, et al. Kinetics of reversible reductive carbonylation of heme in human cystathionine β-synthase. Biochemistry. 2013;52:4553–4562. doi: 10.1021/bi4004556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro AA, Adlong LW, Crocker SJ, Gardner MW, Luikart EF, Gron LU. Effect of garlic-derived organosulfur compounds on mitochondrial function and integrity in isolated mouse liver mitochondria. Toxicol Lett. 2012;214:166–174. doi: 10.1016/j.toxlet.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay M, Kodela R, Olson KR, Kashfi K. NOSH-aspirin (NBS-1120), a novel nitric oxide-and hydrogen sulfide-releasing hybrid is a potent inhibitor of colon cancer cell growth in vitro and in a xenograft mouse model. Biochem Biophys Res Commun. 2012a;419:523–528. doi: 10.1016/j.bbrc.2012.02.051. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay M, Kodela R, Nath N, Dastagirzada YM, Velázquez-Martínez CA, Boring D, et al. Hydrogen sulfide-releasing NSAIDs inhibit the growth of human cancer cells: a general property and evidence of a tissue type-independent effect. Biochem Pharmacol. 2012b;83:715–722. doi: 10.1016/j.bcp.2011.12.018. [DOI] [PubMed] [Google Scholar]
- Cheang WS, Wong WT, Shen B, Lau CW, Tian XY, Tsang SY, et al. 4-aminopyridine-sensitive K+ channels contributes to NaHS-induced membrane hyperpolarization and relaxation in the rat coronary artery. Vascul Pharmacol. 2010;53:94–98. doi: 10.1016/j.vph.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Ndisang JF, Tang G, Cao K, Wang R. Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am J Physiol Heart Circ Physiol. 2004;287:H2316–H2323. doi: 10.1152/ajpheart.00331.2004. [DOI] [PubMed] [Google Scholar]
- Chwatko G, Forma E, Wilkosz J, Głowacki R, Józwiak P, Różański W, et al. Thiosulfate in urine as a facilitator in the diagnosis of prostate cancer for patients with prostate-specific antigen less or equal 10 ng/mL. Clin Chem Lab Med. 2013;6:1–7. doi: 10.1515/cclm-2013-0069. [DOI] [PubMed] [Google Scholar]
- Coletta C, Szabo C. Potential role of hydrogen sulfide in the pathogenesis of vascular dysfunction in septic shock. Curr Vasc Pharmacol. 2013;11:208–221. [PubMed] [Google Scholar]
- Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Módis K, Panopoulos P, et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc Natl Acad Sci U S A. 2012;109:9161–9166. doi: 10.1073/pnas.1202916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper CE, Brown GC. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J Bioenerg Biomembr. 2008;40:533–539. doi: 10.1007/s10863-008-9166-6. [DOI] [PubMed] [Google Scholar]
- Dang CV, Hamaker M, Sun P, Le A, Gao P. Therapeutic targeting of cancer cell metabolism. J Mol Med (Berl) 2011;89:205–212. doi: 10.1007/s00109-011-0730-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rio R, Marcus NJ, Schultz HD. Inhibition of hydrogen sulfide restores normal breathing stability and improves autonomic control during experimental heart failure. J Appl Physiol. 2013;114:1141–1150. doi: 10.1152/japplphysiol.01503.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deplancke B, Gaskins HR. Hydrogen sulfide induces serum-independent cell cycle entry in nontransformed rat intestinal epithelial cells. FASEB J. 2003;17:1310–1312. doi: 10.1096/fj.02-0883fje. [DOI] [PubMed] [Google Scholar]
- Derwall M, Westerkamp M, Löwer C, Deike-Glindemann J, Schnorrenberger NK, Coburn M, et al. Hydrogen sulfide does not increase resuscitability in a porcine model of prolonged cardiac arrest. Shock. 2010;34:190–195. doi: 10.1097/SHK.0b013e3181d0ee3d. [DOI] [PubMed] [Google Scholar]
- Derwall M, Francis RC, Kida K, Bougaki M, Crimi E, Adrie C, et al. Administration of hydrogen sulfide via extracorporeal membrane lung ventilation in sheep with partial cardiopulmonary bypass perfusion: a proof of concept study on metabolic and vasomotor effects. Crit Care. 2011;15:R51. doi: 10.1186/cc10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drabek T, Kochanek PM, Stezoski J, Wu X, Bayir H, Morhard RC, et al. Intravenous hydrogen sulfide does not induce hypothermia or improve survival from hemorrhagic shock in pigs. Shock. 2011;35:67–73. doi: 10.1097/SHK.0b013e3181e86f49. [DOI] [PubMed] [Google Scholar]
- Du X, Matsumura T, Edelstein D, Rossetti L, Zsengellér Z, Szabo C, et al. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest. 2003;112:1049–1057. doi: 10.1172/JCI18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci U S A. 2007;104:15560–15565. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsey DJ, Fowkes RC, Baxter GF. Regulation of cardiovascular cell function by hydrogen sulfide (H2S) Cell Biochem Funct. 2010;28:95–106. doi: 10.1002/cbf.1618. [DOI] [PubMed] [Google Scholar]
- Esechie A, Enkhbaatar P, Traber DL, Jonkam C, Lange M, Hamahata A, et al. Beneficial effect of a hydrogen sulphide donor (sodium sulphide) in an ovine model of burn-and smoke-induced acute lung injury. Br J Pharmacol. 2009;158:1442–1453. doi: 10.1111/j.1476-5381.2009.00411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink MP. Cytopathic hypoxia. Mitochondrial dysfunction as mechanism contributing to organ dysfunction in sepsis. Crit Care Clin. 2001;17:219–237. doi: 10.1016/s0749-0704(05)70161-5. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Distrutti E, Cirino G, Wallace JL. The emerging roles of hydrogen sulfide in the gastrointestinal tract and liver. Gastroenterology. 2006;131:259–271. doi: 10.1053/j.gastro.2006.02.033. [DOI] [PubMed] [Google Scholar]
- Florian B, Vintilescu R, Balseanu AT, Buga A-M, Grisk O, Walker LC. Long-term hypothermia reduces infarct volume in aged rats after focal ischemia. Neurosci Lett. 2008;438:180–185. doi: 10.1016/j.neulet.2008.04.020. [DOI] [PubMed] [Google Scholar]
- Gadalla MM, Snyder SH. Hydrogen sulfide as a gasotransmitter. J Neurochem. 2010;113:14–26. doi: 10.1111/j.1471-4159.2010.06580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaill F. Aspects of life development at deep sea hydrothermal vents. FASEB J. 1993;7:558–565. doi: 10.1096/fasebj.7.6.8472894. [DOI] [PubMed] [Google Scholar]
- Gao Y, Yao X, Zhang Y, Li W, Kang K, Sun L, et al. The protective role of hydrogen sulfide in myocardial ischemia-reperfusion-induced injury in diabetic rats. Int J Cardiol. 2011;152:177–183. doi: 10.1016/j.ijcard.2010.07.012. [DOI] [PubMed] [Google Scholar]
- Garcia Soriano F, Virág L, Jagtap P, Szabo E, Mabley JG, Liaudet L, et al. Diabetic endothelial dysfunction: the role of poly(ADP-ribose) polymerase activation. Nat Med. 2001;7:108–113. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]
- George TJ, Arnaoutakis GJ, Beaty CA, Jandu SK, Santhanam L, Berkowitz DE, et al. Inhaled hydrogen sulfide improves graft function in an experimental model of lung transplantation. J Surg Res. 2012;178:593–600. doi: 10.1016/j.jss.2012.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerö D, Szoleczky P, Suzuki K, Módis K, Oláh G, Coletta C, et al. Cell-based screening identifies paroxetine as an inhibitor of diabetic endothelial dysfunction. Diabetes. 2013;62:953–964. doi: 10.2337/db12-0789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goslar T, Marš T, Podbregar M. Total plasma sulfide as a marker of shock severity in nonsurgical adult patients. Shock. 2011;36:350–355. doi: 10.1097/SHK.0b013e31822bcfd0. [DOI] [PubMed] [Google Scholar]
- Goss SJ. Characterization of cystathionine synthase as a selectable, liver-specific trait in rat hepatomas. J Cell Sci. 1986;82:309–320. doi: 10.1242/jcs.82.1.309. [DOI] [PubMed] [Google Scholar]
- Goubern M, Andriamihaja M, Nubel T, Blachier F, Bouillaud F. Sulfide, the first inorganic substrate for human cells. FASEB J. 2007;21:1699–1706. doi: 10.1096/fj.06-7407com. [DOI] [PubMed] [Google Scholar]
- Greenhouse WV, Lehninger AL. Occurrence of the malate-aspartate shuttle in various tumor types. Cancer Res. 1976;36:1392–1396. [PubMed] [Google Scholar]
- Greenhouse WV, Lehninger AL. Magnitude of malate-aspartate reduced nicotinamide adenine dinucleotide shuttle activity in intact respiring tumor cells. Cancer Res. 1977;37:4173–4181. [PubMed] [Google Scholar]
- Groeger M, Wagner F, Baumgart K, Huber-Lang M, Knoeferl M, Georgieff M, et al. Mitochondrial respiration and cytochrome c inhibition by sulfide in peritoneal macrophages in vitro: effects of temperature and pH. Crit Care. 2010;14:P6. [Google Scholar]
- Groeger M, Matallo J, McCook O, Wagner F, Wachter U, Bastian O, Gierer S, Reich V, Stahl B, Huber-Lang M, Szabó C, Georgieff M, Radermacher P, Calzia E, Wagner K. Temperature and cell-type dependency of sulfide effects on mitochondrial respiration. Shock. 2012;38:367–374. doi: 10.1097/SHK.0b013e3182651fe6. [DOI] [PubMed] [Google Scholar]
- Guan Q, Zhang Y, Yu C, Liu Y, Gao L, Zhao J. Hydrogen sulfide protects against high-glucose-induced apoptosis in endothelial cells. J Cardiovasc Pharmacol. 2012;59:188–193. doi: 10.1097/FJC.0b013e31823b4915. [DOI] [PubMed] [Google Scholar]
- Guan Q, Wang X, Gao L, Chen J, Liu Y, Yu C, et al. Hydrogen sulfide suppresses high glucose-induced expression of intercellular adhesion molecule-1 (ICAM-1) in endothelial cells. J Cardiovasc Pharmacol. 2013 doi: 10.1097/FJC.0b013e31829875ef. (in press) [DOI] [PubMed] [Google Scholar]
- Guo H, Gai JW, Wang Y, Jin HF, Du JB, Jin J. Characterization of hydrogen sulfide and its synthases, cystathionine β-synthase and cystathionine γ-lyase, in human prostatic tissue and cells. Urology. 2012a;79:483.e1–483.e5. doi: 10.1016/j.urology.2011.10.013. [DOI] [PubMed] [Google Scholar]
- Guo W, Kan JT, Cheng ZY, Chen JF, Shen YQ, Xu J, et al. Hydrogen sulfide as an endogenous modulator in mitochondria and mitochondria dysfunction. Oxid Med Cell Longev. 2012b;2012:878052. doi: 10.1155/2012/878052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Clarke SJ, Khaliulin I. The role of mitochondria in protection of the heart by preconditioning. Biochim Biophys Acta. 2007;1767:1007–1031. doi: 10.1016/j.bbabio.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YF, Huang X, Guo X, Wu YS, Liu DH, Lu HL, et al. Evidence that endogenous hydrogen sulfide exerts an excitatory effect on gastric motility in mice. Eur J Pharmacol. 2011;673:85–95. doi: 10.1016/j.ejphar.2011.10.018. [DOI] [PubMed] [Google Scholar]
- Haouzi P. Murine models in critical care research. Crit Care Med. 2011;39:2290–2293. doi: 10.1097/CCM.0b013e3182227550. [DOI] [PubMed] [Google Scholar]
- Haouzi P. Ventilatory and metabolic effects of exogenous hydrogen sulfide. Respir Physiol Neurobiol. 2012;184:170–177. doi: 10.1016/j.resp.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Haouzi P, Notet V, Chenuel B, Chalon B, Sponne I, Ogier V, et al. H2S induced hypometabolism in mice is missing in sedated sheep. Respir Physiol Neurobiol. 2008;160:109–115. doi: 10.1016/j.resp.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Haouzi P, Bell HJ, Notet V, Bihain B. Comparison of the metabolic and ventilatory response to hypoxia and H2S in unsedated mice and rats. Respir Physiol Neurobiol. 2009;167:316–322. doi: 10.1016/j.resp.2009.06.006. [DOI] [PubMed] [Google Scholar]
- Henderson PW, Singh SP, Belkin D, Nagineni V, Weinstein AL, Weissich J, et al. Hydrogen sulfide protects against ischemia-reperfusion injury in an in vitro model of cutaneous tissue transplantation. J Surg Res. 2010;159:451–455. doi: 10.1016/j.jss.2009.05.010. [DOI] [PubMed] [Google Scholar]
- Horváth EM, Benko R, Kiss L, Murányi M, Pék T, Fekete K, et al. Rapid ‘glycaemic swings’ induce nitrosative stress, activate poly(ADP-ribose) polymerase and impair endothelial function in a rat model of diabetes mellitus. Diabetologia. 2009;52:952–961. doi: 10.1007/s00125-009-1304-0. [DOI] [PubMed] [Google Scholar]
- Hosgood SA, Nicholson ML. Hydrogen sulphide ameliorates ischaemia-reperfusion injury in an experimental model of non-heart-beating donor kidney transplantation. Br J Surg. 2010;97:202–209. doi: 10.1002/bjs.6856. [DOI] [PubMed] [Google Scholar]
- Hu LF, Lu M, Wu ZY, Wong PT, Bian JS. Hydrogen sulfide inhibits rotenone-induced apoptosis via preservation of mitochondrial function. Mol Pharmacol. 2009;75:27–34. doi: 10.1124/mol.108.047985. [DOI] [PubMed] [Google Scholar]
- Hu LF, Lu M, Tiong CX, Dawe GS, Hu G, Bian JS. Neuroprotective effects of hydrogen sulfide on Parkinson's disease rat models. Aging Cell. 2010;9:135–146. doi: 10.1111/j.1474-9726.2009.00543.x. [DOI] [PubMed] [Google Scholar]
- Hu LF, Li Y, Neo KL, Yong QC, Lee SW, Tan BK, et al. Hydrogen sulfide regulates Na+/H+ exchanger activity via stimulation of phosphoinositide 3-kinase/Akt and protein kinase G pathways. J Pharmacol Exp Ther. 2011;339:726–735. doi: 10.1124/jpet.111.184754. [DOI] [PubMed] [Google Scholar]
- Hu X, Li T, Bi S, Jin Z, Zhou G, Bai C, et al. Possible role of hydrogen sulfide on the preservation of donor rat hearts. Transplant Proc. 2007;39:3024–3029. doi: 10.1016/j.transproceed.2007.05.086. [DOI] [PubMed] [Google Scholar]
- Hu Y, Chen X, Pan TT, Neo KL, Lee SW, Khin ES, et al. Cardioprotection induced by hydrogen sulfide preconditioning involves activation of ERK and PI3K/Akt pathways. Pflugers Arch. 2008;455:607–616. doi: 10.1007/s00424-007-0321-4. [DOI] [PubMed] [Google Scholar]
- Hunter JP, Hosgood SA, Patel M, Rose R, Read K, Nicholson ML. Effects of hydrogen sulphide in an experimental model of renal ischaemia-reperfusion injury. Br J Surg. 2012;99:1665–1671. doi: 10.1002/bjs.8956. [DOI] [PubMed] [Google Scholar]
- Insko MA, Deckwerth TL, Hill P, Toombs CF, Szabo C. Detection of exhaled hydrogen sulphide gas in rats exposed to intravenous sodium sulphide. Br J Pharmacol. 2009;157:944–951. doi: 10.1111/j.1476-5381.2009.00248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain SK, Bull R, Rains JL, Bass PF, Levine SN, Reddy S, et al. Low levels of hydrogen sulfide in the blood of diabetes patients and streptozotocin-treated rats causes vascular inflammation? Antioxid Redox Signal. 2010;12:1333–1337. doi: 10.1089/ars.2009.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julian D, Statile JL, Wohlgemuth SE, Arp AJ. Enzymatic hydrogen sulfide production in marine invertebrate tissues. Comp Biochem Physiol A Mol Integr Physiol. 2002;133:105–115. doi: 10.1016/s1095-6433(02)00122-8. [DOI] [PubMed] [Google Scholar]
- Kamoun P, Belardinelli MC, Chabli A, Lallouchi K, Chadefaux-Vekemans B. Endogenous hydrogen sulfide overproduction in Down syndrome. Am J Med Genet A. 2003;116A:310–311. doi: 10.1002/ajmg.a.10847. [DOI] [PubMed] [Google Scholar]
- Kashfi K, Olson KR. Biology and therapeutic potential of hydrogen sulfide and hydrogen sulfide-releasing chimeras. Biochem Pharmacol. 2013;85:689–703. doi: 10.1016/j.bcp.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AA, Schuler MM, Prior MG, Yong S, Coppock RW, Florence LZ, et al. Effects of hydrogen sulfide exposure on lung mitochondrial respiratory chain enzymes in rats. Toxicol Appl Pharmacol. 1990;103:482–490. doi: 10.1016/0041-008x(90)90321-k. [DOI] [PubMed] [Google Scholar]
- Khan AA, Yong S, Prior MG, Lillie LE. Cytotoxic effects of hydrogen sulfide on pulmonary alveolar macrophages in rats. J Toxicol Environ Health. 1991;33:57–64. doi: 10.1080/15287399109531505. [DOI] [PubMed] [Google Scholar]
- Kimura H. Hydrogen sulfide: from brain to gut. Antioxid Redox Signal. 2010;12:1111–1123. doi: 10.1089/ars.2009.2919. [DOI] [PubMed] [Google Scholar]
- Kimura H. Production and physiological effects of hydrogen sulfide. Antioxid Redox Signal. 2013 doi: 10.1089/ars.2013.5309. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Shibuya N, Kimura Y. Hydrogen sulfide is a signaling molecule and a cytoprotectant. Antioxid Redox Signal. 2012;17:45–57. doi: 10.1089/ars.2011.4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, et al. H2S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation. 2013;127:1116–1127. doi: 10.1161/CIRCULATIONAHA.112.000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korvald C, Elvenes OP, Myrmel T. Myocardial substrate metabolism influences left ventricular energetics in vivo. Am J Physiol Heart Circ Physiol. 2000;278:H1345–H1351. doi: 10.1152/ajpheart.2000.278.4.H1345. [DOI] [PubMed] [Google Scholar]
- Krishnan N, Fu C, Pappin DJ, Tonks NK. H2S-induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci Signal. 2011;4:ra86. doi: 10.1126/scisignal.2002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Huang J, Cushnir JR, Spanel P, Smith D, Hanna GB. Selected ion flow tube-MS analysis of headspace vapor from gastric content for the diagnosis of gastro-esophageal cancer. Anal Chem. 2012;84:9550–9557. doi: 10.1021/ac302409a. [DOI] [PubMed] [Google Scholar]
- Kundu S, Pushpakumar SB, Tyagi A, Coley D, Sen U. Hydrogen sulfide deficiency and diabetic renal remodeling: role of matrix metalloproteinase-9. Am J Physiol Endocrinol Metab. 2013 doi: 10.1152/ajpendo.00604.2012. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov AV, Schneeberger S, Seiler R, Brandacher G, Mark W, Steurer W, et al. Mitochondrial defects and heterogeneous cytochrome c release after cardiac cold ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2004;286:H1633–H1641. doi: 10.1152/ajpheart.00701.2003. [DOI] [PubMed] [Google Scholar]
- Lagoutte E, Mimoun S, Andriamihaja M, Chaumontet C, Blachier F, Bouillaud F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim Biophys Acta. 2010;1797:1500–1511. doi: 10.1016/j.bbabio.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Lane N. Power games. Nature. 2006;443:901–903. doi: 10.1038/443901a. [DOI] [PubMed] [Google Scholar]
- LaRosa SP, Opal SM. Sepsis strategies in development. Clin Chest Med. 2008;29:735–747. doi: 10.1016/j.ccm.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15:110–121. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Mariappan MM, Feliers D, Cavaglieri RC, Sataranatarajan K, Abboud HE, et al. Hydrogen sulfide inhibits high glucose-induced matrix protein synthesis by activating AMP-activated protein kinase in renal epithelial cells. J Biol Chem. 2012;287:4451–4461. doi: 10.1074/jbc.M111.278325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ZW, Zhou J, Chen CS, Zhao Y, Tan CH, Li L, et al. The slow-releasing hydrogen sulfide donor, GYY4137, exhibits novel anti-cancer effects in vitro and in vivo. PLoS ONE. 2011;6:e21077. doi: 10.1371/journal.pone.0021077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leschelle X, Goubern M, Andriamihaja M, Blottiere HM, Couplan E, Gonzalez-Barroso MD, et al. Adaptative metabolic response of human colonic epithelial cells to the adverse effects of the luminal compound sulfide. Biochim Biophys Acta. 2005;1725:201–212. doi: 10.1016/j.bbagen.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Leslie M. Nothing rotten about hydrogen sulfide's medical promise. Science. 2008;320:1155–1157. doi: 10.1126/science.320.5880.1155. [DOI] [PubMed] [Google Scholar]
- Leverve XM. Mitochondrial function and substrate availability. Crit Care Med. 2007;35:S454–S460. doi: 10.1097/01.CCM.0000278044.19217.73. [DOI] [PubMed] [Google Scholar]
- Levy RJ. Mitochondrial dysfunction, bioenergetic impairment, and metabolic down-regulation in sepsis. Shock. 2007;28:24–28. doi: 10.1097/01.shk.0000235089.30550.2d. [DOI] [PubMed] [Google Scholar]
- Li J, Zhang G, Cai S, Redington AN. Effect of inhaled hydrogen sulfide on metabolic responses in anesthetized, paralyzed, and mechanically ventilated piglets. Pediatr Crit Care Med. 2008;9:110–112. doi: 10.1097/01.PCC.0000298639.08519.0C. [DOI] [PubMed] [Google Scholar]
- Li RQ, McKinstry AR, Moore JT, Caltagarone BM, Eckenhoff MF, Eckenhoff RG. Is hydrogen sulfide-induced suspended animation general anesthesia? J Pharmacol Exp Ther. 2012;341:735–742. doi: 10.1124/jpet.111.187237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SJ, Lee E, Lee EH, Kim SY, Cha JH, Choi H, et al. Docosahexaenoic acid sensitizes colon cancer cells to sulindac sulfide-induced apoptosis. Oncol Rep. 2012;27:2023–2030. doi: 10.3892/or.2012.1706. [DOI] [PubMed] [Google Scholar]
- Lloyd D. Hydrogen sulfide: clandestine microbial messenger? Trends Microbiol. 2006;14:456–462. doi: 10.1016/j.tim.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Lobb I, Mok A, Lan Z, Liu W, Garcia B, Sener A. Supplemental hydrogen sulphide protects transplant kidney function and prolongs recipient survival after prolonged cold ischaemia-reperfusion injury by mitigating renal graft apoptosis and inflammation. BJU Int. 2012;110:E1187–E1195. doi: 10.1111/j.1464-410X.2012.11526.x. [DOI] [PubMed] [Google Scholar]
- Lu F, Xing J, Zhang X, Dong S, Zhao Y, Wang L, et al. Exogenous hydrogen sulfide prevents cardiomyocyte apoptosis from cardiac hypertrophy induced by isoproterenol. Mol Cell Biochem. 2013 doi: 10.1007/s11010-013-1686-7. (in press) [DOI] [PubMed] [Google Scholar]
- Maclean KN, Janosík M, Kraus E, Kozich V, Allen RH, Raab BK, et al. Cystathionine beta-synthase is coordinately regulated with proliferation through a redox-sensitive mechanism in cultured human cells and Saccharomyces cerevisiae. J Cell Physiol. 2002;192:81–92. doi: 10.1002/jcp.10118. [DOI] [PubMed] [Google Scholar]
- Makarenko VV, Nanduri J, Raghuraman G, Fox AP, Gadalla MM, Kumar GK, et al. Endogenous H2S is required for hypoxic sensing by carotid body glomus cells. Am J Physiol Cell Physiol. 2012;303:C916–C923. doi: 10.1152/ajpcell.00100.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna P, Jain SK. Hydrogen sulfide and L-cysteine increase phosphatidylinositol 3,4,5-trisphosphate (PIP3) and glucose utilization by inhibiting phosphatase and tensin homolog (PTEN) protein and activating phosphoinositide 3-kinase (PI3K)/serine/threonine protein kinase (AKT)/protein kinase Cζ/λ (PKCζ/λ) in 3T3l1 adipocytes. J Biol Chem. 2011;286:39848–39859. doi: 10.1074/jbc.M111.270884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna P, Jain SK. L-cysteine and hydrogen sulfide increase PIP3 and AMPK/PPARγ expression and decrease ROS and vascular inflammation markers in high glucose treated human U937 monocytes. J Cell Biochem. 2013 doi: 10.1002/jcb.24578. (in press) [DOI] [PubMed] [Google Scholar]
- Minamishima S, Bougaki M, Sips PY, Yu JD, Minamishima YA, Elrod JW, et al. Hydrogen sulfide improves survival after cardiac arrest and cardiopulmonary resuscitation via a nitric oxide synthase 3-dependent mechanism in mice. Circulation. 2009;120:888–896. doi: 10.1161/CIRCULATIONAHA.108.833491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Módis K, Gero D, Nagy N, Szoleczky P, Tóth ZD, Szabo C. Cytoprotective effects of adenosine and inosine in an in vitro model of acute tubular necrosis. Br J Pharmacol. 2009;158:1565–1578. doi: 10.1111/j.1476-5381.2009.00432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Módis K, Coletta C, Erdélyi K, Papapetropoulos A, Szabo C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013a;27:601–611. doi: 10.1096/fj.12-216507. [DOI] [PubMed] [Google Scholar]
- Módis K, Panopoluos P, Olah G, Coletta C, Papapetropoulos A, Szabo C. Role of phosphodiesterase inhibition and modulation of mitochondrial cAMP levels in the bioenergetic effect of hydrogen sulfide in isolated mitochondria. Nitric Oxide. 2013b (in press) [Google Scholar]
- Módis K, Asimakopoulou A, Coletta C, Papapetropoulos A, Szabo C. Oxidative stress suppresses the cellular bioenergetic effect of the 3-mercaptopyruvate sulfurtransferase/hydrogen sulfide pathway. Biochem Biophys Res Commun. 2013c;433:401–407. doi: 10.1016/j.bbrc.2013.02.131. [DOI] [PubMed] [Google Scholar]
- Módis K, Wolanska K, Wozdek R. Hydrogen sulfide in cell signaling, signal transduction, cellular bioenergetics and physiology in C. elegans. Gen Physiol Biophys. 2013d;32:1–22. doi: 10.4149/gpb_2013001. [DOI] [PubMed] [Google Scholar]
- Morrison ML, Blackwood JE, Lockett SL, Iwata A, Winn RK, Roth MB. Surviving blood loss using hydrogen sulfide. J Trauma. 2008;65:183–188. doi: 10.1097/TA.0b013e3181507579. [DOI] [PubMed] [Google Scholar]
- Mortola JP. Implications of hypoxic hypometabolism during mammalian ontogenesis. Respir Physiol Neurobiol. 2004;141:345–356. doi: 10.1016/j.resp.2004.01.011. [DOI] [PubMed] [Google Scholar]
- Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, et al. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara N, Nagano M, Ito T, Shimamura K, Akimoto T, Suzuki H. Antioxidant enzyme, 3-mercaptopyruvate sulfurtransferase-knockout mice exhibit increased anxiety-like behaviors: a model for human mercaptolactate-cysteine disulfiduria. Sci Rep. 2013;3:1986. doi: 10.1038/srep01986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- Olson KR. A practical look at the chemistry and biology of hydrogen sulfide. Antioxid Redox Signal. 2012;17:32–44. doi: 10.1089/ars.2011.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson KR. Hydrogen sulfide as an oxygen sensor. Clin Chem Lab Med. 2013a;51:623–632. doi: 10.1515/cclm-2012-0551. [DOI] [PubMed] [Google Scholar]
- Olson KR. Hydrogen sulfide: both feet on the gas and none on the brake? Front Physiol. 2013b;4:2. doi: 10.3389/fphys.2013.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson KR. A theoretical examination of hydrogen sulfide metabolism and its potential in autocrine/paracrine oxygen sensing. Respir Physiol Neurobiol. 2013c;186:173–179. doi: 10.1016/j.resp.2013.01.010. [DOI] [PubMed] [Google Scholar]
- Olson KR, Whitfield NL. Hydrogen sulfide and oxygen sensing in the cardiovascular system. Antioxid Redox Signal. 2010;12:1219–1234. doi: 10.1089/ars.2009.2921. [DOI] [PubMed] [Google Scholar]
- Osipov RM, Robich MP, Feng J, Liu Y, Clements RT, Glazer HP, et al. Effect of hydrogen sulfide in a porcine model of myocardial ischemia-reperfusion: comparison of different administration regimens and characterization of the cellular mechanisms of protection. J Cardiovasc Pharmacol. 2009;54:287–297. doi: 10.1097/FJC.0b013e3181b2b72b. [DOI] [PubMed] [Google Scholar]
- Osipov RM, Robich MP, Feng J, Chan V, Clements RT, Deyo RJ, et al. Effect of hydrogen sulfide on myocardial protection in the setting of cardioplegia and cardiopulmonary bypass. Interact Cardiovasc Thorac Surg. 2010;10:506–512. doi: 10.1510/icvts.2009.219535. [DOI] [PubMed] [Google Scholar]
- Pacher P, Szabo C. Role of peroxynitrite in the pathogenesis of cardiovascular complications of diabetes. Curr Opin Pharmacol. 2006;6:136–141. doi: 10.1016/j.coph.2006.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangare M, Makino A. Mitochondrial function in vascular endothelial cell in diabetes. J Smooth Muscle Res. 2012;48:1–26. doi: 10.1540/jsmr.48.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetropoulos A, Pyriochou A, Altaany Z, Yang G, Marazioti A, Zhou Z, et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci U S A. 2009;106:21972–21977. doi: 10.1073/pnas.0908047106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania D, Millard M, Neamati N. Opportunities in discovery and delivery of anticancer drugs targeting mitochondria and cancer cell metabolism. Adv Drug Deliv Rev. 2009;61:1250–1275. doi: 10.1016/j.addr.2009.05.010. [DOI] [PubMed] [Google Scholar]
- Paul BD, Snyder SH. H2S signalling through protein sulfhydration and beyond. Nat Rev Mol Cell Biol. 2012;13:499–507. doi: 10.1038/nrm3391. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Nanduri J, Raghuraman G, Souvannakitti D, Gadalla MM, Kumar GK, et al. H2S mediates O2 sensing in the carotid body. Proc Natl Acad Sci U S A. 2010;107:10719–10724. doi: 10.1073/pnas.1005866107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polhemus D, Kondo K, Bhushan S, Bir SC, Kevil CG, Murohara T, et al. Hydrogen sulfide attenuates cardiac dysfunction following heart failure via induction of angiogenesis. Circ Heart Fail. 2013 doi: 10.1161/CIRCHEARTFAILURE.113.000299. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell MA, Somero GN. Hydrogen sulfide oxidation is coupled to oxidative phosphorylation in mitochondria of Solemya reidi. Science. 1986;233:563–566. doi: 10.1126/science.233.4763.563. [DOI] [PubMed] [Google Scholar]
- Predmore BL, Lefer DJ. Development of hydrogen sulfide-based therapeutics for cardiovascular disease. J Cardiovasc Transl Res. 2010;3:487–498. doi: 10.1007/s12265-010-9201-y. [DOI] [PubMed] [Google Scholar]
- Pun PB, Lu J, Kan EM, Moochhala S. Gases in the mitochondria. Mitochondrion. 2010;10:83–93. doi: 10.1016/j.mito.2009.12.142. [DOI] [PubMed] [Google Scholar]
- Ramsay EE, Hogg PJ, Dilda PJ. Mitochondrial metabolism inhibitors for cancer therapy. Pharm Res. 2011;28:2731–2744. doi: 10.1007/s11095-011-0584-5. [DOI] [PubMed] [Google Scholar]
- Rapozzi V, Della Pietra E, Zorzet S, Zacchigna M, Bonavida B, Xodo LE. Nitric oxide-mediated activity in anti-cancer photodynamic therapy. Nitric Oxide. 2013;30:26–35. doi: 10.1016/j.niox.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Rashid S, Heer JK, Garle MJ, Alexander SP, Roberts RE. Hydrogen sulphide-induced relaxation of porcine peripheral bronchioles. Br J Pharmacol. 2013;168:1902–1910. doi: 10.1111/bph.12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert K, Chassé JF, Santiard-Baron D, Vayssettes C, Chabli A, Aupetit J, et al. Altered gene expression in liver from a murine model of hyperhomocysteinemia. J Biol Chem. 2003;278:31504–31511. doi: 10.1074/jbc.M213036200. [DOI] [PubMed] [Google Scholar]
- Rosenson RS, Fioretto P, Dodson PM. Does microvascular disease predict macrovascular events in type 2 diabetes? Atherosclerosis. 2011;218:13–18. doi: 10.1016/j.atherosclerosis.2011.06.029. [DOI] [PubMed] [Google Scholar]
- Sadat U. Signaling pathways of cardioprotective ischemic preconditioning. Int J Surg. 2009;7:490–498. doi: 10.1016/j.ijsu.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491:364–373. doi: 10.1038/nature11706. [DOI] [PubMed] [Google Scholar]
- Seitz DH, Fröba JS, Niesler U, Palmer A, Veltkamp HA, Braumüller ST. Inhaled hydrogen sulfide induces suspended animation, but does not alter the inflammatory response after blunt chest trauma. Shock. 2012;37:197–204. doi: 10.1097/SHK.0b013e31823f19a0. [DOI] [PubMed] [Google Scholar]
- Shin D, Pregenzer G, Jr, Gardin JM. Erectile dysfunction: a disease marker for cardiovascular disease. Cardiol Rev. 2011;19:5–11. doi: 10.1097/CRD.0b013e3181fb7eb8. [DOI] [PubMed] [Google Scholar]
- Shin HA, Cha YY, Park MS, Kim JM, Lim YC. Diallyl sulfide induces growth inhibition and apoptosis of anaplastic thyroid cancer cells by mitochondrial signaling pathway. Oral Oncol. 2010;46:e15–e18. doi: 10.1016/j.oraloncology.2009.10.012. [DOI] [PubMed] [Google Scholar]
- Si YF, Wang J, Guan J, Zhou L, Sheng Y, Zhao J. Treatment with hydrogen sulfide alleviates streptozotocin-induced diabetic retinopathy in rats. Br J Pharmacol. 2013;169:619–631. doi: 10.1111/bph.12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon F, Giudici R, Duy CN, Schelzig H, Oter S, Gröger M, et al. Hemodynamic and metabolic effects of hydrogen sulfide during porcine ischemia/reperfusion injury. Shock. 2008;30:359–364. doi: 10.1097/SHK.0b013e3181674185. [DOI] [PubMed] [Google Scholar]
- Simon F, Scheuerle A, Gröger M, Stahl B, Wachter U, Vogt J, et al. Effects of intravenous sulfide during porcine aortic occlusion-induced kidney ischemia/reperfusion injury. Shock. 2011;35:156–163. doi: 10.1097/SHK.0b013e3181f0dc91. [DOI] [PubMed] [Google Scholar]
- Siriussawakul A, Chen LI, Lang JD. Medical gases: a novel strategy for attenuating ischemia-reperfusion injury in organ transplantation? J Transplant. 2012;2012:819382. doi: 10.1155/2012/819382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivitz WI, Yorek MA. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid Redox Signal. 2010;12:537–577. doi: 10.1089/ars.2009.2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder PM, de Boer RA, Bos EM, van den Born JC, Ruifrok WP, Vreeswijk-Baudoin I, et al. Gaseous hydrogen sulfide protects against myocardial ischemia-reperfusion injury in mice partially independent from hypometabolism. PLoS ONE. 2013;8:e63291. doi: 10.1371/journal.pone.0063291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodha NR, Clements RT, Feng J, Liu Y, Bianchi C, Horvath EM, et al. The effects of therapeutic sulfide on myocardial apoptosis in response to ischemia-reperfusion injury. Eur J Cardiothorac Surg. 2008;33:906–913. doi: 10.1016/j.ejcts.2008.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodha NR, Clements RT, Feng J, Liu Y, Bianchi C, Horvath EM, et al. Hydrogen sulfide therapy attenuates the inflammatory response in a porcine model of myocardial ischemia/reperfusion injury. J Thorac Cardiovasc Surg. 2009;138:977–984. doi: 10.1016/j.jtcvs.2008.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitt AW, Lois N, Medina RJ, Adamson P, Curtis TM. Advances in our understanding of diabetic retinopathy. Clin Sci (Lond) 2013;125:1–17. doi: 10.1042/CS20120588. [DOI] [PubMed] [Google Scholar]
- Sun WH, Liu F, Chen Y, Zhu YC. Hydrogen sulfide decreases the levels of ROS by inhibiting mitochondrial Complex IV and increasing SOD activities in cardiomyocytes under ischemia/reperfusion. Biochem Biophys Res Commun. 2012;421:164–169. doi: 10.1016/j.bbrc.2012.03.121. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Olah G, Módis K, Coletta C, Kulp G, Gerö D, et al. Hydrogen sulfide replacement therapy protects the vascular endothelium in hyperglycemia by preserving mitochondrial function. Proc Natl Acad Sci U S A. 2011;108:13829–13834. doi: 10.1073/pnas.1105121108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C. Roles of poly(ADP-ribose) polymerase activation in the pathogenesis of diabetes mellitus and its complications. Pharmacol Res. 2005;52:60–71. doi: 10.1016/j.phrs.2005.02.015. [DOI] [PubMed] [Google Scholar]
- Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- Szabo C. Role of nitrosative stress in the pathogenesis of diabetic vascular dysfunction. Br J Pharmacol. 2009;156:713–727. doi: 10.1111/j.1476-5381.2008.00086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C. Gaseotransmitters: new frontiers for translational science. Sci Transl Med. 2010;2:59ps54. doi: 10.1126/scitranslmed.3000721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C. Roles of hydrogen sulfide in the pathogenesis of diabetes mellitus and its complications. Antioxid Redox Signal. 2012;17:68–80. doi: 10.1089/ars.2011.4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C, Papapetropoulos A. Hydrogen sulphide and angiogenesis: mechanisms and applications. Br J Pharmacol. 2011;164:853–865. doi: 10.1111/j.1476-5381.2010.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C, Módis K, Ransy C, Andriamihaja M, Murghes B, Coletta C, et al. Regulation of mitochondrial function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br J Pharmacol. 2013a doi: 10.1111/bph.12369. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C, Coletta C, Chao C, Módis K, Szczeszny B, Papapetropoulos A, et al. Tumor-derived H2S, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation and angiogenesis in colon cancer. Proc Natl Acad Sci U S A. 2013b;110:12474–12479. doi: 10.1073/pnas.1306241110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo G, Veres G, Radovits T, Gero D, Módis K, Miesel-Gröschel C, et al. Cardioprotective effects of hydrogen sulfide. Nitric Oxide. 2011;25:201–210. doi: 10.1016/j.niox.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szoleczky P, Módis K, Nagy N, Dóri Tóth Z, DeWitt D, Szabo C, et al. Identification of agents that reduce renal hypoxia-reoxygenation injury using cellbased screening: purine nucleosides are alternative energy sources in LLC-PK1 cells during hypoxia. Arch Biochem Biophys. 2012;517:53–70. doi: 10.1016/j.abb.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang XQ, Ren YK, Zhou CF, Yang CT, Gu HF, He JQ, et al. Hydrogen sulfide prevents formaldehyde-induced neurotoxicity to PC12 cells by attenuation of mitochondrial dysfunction and pro-apoptotic potential. Neurochem Int. 2012;61:16–24. doi: 10.1016/j.neuint.2012.04.011. [DOI] [PubMed] [Google Scholar]
- Teng H, Wu B, Zhao K, Yang G, Wu L, Wang R. Oxygen-sensitive mitochondrial accumulation of cystathione β-synthase mediated by Lon protease. Proc Natl Acad Sci U S A. 2013 doi: 10.1073/pnas.1308487110. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RW, Valentine HL, Valentine WM. Cytotoxic mechanisms of hydrosulfide anion and cyanide anion in primary rat hepatocyte cultures. Toxicology. 2003;188:149–159. doi: 10.1016/s0300-483x(03)00079-9. [DOI] [PubMed] [Google Scholar]
- Thornburg JM, Nelson KK, Clem BF, Lane AN, Arumugam S, Simmons A, et al. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res. 2008;10:R84. doi: 10.1186/bcr2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiranti V, Zeviani M. Altered sulfide (H2S) metabolism in ethylmalonic encephalopathy. Cold Spring Harb Perspect Biol. 2013;5:a011437. doi: 10.1101/cshperspect.a011437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiranti V, Viscomi C, Hildebrandt T, Di Meo I, Mineri R, Tiveron C, et al. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat Med. 2009;15:200–205. doi: 10.1038/nm.1907. [DOI] [PubMed] [Google Scholar]
- Tokuda K, Kida K, Marutani E, Crimi E, Bougaki M, Khatri A, et al. Inhaled hydrogen sulfide prevents endotoxin-induced systemic inflammation and improves survival by altering sulfide metabolism in mice. Antioxid Redox Signal. 2012;17:11–21. doi: 10.1089/ars.2011.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaskova Z, Cacanyiova S, Benco A, Kristek F, Dugovicova L, Hrbac J, et al. Lipids modulate H2S/HS- induced NO release from S-nitrosoglutathione. Biochem Biophys Res Commun. 2009;390:1241–1244. doi: 10.1016/j.bbrc.2009.10.128. [DOI] [PubMed] [Google Scholar]
- Toombs CF, Insko MA, Wintner EA, Deckwerth TL, Usansky H, Jamil K, et al. Detection of exhaled hydrogen sulphide gas in healthy human volunteers during intravenous administration of sodium sulphide. Br J Clin Pharmacol. 2010;69:626–636. doi: 10.1111/j.1365-2125.2010.03636.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong DH, Eghbal MA, Hindmarsh W, Roth SH, O'Brien PJ. Molecular mechanisms of hydrogen sulfide toxicity. Drug Metab Rev. 2006;38:733–744. doi: 10.1080/03602530600959607. [DOI] [PubMed] [Google Scholar]
- Tsubura A, Lai YC, Kuwata M, Uehara N, Yoshizawa K. Anticancer effects of garlic and garlic-derived compounds for breast cancer control. Anticancer Agents Med Chem. 2011;11:249–253. doi: 10.2174/187152011795347441. [DOI] [PubMed] [Google Scholar]
- Van de Louw A, Haouzi P. Oxygen deficit and H2S in hemorrhagic shock in rats. Crit Care. 2012;16:R178. doi: 10.1186/cc11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Marken Lichtenbelt WD, Schrauwen P. Implications of nonshivering thermogenesis for energy balance regulation in humans. Am J Physiol Regul Integr Comp Physiol. 2011;301:R285–R296. doi: 10.1152/ajpregu.00652.2010. [DOI] [PubMed] [Google Scholar]
- VanderHeiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkel S, Grieshaber M. Mitochondrial sulfide oxidation in Arenicola marina. Eur J Biochem. 1996;235:231–237. doi: 10.1111/j.1432-1033.1996.00231.x. [DOI] [PubMed] [Google Scholar]
- Volpato GP, Searles R, Yu B, Scherrer-Crosbie M, Bloch KD, Ichinose F, et al. Inhaled hydrogen sulfide: a rapidly reversible inhibitor of cardiac and metabolic function in the mouse. Anesthesiology. 2008;108:659–668. doi: 10.1097/ALN.0b013e318167af0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vozdek R, Hnízda A, Krijt J, Kostrouchová M, Kožich V. Novel structural arrangement of nematode cystathionine β-synthases: characterization of Caenorhabditis elegans CBS-1. Biochem J. 2012;443:535–547. doi: 10.1042/BJ20111478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner F, Asfar P, Calzia E, Radermacher P, Szabo C. Bench-to-bedside review: hydrogen sulfide – the third gaseous transmitter: applications for critical care. Crit Care. 2009;13:213. doi: 10.1186/cc7700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner F, Wagner K, Weber S, Stahl B, Knöferl MW, Huber-Lang M, et al. Inflammatory effects of hypothermia and inhaled H2S during resuscitated, hyperdynamic murine septic shock. Shock. 2011;35:396–402. doi: 10.1097/SHK.0b013e3181ffff0e. [DOI] [PubMed] [Google Scholar]
- Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- Wang Z, Liu DX, Wang FW, Zhang Q, Du ZX, Zhan JM, et al. L-Cysteine promotes the proliferation and differentiation of neural stem cells via the CBS/H2S pathway. Neuroscience. 2013;237:106–117. doi: 10.1016/j.neuroscience.2012.12.057. [DOI] [PubMed] [Google Scholar]
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen YD, Wang H, Kho SH, Rinkiko S, Sheng X, Shen HM, et al. Hydrogen sulfide protects HUVECs against hydrogen peroxide induced mitochondrial dysfunction and oxidative stress. PLoS ONE. 2013;8:e53147. doi: 10.1371/journal.pone.0053147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman M, Winyard PG. Hydrogen sulfide and inflammation: the good, the bad, the ugly and the promising. Expert Rev Clin Pharmacol. 2011;4:13–32. doi: 10.1586/ecp.10.134. [DOI] [PubMed] [Google Scholar]
- Whiteman M, Li L, Kostetski I, Chu SH, Siau JL, Bhatia M, et al. Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide. Biochem Biophys Res Commun. 2006;343:303–310. doi: 10.1016/j.bbrc.2006.02.154. [DOI] [PubMed] [Google Scholar]
- Whiteman M, Gooding KM, Whatmore JL, Ball CI, Mawson D, Skinner K, et al. Adiposity is a major determinant of plasma levels of the novel vasodilator hydrogen sulphide. Diabetologia. 2010;53:1722–1726. doi: 10.1007/s00125-010-1761-5. [DOI] [PubMed] [Google Scholar]
- Whiteman M, Le Trionnaire S, Chopra M, Fox B, Whatmore J. Emerging role of hydrogen sulfide in health and disease: critical appraisal of biomarkers and pharmacological tools. Clin Sci (Lond) 2011;121:459–488. doi: 10.1042/CS20110267. [DOI] [PubMed] [Google Scholar]
- Whitfield NL, Kreimier EL, Verdial FC, Skovgaard N, Olson KR. Reappraisal of H2S/sulfide concentration in vertebrate blood and its potential significance in ischemic preconditioning and vascular signaling. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1930–R1937. doi: 10.1152/ajpregu.00025.2008. [DOI] [PubMed] [Google Scholar]
- Wintner EA, Deckwerth TL, Langston W, Bengtsson A, Leviten D, Hill P, et al. A monobromobimane-based assay to measure the pharmacokinetic profile of reactive sulphide species in blood. Br J Pharmacol. 2010;160:941–957. doi: 10.1111/j.1476-5381.2010.00704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Wei J, You X, Chen X, Zhu H, Zhu X, et al. Inhibition of hydrogen sulfide generation contributes to lung injury after experimental orthotopic lung transplantation. J Surg Res. 2013;182:e25–e33. doi: 10.1016/j.jss.2012.09.028. [DOI] [PubMed] [Google Scholar]
- Wu YC, Wang XJ, Yu L, Chan FK, Cheng AS, Yu J, et al. Hydrogen sulfide lowers proliferation and induces protective autophagy in colon epithelial cells. PLoS ONE. 2012;7:e37572. doi: 10.1371/journal.pone.0037572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Sun A, Zhu W, Huang Z, Hu X, Jia J, et al. Transplantation of mesenchymal stem cells preconditioned with hydrogen sulfide enhances repair of myocardial infarction in rats. Tohoku J Exp Med. 2012;226:29–36. doi: 10.1620/tjem.226.29. [DOI] [PubMed] [Google Scholar]
- Yamagishi K, Onuma K, Chiba Y, Yagi S, Aoki S, Sato T, et al. Generation of gaseous sulfur-containing compounds in tumour tissue and suppression of gas diffusion as an antitumour treatment. Gut. 2012;61:554–561. doi: 10.1136/gutjnl-2011-300721. [DOI] [PubMed] [Google Scholar]
- Yamamoto J, Sato W, Kosugi T, Yamamoto T, Kimura T, Taniguchi S, et al. Distribution of hydrogen sulfide (H2S)-producing enzymes and the roles of the H2S donor sodium hydrosulfide in diabetic nephropathy. Clin Exp Nephrol. 2013;17:32–40. doi: 10.1007/s10157-012-0670-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Tang G, Zhang L, Wu L, Wang R. The pathogenic role of cystathionine γ-lyase/hydrogen sulfide in streptozotocin-induced diabetes in mice. Am J Pathol. 2011;179:869–879. doi: 10.1016/j.ajpath.2011.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Cohen MV, Downey JM. Mechanism of cardioprotection by early ischemic preconditioning. Cardiovasc Drugs Ther. 2010;24:225–234. doi: 10.1007/s10557-010-6236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao LL, Huang X-W, Wang Y-G, Cao Y-X, Zhang C-C, Zhu YZ. Hydrogen sulfide protects cardiomyocytes from hypoxia/reoxygenation-induced apoptosis by preventing GSK-3beta-dependent opening of mPTP. Am J Physiol Heart Circ Physiol. 2010;298:H1310–H1319. doi: 10.1152/ajpheart.00339.2009. [DOI] [PubMed] [Google Scholar]
- Yuan P, Xue H, Zhou L, Qu L, Li C, Wang Z, et al. Rescue of mesangial cells from high glucose-induced over-proliferation and extracellular matrix secretion by hydrogen sulfide. Nephrol Dial Transplant. 2011;26:2119–2126. doi: 10.1093/ndt/gfq749. [DOI] [PubMed] [Google Scholar]
- Yusof M, Kamada K, Kalogeris T, Gaskin FS, Korthuis RJ. Hydrogen sulfide triggers late-phase preconditioning in postischemic small intestine by an NO-and p38 MAPK-dependent mechanism. Am J Physiol Heart Circ Physiol. 2009;296:H868–H876. doi: 10.1152/ajpheart.01111.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanardo RCO, Brancaleone V, Distrutti E, Fiorucci S, Cirino G, Wallace JL. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006;20:2118–2120. doi: 10.1096/fj.06-6270fje. [DOI] [PubMed] [Google Scholar]
- Zhang W, Braun A, Bauman Z, Olteanu H, Madzelan P, Banerjee R. Expression profiling of homocysteine junction enzymes in the NCI60 panel of human cancer cell lines. Cancer Res. 2005;65:1554–1560. doi: 10.1158/0008-5472.CAN-04-1554. [DOI] [PubMed] [Google Scholar]
- Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X, Wang L, Wang Y, Dong S, Leng X, Jia J, et al. Exogenous hydrogen sulfide attenuates diabetic myocardial injury through cardiac mitochondrial protection. Mol Cell Biochem. 2012;371:187–198. doi: 10.1007/s11010-012-1435-3. [DOI] [PubMed] [Google Scholar]
- Zhou X, Lu X. Hydrogen sulfide inhibits high-glucose-induced apoptosis in neonatal rat cardiomyocytes. Exp Biol Med. 2013;238:370–374. doi: 10.1177/1535370213477989. [DOI] [PubMed] [Google Scholar]
- Zhu JX, Kalbfleisch M, Yang YX, Bihari R, Lobb I, Davison M, et al. Detrimental effects of prolonged warm renal ischaemia-reperfusion injury are abrogated by supplemental hydrogen sulphide: an analysis using real-time intravital microscopy and polymerase chain reaction. BJU Int. 2012;110:E1218–E1227. doi: 10.1111/j.1464-410X.2012.11555.x. [DOI] [PubMed] [Google Scholar]
- Zuckerbraun BS, Chin BY, Bilban M, d'Avila JC, Rao J, Billiar TR, et al. Carbon monoxide signals via inhibition of cytochrome c oxidase and generation of mitochondrial reactive oxygen species. FASEB J. 2007;21:1099–1106. doi: 10.1096/fj.06-6644com. [DOI] [PubMed] [Google Scholar]