

Abstract
Transition metals are critical for enzyme function and protein folding, but in excess can mediate neurotoxic oxidative processes. As mitochondria are particularly vulnerable to oxidative damage due to radicals generated during ATP production, mitochondrial biometal homeostasis must therefore be tightly controlled to safely harness the redox potential of metal enzyme cofactors. Dysregulation of metal functions is evident in numerous neurological disorders including Alzheimer's disease, stroke, Parkinson's disease, Huntington's disease, amyotrophic lateral sclerosis and Friedrich's ataxia. This review describes the mitochondrial metal defects in these disorders and highlights novel metal-based therapeutic approaches that target mitochondrial metal homeostasis in neurological disorders.
Linked Articles
This article is part of a themed issue on Mitochondrial Pharmacology: Energy, Injury & Beyond. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2014.171.issue-8
Keywords: mitochondria, biometal homeostasis, neurodegeneration, stroke, Cu(atsm), Mn porphyrins, curcumin, deferiprone
Mitochondrial metal homeostasis
Transition metals are crucial for enzyme activity and correct folding and conformation of proteins. Due to the central role for mitochondria in energy production, which generates reactive oxygen intermediates as by-products, transition metals are required for ATP generation and control of potential oxidative damage in mitochondria. Copper, zinc, iron and manganese are all localized to various compartments in the mitochondria and their regulation is controlled by a diverse range of cofactors, chaperones and ion pumps (Pierrel et al., 2007; Rines and Ardehali, 2013). Mitochondrial control of Cu homeostasis has been well characterized in recent years, although predominantly in yeast rather than mammalian cells (reviewed in Pierrel et al., 2007); however, far less is known of the transport and chaperone processes involved in regulation of other transition metals. This review will describe the functions of transition metals in the mitochondria and highlight a number of neurological diseases where perturbations to mitochondrial metal homeostasis contribute to pathology. We will discuss the potential of novel therapeutics including metallo-complexes and metal chelators that target mitochondrial dysfunction and/or correct mitochondrial metal disturbances through metal delivery, sequestration or antioxidant properties.
Iron has an essential role in mitochondrial iron-sulfur clusters and haeme-containing proteins, which are crucial for the function of several mitochondrial and cytosolic enzymes including elements of the electron transport chain (ETC) associated with ATP energy production (Atamna et al., 2002; Pierrel et al., 2007). The mechanism of iron import into the intermembrane space (IMS) is unknown; however, mitoferrins pump iron into the matrix (Richardson et al., 2010; Huang et al., 2011).
Cu is an essential element of complex IV (cytochrome c oxidase) in the mitochondria. The Cu is delivered into the IMS from cytosolic pools via Cox17 to additional carriers, Sco1 or Cox11 and subsequently to Cox1 or Cox2 (Kim et al., 2008). Although Cu levels in the mitochondria are relatively low compared with iron levels, Cu exists at least an order of magnitude higher than required for cytochrome c oxidase function, mostly as a low molecular weight soluble ligand, suggesting that there are additional critical functions for Cu in the mitochondria that have yet to be determined.
Manganese has a central role in maintaining antioxidant enzyme function in the mitochondria as the key element of manganese superoxide dismutase (MnSOD also known as SOD2) located in the mitochondrial matrix and has additional enzymatic roles in arginine synthesis (Aschner et al., 2009). MnSOD is essential for removal of superoxide anions released during mitochondrial respiration that are precursors of damaging reactive oxygen species (ROS). In fact, the mitochondria are the primary site for manganese accumulation in the cell and manganese is delivered from the cytosol by a Ca2+ uniporter (Gunter and Puskin, 1972; Liccione and Maines, 1988). Interestingly, despite the close similarity between manganese-induced and Parkinson's disease-associated neurotoxicity (see below/see Parkinson's disease section), little is known of manganese handling in the mitochondria. There is also limited information on mitochondrial control of zinc homeostasis. A mitochondrial zinc pool exists in the matrix and is critical for maintenance of zinc-binding metallo-enzymes. A pool can also be mobilized during stress, although the mechanisms are unclear (Sensi et al., 2003). Zinc, along with copper has an essential antioxidant function in Cu/Zn superoxide dismutase 1 (SOD1) that exists in the cytosol and mitochondrial IMS (Field et al., 2003).
Despite the lack of understanding regarding regulation of mitochondrial transition metal pools, particularly in the brain, considerable evidence supports perturbation to these pools in several neurodegenerative disorders (summarized in Table 1). As reviewed below, loss or accumulation of essential transition metal pools in specific compartments of the mitochondria feature strongly in neurodegenerative diseases and may have a critical role in leading to impaired energy production, especially critical for the high energy demands of neurons and/or generation of reactive oxygen species (ROS) and activation of mitochondrial-dependent apoptosis. This review describes the mitochondrial metal defects in amyotrophic lateral sclerosis (ALS), Parkinson's disease (PD), stroke, Friedrich's ataxia (FRDA) and Huntington's disease (HD). Mitochondrial metal homeostasis is also altered in AD, but this is reviewed in detail elsewhere (Eckert et al., 2012) and will not be covered in the present review. Importantly, the abnormal metal handling of mitochondria provides a unique set of targets for therapeutic intervention as covered in the final section of this review.
Table 1.
A summary of the cell and animal models, metal distribution and mitochondrial dysfunction in neurological disorders with altered mitochondrial function
| Disorder | In vitro/in vivo models | Aberrant metal distribution | Mito protein dysfunction |
|---|---|---|---|
| Amyotrophic lateral sclerosis | Most common models: SOD1G37R, SOD1G93A transgenic mice | Mitochondrial Fe accumulation | Aberrant mitochondrial localization of SOD1 Deregulation of Fe-S proteins Elevated MnSOD levels |
| Parkinson's disease | Complex I inhibitors (e.g. rotenone, 6OHDA, paraquat, MPP+, MPTP) Mn elicits similar symptoms |
Mitochondrial Fe accumulation Loss of Cu |
Complex I inhibition Inhibitors increase mitochondrial Fe |
| Stroke | Cerebral artery occlusion ischaemia/reperfusion injury Subarachnoid haemorrhage Rabbit small clot embolic stroke |
Possible aberrant Zn redistribution Lysosomal Fe transport to mitochondria |
|
| Friedrich's ataxia | Frataxin knockdown cell models Frataxin knockdown Drosophila, C. elegans models Heterozygous frataxin knockout mice |
Mitochondrial Fe accumulation Cytosolic depletion of Fe |
Possible impaired Fe-S cluster biogenesis Reduced frataxin expression, overexpression of mitoferrin |
| Huntington's disease | Complex II inhibitors (e.g. 3NP), CAG140 knock-in mice | Accumulation of Fe, Cu, Zn Possible mito Zn |
Complex II/III inhibition Fe-S subunit in complex II |
Amyotrophic lateral sclerosis
ALS is a fatal adult-onset neurodegenerative disease in which motor neurons in the spinal cord and brain progressively deteriorate (Bradley, 1995). ALS is characterized by progressive paralysis and wasting of skeletal muscles. The average age of onset is around 50–60 years and patients rarely survive more than 3–5 years after diagnosis, with respiratory failure the most common cause of death (Al-Chalabi et al., 2012). About 5–20% of ALS cases are familial, caused by mutations in SOD1, Tar DNA-binding protein (TDP-43), fused in sarcoma, C9orf72 and multiple other genes, and the remainder of cases are sporadic (Al-Chalabi et al., 2012). Mitochondrial dysfunction is evident in ALS (reviewed in Shi et al., 2010), including mitochondrial abnormalities in the spinal cord of sporadic ALS patients (Sasaki and Iwata, 1996), and impaired mitochondrial function in ALS patients (Bowling et al., 1993) and transgenic mice (Dupuis et al., 2011; Panov et al., 2011).
A portion of the mainly cytosolic Cu/Zn antioxidant protein SOD1 is localized to mitochondria, predominantly in the IMS (Kawamata and Manfredi, 2010), although a portion of mutated SOD1 has been found bound to the mitochondrial outer membrane (Vande Velde et al., 2008). IMS localization of SOD1 is promoted by the copper chaperone for SOD1 (CCS), which delivers copper to SOD1 (Kawamata and Manfredi, 2010). Enhanced delivery of copper to SOD1 by overexpression of CCS increases aggregation of mitochondrial SOD1G93A and induces toxicity in cultured neuronal cells (Cozzolino et al., 2009). Furthermore, increasing copper delivery to mutant SOD1 in vivo in double transgenic mice overexpressing SOD1G93A and CCS greatly accelerated the disease course, increased mitochondrial mutant SOD1 content and caused severe mitochondrial pathology (Son et al., 2007). In addition, increasing mitochondrial mutated SOD1 content by genetically introducing an IMS targeting signal to SOD1 causes toxicity in cultured motor neuron cells (Magrane et al., 2009) and, in vivo, transgenic mice expressing mitochondrially targeted mutant SOD1G93A develop mitochondrial dysfunction, motor impairment and motor neuron death. However, the abnormalities of mutant SOD1 in mitochondria only partially epitomize the ALS disease phenotype, with no muscle denervation. Hence mitochondrial localization of mutant SOD1 causes many but not all features of ALS, and mutant SOD1 localized to compartments other than mitochondria may contribute to disease (Igoudjil et al., 2011). These studies indicate that increasing the mitochondrial content of ALS-associated mutated forms of SOD1 may contribute to the mitochondrial aberrations evident in ALS.
Perturbations in iron are also evident in ALS. Iron accumulation has been reported in the CNS of both familial and sporadic forms of ALS patients (Oba et al., 1993; Kasarskis et al., 1995) and in mitochondria in the spinal cord of transgenic SOD1G37R ALS model mice (Jeong et al., 2009). Dysregulation of mitochondrial iron proteins including iron-sulfur cluster scaffold protein, mitoferrin and frataxin are evident in in vivo and in vitro models of ALS (Jeong et al., 2009; Hadzhieva et al., 2013).
A subset of apparently sporadic ALS patients exhibit elevated mitochondrial MnSOD (McEachern et al., 2000). Overexpression of MnSOD rescues neuroblastoma cells in the presence of several different ALS-associated SOD1 mutations (Zimmerman et al., 2007). Selenium exposure induces toxicity, increases MnSOD and induces translocation of SOD1 into mitochondria in neuroblastoma cells (Maraldi et al., 2011). These studies suggest MnSOD responds to ALS-associated mitochondrial stress, but it is unclear whether it helps or contributes to dysfunction.
Stroke
Stroke, or ischaemia, caused by blockage of blood vessels in the brain is a leading cause of adult death worldwide. Blockage of major blood vessels due to thrombosis or embolism results in deprivation of oxygen and nutrients to local brain tissue. In ischaemia-reperfusion injury, the initial blockage is followed by a potentially more damaging period of re-oxygenation after removal of the blockage. The process results in loss of energy production in neurons and subsequent neuronal cell death and, during reperfusion, this is exacerbated by high levels of oxidative damage from returning oxygen. Although the pathways of neuronal damage are complex, one of the major factors is dysregulated Ca2+ and/or zinc influx via glutamate receptors (Sensi and Jeng, 2004), leading to activation of mitochondrial-associated cell death pathways (reviewed in detail in this special issue by Duchen, 2012). In fact, the mitochondria are thought to be the primary target of altered cellular metabolism associated with ischaemia and ischaemia-reperfusion injury (Jordan et al., 2011).
There is currently little understanding of how mitochondrial transition metal homeostasis is altered during stroke. However, it is thought that zinc is released from both mitochondrial and cytosolic stores in response to ischaemia-reperfusion injury, in addition to potential influx via glutamate receptors (Sensi and Jeng, 2004). This leads to generation of ROS and subsequent oxidative damage to neurons. These effects appear to be very potent, occurring at zinc concentrations lower than the concentrations required for Ca2+-mediated neurotoxicity (Jiang et al., 2001; Dineley et al., 2005). The importance of abnormal mitochondrial zinc accumulation in ischaemic damage has been supported by studies demonstrating that depletion of zinc reduces mitochondrial cytochrome c release and activation of downstream caspases (Calderone et al., 2004). The role of additional transition metals in mitochondria during ischaemia-reperfusion is even less well understood than zinc. There are currently very few studies on the distribution of copper, iron or manganese pools in mitochondria during ischaemia-reperfusion. Zhang and Lemasters observed that during ischaemia in rat hepatocytes, iron released from lysosomes was transported to mitochondria (Zhang and Lemasters, 2013). However, similar studies in neurons have not been reported. Studies supporting a role for mitochondrial transition metals in the regulation of mitochondrial function during ischaemia have instead come largely from studies on MnSOD. Several reports have described a central role for the mitochondrial manganese-containing MnSOD in neuroprotection against ischaemia-reperfusion injury in gerbils, rats and mice (Wakade et al., 2008; Jung et al., 2009). This protective action is probably related to the essential role of the enzyme in removal of superoxide radicals in mitochondria, and is supported by the therapeutic action of manganese porphyrins as discussed below (Saba et al., 2007). No studies have yet reported changes to mitochondrial copper pools during ischaemia.
Friedrich's ataxia
FRDA is a progressive neurodegenerative disease clinically characterized by progressive ataxia (lack of voluntary muscle coordination), dysarthria (speech impairment), areflexia and progressive motor weakness. Patients also develop hypertrophic cardiomyopathy and diabetes. Symptoms appear in childhood and progressively worsen through adulthood, commonly fatal in mid-adulthood. Neuropathologically, FDRA involves the degeneration of the dentate nuclei in the cerebellum and the sensory and motor tracts of the spinal cord (Marmolino, 2011).
FDRA is caused by an autosomal recessive inherited GAA repeat expansion in the FXN gene, impairing transcription of the protein frataxin. Frataxin is a small ∼210 amino acid mitochondrial protein. In FDRA, frataxin expression is impaired but not absent and is required for survival: full FXN deletion in mice results in embryonic lethality (Cossee et al., 2000).
Although the exact function of frataxin is still being elucidated, extensive evidence links frataxin to iron homeostasis. Genetic models of FRDA are characterized by mitochondrial iron accumulation and cytosolic iron depletion. Loss of frataxin increases the mitochondrial iron importer mitoferrin 2 (Huang et al., 2009), and reintroduction of frataxin to frataxin-deficient yeast cells corrects the iron imbalance between mitochondria and cytosol (Radisky et al., 1999). The FXN gene appears to be regulated by iron, with frataxin expression increased with iron loading and decreased with iron restriction (Sarsero et al., 2003; Li et al., 2008).
Mitochondria are the site of iron-sulfur cluster biogenesis. Loss of frataxin impairs iron-sulfur cluster-dependent proteins, including mitochondrial respiratory chain complexes and aconitase (Rotig et al., 1997; Wilson and Roof, 1997; Anderson et al., 2005; Long et al., 2008). This disruption of iron-sulfur cluster proteins occurs before mitochondrial iron accumulation (Puccio et al., 2001), suggesting that frataxin is involved in iron-sulfur cluster biogenesis (Muhlenhoff et al., 2002). Lack of frataxin is correlated to oxidative stress (Wong et al., 1999), which probably occurs via reactions of excess iron in mitochondria with superoxide.
The above studies provide evidence for a role of frataxin in mitochondrial iron homeostasis. However, these findings were obtained from experimental genetic models. In human FDRA patients, no neurological iron overload has been detected: iron deposits cannot be histologically detected in the affected regions in the CNS (Bradley et al., 2000; Koeppen et al., 2007), and no difference was found in total iron and ferritin between patients and controls in dentate nuclei in cerebellar sections (Koeppen et al., 2007). Nevertheless, cells cultured from FRDA patients and mouse models do show a modest increase in mitochondrial iron (Wong et al., 1999; Puccio et al., 2001; Tan et al., 2001; Calmels et al., 2009), and derangements in mitochondrial iron are evident in cardiac tissue of FRDA patients (Rotig et al., 1997; Bradley et al., 2000).
Parkinson's disease
PD is a progressive adult-onset neurodegenerative disease characterized by resting tremor, bradykinesia and rigidity, as well as cognitive impairment (Litvan et al., 2012). PD is the second most common neurodegenerative disease behind Alzheimer's disease. Pathologically, PD is defined by a selective loss of dopaminergic neurons in the substantia nigra (SN) region of the brain and more widespread aggregation of the protein α-synuclein in Lewy bodies (Spillantini et al., 1997; Goedert, 2001). The incidence of PD is associated with exposure to mitochondrial complex I inhibitors (Tanner et al., 2011), and the inhibition of complex I specifically in the SN (Reichmann et al., 1995; Subramaniam and Chesselet, 2013).
A key pathological hallmark of PD is the accumulation of iron in the SN (Sian-Hulsmann et al., 2011). Concomitant with this characteristic increase in iron is a decrease in copper content (Dexter et al., 1989; 1991; Loeffler et al., 1996; Popescu et al., 2009). Although the elevated iron is most likely glial – iron accumulation is evident in astrocyte mitochondria in aging rat SN (Schipper et al., 1998) – an increase in iron has been detected specifically in dopaminergic neurons in the SN in PD (Oakley et al., 2007). Neuronal mitochondrial iron accumulation can be induced by exposure to complex I inhibitors such as rotenone, paraquat, 1-methyl-4-phenylpyridinium (MPP+), and its precursor MPTP, via up-regulation of iron import proteins such as transferrin receptor-2 and mitoferrin-2 (Mastroberardino et al., 2009; Carroll et al., 2011). This accumulation of iron is thought to induce oxidative stress, which contributes to dopaminergic cell death (Mochizuki and Yasuda, 2012). For example, iron induces mitochondrial damage in SK-N-SH dopaminergic cells (Kooncumchoo et al., 2006), whereas genetic or pharmacological iron chelation improves dopaminergic neuron survival in response to MPTP in rats (Kaur et al., 2003).
Although the mechanism of iron accumulation in dopaminergic neurons in PD is not yet known, one hypothesis suggests that iron-sulfur clusters may serve as sensitive mitochondrial iron sensors. When complex I is inhibited, iron-sulfur clusters are oxidized, preventing the binding of iron, and promoting the unchecked influx of iron into mitochondria (Mastroberardino et al., 2009). An alternative possibility is that the characteristic depletion of glutathione evident in PD (Sian et al., 1994; Pearce et al., 1997; Martin and Teismann, 2009) inhibits mitochondrial glutaredoxin 2 (Grx2). As Grx2 is involved in iron-sulfur cluster generation, inhibition of Grx2 leads to impaired iron-sulfur cluster biogenesis and impaired function of iron-sulfur cluster-dependent proteins such as complex I and aconitase (Lee et al., 2009).
Environmental exposure to manganese has been postulated to be a risk factor for PD as accumulation of manganese in the brain causes manganism, a condition that shares many symptoms with PD. However, manganism and PD differ in several neuropathological aspects and could be considered as separate entities (Caudle et al., 2012). Regardless, accumulation of manganese in the brain causes mitochondrial impairment, which contributes to the motor impairments reminiscent of PD (reviewed in Aschner et al., 2009).
The manganese-containing mitochondrial enzyme MnSOD has been studied in relation to PD. MnSOD is up-regulated in genetic models of PD (Andres-Mateos et al., 2007; Xun et al., 2008). In contrast, transgenic mice overexpressing MnSOD are resistant to 6-hydroxydopamine (6-OHDA) toxicity (Callio et al., 2005). Furthermore, dopamine quinones inhibit MnSOD, increasing mitochondrial ROS production (Belluzzi et al., 2012). These studies suggest MnSOD activity is important to prevent oxidative stress in models of PD.
Huntington's disease
HD is an inherited autosomal dominant neurodegenerative disease caused by a CAG expansion in the huntingtin gene (Anonymous, 1993). Disease onset and severity is directly correlated to the number of CAG repeats (Andrew et al., 1993). HD causes progressive chorea and cognitive impairment, and is pathologically characterized by the selective loss of striatal neurons and aggregation of the mutant huntingtin protein. While the function of huntingtin is yet to be fully elucidated, deletion is embryonically lethal (Nasir et al., 1995; Zeitlin et al., 1995) and heterozygotes display severe cognitive deficits and neuronal loss in the subthalamic nucleus (Nasir et al., 1995).
HD has long been associated with mitochondrial deficits (reviewed in Oliveira, 2010). Expression of mitochondrial complexes II and III have been found to be reduced in the striatum of HD patients (Brennan et al., 1985; Gu et al., 1996; Benchoua et al., 2006), and transgenic mouse models of HD (Damiano et al., 2013), while inhibitors of complex II such as 3-nitroproprionic acid (3NP) recapitulate many features of HD (Beal et al., 1993). Overexpression of the iron-sulfur cluster subunit of complex II is correlated with attenuations of symptoms, suggesting the deficit in the iron-sulfur subunit of complex II may be central to the pathogenesis of HD (Damiano et al., 2013). Further to this, huntingtin appears to be involved in iron regulation: huntingtin-deficit zebrafish display impaired cellular iron uptake (Lumsden et al., 2007) and desferrioxamine treatment increases huntingtin expression in embryonic stem cells (Hilditch-Maguire et al., 2000). Furthermore, iron content is elevated in HD-affected brain regions (Dexter et al., 1991; Bartzokis et al., 1999; Dumas et al., 2012; Dusek et al., 2012; Rosas et al., 2012), and this iron accumulation appears to precede symptom onset (Rosas et al., 2012).
Striatal elevation in copper content has also been reported in HD brains (Dexter et al., 1991), although a more recent study found no change in copper content (Rosas et al., 2012). An elevation in copper may be of importance as copper has been shown to promote huntingtin aggregation (Fox et al., 2007); however, it is currently unclear whether aberrant copper distribution interacts with mitochondria.
An elevation in striatal zinc content has been reported in HD brains (Rosas et al., 2012). Elevated zinc has also been associated with exposure to the complex II inhibitor 3NP (Sheline et al., 2013). Interestingly, inhibiting zinc accumulation attenuates 3NP-induced toxicity (Sheline et al., 2013), suggesting a role for zinc in 3NP toxicity and mitochondrial inhibition in HD.
In summary, a key feature of neurological diseases with impaired mitochondrial functions include a propensity for accumulation of mitochondrial biometals, particularly iron, zinc, copper and manganese, which overwhelm key antioxidant systems and result in oxidative stress-dependent neurotoxicity (Table 1). Therefore, restoration of antioxidant functions through modulation of biometal homeostasis is an attractive therapeutic target for these disorders. This review describes several novel therapeutic approaches to correct biometal homeostasis through metallo-complexes and metal chelating agents, which can be pharmacologically manipulated to improve brain delivery, mitochondrial targeting and antioxidant functionality.
Therapeutic mitochondrial targeting
Lipophilic cations
Lipophilic cations are ideal mitochondrial targeting agents. Due to lipophilic shielding of the positive charge over the surface of the molecule, these compounds readily cross membranes without requiring specific transporters or energy expenditure. The high membrane potential of mitochondria (∼180–200 mV in vitro, 130–150 mV in vivo) compared with plasma membrane potentials, which are typically in the range of 30–60 mV, results in selective targeting of lipophilic cations to mitochondria (Chen, 1988). To date, the most effective and well-studied metal-based neurotherapeutics belong to two classes of complexes – bis thio semicarbazones (btsc) and pyridyl-porphyrins containing a central metal ligand (for a summary of neuroprotective actions, see Figure 1).
Figure 1.
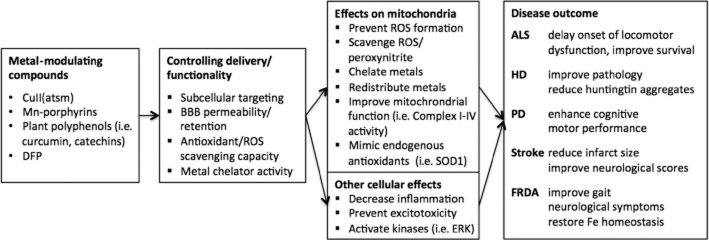
Metal-modulating compounds improve mitochondrial function and disease outcome of neurological disorders. Novel metal-based therapeutics, metal chelators and natural plant-based polyphenolic antioxidants can be chemically modified for subtle control of delivery and functionality. These compounds exert their actions through mitochondrial and other cellular pathways that restore mitochondrial metal homeostasis and lead to therapeutic outcomes in animal models of ALS, PD, HD, stroke and FRDA.
CuII(atsm)
CuII(atsm) is a btsc complex, which has been tested as a neurotherapeutic for several diseases where mitochondrial dysfunction contributes to pathology, including ALS and PD (Soon et al., 2011; Hung et al., 2012). In four PD mouse models, including both genetic and neurotoxin-lesioned models, CuII(astm) was reported to enhance cognitive and motor performance, even when administered after onset of toxin-mediated cell death (Hung et al., 2012). Moreover, CuII(atsm) delayed onset of locomotor dysfunction, prevented motor neuron loss and improved survival when administered both to presymptomatic and symptomatic SOD1G93A and SOD1G37R ALS model mice (Soon et al., 2011; McAllum et al., 2013). The mechanism of this therapeutic action is still uncertain but evidence indicates a role for mitochondria in its action.
Therapeutic strategies for diseases of mitochondrial dysfunction may promote indirect benefits to mitochondrial energy metabolism or downstream cellular pathways. Indeed, some controversy exists regarding subcellular trafficking of CuII(atsm), reported to localize to cytosolic (Pascu et al., 2010), lysosomal (Price et al., 2011) and microsomal (Obata et al., 2001) compartments. A fluorescent pyrenylated derivative of CuII(atsm), CuII(L1) showed punctate staining in M17 neuroblastoma cells with partial colocalization with lysosomal and endoplasmic reticulum, but not mitochondrial organelle markers (Price et al., 2011). However, subcellular targeting of copper complexes may be cell type specific, as CuII(L1) revealed lysosomal localization in HeLa epithelial cells (Lim et al., 2010). Alternatively, it is possible that addition of a pyrene group may affect the cellular trafficking of CuII(atsm), as modification of the btsc backbone is known to subtly alter metal delivery properties of different CuII(btsc) complexes (Xiao et al., 2008). Moreover, the localization may be concentration-dependent, as higher concentrations of fluorescently labelled copper compounds can lead to complex aggregation, colloid formation and subsequent localization in lysosomes (Morgan et al., 2011). The identity of the metal ligand also contributes to intracellular targeting, as Zn(btsc) complexes were reported to localize to lysosomes and mitochondria (Pascu et al., 2008). The rapidly increasing resolution of synchrotron X-ray fluorescence microscopy and laser ablation inductively coupled plasma MS elemental imaging is likely to provide crucial insights into the subcellular distribution of unlabeled CuII(btsc) complexes (New, 2013).
Intracellular copper retention of Cu(II) delivered by the btsc coordinate is controlled by cellular reductants, which catalyse reductive transfer of Cu(I) to high-affinity cuproproteins. Structural variations in the btsc backbone, specifically of the R1 and R2 ligands, yields complexes with vastly different cellular copper retention properties, determined by the relative copper-binding affinities of the complex compared with that of cellular reductants. Copper transfer from CuII(atsm) to the high copper-affinity proteins, Atx1 and Ctr1 purified from yeast, was detected only in the presence of a strong reductant, dithionite, whereas Apo-Ctr1 alone was capable of irreversible trapping of CuII(gtsm), another btsc compound that delivers copper to all cells (Xiao et al., 2008). In vivo, multiple reductants may be responsible for copper retention from CuII(atsm). NADH-cytochrome b5 reductase and NADPH-cytochrome P450 reductase were reported to catalyse copper retention in Ehrlich ascites hypoxic brain tumour cells (Obata et al., 2001). Recent work by our group and supported by others implicates impaired mitochondrial function as a prerequisite for copper retention from CuII(atsm) (Donnelly et al., 2012; Yoshii et al., 2012). Indeed, dissociation of the coordinate occurred only in hypoxic cells or in mitochondrial xenocybrid cells that encode incompatible ETC components on interspecies mitochondrial and nuclear chromosomes, and hence exhibit overactive glycolytic ATP production and NADH accumulation due to defective ETC function (Donnelly et al., 2012). NADH accumulation in hypoxic cells was also demonstrated to play a role in ligand dissociation from the complex (Donnelly et al., 2012).
Peroxynitrite has long been known to inhibit components of the mitochondrial ETC (Radi et al., 1994) and induce mitochondrial calcium release (Packer and Murphy, 1994), and selectively induce neuronal mitochondrial dysfunction (Bolanos et al., 1995). A proposed therapeutic mechanism of action of CuII(atsm) is the ability to rescue peroxynitrite-dependent toxicity and inhibit nitrate-driven α-synuclein-aggregation (Hung et al., 2012). Moreover, paraquat-induced apoptosis, which involves superoxide generation and mitochondrial cytochrome c and apoptosis inducing factor release (Czerniczyniec et al., 2013), is corrected in SH-SY5Y cells by CuII(atsm) treatment (Parker et al., 2012). CuII(atsm) also induced phosphorylation of the ERK by a mechanism involving copper release (Donnelly et al., 2012). While it is not known whether the complexes directly target metal delivery to mitochondria in these models, the dramatic effect on mitochondrial function may be attributed to these antioxidant mechanisms, as well as modulation of kinase phosphorylation profiles, leading to reduced neuropathology including TDP-43 aggregate accumulation within stress granules (Parker et al., 2012) and mouse tissues (Soon et al., 2011). CuII(atsm) is therefore potentially an ideal self-targeting drug for mitochondrial dysfunction disorders, as this compound specifically depends on mitochondrial impairment for its molecular therapeutic action.
Manganese porphyrins
Considerable attention has been paid in recent years to investigation of the neuroprotective effects of manganese porphyrins in numerous CNS disease models including in vitro and in vivo models of stroke, spinal cord injury, subarachnoid haemorrhage, cerebral palsy, ALS, PD and oxygen and glucose deprivation (Mackensen et al., 2001; Saba et al., 2007; Sheng et al., 2009; 2011; Wise-Faberowski et al., 2009; Batinic-Haberle et al., 2011). Manganese has been utilized as the ligand in these complexes, due to its capacity to participate in cellular redox cycling reactions as it exists in numerous oxidation states. To different extents, strongly influenced by the porphyrin backbone, these compounds have been designed to be blood–brain barrier (BBB)-permeable, preferentially accumulate in mitochondria and possess strong antioxidant SOD surrogate activity. Lipophilicity and hence mitochondrial accumulation of the complexes can be enhanced by transfer of pyridyl groups to the meta position (Mackensen et al., 2001), elongation of the alkyl chains (Tovmasyan et al., 2011) or reduction of Mn(III) to Mn(II) (reviewed in Batinic-Haberle et al., 2011). For instance, the higher redox potential of Mn(III) meso-tetrakis (N-ethylpyridinium-2-yl)porphyrin (MnTE-2-PyP5+) results in more robust antioxidant activity than its para-isomer MnTE-4-PyP5+ (Mackensen et al., 2001). Mild toxicity can also be reduced by modulation of the steric properties of the complexes to prevent interactions with DNA.
Given that in ischaemia, superoxide radicals are primarily generated by the mitochondrial ETC (Piantadosi and Zhang, 1996), manganese-pyridyl porphyrins, possessing inherent SOD-like antioxidant and peroxynitrite-scavenging properties (Pasternack et al., 1981) were hypothesized to be ideal therapeutic targets for oxidative mitochondrial dysfunction. To date, the most commonly tested complexes in vivo are MnTE-2-PyP5+ and MnTnHex-2-PyP5+.
MnTE-2-PyP5+ significantly reduced neuropathology including cerebral infarct size and substantially enhanced neurological scores in a rat model of ischaemia-reperfusion injury, even when administered 6 h following reperfusion (Mackensen et al., 2001). Moreover, improved mitochondrial function was observed by elevated mitochondrial aconitase activity and reduced oxidative DNA modification in MnTE-2-PyP5+-treated rats following reperfusion (Mackensen et al., 2001). However, this molecule failed to have an effect in trials in a rabbit cerebral palsy uterus ischaemia model (Yu et al., 2010).
The hexyl substituted, Mn(III) ortho N-n-hexylpyridylporphyrin, (MnTnHex-2-PyP5+), was reported to be 13 500-fold more lipophilic than its ethyl analogue and thus exhibits stronger mitochondrial accumulation, a greater propensity to be reduced (Keir et al., 2011) and enhanced tissue retention, even at fivefold lower doses (Weitner et al., 2013). This CNS-bioavailable complex was also reported to improve multiple disease indicators in rat stroke models of cerebral artery occlusion, subarachnoid haemorrhage (Sheng et al., 2011) and in a cerebral palsy rabbit uterus ischaemia model (Yu et al., 2010). The substantial benefits afforded by MnTnHex-2-PyP5+ in a stroke model may be attributed to the high brain retention of the complex, coupled with robust antioxidant activity (Sheng et al., 2011). Similarly, extension of the alkyl chain in MnTnOct-2-PyP5+ resulted in improved bioavailability compared with the ethyl analogue and three orders of magnitude reduction in the effective dose required to protect rat mixed neuronal and glial cultures from oxygen and glucose starvation (Wise-Faberowski et al., 2009).
The related imidazolium complex, Mn(III) tetrakis[N-N′-diethylimidazolium-2-yl]porphyrin (MnIIITDE-ImP(2)), improved survival, reduced inflammatory, oxidative and nitrosative pathological damage in the SOD1G93A ALS mouse model (Crow et al., 2005). Another mitochondrial SOD mimic, Mn(III) 1,4,8,11-tetraazacyclodecane (Mn-cyclam), and to a lesser extent its Ni-coordinate, inhibited mitochondrial membrane depolarization and peroxynitrite-mediated cell death in glucose-starved, activated astrocytes (Choi et al., 2003).
Aside from their antioxidant function, investigation of the cellular mechanism of action of this class of compounds has been limited. Manganese porphyrins were reported to inhibit activation of inflammatory transcriptional programmes, including NF-κB and AP-1 (Sheng et al., 2009; 2011). Recent evidence implicated a pro-oxidant mechanism involving direct oxidation of sulfhydryl groups on p50 NF-κB, AEP1/Ref-1 or thioredoxin transcription factor subunits. Another mechanism of neuroprotection was proposed whereby metalloporphyrins are able to suppress excitotoxicity and the associated mitochondrial Ca2+ influx by preventing postsynaptic NMDA receptor-dependent increases in Ca2+ (Tauskela et al., 2005). This activity did not correlate to SOD1 antioxidant properties, indicating that alternative synthesis approaches should also be considered (Tauskela et al., 2005). Modification of the complexes to enhance Ca2+/NMDA-mediated neuroprotection could therefore be a useful strategy for disorders, such as ALS, where excitotoxicity significantly contributes to their neuropathology (Ferraiuolo et al., 2011).
Metal chelators
Deferiprone
Another class of therapeutics with metal-dependent antioxidant activity are metal chelators. The BBB-permeable (Fredenburg et al., 1996) iron siderophore, deferiprone (DFP), has been investigated as a therapeutic for FRDA in multiple cell and animal models, as well as two clinical trials. DFP treatment of HEK-293 cells stably transfected with a tetracycline-inducible frataxin shRNA, resulted in restoration of mitochondrial functions including decreased mitochondrial redox potential, ROS production and labile iron pools (Kakhlon et al., 2008). DFP also increased survival and improved motor climbing ability in a frataxin-knockdown Drosophila FRDA model (Soriano et al., 2013).
A 6 month clinical trial involving daily DFP administration in young FRDA patients at 20–30 mg·kg−1 improved gait and other neurological signs in all nine patients, and induced a significant progressive reduction in dentate nuclei-localized iron accumulation in eight patients (Boddaert et al., 2007). However, a subsequent 11-month study of combination therapy with DFP and idebenone (IDE), a synthetic analogue of the antioxidant coenzyme Q10, on a cohort of 20 FRDA patients did not show an overall significant improvement in ataxia neurological function scores (Velasco-Sanchez et al., 2011). Nonetheless, as neurological scores are expected to decline with disease progression over the long treatment period (Velasco-Sanchez et al., 2011), it could be inferred that the trial was moderately successful. Moreover, a significant reduction in serum ferritin concomitant with increased transferrin was observed in DFP and IDE co-treated patients (Velasco-Sanchez et al., 2011), suggesting that restoration of iron homeostasis could be the mechanism involved. However, care must be taken with administration of non-selective iron chelators, as these can often have toxic consequences. For example, deferasirox (DFX) is highly toxic in people without iron overload (Singh et al., 1995), and oral administration of dipyridyl is toxic to rats (Groce and Kimbrough, 1982).
Interestingly, the iron affinity of the chelators, DFP, DFX and alicylaldehyde isonicotinoyl hydrazone, was inversely correlated with their ability to improve mitochondrial function in frataxin knockdown cells (Kakhlon et al., 2008). It has been hypothesized that the reason DFP is more effective than other iron chelators (Kakhlon et al., 2008) is its ability to act as a frataxin mimetic ionophore, with functions both in chelation of excess iron and redistribution to iron-deficient subcellular sites (Goncalves et al., 2008). Consistent with this, DFP shuttled iron from iron donor to iron acceptor probes in multiple cellular compartments, including extracellular media, nuclei, mitochondria and endosomes (Sohn et al., 2008). This indicates that DFP is capable of iron mobilization from accumulated stores to sites of low iron bioavailability and therefore acts as a membrane permeable multidirectional iron transporter. Indeed, DFP increased soluble mitochondrial iron and prevented aggregation of insoluble Fe3+ in a frataxin-knockdown Drosophila model (Soriano et al., 2013). However, caution must be exercised with the use of DFP, as excessive depletion of labile mitochondrial iron pools may promote adverse effects including loss of mitochondrial aconitase activity and growth inhibition, as reported with high dose treatments in control and FRDA patient fibroblasts (Goncalves et al., 2008).
Natural plant-based polyphenols
The neuroprotective role of various natural plant-based polyphenolic compounds has been examined in numerous cell and animal models of neurodegeneration. These include baicalein (from skullcap flowers), tocotrienol (from palm oil), epigallocatechin gallate (EGCG; from green tea), rosmarinic acid (from rosemary), tannic acid (from various plant sources), nordihydroguaiaretic acid (from the creosote bush), curcumin (from turmeric), myricetin (from numerous sources including berries, nuts, fruit, vegetables) and kaempferol (from various sources including tea, berries, fruit and vegetables). These compounds are potent metal chelators and free radical scavengers and have been reported to restore multiple parameters of mitochondrial function including mitochondrial lipid peroxidation, membrane potential, mitochondrial swelling and cytochrome c release (Kamat and Devasagayam, 1995; Mandel et al., 2005; Ono and Yamada, 2006; Cheng et al., 2008; He et al., 2009; Mu et al., 2009; Caruana et al., 2011). This review will focus on two of these: the green tea catechin, EGCG and curcumin.
Green tea catechins
Green tea, which has long been recognized for numerous health benefits, contains a number of polyphenolic flavonoids called catechins, including EGCG, epigallocatechin (EGC), (–)-epicatechin (EC) and (–)-epicatechin-3-gallate (ECG), in order of abundance. These natural compounds are strong antioxidants and have been reported to chelate iron with a similar efficacy as DFP (for a detailed review, see Mandel et al., 2005). Green tea consumption has been linked to increased cognitive performance and is reported to significantly lower the risk of developing PD (Kuriyama et al., 2006; Hu et al., 2007). ECGG is implicated in hippocampal neuroprotection in a gerbil model of ischaemia/reperfusion injury by carotid artery occlusion (Lee et al., 2000), is shown to prevent 6-OHDA-induced neurotoxicity in vitro (Levites et al., 2002) and dopaminergic neuron loss in the SN in the MPP+-induced mouse model of PD (Levites et al., 2001). Moreover, EGCG can both attenuate fibrillar α-synuclein aggregation and induce dissociation of formed fibrils (Ono and Yamada, 2006). Similar to the other antioxidant metal-modulating agents described above, catechins exhibit a biphasic action, where lower concentrations mediate neuroprotection and higher doses induce apoptosis, suggestive of promising therapeutic potential for both neurodegeneration and cancer (Weinreb et al., 2003).
Of the green tea polyphenols, EGCG displays the most robust neuroprotective activity, and hence is the best studied of the catechins (Nie et al., 2002). The molecular mechanism of action of EGCG involves direct antioxidant activity through free radical scavenging (Salah et al., 1995; Nanjo et al., 1996), as well as through potent induction of endogenous antioxidant systems such SOD1 and catalase (Levites et al., 2001). In a study examining the free radical scavenging capacity of EGCG and closely related molecules, it was determined that the ortho-trihydroxyl group on the aromatic ring and the galloyl substituent are primarily responsible for the potent redox properties of EGCG (Nanjo et al., 1996). In the mitochondria, EGCG inhibited 6-OHDA-mediated mitochondrial membrane depolarization and prevented impaired mitochondrial complex I-V function following a hypoxic insult in rats (Sutherland et al., 2005). Moreover, EGCG preferentially targets the mitochondria and displays specific neuroprotective activity to mitochondrial toxic stimuli, but not other apoptosis-inducing agents in rat cerebellar neurons (Schroeder et al., 2009).
In addition to direct effects on mitochondrial function, EGCG activates neuroprotective cellular signalling pathways including the antioxidant response through nrf2-dependent transcription of haeme oxygenase-1 (Romeo et al., 2009). Moreover, in neuronal SH-SY5Y cells, EGCG stimulated the PKC phospho cascades, and attenuated the expression of the pro-apoptotic genes activated by 6-OHDA (Levites et al., 2002). As described for other metal-modulating compounds, EGCG has also been reported to induce ERK activation (Levites et al., 2002; Schroeter et al., 2007), which may contribute to neuroprotection.
Curcumin
Curcumin, a natural plant polyphenol derived from the spice, turmeric, has been shown to exert potent anti-cancer activity. Curcumin is able to bind metal ions and act as a metal ionophore (Dairam et al., 2008; Garcia et al., 2012). Curcumin was demonstrated to exert pleiotropic cellular effects, which include antioxidant activity and direct detoxification of ROS and peroxynitrite (Iwunze and McEwan, 2004). Hence, the neuroprotective efficacy of curcumin has been assessed in various diseases of mitochondrial metal dysfunction including AD, PD and stroke models. For a detailed review on mitochondrial dysfunction and curcumin treatment in AD models see Eckert et al. (2012). Although curcumin is able to cross the BBB, its rapid elimination from the brain has resulted in several drug design approaches to improve bioavailability. Recently, modification of natural compounds such as curcumin by combining individual neuroprotective functionalities into multi-target complexes based on two scaffolds is gaining increasing support. Diester coupling of amino acid moieties has been used as a delivery platform to improve bioavailability, as amino acid transport systems facilitate prodrug influx and intracellular esterase-mediated reactions liberate functional curcumin molecules (Dubey et al., 2008). Naturally, curcumin derivatives display altered metal-binding kinetics, largely dependent on the nature of the aromatic ring substituents (Ferrari et al., 2013).
Knowledge regarding the cellular localization of curcumin is limited. One study demonstrated that curcumin accumulates primarily in the membrane with some nuclear localization (Kunwar et al., 2008), but more research is required to determine the intracellular targeting of curcumin and its derivatives in brain cells. Curcumin is a potent iron chelator (Jiao et al., 2006; 2009) and induces up-regulation of iron regulatory proteins and the transferrin receptor (Jiao et al., 2006), indicative of systemic iron deficiency. Curcumin has been shown to bind Fe2+ ions, which prevents iron-mediated hydroxyl radical production (Dairam et al., 2008). Curcumin also strongly binds and stabilizes Fe3+ (Dairam et al., 2008; Garcia et al., 2012), thereby inhibiting iron redox cycling.
Curcumin and its derivatives have been tested in numerous cell and animal models of PD. Treatment of N27 dopaminergic neurons with a glutamic acid substituted curcumin derivative-induced neuroprotection involving increased GSH synthesis and a reduction in oxidative stress, lipid peroxidation and H2O2 production (Harish et al., 2010). N27 neurons were also protected from MPP+-induced cytotoxicity by Di-glutamoyl curcumin (Mythri et al., 2011). Furthermore, curcumin improved survival of dopaminergic neurons in the SN in MPTP-lesioned C57BL/6 mice via JNK-dependent inhibition of mitochondrial swelling and cytochrome c release (Pan et al., 2012). Curcumin-fed rats that had been 6-OHDA-lesioned displayed elevated striatal dopamine levels and had a decreased number of iron-positive cells in the SN, suggesting that curcumin-mediated iron chelation may be responsible for protection of dopaminergic neurons from Fenton chemistry-dependent neurodegeneration (Du et al., 2012). Curcumin has also been reported to reduce huntingtin accumulation and improve rearing behaviour in HD CAG140 knock-in mice (Hickey et al., 2012).
Similar to CuII(atsm), the cellular mechanism of action of curcumin involves activation of kinase signalling pathways. Pharmacological inhibition of the Akt, but not MAPK, pathways decreased curcumin-stimulated Nrf2 activation and Nrf2-dependent gene expression in primary rat cultured cortical neurons (Wu et al., 2013). Moreover, blocking Akt prevented curcumin-mediated reduction in infarct size and oxidative stress in a rat transient middle cerebral artery occlusion stroke model (Wu et al., 2013). Nrf2 activation was also reported to contribute to curcumin-mediated amelioration of mitochondrial dysfunction in spinal cord astrocytes treated with H2O2 (Jiang et al., 2011).
As mentioned earlier, limitations with bioavailability of curcumin have been overcome by the development of multifunctional drug candidates including CNB-001, which combines the neuroprotective actions of curcumin and cyclohexyl bisphenol A, and was detected in the mouse brain 6 days after a single oral dose (Liu et al., 2008). CNB-001, but not curcumin, was reported to protect PC12 cells from excitotoxicity and glucose starvation, despite a 20-fold lower antioxidant activity (Liu et al., 2008). CNB-001 also restored ATP levels and protected HT22 cells from glutamate toxicity (Lapchak et al., 2011). The in vivo action of CNB-001 in a rabbit small clot embolic stroke model was also associated with induction of the neuroprotective BDNF, PI3K, Akt and calcium-calmodulin-dependent kinase signalling pathways (Lapchak et al., 2011).
Curcumin is widely reported to reduce cellular ROS production (Wang et al., 2010; Jiang et al., 2011; Liu et al., 2011) and prevent toxicity associated with pathological protein aggregation, including both the PD-associated α-synuclein and ALS-associated TDP-43 aggregates (Wang et al., 2010; Lu et al., 2012). Moreover, curcumin and its derivatives exhibit numerous protective effects on mitochondria. Curcumin improved multiple indicators of mitochondrial function, including cytochrome c release, mitochondrial membrane depolarization and caspase activation in a PC12 neuroblastoma cell model of A53T α-synuclein toxicity (Liu et al., 2011). Mitochondrial dysfunction was also attenuated in a model of aluminium-induced cytotoxicity, as demonstrated by enhanced activity of the mitochondrial NADH dehydrogenase, succinic dehydrogenase and cytochrome c oxidase complexes (Sood et al., 2011). Addition of various substituents on the aromatic rings appears to yield protection against mitochondrial dysfunction. Indeed, mitochondrial swelling, membrane potential and NADH dehydrogenase activity of mice isolated brain mitochondria were substantially improved by a glutamoyl diester of curcumin, as compared with curcumin alone (Mythri et al., 2011). Similarly, a dimethoxy curcumin derivative also attenuated TDP-43-induced loss of mitochondrial membrane potential and mitochondrial NADH dehydrogenase activity (Lu et al., 2012).
Conclusion
The therapeutic approaches presented above demonstrate immense promise in treating neurological diseases of mitochondrial dysfunction by restoration of aberrant mitochondrial metal homeostasis. The common theme from the complexes presented in this review is that the protective action on mitochondrial function is mediated through antioxidant mechanisms that prevent toxic build-up of redox active metals in the vicinity of ETC components (Figure 1). Derivatives of these prototype complexes can therefore be engineered to improve specific molecular properties such as BBB and organelle targeting, antioxidant function and metal binding and release properties. In vitro disease models with high throughput screening capabilities are required to examine various functionalities of derivative and multi-scaffold molecules for targeted design of novel multifunctional metal-based therapeutics for mitochondrial dysfunction. Together with an improved understanding of the fundamental molecular mechanism(s) of action of the complexes, these strategies will lead to improved outcomes in animal and clinical trials.
Acknowledgments
A. R. W. is a recipient of an ARC Future Fellowship and J. R. L. is the recipient of an NHMRC Biomedical Fellowship.
Glossary
- 3NP
3-nitroproprionic acid
- 6-OHDA
6-hydroxydopamine
- ALS
amyotrophic lateral sclerosis
- BBB
blood–brain barrier
- Cox
cytochrome C oxidase
- DFP
deferiprone
- DFX
deferasirox
- EC
(–)-epicatechin
- ECG
(–)-epicatechin-3-gallate
- EGC
epigallocatechin
- EGCG
epigallocatechin gallate
- ETC
electron transport chain
- FRDA
Friedrich's ataxia
- Grx2
glutaredoxin 2
- HD
Huntington's disease
- IDE
idebenone
- IMS
intermembrane space
- MnSOD
(SOD2) manganese superoxide dismutase
- MPP+
1-methyl-4-phenylpyridinium
- PD
Parkinson's disease
- ROS
reactive oxygen species
- SN
substantia nigra
- SOD1
superoxide dismutase 1
- TDP-43
Tar DNA-binding protein
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Al-Chalabi A, Jones A, Troakes C, King A, Al-Sarraj S, Van Den Berg LH. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol (Berl) 2012;124:339–352. doi: 10.1007/s00401-012-1022-4. [DOI] [PubMed] [Google Scholar]
- Anderson PR, Kirby K, Hilliker AJ, Phillips JP. RNAi-mediated suppression of the mitochondrial iron chaperone, frataxin, in Drosophila. Hum Mol Genet. 2005;14:3397–3405. doi: 10.1093/hmg/ddi367. [DOI] [PubMed] [Google Scholar]
- Andres-Mateos E, Perier C, Zhang L, Blanchard-Fillion B, Greco TM, Thomas B, et al. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc Natl Acad Sci U S A. 2007;104:14807–14812. doi: 10.1073/pnas.0703219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew SE, Goldberg YP, Kremer B, Telenius H, Theilmann J, Adam S, et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nat Genet. 1993;4:398–403. doi: 10.1038/ng0893-398. [DOI] [PubMed] [Google Scholar]
- Anonymous. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Aschner M, Erikson KM, Herrero Hernandez E, Tjalkens R. Manganese and its role in Parkinson's disease: from transport to neuropathology. Neuromolecular Med. 2009;11:252–266. doi: 10.1007/s12017-009-8083-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atamna H, Walter PB, Ames BN. The role of heme and iron-sulfur clusters in mitochondrial biogenesis, maintenance, and decay with age. Arch Biochem Biophys. 2002;397:345–353. doi: 10.1006/abbi.2001.2671. [DOI] [PubMed] [Google Scholar]
- Bartzokis G, Cummings J, Perlman S, Hance DB, Mintz J. Increased basal ganglia iron levels in Huntington disease. Arch Neurol. 1999;56:569–574. doi: 10.1001/archneur.56.5.569. [DOI] [PubMed] [Google Scholar]
- Batinic-Haberle I, Rajic Z, Tovmasyan A, Reboucas JS, Ye X, Leong KW, et al. Diverse functions of cationic Mn(III) N-substituted pyridylporphyrins, recognized as SOD mimics. Free Radic Biol Med. 2011;51:1035–1053. doi: 10.1016/j.freeradbiomed.2011.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, et al. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belluzzi E, Bisaglia M, Lazzarini E, Tabares LC, Beltramini M, Bubacco L. Human SOD2 modification by dopamine quinones affects enzymatic activity by promoting its aggregation: possible implications for Parkinson's disease. PLoS ONE. 2012;7:e38026. doi: 10.1371/journal.pone.0038026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benchoua A, Trioulier Y, Zala D, Gaillard MC, Lefort N, Dufour N, et al. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol Biol Cell. 2006;17:1652–1663. doi: 10.1091/mbc.E05-07-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddaert N, Le Quan Sang KH, Rotig A, Leroy-Willig A, Gallet S, Brunelle F, et al. Selective iron chelation in Friedreich ataxia: biologic and clinical implications. Blood. 2007;110:401–408. doi: 10.1182/blood-2006-12-065433. [DOI] [PubMed] [Google Scholar]
- Bolanos JP, Heales SJ, Land JM, Clark JB. Effect of peroxynitrite on the mitochondrial respiratory chain: differential susceptibility of neurones and astrocytes in primary culture. J Neurochem. 1995;64:1965–1972. doi: 10.1046/j.1471-4159.1995.64051965.x. [DOI] [PubMed] [Google Scholar]
- Bowling AC, Schulz JB, Brown RH, Jr, Beal MF. Superoxide dismutase activity, oxidative damage, and mitochondrial energy metabolism in familial and sporadic amyotrophic lateral sclerosis. J Neurochem. 1993;61:2322–2325. doi: 10.1111/j.1471-4159.1993.tb07478.x. [DOI] [PubMed] [Google Scholar]
- Bradley JL, Blake JC, Chamberlain S, Thomas PK, Cooper JM, Schapira AH. Clinical, biochemical and molecular genetic correlations in Friedreich's ataxia. Hum Mol Genet. 2000;9:275–282. doi: 10.1093/hmg/9.2.275. [DOI] [PubMed] [Google Scholar]
- Bradley WG. Overview of motor neuron disease: classification and nomenclature. Clin Neurosci. 1995;3:323–326. [PubMed] [Google Scholar]
- Brennan WA, Jr, Bird ED, Aprille JR. Regional mitochondrial respiratory activity in Huntington's disease brain. J Neurochem. 1985;44:1948–1950. doi: 10.1111/j.1471-4159.1985.tb07192.x. [DOI] [PubMed] [Google Scholar]
- Calderone A, Jover T, Mashiko T, Noh KM, Tanaka H, Bennett MV, et al. Late calcium EDTA rescues hippocampal CA1 neurons from global ischemia-induced death. J Neurosci. 2004;24:9903–9913. doi: 10.1523/JNEUROSCI.1713-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callio J, Oury TD, Chu CT. Manganese superoxide dismutase protects against 6-hydroxydopamine injury in mouse brains. J Biol Chem. 2005;280:18536–18542. doi: 10.1074/jbc.M413224200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calmels N, Schmucker S, Wattenhofer-Donze M, Martelli A, Vaucamps N, Reutenauer L, et al. The first cellular models based on frataxin missense mutations that reproduce spontaneously the defects associated with Friedreich ataxia. PLoS ONE. 2009;4:e6379. doi: 10.1371/journal.pone.0006379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CB, Zeissler ML, Chadborn N, Gibson K, Williams G, Zajicek JP, et al. Changes in iron-regulatory gene expression occur in human cell culture models of Parkinson's disease. Neurochem Int. 2011;59:73–80. doi: 10.1016/j.neuint.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Caruana M, Hogen T, Levin J, Hillmer A, Giese A, Vassallo N. Inhibition and disaggregation of alpha-synuclein oligomers by natural polyphenolic compounds. FEBS Lett. 2011;585:1113–1120. doi: 10.1016/j.febslet.2011.03.046. [DOI] [PubMed] [Google Scholar]
- Caudle WM, Guillot TS, Lazo CR, Miller GW. Industrial toxicants and Parkinson's disease. Neurotoxicology. 2012;33:178–188. doi: 10.1016/j.neuro.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LB. Mitochondrial membrane potential in living cells. Annu Rev Cell Biol. 1988;4:155–181. doi: 10.1146/annurev.cb.04.110188.001103. [DOI] [PubMed] [Google Scholar]
- Cheng Y, He G, Mu X, Zhang T, Li X, Hu J, et al. Neuroprotective effect of baicalein against MPTP neurotoxicity: behavioral, biochemical and immunohistochemical profile. Neurosci Lett. 2008;441:16–20. doi: 10.1016/j.neulet.2008.05.116. [DOI] [PubMed] [Google Scholar]
- Choi MY, Pollard JA, Webb MA, McHale JL. Counterion-dependent excitonic spectra of tetra(p-carboxyphenyl)porphyrin aggregates in acidic aqueous solution. J Am Chem Soc. 2003;125:810–820. doi: 10.1021/ja0274397. [DOI] [PubMed] [Google Scholar]
- Cossee M, Puccio H, Gansmuller A, Koutnikova H, Dierich A, Lemeur M, et al. Inactivation of the Friedreich ataxia mouse gene leads to early embryonic lethality without iron accumulation. Hum Mol Genet. 2000;9:1219–1226. doi: 10.1093/hmg/9.8.1219. [DOI] [PubMed] [Google Scholar]
- Cozzolino M, Pesaresi MG, Amori I, Crosio C, Ferri A, Nencini M, et al. Oligomerization of mutant SOD1 in mitochondria of motoneuronal cells drives mitochondrial damage and cell toxicity. Antioxid Redox Signal. 2009;11:1547–1558. doi: 10.1089/ars.2009.2545. [DOI] [PubMed] [Google Scholar]
- Crow JP, Calingasan NY, Chen J, Hill JL, Beal MF. Manganese porphyrin given at symptom onset markedly extends survival of ALS mice. Ann Neurol. 2005;58:258–265. doi: 10.1002/ana.20552. [DOI] [PubMed] [Google Scholar]
- Czerniczyniec A, Lores-Arnaiz S, Bustamante J. Mitochondrial susceptibility in a model of paraquat neurotoxicity. Free Radic Res. 2013;47:614–623. doi: 10.3109/10715762.2013.806797. [DOI] [PubMed] [Google Scholar]
- Dairam A, Fogel R, Daya S, Limson JL. Antioxidant and iron-binding properties of curcumin, capsaicin, and S-allylcysteine reduce oxidative stress in rat brain homogenate. J Agric Food Chem. 2008;56:3350–3356. doi: 10.1021/jf0734931. [DOI] [PubMed] [Google Scholar]
- Damiano M, Diguet E, Malgorn C, D'aurelio M, Galvan L, Petit F, et al. A role of mitochondrial complex II defects in genetic models of Huntington's disease expressing N-terminal fragments of mutant huntingtin. Hum Mol Genet. 2013;22:3869–3882. doi: 10.1093/hmg/ddt242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dexter DT, Wells FR, Lees AJ, Agid F, Agid Y, Jenner P, et al. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson's disease. J Neurochem. 1989;52:1830–1836. doi: 10.1111/j.1471-4159.1989.tb07264.x. [DOI] [PubMed] [Google Scholar]
- Dexter DT, Carayon A, Javoy-Agid F, Agid Y, Wells FR, Daniel SE, et al. Alterations in the levels of iron, ferritin and other trace metals in Parkinson's disease and other neurodegenerative diseases affecting the basal ganglia. Brain. 1991;114(Pt 4):1953–1975. doi: 10.1093/brain/114.4.1953. [DOI] [PubMed] [Google Scholar]
- Dineley KE, Richards LL, Votyakova TV, Reynolds IJ. Zinc causes loss of membrane potential and elevates reactive oxygen species in rat brain mitochondria. Mitochondrion. 2005;5:55–65. doi: 10.1016/j.mito.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Donnelly PS, Liddell JR, Lim S, Paterson BM, Cater MA, Savva MS, et al. An impaired mitochondrial electron transport chain increases retention of the hypoxia imaging agent diacetylbis (4-methylthiosemicarbazonato)copperII. Proc Natl Acad Sci U S A. 2012;109:47–52. doi: 10.1073/pnas.1116227108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du XX, Xu HM, Jiang H, Song N, Wang J, Xie JX. Curcumin protects nigral dopaminergic neurons by iron-chelation in the 6-hydroxydopamine rat model of Parkinson's disease. Neurosci Bull. 2012;28:253–258. doi: 10.1007/s12264-012-1238-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey SK, Sharma AK, Narain U, Misra K, Pati U. Design, synthesis and characterization of some bioactive conjugates of curcumin with glycine, glutamic acid, valine and demethylenated piperic acid and study of their antimicrobial and antiproliferative properties. Eur J Med Chem. 2008;43:1837–1846. doi: 10.1016/j.ejmech.2007.11.027. [DOI] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria, calcium-dependent neuronal death and neurodegenerative disease. Pflugers Arch. 2012;464:111–121. doi: 10.1007/s00424-012-1112-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas EM, Versluis MJ, Van Den Bogaard SJ, Van Osch MJ, Hart EP, Van Roon-Mom WM, et al. Elevated brain iron is independent from atrophy in Huntington's disease. Neuroimage. 2012;61:558–564. doi: 10.1016/j.neuroimage.2012.03.056. [DOI] [PubMed] [Google Scholar]
- Dupuis L, Pradat PF, Ludolph AC, Loeffler JP. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. 2011;10:75–82. doi: 10.1016/S1474-4422(10)70224-6. [DOI] [PubMed] [Google Scholar]
- Dusek P, Jankovic J, Le W. Iron dysregulation in movement disorders. Neurobiol Dis. 2012;46:1–18. doi: 10.1016/j.nbd.2011.12.054. [DOI] [PubMed] [Google Scholar]
- Eckert GP, Renner K, Eckert SH, Eckmann J, Hagl S, Abdel-Kader RM, et al. Mitochondrial dysfunction – a pharmacological target in Alzheimer's disease. Mol Neurobiol. 2012;46:136–150. doi: 10.1007/s12035-012-8271-z. [DOI] [PubMed] [Google Scholar]
- Ferraiuolo L, Kirby J, Grierson AJ, Sendtner M, Shaw PJ. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;7:616–630. doi: 10.1038/nrneurol.2011.152. [DOI] [PubMed] [Google Scholar]
- Ferrari E, Asti M, Benassi R, Pignedoli F, Saladini M. Metal binding ability of curcumin derivatives: a theoretical vs. experimental approach. Dalton Trans. 2013;42:5304–5313. doi: 10.1039/c3dt33072a. [DOI] [PubMed] [Google Scholar]
- Field LS, Furukawa Y, O'halloran TV, Culotta VC. Factors controlling the uptake of yeast copper/zinc superoxide dismutase into mitochondria. J Biol Chem. 2003;278:28052–28059. doi: 10.1074/jbc.M304296200. [DOI] [PubMed] [Google Scholar]
- Fox JH, Kama JA, Lieberman G, Chopra R, Dorsey K, Chopra V, et al. Mechanisms of copper ion mediated Huntington's disease progression. PLoS ONE. 2007;2:e334. doi: 10.1371/journal.pone.0000334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredenburg AM, Sethi RK, Allen DD, Yokel RA. The pharmacokinetics and blood-brain barrier permeation of the chelators 1,2 dimethly-, 1,2 diethyl-, and 1-[ethan-1'ol]-2-methyl-3-hydroxypyridin-4-one in the rat. Toxicology. 1996;108:191–199. doi: 10.1016/0300-483x(95)03301-u. [DOI] [PubMed] [Google Scholar]
- Garcia CR, Angele-Martinez C, Wilkes JA, Wang HC, Battin EE, Brumaghim JL. Prevention of iron-and copper-mediated DNA damage by catecholamine and amino acid neurotransmitters, L-DOPA, and curcumin: metal binding as a general antioxidant mechanism. Dalton Trans. 2012;41:6458–6467. doi: 10.1039/c2dt30060e. [DOI] [PubMed] [Google Scholar]
- Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosc. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- Goncalves S, Paupe V, Dassa EP, Rustin P. Deferiprone targets aconitase: implication for Friedreich's ataxia treatment. BMC Neurol. 2008;8:20. doi: 10.1186/1471-2377-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groce DF, Kimbrough RD. Acute and subacute toxicity in Sherman strain rats exposed to 4,4′-and 2,2′-dipyridyl. J Toxicol Environ Health. 1982;10:363–372. doi: 10.1080/15287398209530259. [DOI] [PubMed] [Google Scholar]
- Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH. Mitochondrial defect in Huntington's disease caudate nucleus. Ann Neurol. 1996;39:385–389. doi: 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Puskin JS. Manganous ion as a spin label in studies of mitochondrial uptake of manganese. Biophys J. 1972;12:625–635. doi: 10.1016/S0006-3495(72)86108-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadzhieva M, Kirches E, Wilisch-Neumann A, Pachow D, Wallesch M, Schoenfeld P, et al. Dysregulation of iron protein expression in the G93A model of amyotrophic lateral sclerosis. Neuroscience. 2013;230:94–101. doi: 10.1016/j.neuroscience.2012.11.021. [DOI] [PubMed] [Google Scholar]
- Harish G, Venkateshappa C, Mythri RB, Dubey SK, Mishra K, Singh N, et al. Bioconjugates of curcumin display improved protection against glutathione depletion mediated oxidative stress in a dopaminergic neuronal cell line: implications for Parkinson's disease. Bioorg Med Chem. 2010;18:2631–2638. doi: 10.1016/j.bmc.2010.02.029. [DOI] [PubMed] [Google Scholar]
- He XL, Wang YH, Gao M, Li XX, Zhang TT, Du GH. Baicalein protects rat brain mitochondria against chronic cerebral hypoperfusion-induced oxidative damage. Brain Res. 2009;1249:212–221. doi: 10.1016/j.brainres.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Hickey MA, Zhu C, Medvedeva V, Lerner RP, Patassini S, Franich NR, et al. Improvement of neuropathology and transcriptional deficits in CAG 140 knock-in mice supports a beneficial effect of dietary curcumin in Huntington's disease. Mol Neurodegener. 2012;7:12. doi: 10.1186/1750-1326-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilditch-Maguire P, Trettel F, Passani LA, Auerbach A, Persichetti F, Macdonald ME. Huntingtin: an iron-regulated protein essential for normal nuclear and perinuclear organelles. Hum Mol Genet. 2000;9:2789–2797. doi: 10.1093/hmg/9.19.2789. [DOI] [PubMed] [Google Scholar]
- Hu G, Bidel S, Jousilahti P, Antikainen R, Tuomilehto J. Coffee and tea consumption and the risk of Parkinson's disease. Mov Disord. 2007;22:2242–2248. doi: 10.1002/mds.21706. [DOI] [PubMed] [Google Scholar]
- Huang ML, Becker EM, Whitnall M, Suryo Rahmanto Y, Ponka P, Richardson DR. Elucidation of the mechanism of mitochondrial iron loading in Friedreich's ataxia by analysis of a mouse mutant. Proc Natl Acad Sci U S A. 2009;106:16381–16386. doi: 10.1073/pnas.0906784106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ML, Lane DJ, Richardson DR. Mitochondrial mayhem: the mitochondrion as a modulator of iron metabolism and its role in disease. Antioxid Redox Signal. 2011;15:3003–3019. doi: 10.1089/ars.2011.3921. [DOI] [PubMed] [Google Scholar]
- Hung LW, Villemagne VL, Cheng L, Sherratt NA, Ayton S, White AR, et al. The hypoxia imaging agent CuII(atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson's disease. J Exp Med. 2012;209:837–854. doi: 10.1084/jem.20112285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igoudjil A, Magrane J, Fischer LR, Kim HJ, Hervias I, Dumont M, et al. In vivo pathogenic role of mutant SOD1 localized in the mitochondrial intermembrane space. J Neurosci. 2011;31:15826–15837. doi: 10.1523/JNEUROSCI.1965-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwunze MO, McEwan D. Peroxynitrite interaction with curcumin solubilized in ethanolic solution. Cell Mol Biol (Noisy-le-grand) 2004;50:749–752. [PubMed] [Google Scholar]
- Jeong SY, Rathore KI, Schulz K, Ponka P, Arosio P, David S. Dysregulation of iron homeostasis in the CNS contributes to disease progression in a mouse model of amyotrophic lateral sclerosis. J Neurosci. 2009;29:610–619. doi: 10.1523/JNEUROSCI.5443-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Sullivan PG, Sensi SL, Steward O, Weiss JH. Zn(2+) induces permeability transition pore opening and release of pro-apoptotic peptides from neuronal mitochondria. J Biol Chem. 2001;276:47524–47529. doi: 10.1074/jbc.M108834200. [DOI] [PubMed] [Google Scholar]
- Jiang H, Tian X, Guo Y, Duan W, Bu H, Li C. Activation of nuclear factor erythroid 2-related factor 2 cytoprotective signaling by curcumin protect primary spinal cord astrocytes against oxidative toxicity. Biol Pharm Bull. 2011;34:1194–1197. doi: 10.1248/bpb.34.1194. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Wilkinson JT, Christine Pietsch E, Buss JL, Wang W, Planalp R, et al. Iron chelation in the biological activity of curcumin. Free Radic Biol Med. 2006;40:1152–1160. doi: 10.1016/j.freeradbiomed.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Wilkinson JT, Di X, Wang W, Hatcher H, Kock ND, et al. Curcumin, a cancer chemopreventive and chemotherapeutic agent, is a biologically active iron chelator. Blood. 2009;113:462–469. doi: 10.1182/blood-2008-05-155952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan J, De Groot PW, Galindo MF. Mitochondria: the headquarters in ischemia-induced neuronal death. Cent Nerv Syst Agents Med Chem. 2011;11:98–106. doi: 10.2174/187152411796011358. [DOI] [PubMed] [Google Scholar]
- Jung JE, Kim GS, Narasimhan P, Song YS, Chan PH. Regulation of Mn-superoxide dismutase activity and neuroprotection by STAT3 in mice after cerebral ischemia. J Neurosci. 2009;29:7003–7014. doi: 10.1523/JNEUROSCI.1110-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakhlon O, Manning H, Breuer W, Melamed-Book N, Lu C, Cortopassi G, et al. Cell functions impaired by frataxin deficiency are restored by drug-mediated iron relocation. Blood. 2008;112:5219–5227. doi: 10.1182/blood-2008-06-161919. [DOI] [PubMed] [Google Scholar]
- Kamat JP, Devasagayam TP. Tocotrienols from palm oil as potent inhibitors of lipid peroxidation and protein oxidation in rat brain mitochondria. Neurosci Lett. 1995;195:179–182. doi: 10.1016/0304-3940(95)11812-b. [DOI] [PubMed] [Google Scholar]
- Kasarskis EJ, Tandon L, Lovell MA, Ehmann WD. Aluminum, calcium, and iron in the spinal cord of patients with sporadic amyotrophic lateral sclerosis using laser microprobe mass spectroscopy: a preliminary study. J Neurol Sci. 1995;130:203–208. doi: 10.1016/0022-510x(95)00037-3. [DOI] [PubMed] [Google Scholar]
- Kaur D, Yantiri F, Rajagopalan S, Kumar J, Mo JQ, Boonplueang R, et al. Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson's disease. Neuron. 2003;37:899–909. doi: 10.1016/s0896-6273(03)00126-0. [DOI] [PubMed] [Google Scholar]
- Kawamata H, Manfredi G. Import, maturation, and function of SOD1 and its copper chaperone CCS in the mitochondrial intermembrane space. Antioxid Redox Signal. 2010;13:1375–1384. doi: 10.1089/ars.2010.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keir ST, Dewhirst MW, Kirkpatrick JP, Bigner DD, Batinic-Haberle I. Cellular redox modulator, ortho Mn(III) meso-tetrakis(N-n-hexylpyridinium-2-yl)porphyrin, MnTnHex-2-PyP(5+) in the treatment of brain tumors. Anticancer Agents Med Chem. 2011;11:202–212. doi: 10.2174/187152011795255957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. 2008;4:176–185. doi: 10.1038/nchembio.72. [DOI] [PubMed] [Google Scholar]
- Koeppen AH, Michael SC, Knutson MD, Haile DJ, Qian J, Levi S, et al. The dentate nucleus in Friedreich's ataxia: the role of iron-responsive proteins. Acta Neuropathol (Berl) 2007;114:163–173. doi: 10.1007/s00401-007-0220-y. [DOI] [PubMed] [Google Scholar]
- Kooncumchoo P, Sharma S, Porter J, Govitrapong P, Ebadi M. Coenzyme Q(10) provides neuroprotection in iron-induced apoptosis in dopaminergic neurons. J Mol Neurosci. 2006;28:125–141. doi: 10.1385/JMN:28:2:125. [DOI] [PubMed] [Google Scholar]
- Kunwar A, Barik A, Mishra B, Rathinasamy K, Pandey R, Priyadarsini KI. Quantitative cellular uptake, localization and cytotoxicity of curcumin in normal and tumor cells. Biochim Biophys Acta. 2008;1780:673–679. doi: 10.1016/j.bbagen.2007.11.016. [DOI] [PubMed] [Google Scholar]
- Kuriyama S, Hozawa A, Ohmori K, Shimazu T, Matsui T, Ebihara S, et al. Green tea consumption and cognitive function: a cross-sectional study from the Tsurugaya Project 1. Am J Clin Nutr. 2006;83:355–361. doi: 10.1093/ajcn/83.2.355. [DOI] [PubMed] [Google Scholar]
- Lapchak PA, Schubert DR, Maher PA. Delayed treatment with a novel neurotrophic compound reduces behavioral deficits in rabbit ischemic stroke. J Neurochem. 2011;116:122–131. doi: 10.1111/j.1471-4159.2010.07090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DW, Kaur D, Chinta SJ, Rajagopalan S, Andersen JK. A disruption in iron-sulfur center biogenesis via inhibition of mitochondrial dithiol glutaredoxin 2 may contribute to mitochondrial and cellular iron dysregulation in mammalian glutathione-depleted dopaminergic cells: implications for Parkinson's disease. Antioxid Redox Signal. 2009;11:2083–2094. doi: 10.1089/ars.2009.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Suh S, Kim S. Protective effects of the green tea polyphenol (-)-epigallocatechin gallate against hippocampal neuronal damage after transient global ischemia in gerbils. Neurosci Lett. 2000;287:191–194. doi: 10.1016/s0304-3940(00)01159-9. [DOI] [PubMed] [Google Scholar]
- Levites Y, Weinreb O, Maor G, Youdim MB, Mandel S. Green tea polyphenol (-)-epigallocatechin-3-gallate prevents N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced dopaminergic neurodegeneration. J Neurochem. 2001;78:1073–1082. doi: 10.1046/j.1471-4159.2001.00490.x. [DOI] [PubMed] [Google Scholar]
- Levites Y, Amit T, Youdim MB, Mandel S. Involvement of protein kinase C activation and cell survival/ cell cycle genes in green tea polyphenol (-)-epigallocatechin 3-gallate neuroprotective action. J Biol Chem. 2002;277:30574–30580. doi: 10.1074/jbc.M202832200. [DOI] [PubMed] [Google Scholar]
- Li K, Besse EK, Ha D, Kovtunovych G, Rouault TA. Iron-dependent regulation of frataxin expression: implications for treatment of Friedreich ataxia. Hum Mol Genet. 2008;17:2265–2273. doi: 10.1093/hmg/ddn127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liccione JJ, Maines MD. Selective vulnerability of glutathione metabolism and cellular defense mechanisms in rat striatum to manganese. J Pharmacol Exp Ther. 1988;247:156–161. [PubMed] [Google Scholar]
- Lim S, Price KA, Chong SF, Paterson BM, Caragounis A, Barnham KJ, et al. Copper and zinc bis(thiosemicarbazonato) complexes with a fluorescent tag: synthesis, radiolabelling with copper-64, cell uptake and fluorescence studies. J Biol Inorg Chem. 2010;15:225–235. doi: 10.1007/s00775-009-0587-4. [DOI] [PubMed] [Google Scholar]
- Litvan I, Goldman JG, Troster AI, Schmand BA, Weintraub D, Petersen RC, et al. Diagnostic criteria for mild cognitive impairment in Parkinson's disease: Movement Disorder Society Task Force guidelines. Mov Disord. 2012;27:349–356. doi: 10.1002/mds.24893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Dargusch R, Maher P, Schubert D. A broadly neuroprotective derivative of curcumin. J Neurochem. 2008;105:1336–1345. doi: 10.1111/j.1471-4159.2008.05236.x. [DOI] [PubMed] [Google Scholar]
- Liu Z, Yu Y, Li X, Ross CA, Smith WW. Curcumin protects against A53T alpha-synuclein-induced toxicity in a PC12 inducible cell model for Parkinsonism. Pharmacol Res. 2011;63:439–444. doi: 10.1016/j.phrs.2011.01.004. [DOI] [PubMed] [Google Scholar]
- Loeffler DA, Lewitt PA, Juneau PL, Sima AA, Nguyen HU, Demaggio AJ, et al. Increased regional brain concentrations of ceruloplasmin in neurodegenerative disorders. Brain Res. 1996;738:265–274. doi: 10.1016/s0006-8993(96)00782-2. [DOI] [PubMed] [Google Scholar]
- Long S, Jirku M, Mach J, Ginger ML, Sutak R, Richardson D, et al. Ancestral roles of eukaryotic frataxin: mitochondrial frataxin function and heterologous expression of hydrogenosomal Trichomonas homologues in trypanosomes. Mol Microbiol. 2008;69:94–109. doi: 10.1111/j.1365-2958.2008.06260.x. [DOI] [PubMed] [Google Scholar]
- Lu J, Duan W, Guo Y, Jiang H, Li Z, Huang J, et al. Mitochondrial dysfunction in human TDP-43 transfected NSC34 cell lines and the protective effect of dimethoxy curcumin. Brain Res Bull. 2012;89:185–190. doi: 10.1016/j.brainresbull.2012.09.005. [DOI] [PubMed] [Google Scholar]
- Lumsden AL, Henshall TL, Dayan S, Lardelli MT, Richards RI. Huntingtin-deficient zebrafish exhibit defects in iron utilization and development. Hum Mol Genet. 2007;16:1905–1920. doi: 10.1093/hmg/ddm138. [DOI] [PubMed] [Google Scholar]
- McAllum EJ, Lim NK, Hickey JL, Paterson BM, Donnelly PS, Li QX, et al. Therapeutic effects of Cu(II)(atsm) in the SOD1-G37R mouse model of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:586–590. doi: 10.3109/21678421.2013.824000. [DOI] [PubMed] [Google Scholar]
- McEachern G, Kassovska-Bratinova S, Raha S, Tarnopolsky MA, Turnbull J, Bourgeois J, et al. Manganese superoxide dismutase levels are elevated in a proportion of amyotrophic lateral sclerosis patient cell lines. Biochem Biophys Res Commun. 2000;273:359–363. doi: 10.1006/bbrc.2000.2933. [DOI] [PubMed] [Google Scholar]
- Mackensen GB, Patel M, Sheng H, Calvi CL, Batinic-Haberle I, Day BJ, et al. Neuroprotection from delayed postischemic administration of a metalloporphyrin catalytic antioxidant. J Neurosci. 2001;21:4582–4592. doi: 10.1523/JNEUROSCI.21-13-04582.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane J, Hervias I, Henning MS, Damiano M, Kawamata H, Manfredi G. Mutant SOD1 in neuronal mitochondria causes toxicity and mitochondrial dynamics abnormalities. Hum Mol Genet. 2009;18:4552–4564. doi: 10.1093/hmg/ddp421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel SA, Avramovich-Tirosh Y, Reznichenko L, Zheng H, Weinreb O, Amit T, et al. Multifunctional activities of green tea catechins in neuroprotection. Modulation of cell survival genes, iron-dependent oxidative stress and PKC signaling pathway. Neurosignals. 2005;14:46–60. doi: 10.1159/000085385. [DOI] [PubMed] [Google Scholar]
- Maraldi T, Riccio M, Zambonin L, Vinceti M, De Pol A, Hakim G. Low levels of selenium compounds are selectively toxic for a human neuron cell line through ROS/RNS increase and apoptotic process activation. Neurotoxicology. 2011;32:180–187. doi: 10.1016/j.neuro.2010.10.008. [DOI] [PubMed] [Google Scholar]
- Marmolino D. Friedreich's ataxia: past, present and future. Brain Res Rev. 2011;67:311–330. doi: 10.1016/j.brainresrev.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Martin HL, Teismann P. Glutathione – a review on its role and significance in Parkinson's disease. FASEB J. 2009;23:3263–3272. doi: 10.1096/fj.08-125443. [DOI] [PubMed] [Google Scholar]
- Mastroberardino PG, Hoffman EK, Horowitz MP, Betarbet R, Taylor G, Cheng D, et al. A novel transferrin/TfR2-mediated mitochondrial iron transport system is disrupted in Parkinson's disease. Neurobiol Dis. 2009;34:417–431. doi: 10.1016/j.nbd.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki H, Yasuda T. Iron accumulation in Parkinson's disease. J Neural Transm. 2012;119:1511–1514. doi: 10.1007/s00702-012-0905-9. [DOI] [PubMed] [Google Scholar]
- Morgan MT, Bagchi P, Fahrni CJ. Designed to dissolve: suppression of colloidal aggregation of Cu(I)-selective fluorescent probes in aqueous buffer and in-gel detection of a metallochaperone. J Am Chem Soc. 2011;133:15906–15909. doi: 10.1021/ja207004v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu X, He G, Cheng Y, Li X, Xu B, Du G. Baicalein exerts neuroprotective effects in 6-hydroxydopamine-induced experimental parkinsonism in vivo and in vitro. Pharmacol Biochem Behav. 2009;92:642–648. doi: 10.1016/j.pbb.2009.03.008. [DOI] [PubMed] [Google Scholar]
- Muhlenhoff U, Richhardt N, Ristow M, Kispal G, Lill R. The yeast frataxin homolog Yfh1p plays a specific role in the maturation of cellular Fe/S proteins. Hum Mol Genet. 2002;11:2025–2036. doi: 10.1093/hmg/11.17.2025. [DOI] [PubMed] [Google Scholar]
- Mythri RB, Harish G, Dubey SK, Misra K, Bharath MM. Glutamoyl diester of the dietary polyphenol curcumin offers improved protection against peroxynitrite-mediated nitrosative stress and damage of brain mitochondria in vitro: implications for Parkinson's disease. Mol Cell Biochem. 2011;347:135–143. doi: 10.1007/s11010-010-0621-4. [DOI] [PubMed] [Google Scholar]
- Nanjo F, Goto K, Seto R, Suzuki M, Sakai M, Hara Y. Scavenging effects of tea catechins and their derivatives on 1,1-diphenyl-2-picrylhydrazyl radical. Free Radic Biol Med. 1996;21:895–902. doi: 10.1016/0891-5849(96)00237-7. [DOI] [PubMed] [Google Scholar]
- Nasir J, Floresco SB, O'Kusky JR, Diewert VM, Richman JM, Zeisler J, et al. Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- New EJ. Tools to study distinct metal pools in biology. Dalton Trans. 2013;42:3210–3219. doi: 10.1039/c2dt31933k. [DOI] [PubMed] [Google Scholar]
- Nie G, Jin C, Cao Y, Shen S, Zhao B. Distinct effects of tea catechins on 6-hydroxydopamine-induced apoptosis in PC12 cells. Arch Biochem Biophys. 2002;397:84–90. doi: 10.1006/abbi.2001.2636. [DOI] [PubMed] [Google Scholar]
- Oakley AE, Collingwood JF, Dobson J, Love G, Perrott HR, Edwardson JA, et al. Individual dopaminergic neurons show raised iron levels in Parkinson disease. Neurology. 2007;68:1820–1825. doi: 10.1212/01.wnl.0000262033.01945.9a. [DOI] [PubMed] [Google Scholar]
- Oba H, Araki T, Ohtomo K, Monzawa S, Uchiyama G, Koizumi K, et al. Amyotrophic lateral sclerosis: T2 shortening in motor cortex at MR imaging. Radiology. 1993;189:843–846. doi: 10.1148/radiology.189.3.8234713. [DOI] [PubMed] [Google Scholar]
- Obata A, Yoshimi E, Waki A, Lewis JS, Oyama N, Welch MJ, et al. Retention mechanism of hypoxia selective nuclear imaging/radiotherapeutic agent cu-diacetyl-bis(N4-methylthiosemicarbazone) (Cu-ATSM) in tumor cells. Ann Nucl Med. 2001;15:499–504. doi: 10.1007/BF02988502. [DOI] [PubMed] [Google Scholar]
- Oliveira JM. Nature and cause of mitochondrial dysfunction in Huntington's disease: focusing on huntingtin and the striatum. J Neurochem. 2010;114:1–12. doi: 10.1111/j.1471-4159.2010.06741.x. [DOI] [PubMed] [Google Scholar]
- Ono K, Yamada M. Antioxidant compounds have potent anti-fibrillogenic and fibril-destabilizing effects for alpha-synuclein fibrils in vitro. J Neurochem. 2006;97:105–115. doi: 10.1111/j.1471-4159.2006.03707.x. [DOI] [PubMed] [Google Scholar]
- Packer MA, Murphy MP. Peroxynitrite causes calcium efflux from mitochondria which is prevented by cyclosporin A. FEBS Lett. 1994;345:237–240. doi: 10.1016/0014-5793(94)00461-7. [DOI] [PubMed] [Google Scholar]
- Pan J, Li H, Ma JF, Tan YY, Xiao Q, Ding JQ, et al. Curcumin inhibition of JNKs prevents dopaminergic neuronal loss in a mouse model of Parkinson's disease through suppressing mitochondria dysfunction. Transl Neurodegener. 2012;1:16. doi: 10.1186/2047-9158-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panov A, Kubalik N, Zinchenko N, Hemendinger R, Dikalov S, Bonkovsky HL. Respiration and ROS production in brain and spinal cord mitochondria of transgenic rats with mutant G93a Cu/Zn-superoxide dismutase gene. Neurobiol Dis. 2011;44:53–62. doi: 10.1016/j.nbd.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Parker SJ, Meyerowitz J, James JL, Liddell JR, Nonaka T, Hasegawa M, et al. Inhibition of TDP-43 accumulation by bis(thiosemicarbazonato)-copper complexes. PLoS ONE. 2012;7:e42277. doi: 10.1371/journal.pone.0042277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascu SI, Waghorn PA, Conry TD, Lin B, Betts HM, Dilworth JR, et al. Cellular confocal fluorescence studies and cytotoxic activity of new Zn(II) bis(thiosemicarbazonato) complexes. Dalton Trans. 2008;2008:2107–2110. doi: 10.1039/b802806k. [DOI] [PubMed] [Google Scholar]
- Pascu SI, Waghorn PA, Kennedy BW, Arrowsmith RL, Bayly SR, Dilworth JR, et al. Fluorescent copper(II) bis(thiosemicarbazonates): synthesis, structures, electron paramagnetic resonance, radiolabeling, in vitro cytotoxicity and confocal fluorescence microscopy studies. Chem Asian J. 2010;5:506–519. doi: 10.1002/asia.200900446. [DOI] [PubMed] [Google Scholar]
- Pasternack RF, Banth A, Pasternack JM, Johnson CS. Catalysis of the disproportionation of superoxide by metalloporphyrins. III. J Inorg Biochem. 1981;15:261–267. doi: 10.1016/s0162-0134(00)80161-0. [DOI] [PubMed] [Google Scholar]
- Pearce RK, Owen A, Daniel S, Jenner P, Marsden CD. Alterations in the distribution of glutathione in the substantia nigra in Parkinson's disease. J Neural Transm. 1997;104:661–677. doi: 10.1007/BF01291884. [DOI] [PubMed] [Google Scholar]
- Piantadosi CA, Zhang J. Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke. 1996;27:327–331. doi: 10.1161/01.str.27.2.327. discussion 332. [DOI] [PubMed] [Google Scholar]
- Pierrel F, Cobine PA, Winge DR. Metal ion availability in mitochondria. Biometals. 2007;20:675–682. doi: 10.1007/s10534-006-9052-9. [DOI] [PubMed] [Google Scholar]
- Popescu BF, George MJ, Bergmann U, Garachtchenko AV, Kelly ME, McCrea RP, et al. Mapping metals in Parkinson's and normal brain using rapid-scanning X-ray fluorescence. Phys Med Biol. 2009;54:651–663. doi: 10.1088/0031-9155/54/3/012. [DOI] [PubMed] [Google Scholar]
- Price KA, Crouch PJ, Lim S, Paterson BM, Liddell JR, Donnelly PS, et al. Subcellular localization of a fluorescent derivative of CuII(atsm) offers insight into the neuroprotective action of CuII(atsm) Metallomics. 2011;3:1280–1290. doi: 10.1039/c1mt00092f. [DOI] [PubMed] [Google Scholar]
- Puccio H, Simon D, Cossee M, Criqui-Filipe P, Tiziano F, Melki J, et al. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet. 2001;27:181–186. doi: 10.1038/84818. [DOI] [PubMed] [Google Scholar]
- Radi R, Rodriguez M, Castro L, Telleri R. Inhibition of mitochondrial electron transport by peroxynitrite. Arch Biochem Biophys. 1994;308:89–95. doi: 10.1006/abbi.1994.1013. [DOI] [PubMed] [Google Scholar]
- Radisky DC, Babcock MC, Kaplan J. The yeast frataxin homologue mediates mitochondrial iron efflux. Evidence for a mitochondrial iron cycle. J Biol Chem. 1999;274:4497–4499. doi: 10.1074/jbc.274.8.4497. [DOI] [PubMed] [Google Scholar]
- Reichmann H, Janetzky B, Riederer P. Iron-dependent enzymes in Parkinson's disease. J Neural Transm Suppl. 1995;46:157–164. [PubMed] [Google Scholar]
- Richardson DR, Lane DJ, Becker EM, Huang ML, Whitnall M, Suryo Rahmanto Y, et al. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc Natl Acad Sci U S A. 2010;107:10775–10782. doi: 10.1073/pnas.0912925107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rines AK, Ardehali H. Transition metals and mitochondrial metabolism in the heart. J Mol Cell Cardiol. 2013;55:50–57. doi: 10.1016/j.yjmcc.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo L, Intrieri M, D'agata V, Mangano NG, Oriani G, Ontario ML, et al. The major green tea polyphenol, (-)-epigallocatechin-3-gallate, induces heme oxygenase in rat neurons and acts as an effective neuroprotective agent against oxidative stress. J Am Coll Nutr. 2009;28:492S–499S. doi: 10.1080/07315724.2009.10718116. (Suppl) [DOI] [PubMed] [Google Scholar]
- Rosas HD, Chen YI, Doros G, Salat DH, Chen NK, Kwong KK, et al. Alterations in brain transition metals in Huntington disease: an evolving and intricate story. Arch Neurol. 2012;69:887–893. doi: 10.1001/archneurol.2011.2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotig A, De Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, et al. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet. 1997;17:215–217. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- Saba H, Batinic-Haberle I, Munusamy S, Mitchell T, Lichti C, Megyesi J, et al. Manganese porphyrin reduces renal injury and mitochondrial damage during ischemia/reperfusion. Free Radic Biol Med. 2007;42:1571–1578. doi: 10.1016/j.freeradbiomed.2007.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salah N, Miller NJ, Paganga G, Tijburg L, Bolwell GP, Rice-Evans C. Polyphenolic flavanols as scavengers of aqueous phase radicals and as chain-breaking antioxidants. Arch Biochem Biophys. 1995;322:339–346. doi: 10.1006/abbi.1995.1473. [DOI] [PubMed] [Google Scholar]
- Sarsero JP, Li L, Wardan H, Sitte K, Williamson R, Ioannou PA. Upregulation of expression from the FRDA genomic locus for the therapy of Friedreich ataxia. J Gene Med. 2003;5:72–81. doi: 10.1002/jgm.320. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Iwata M. Ultrastructural study of synapses in the anterior horn neurons of patients with amyotrophic lateral sclerosis. Neurosci Lett. 1996;204:53–56. doi: 10.1016/0304-3940(96)12314-4. [DOI] [PubMed] [Google Scholar]
- Schipper HM, Vininsky R, Brull R, Small L, Brawer JR. Astrocyte mitochondria: a substrate for iron deposition in the aging rat substantia nigra. Exp Neurol. 1998;152:188–196. doi: 10.1006/exnr.1998.6854. [DOI] [PubMed] [Google Scholar]
- Schroeder EK, Kelsey NA, Doyle J, Breed E, Bouchard RJ, Loucks FA, et al. Green tea epigallocatechin 3-gallate accumulates in mitochondria and displays a selective antiapoptotic effect against inducers of mitochondrial oxidative stress in neurons. Antioxid Redox Signal. 2009;11:469–480. doi: 10.1089/ars.2008.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeter H, Bahia P, Spencer JP, Sheppard O, Rattray M, Cadenas E, et al. (-)Epicatechin stimulates ERK-dependent cyclic AMP response element activity and up-regulates GluR2 in cortical neurons. J Neurochem. 2007;101:1596–1606. doi: 10.1111/j.1471-4159.2006.04434.x. [DOI] [PubMed] [Google Scholar]
- Sensi SL, Jeng JM. Rethinking the excitotoxic ionic milieu: the emerging role of Zn(2+) in ischemic neuronal injury. Curr Mol Med. 2004;4:87–111. doi: 10.2174/1566524043479211. [DOI] [PubMed] [Google Scholar]
- Sensi SL, Ton-That D, Sullivan PG, Jonas EA, Gee KR, Kaczmarek LK, et al. Modulation of mitochondrial function by endogenous Zn2+ pools. Proc Natl Acad Sci U S A. 2003;100:6157–6162. doi: 10.1073/pnas.1031598100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline CT, Zhu J, Zhang W, Shi C, Cai AL. Mitochondrial inhibitor models of Huntington's disease and Parkinson's disease induce zinc accumulation and are attenuated by inhibition of zinc neurotoxicity in vitro or in vivo. Neurodegener Dis. 2013;11:49–58. doi: 10.1159/000336558. [DOI] [PubMed] [Google Scholar]
- Sheng H, Yang W, Fukuda S, Tse HM, Paschen W, Johnson K, et al. Long-term neuroprotection from a potent redox-modulating metalloporphyrin in the rat. Free Radic Biol Med. 2009;47:917–923. doi: 10.1016/j.freeradbiomed.2009.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng H, Spasojevic I, Tse HM, Jung JY, Hong J, Zhang Z, et al. Neuroprotective efficacy from a lipophilic redox-modulating Mn(III) N-Hexylpyridylporphyrin, MnTnHex-2-PyP: rodent models of ischemic stroke and subarachnoid hemorrhage. J Pharmacol Exp Ther. 2011;338:906–916. doi: 10.1124/jpet.110.176701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi P, Gal J, Kwinter DM, Liu X, Zhu H. Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochim Biophys Acta. 2010;1802:45–51. doi: 10.1016/j.bbadis.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, et al. Alterations in glutathione levels in Parkinson's disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36:348–355. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- Sian-Hulsmann J, Mandel S, Youdim MB, Riederer P. The relevance of iron in the pathogenesis of Parkinson's disease. J Neurochem. 2011;118:939–957. doi: 10.1111/j.1471-4159.2010.07132.x. [DOI] [PubMed] [Google Scholar]
- Singh S, Khodr H, Taylor MI, Hider RC. Therapeutic iron chelators and their potential side-effects. Biochem Soc Symp. 1995;61:127–137. doi: 10.1042/bss0610127. [DOI] [PubMed] [Google Scholar]
- Sohn YS, Breuer W, Munnich A, Cabantchik ZI. Redistribution of accumulated cell iron: a modality of chelation with therapeutic implications. Blood. 2008;111:1690–1699. doi: 10.1182/blood-2007-07-102335. [DOI] [PubMed] [Google Scholar]
- Son M, Puttaparthi K, Kawamata H, Rajendran B, Boyer PJ, Manfredi G, et al. Overexpression of CCS in G93A-SOD1 mice leads to accelerated neurological deficits with severe mitochondrial pathology. Proc Natl Acad Sci U S A. 2007;104:6072–6077. doi: 10.1073/pnas.0610923104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood PK, Nahar U, Nehru B. Curcumin attenuates aluminum-induced oxidative stress and mitochondrial dysfunction in rat brain. Neurotox Res. 2011;20:351–361. doi: 10.1007/s12640-011-9249-8. [DOI] [PubMed] [Google Scholar]
- Soon CP, Donnelly PS, Turner BJ, Hung LW, Crouch PJ, Sherratt NA, et al. Diacetylbis(N(4)-methylthiosemicarbazonato) copper(II) (CuII(atsm)) protects against peroxynitrite-induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model. J Biol Chem. 2011;286:44035–44044. doi: 10.1074/jbc.M111.274407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano S, Llorens JV, Blanco-Sobero L, Gutierrez L, Calap-Quintana P, Morales MP, et al. Deferiprone and idebenone rescue frataxin depletion phenotypes in a Drosophila model of Friedreich's ataxia. Gene. 2013;521:274–281. doi: 10.1016/j.gene.2013.02.049. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Subramaniam SR, Chesselet MF. Mitochondrial dysfunction and oxidative stress in Parkinson's disease. Prog Neurobiol. 2013;106–107:17–32. doi: 10.1016/j.pneurobio.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland BA, Shaw OM, Clarkson AN, Jackson DN, Sammut IA, Appleton I. Neuroprotective effects of (-)-epigallocatechin gallate following hypoxia-ischemia-induced brain damage: novel mechanisms of action. FASEB J. 2005;19:258–260. doi: 10.1096/fj.04-2806fje. [DOI] [PubMed] [Google Scholar]
- Tan G, Chen LS, Lonnerdal B, Gellera C, Taroni FA, Cortopassi GA. Frataxin expression rescues mitochondrial dysfunctions in FRDA cells. Hum Mol Genet. 2001;10:2099–2107. doi: 10.1093/hmg/10.19.2099. [DOI] [PubMed] [Google Scholar]
- Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, et al. Rotenone, paraquat, and Parkinson's disease. Environ Health Perspect. 2011;119:866–872. doi: 10.1289/ehp.1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauskela JS, Brunette E, O'reilly N, Mealing G, Comas T, Gendron TF, et al. An alternative Ca2+-dependent mechanism of neuroprotection by the metalloporphyrin class of superoxide dismutase mimetics. FASEB J. 2005;19:1734–1736. doi: 10.1096/fj.05-3795fje. [DOI] [PubMed] [Google Scholar]
- Tovmasyan AG, Rajic Z, Spasojevic I, Reboucas JS, Chen X, Salvemini D, et al. Methoxy-derivatization of alkyl chains increases the in vivo efficacy of cationic Mn porphyrins. Synthesis, characterization, SOD-like activity, and SOD-deficient E. coli study of meta Mn(III) N-methoxyalkylpyridylporphyrins. Dalton Trans. 2011;40:4111–4121. doi: 10.1039/c0dt01321h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Velde C, Miller TM, Cashman NR, Cleveland DW. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc Natl Acad Sci U S A. 2008;105:4022–4027. doi: 10.1073/pnas.0712209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco-Sanchez D, Aracil A, Montero R, Mas A, Jimenez L, O'callaghan M, et al. Combined therapy with idebenone and deferiprone in patients with Friedreich's ataxia. Cerebellum. 2011;10:1–8. doi: 10.1007/s12311-010-0212-7. [DOI] [PubMed] [Google Scholar]
- Wakade C, Khan MM, De Sevilla LM, Zhang QG, Mahesh VB, Brann DW. Tamoxifen neuroprotection in cerebral ischemia involves attenuation of kinase activation and superoxide production and potentiation of mitochondrial superoxide dismutase. Endocrinology. 2008;149:367–379. doi: 10.1210/en.2007-0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MS, Boddapati S, Emadi S, Sierks MR. Curcumin reduces alpha-synuclein induced cytotoxicity in Parkinson's disease cell model. BMC Neurosci. 2010;11:57. doi: 10.1186/1471-2202-11-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb O, Mandel S, Youdim MB. cDNA gene expression profile homology of antioxidants and their antiapoptotic and proapoptotic activities in human neuroblastoma cells. FASEB J. 2003;17:935–937. doi: 10.1096/fj.02-0712fje. [DOI] [PubMed] [Google Scholar]
- Weitner T, Kos I, Sheng H, Tovmasyan A, Reboucas JS, Fan P, et al. Comprehensive pharmacokinetic studies and oral bioavailability of two Mn porphyrin-based SOD mimics, MnTE-2-PyP5+ and MnTnHex-2-PyP5+ Free Radic Biol Med. 2013;58:73–80. doi: 10.1016/j.freeradbiomed.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RB, Roof DM. Respiratory deficiency due to loss of mitochondrial DNA in yeast lacking the frataxin homologue. Nat Genet. 1997;16:352–357. doi: 10.1038/ng0897-352. [DOI] [PubMed] [Google Scholar]
- Wise-Faberowski L, Warner DS, Spasojevic I, Batinic-Haberle I. Effect of lipophilicity of Mn (III) ortho N-alkylpyridyl-and diortho N, N′-diethylimidazolylporphyrins in two in-vitro models of oxygen and glucose deprivation-induced neuronal death. Free Radic Res. 2009;43:329–339. doi: 10.1080/10715760902736283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A, Yang J, Cavadini P, Gellera C, Lonnerdal B, Taroni F, et al. The Friedreich's ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum Mol Genet. 1999;8:425–430. doi: 10.1093/hmg/8.3.425. [DOI] [PubMed] [Google Scholar]
- Wu J, Li Q, Wang X, Yu S, Li L, Wu X, et al. Neuroprotection by curcumin in ischemic brain injury involves the Akt/Nrf2 pathway. PLoS ONE. 2013;8:e59843. doi: 10.1371/journal.pone.0059843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Donnelly PS, Zimmermann M, Wedd AG. Transfer of copper between bis(thiosemicarbazone) ligands and intracellular copper-binding proteins. Insights into mechanisms of copper uptake and hypoxia selectivity. Inorg Chem. 2008;47:4338–4347. doi: 10.1021/ic702440e. [DOI] [PubMed] [Google Scholar]
- Xun Z, Sowell RA, Kaufman TC, Clemmer DE. Quantitative proteomics of a presymptomatic A53T alpha-synuclein Drosophila model of Parkinson disease. Mol Cell Proteomics. 2008;7:1191–1203. doi: 10.1074/mcp.M700467-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii Y, Yoneda M, Ikawa M, Furukawa T, Kiyono Y, Mori T, et al. Radiolabeled Cu-ATSM as a novel indicator of overreduced intracellular state due to mitochondrial dysfunction: studies with mitochondrial DNA-less rho0 cells and cybrids carrying MELAS mitochondrial DNA mutation. Nucl Med Biol. 2012;39:177–185. doi: 10.1016/j.nucmedbio.2011.08.008. [DOI] [PubMed] [Google Scholar]
- Yu L, Jia X, Derricka M, Drobyshevsky A, Liu T, Batinic-Haberle I, et al. 2010. Testing new porphyrins in in vivo model systems: effect of Mn porphyrins in animal model of cerebral palsy. 6th International Conference on Porphyrins and Phthalocyanines, Book of Absracts. Albuquerque, USA.
- Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat Genet. 1995;11:155–163. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- Zhang X, Lemasters JJ. Translocation of iron from lysosomes to mitochondria during ischemia predisposes to injury after reperfusion in rat hepatocytes. Free Radic Biol Med. 2013;63:243–253. doi: 10.1016/j.freeradbiomed.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman MC, Oberley LW, Flanagan SW. Mutant SOD1-induced neuronal toxicity is mediated by increased mitochondrial superoxide levels. J Neurochem. 2007;102:609–618. doi: 10.1111/j.1471-4159.2007.04502.x. [DOI] [PubMed] [Google Scholar]