

Abstract
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disease characterized by the selective death of upper and lower motor neurons which ultimately leads to paralysis and ultimately death. Pathological changes in ALS are closely associated with pronounced and progressive changes in mitochondrial morphology, bioenergetics and calcium homeostasis. Converging evidence suggests that impaired mitochondrial function could be pivotal in the rapid neurodegeneration of this condition. In this review, we provide an update of recent advances in understanding mitochondrial biology in the pathogenesis of ALS and highlight the therapeutic value of pharmacologically targeting mitochondrial biology to slow disease progression.
Linked Articles
This article is part of a themed issue on Mitochondrial Pharmacology: Energy, Injury & Beyond. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2014.171.issue-8
Keywords: amyotrophic lateral sclerosis, mitochondria, SOD-1, TDP-43, therapeutic, cell death
Introduction
Amyotrophic lateral sclerosis (ALS) is a lethal disorder of incompletely understood aetiology encompassing a range of clinicopathological entities (Turner et al., 2013). The disease is characterized by selective death of upper motor neurons in the cerebral cortex and of lower motor neurons in the brainstem and spinal cord resulting in paralysis, disability and death within 1 to 5 years from diagnosis. Moreover, there is no cure or effective treatment. Most cases of ALS are sporadic but about 5% are familial (fALS) and 20% of these are caused by mutations in the gene for superoxide dismutase (SOD1) (Robberecht and Philips, 2013). Recently, a number of new genes have been causally linked to the pathogenesis of fALS. These include genes directly linked to RNA metabolism such as TARDBP that encodes for the TAR DNA binding protein TDP-43 (Sreedharan et al., 2008) and the Fused-in-Sarcoma (FUS) encoding gene FUS/TLD (Vance et al., 2009). fALS can also be caused by mutations in genes associated with protein degradation pathways, including ubiquilin-2 (UBQLN2) (Deng et al., 2011), valosin-containing protein (VCP) (Johnson et al., 2010) and the vesicle-associated membrane protein-associated protein B (VAPB) (Nishimura et al., 2004), p62/sequestosome (SQSTM1) (Fecto et al., 2011). More recently, hexanucleotide expansion in the C9ORF72 gene has been found to be a common genetic cause for ALS (Dejesus-Hernandez et al., 2011; Renton et al., 2011). The discovery of new disease-causing genes represents a paradigm shift in the understanding of the pathogenesis of ALS. At present, it is generally believed that dysfunctional RNA metabolism and the subsequent disturbance of protein homeostasis is fundamental to the pathogenesis of this condition.
The histopathological hallmark of ALS is the presence of cytosolic protein aggregates (Lansbury and Lashuel, 2006). Current evidence suggests that these protein aggregates interfere with normal cellular processes, resulting in oxidative stress, excitotoxicity, mitochondrial dysfunction and finally retraction of axons, synaptic disintegration and cell death. These observations have led to the postulation of the ‘dying back’ hypothesis of motor neuron death in ALS (Redler and Dokholyan, 2012). There is also considerable evidence suggesting that the activation of caspase-dependent apoptotic pathways is involved in neurodegeneration in ALS (Muyderman et al., 2009; Martin, 2011; Soo et al., 2012). However, whether caspase-activation represents the end point of other pathological processes or is a key process in ALS remains controversial and pharmacological approaches aimed at inhibiting apoptosis have yielded little or no success.
Mitochondrial dysfunction in ALS
Mitochondrial dysfunction has been implicated as playing a role in motor neuron death in ALS. Fragmentation of mitochondria and changes in mitochondrial morphology and expression of fusion/fission proteins are well described in ALS and have pronounced effects on normal mitochondrial function (Sasaki et al., 2007). Defective mitochondrial transport may be responsible for the accumulation of abnormal mitochondria in motor neuron axons seen in animal models of ALS and also in human patients. Studies in cell culture models and in transgenic animals have demonstrated aberrations in oxidative metabolism linked to changes in electron transport chain (ETC) activity and impaired ATP production (Keep et al., 2001; Mattiazzi et al., 2002; Menzies et al., 2002; Knott et al., 2008). Mitochondria from ALS patients have impaired Ca2+ homeostasis and an increased production of reactive oxygen species (ROS) which is associated with oxidative-related damage including changes in protein carbonylates and tyrosine nitration (Beal et al., 1997; Beal, 2002). Indeed, glutamate-receptor mediated neurotoxicity has been linked to an overload of mitochondrial calcium and ROS production in cultured spinal motor neurons from transgenic ALS animals (Carriedo et al., 2000). Together, these studies demonstrate that changes in mitochondrial function and dynamics are a central and common feature of the pathogenesis in ALS. However, it is not known if mitochondrial dysfunction is a primary or secondary event in these processes.
Mitochondria-linked cell death pathways
Apoptotic cell death is intimately linked to mitochondrial dysfunction and commonly involves activation of caspases via the ‘intrinsic’ or the ‘extrinsic’ pathway (Figure 1) (Kroemer et al., 1997; Kroemer, 2010; Sims and Muyderman, 2010). The intrinsic pathway is dependent on the mitochondrial release of proteins leading to the activation of caspases, particularly caspase-3, thereby initiating the apoptotic cascade that finally results in chromatin condensation and DNA fragmentation. The extrinsic pathway is initiated by the binding of specific ligands to plasma membrane cell death receptors such as FAS and DR6, thereby triggering activation of caspase-8 which in turn activates other ‘executioner’ caspases (Kantari and Walczak, 2011). Executioner caspase activation in the extrinsic pathway can occur without the involvement of mitochondria (Galluzzi et al., 2009; 2012) but may also result in caspase-8-mediated cleavage of Bid to produce truncated Bid, thereby promoting the release of mitochondrial apoptogenic proteins and producing a mixed intrinsic/extrinsic response. In addition, mitochondria-mediated caspase-independent modes of apoptosis have been reported in neurodegenerative diseases involving release of the apoptosis-inducing factor AIF (Jordan et al., 2003; Polster, 2013).
Figure 1.
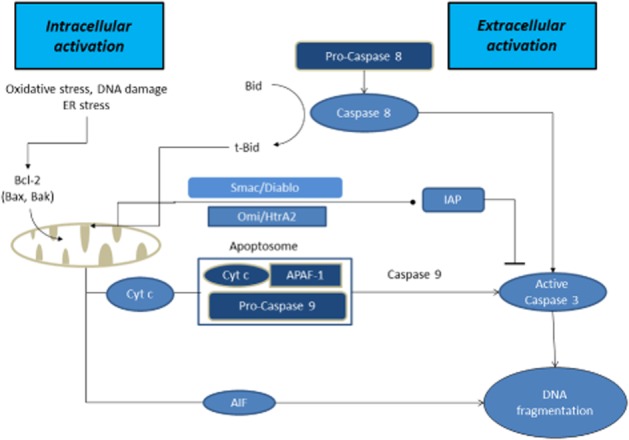
Proteins released from the intermembrane space and their contribution to apoptosis. Release of cytochrome c is a key step in the intrinsic pathway of apoptosis forming a complex known as an apoptosome with the proteins APAF-1 and procaspase-9 and with ATP. Apoptosome formation leads to activation of executioner caspases, particularly caspase-3 resulting in internuclear protein and DNA degradation. The process can be promoted by the release of Smac/DIABLO and/or Omi/HtrA2 that block the IAPs family of proteins that are endogenous inhibitors of caspases. Release of AIF may lead directly to a caspase-independent form of apoptosis after its interaction with cyclophilin A. The extrinsic pathway is initiated by the binding of specific ligands to plasma membrane cell death receptors such as FAS and DR6 triggering activation of caspase-8 which in turn activates other executioner caspases. Executioner caspase activation in the extrinsic pathway can occur without involvement of mitochondria but may also result in caspase-8 mediated cleavage of Bid to produce truncated Bid, promoting release of mitochondrial apoptogenic proteins producing a mixed intrinsic/extrinsic presentation.
Mitochondrial release of large apoptogenic proteins, including cytochrome c, into the cytosol during the development of apoptosis requires a substantial increase in permeability of the outer mitochondrial membrane. Mechanisms underlying protein release from mitochondria are believed to involve the proapoptotic Bcl-2 family of proteins (Galluzzi et al., 2009; 2012; Michels et al., 2013). Although not fully understood, this process seems to involve the translocation of the two proapoptotic proteins Bim and Bax from the cytosol to the mitochondrial membrane, resulting in an increased permeability of the outer membrane. Movement of another Bcl-2 family protein, Bad, from the cytosol to the mitochondria can also contribute to membrane permeabilization under some circumstances and coupled with the activation of caspase-8 via the extrinsic apoptotic pathway can result in the cleavage of yet another Bcl-2 family protein, Bid, thereby promoting the release of proapoptotic proteins. Other members of the Bcl-2 family are anti-apoptotic. For example, the Bcl-2 homolog BCL-XL binds to BAX and BAK, preventing formation of pores in the outer membrane and hence limiting the release of apoptogenic proteins (Michels et al., 2013). An alternative process concerns the formation of the mitochondrial transition pore which leads to permeabilization of the inner membrane. Subsequent swelling of the mitochondria due to water entry into the matrix following pore opening results in disruption of the outer membrane and the release of proteins from the intermembrane space (Brenner and Moulin, 2012). The opening of the transition pore is usually induced by abnormal accumulation of calcium but can be promoted by multiple factors including oxidative stress. The composition of the permeability transition pore is not fully understood but is believed to include an adenine nucleotide translocase in the inner mitochondrial membrane, a voltage-dependent anion carrier in the outer membrane and the matrix protein cyclophilin D (Brenner and Moulin, 2012).
Mutant SOD1, ALS and mitochondria
Significant advances in understanding the mechanisms of motor neuron pathology in ALS have come from studies using transgenic rodents expressing mutant forms of the human SOD1 (mutant SOD1) gene that mimic the familial form of ALS (Gurney et al., 1994; Nagai et al., 2001). At present there are more than 150 known mutations in the SOD1 gene. Animals expressing mutant SOD1 typically display a phenotype that resembles ALS and demonstrate most of the histopathological and biochemical features and also the symptoms of the human disease. Pathology first appears in motor neurons of the spinal cord within 6 weeks of birth and the first motor symptoms appear at 3 months of age resulting in a progressive paralysis similar to that found in humans (Chiu et al., 1995; Julien and Kriz, 2006). Several studies have demonstrated a role for mutant SOD1 in mitochondrial dysfunction in ALS pathogenesis (see Table 1). Mutant SOD1 is often found as aggregates at the outer membrane of mitochondria in motor neurons of various mouse models and in fALS patients (Higgins et al., 2003). Thus, it is believed that disruption of mitochondrial function by the presence of misfolded protein aggregates results in mitochondrial damage, including increased mitochondrial volume and excess superoxide production (Pasinelli et al., 2004; Pickles et al., 2013). Moreover, cells expressing some forms of mutant SOD1 undergo mitochondrial apoptotic signalling (see Figure 2: Table 1) and mutant SOD1 transgenic animals overexpress proapoptotic proteins such as the BH3-only protein Bim and Bax while Bcl-2 and Bcl-Xl have been found to be decreased (Vukosavic et al., 1999; 2000). Overexpressing Bcl-2 delays caspase activation in the mutant SOD1G93A transgenic animal (Vukosavic et al., 2000) and silencing Bim protein expression delays disease onset in other animal models of the disease. A similar anti-apoptotic effect is seen in cell culture models of mutant SOD1 ALS (Hetz et al., 2007; Soo et al., 2012). In mutant SOD1G85R-expressing Neuro2a cells, Bim deletion leads to reduced Bax recruitment to mitochondria and decreased cytochrome c redistribution. As Bim is considered a direct link between endoplasmic reticulum (ER) stress and mitochondrial apoptosis, these studies indicate a clear pathway to cell death mediated by mutant SOD1 involving ER stress (Soo et al., 2012). Mutant SOD1 could also damage mitochondria directly (Figure 2). Mitochondria containing mutant SOD1G93A, but not wild-type SOD1, display changes in volume, aggregation, fragmentation and vacuolization (Higgins et al., 2003; Sasaki et al., 2004). Moreover, findings in human post-mortem or biopsy samples have reported abnormal mitochondria in cell bodies of motor neurons, proximal axons and in intramuscular nerves and skeletal muscle (Chung and Suh, 2002; Echaniz-Laguna et al., 2002; Sasaki and Iwata, 2007). Interestingly, such changes often precede disease onset in the mutant SOD1G93A transgenic mouse and occurs before any other signs of motor neuron degeneration (Kong and Xu, 1998). These early changes are often followed by a substantial increase in mitochondrial vacuolization at the time of symptom onset (Kong and Xu, 1998; Bendotti et al., 2001). Other studies have shown that mutant SODG37R binds directly to a voltage-dependent anion channel in the outer mitochondrial membrane and that this interaction inhibits channel conductance (Israelson et al., 2010). Moreover, deletion of this channel results in decreased lifespan of the G37R mouse. Together these and other studies strongly suggest a direct link between mitochondrial viability and motor neuron degeneration and are further supported by other studies demonstrating sequential activation of caspase-1 and 3, with caspase-1 activation occurring before the onset of symptoms and caspase-3 activation being associated with later motor neuron loss (see Table 1) (Pasinelli et al., 2000). Consistent with these findings, treatment with a broad caspase inhibitor delays onset and slows disease progression in this transgenic mouse (Li et al., 2000).
Table 1.
Mitochondrial dysfunction in ALS
| Mitochondria dysfunctions | Model | Gene mutation | Major findings | References |
|---|---|---|---|---|
| Morphology, fusion/fission and transport | Mouse primary motor neurons | FUS R521G R521H |
Shortened mitochondria in motor neurons expressing mutant FUS. | Sasaki et al., 2007 |
| Tg rats | FUS R521C |
Transgenic rats expressing mutant FUS showed ubiquitinated aggregates positive for the mitochondria marker COXIV | Huang et al., 2011 | |
| SH-SY5Y, NSC-34 | SOD1 G93A |
Mutant SOD1 increased mitochondria Opa1, decreased Drp1 | Ferri et al., 2010 | |
| Tg mice | SOD1 G93A |
Fusion proteins (Mfn1 and Opa1) and fission proteins (Drp1 and Fis1) increased. | Liu et al., 2013 | |
| Tg mice | SOD1 G93A |
Reduced density of mitochondria in motor axons and motor neurons in the triangularis sterni | Marinkovic et al., 2012 | |
| Tg mice | SOD1 G93A |
Decreased mitochondrial length and accumulation of fragmented mitochondria. Arrest in both anterograde and retrograde axonal transport and increased cell death | Song et al., 2013 | |
| Knockout mice | p62−/− (SQSTM1) | Fragmented mitochondria in p62−/− mouse embryonic fibroblast cells. | Lamar Seibenhener et al., 2013 | |
| Tg mice | wt-hTDP-43 | Accumulation of mitochondria in TDP-43-negative cytoplasmic inclusions in motor neurons, lack of mitochondria in motor axon terminals. | Shan et al., 2010 | |
| Tg mice | wt-hTDP-43 | Abnormal juxtanuclear aggregates of mitochondria accompanied by enhanced levels of Fis1 and phosphorylated DLP1; reduction in Mfn 1 expression | Xu et al., 2011 | |
| NSC-34 | TDP-43 Q331K, M337V |
Swollen mitochondria | Lu et al., 2012 | |
| Rat motor neurons Tg mice |
Wt-hTD-P43 Q331K M337V |
Overexpression of Wt and mutant TDP-43 resulted in reduced mitochondrial length and in neurites of primary motor neuron, suppression of TDP-43 resulted in significantly increased mitochondrial length and density in neuritis. Abnormal localization of TDP-43 in cytoplasm induced substantial and widespread abnormal mitochondrial dynamics. Co-expression of mitochondrial fusion protein mitofusin 2 (Mfn2) abolished TDP-43-induced mitochondrial dynamics abnormalities and mitochondrial dysfunction. | Wang et al., 2013 | |
| NSC-34 | Wt-hTDP-43 Q331K M337V C-terminal fragment |
Full length TDP-43 and its C-terminal fragment induced clustered and unevenly distributed mitochondria. | Hong et al., 2012 | |
| Human spine cord samples | NA | Accumulation of mitochondria in the somata, dendrites and proximal axons. | Sasaki et al., 2007 | |
| Rat cortical neurons | VAPB P56S |
VAPBP56S selectively disrupted anterograde axonal transport of mitochondria, disrupted Ca2+ homeostasis and effected the Miro1/kinesin-1 interaction with tubulin. | Morotz et al., 2012 | |
| Knock-in mice | VCP R155H |
Electron microscopic analysis of 19-month-old VCPR155H knock-in mouse muscle exhibited extensive accumulation of abnormal mitochondria and vacuoles in the inter-myofibrillar space. | Nalbandian et al., 2013 | |
| Complex activity, oxidative phosphorylation and redox | NSC-34 | SOD1 | Presence of mutSOD1s resulted in impairment of the respiratory chain and a shift in the mitochondrial redox balance. | Ferri et al., 2006 |
| Tg rats | SOD1 G93A |
Mutant SOD1 up-regulated in the IMS, increased ROS production from the spinal cord at the presymptomatic stage | Ahtoniemi et al., 2008 | |
| Tg mice | SOD1 G93A |
Deficiency in mitochondrial respiration, electron transfer chain, and ATP synthesis. | Mattiazzi et al., 2002 | |
| SH-SY5Y | SOD1 G93A, H80R |
Impaired ATP production | Pesaresi et al., 2011 | |
| Tg mice | SOD1-G93A/ PGC-1α | PGC-1α restores mitochondrial electron transport chain activities in the spinal cord in double transgenic (SOD1-G93A/PGC1-α) mice. | Zhao et al., 2011 | |
| Tg mice | SOD1 G93A |
Mitochondrial ETC activities are decreased in the ventral horn prior to the disease onset and during the course of disease progression | Jung et al., 2002 | |
| Tg rats | SOD1 G93A |
Defective respiratory function in astrocytes: decreased oxygen consumption, lack of ADP-dependent respiratory control and decreased membrane potential. | Cassina et al., 2008 | |
| Knockout mice | p62−/− (SQSTM1) | Mitochondria in p62 −/− cells displayed decreased mitochondrial membrane potential. Impairment in ATP production. | Lamar Seibenhener et al., 2013 | |
| SH-SY5Y | endogenous TDP-43 | Paraquat, an inhibitor of the mitochondrial electron transport chain and inducer of superoxide/peroxynitrite stress, induced cytoplasmic accumulation of TDP-43 including aggregates of TDP-43 resembling RNA stress granules. | Meyerowitz et al., 2011 | |
| NSC-34 | Wt-hTDP-43 Q331K M337V |
Wt-TDP-43 as well as mutant impairs ATP generation. | Hong et al., 2012 | |
| SH-SY5Y Human primary fibroblasts, neurons and astrocytes Mice primary cortical neurons |
VCP R155C R155H R191Q siRNA shRNA |
VCP deficiency is associated with decreased mitochondrial membrane potential, ATP levels and increased mitochondrial respiration and oxygen consumption. | Bartolome et al., 2013 | |
| Human muscle samples | Cytochrome c oxidase deficiency. | Crugnola et al., 2010 | ||
| Calcium buffering | Tg mice | SOD1 G93A, G85R |
Decreased mitochondrial Ca2+ loading capacity in brain and spinal cord prior to onset of motor symptoms. | Damiano et al., 2006 |
| Tg mice | SOD1 G93A G37R, G85R |
Deletion of cyclophilin D results in a chronic increase in mitochondrial buffering of Ca2+ and is associated with improved mitochondrial ATP synthesis, reduced mitochondrial swelling, and retention of normal morphology. | Parone et al., 2013 | |
| Tg mice hSOD1 |
SOD1 G93A/chat-GluR2 |
Cholinergic neuron-specific GluR2 overexpression resulted in reduction of Ca2+-permeable AMPA receptors in spinal motoneurons, delayed cytochrome c-release from mitochondria and reduced gliosis. | Tateno et al., 2004 | |
| Tg mice | SOD1 G93A |
Increased susceptibility to kainate-induced excitotoxicity. SOD1 G93A mutation causes changes in AMPA-receptor expression and function. | Spalloni et al., 2004 | |
| HEK293 | VAPB P56S |
VAPB interacts with the outer mitochondrial membrane protein PTPIP51. Loss of either VAPB or PTPIP51 impaired mitochondrial calcium homeostasis. | De Vos et al., 2012 | |
| Oxidative stress | Human ALS CSF samples | NA | Increase in 3-nitrotyrosine and 3-nitrotyrosine/ tyrosine ratio. | Tohgi et al., 1999 |
| Tg mice Knockout mice |
SOD1 G93A SOD1-/- |
Increased ROS production. | Goldstein et al., 2008 | |
| Tg mice | SOD1 G93A |
Enhanced oxidative damage of mitochondrial proteins. | Mattiazzi et al., 2002 | |
| Tg mice | SOD1 G93A |
Increased oxyradical production, sustained elevations of intracellular calcium levels, and mitochondrial dysfunction. | Kruman et al., 1999 | |
| NSC-34 | SOD1 G93A |
Depletion of mitochondrial glutathione | Muyderman et al., 2009 | |
| NSC-34 | Wt-hTDP-43 Q331K M337V |
Increased ROS production. | Hong et al., 2012 | |
| Yeast | TDP-43 Q337K |
Increased ROS production | Braun et al., 2011 | |
| Apoptosis | NSC-34 | SOD1 G85R, A4V |
Activated caspase 3, Bax and cytochrome c in cells bearing mutant SOD1 inclusions but not in cells expressing dispersed SOD1. | Soo et al., 2009 |
| HEK293, SH-SY5Y | SOD1 G93A | Mutant SOD1 induces mitochondrial morphological changes and compromises mitochondrial membrane integrity leading to release of cytochrome C only in the presence of Bcl-2. | Pedrini et al., 2010 | |
| NSC-34 Tg mice |
SOD1 G93A |
Induced Bcl2-A1 expression via the AP1 transcription factor in motor neuronal cells. Bcl2-A1 interacted with pro-caspase-3, via its C-terminal helix α9, preventing the completion of caspase-3 processing. | Iaccarino et al., 2011 | |
| Tg rats | SOD1 G93A |
Decrease in mitochondrial protein import. | Li et al., 2010 | |
| N2A | SOD1 G93A |
Both Wt and mutant SOD1 bind the anti-apoptotic protein Bcl-2. | Pasinelli et al., 2004 | |
| Tg mice, Tg rats | SOD1 G93A, H46R, G85R, G127X | Misfolded SOD1 deposited onto the cytoplasmic face of the outer mitochondrial membrane. | Vande et al., 2008 | |
| Tg mice G93A-SOD1/BAX-BAK deleted mice | SOD1 G93A |
Neuronal deletion of Bax and Bak halted neuronal loss, prevented axonal degeneration, delayed symptom onset, weight loss and extended survival. | Reyes et al., 2010 | |
| Tg mice | SOD1 G93A |
Bid cleavage at the time for disease onset. | Guegan et al., 2002 | |
| Tg mice | SOD1 G93A |
A broad caspase inhibitor zVAD-fmk delayed disease onset and mortality. | Li et al., 2000 | |
| Tg mice | Wt-TDP-43 | Caspase-3 activation. | Tsai et al., 2010 | |
| Human ALS spinal cord samples | Increased expression of Bax but no change in Bcl-2 and ICH-1 L expressions. Positive for TUNEL staining of motor neurons. | Ekegren et al., 1999 |
Figure 2.
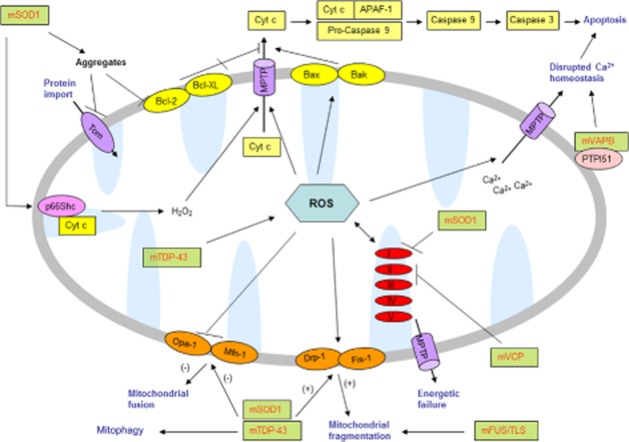
ALS-associated mutant proteins in mitochondrial dysfunction: mutant SOD1 (mSOD) aggregates at the outer membrane of mitochondria, inactivating the anti-apoptosis protein Bcl-2 (Pedrini et al., 2010) resulting in the release of cytochrome c possibly triggering mitochondrial intrinsic apoptosis (Pasinelli et al., 2000; 2004). mutant SOD1 also activates p66Shc at the intermembrane space, inducing cytochrome c oxidation and production of hydrogen peroxide (Pesaresi et al., 2011). mutant SOD1 association with mitochondria also results in other mitochondrial dysfunctions such as: aberrant morphology (Song et al., 2013), impair fusion/fission processes (Liu et al., 2013), impaired protein import (Li et al., 2010), impaired ETC complex activity affecting oxidative phosphorylation (Ferri et al., 2006), impaired calcium homeostasis (Parone et al., 2013) and increased oxidative stress (Mattiazzi et al., 2002; Goldsteins et al., 2008). Abnormal mitochondrial dynamics and fragmentation are also found in mutant TDP-43 (mTDP-43), FUS/TLS (mFUS/TLS) and VCP (mVCP) models (Sasaki et al., 2007; Huang et al., 2011; Xu et al., 2011; Nalbandian et al., 2013; Wang et al., 2013). Both mutant SOD1 and mutant TDP-43 are localized to mitochondria and may induce various degrees of mitochondrial dysfunction (Mattiazzi et al., 2002; Wang et al., 2013). Mutant VAPB (mVAPB) interacting with outer mitochondrial membrane protein PTPIP51 disrupts calcium buffering (De Vos et al., 2012).
Mutant SOD1, mitochondria and oxidative stress
Several recent studies link the presence of mutant SOD to an increased production of free radicals such as peroxynitrite, hydrogen peroxide and hydroxyl radicals (Table 1) (Crow et al., 1997; Beckman et al., 2001; Poon et al., 2005; Rizzardini et al., 2005). It has been suggested that the presence of mutations in SOD1 causes the dissociation of zinc from the enzyme thereby promoting an increase in mitochondrial superoxide production which results in downstream radical formation and oxidative damage (Estevez et al., 1999). Both wild-type and mutant SOD1 also directly interact with the superoxide producing enzyme NADPH oxidase via the RAc1 protein (Harraz et al., 2008; Polymenidou and Cleveland, 2008). However, mutant SOD1 has a higher affinity for RAc1 than wild-type SOD1 possibly resulting in chronic activation of NADPH oxidase. In support of this finding, a more than 10-fold increase in superoxide production has been reported in cells carrying mutant SOD1G93A compared with cells overexpressing the wild-type form of the protein (Harraz et al., 2008).
Cellular defence against oxidative stress involves SOD itself, catalase and glutathione-dependent processes. Glutathione is mainly localized to the cytoplasm but some is transported into the mitochondria and typically accounts for less than 15% of the total (Griffith and Meister, 1985; Meister, 1995; Fernandez-Checa et al., 1998). Glutathione acts by both directly detoxifying ROS and, acting as a substrate for many different peroxidases (Dringen, 2000; Sims et al., 2004; Sims and Muyderman, 2010). However, glutathione is also involved in other important functions in cells including the modification of exogenous molecules via the actions of glutathione S-transferases (Sheehan et al., 2001) and the reversible glutathionylation of proteins (Giustarini et al., 2004; Shelton et al., 2005). It is important to note that mitochondria lack catalase the emzyme that normally detoxifies hydrogen peroxide. Mitochondrial glutathione plays a major role in reactions catalyzed by glutathione peroxidase that are involved in the removal of mitochondrial peroxides. We have previously demonstrated the importance of mitochondrial glutathione in maintaining viability in several neural cell types and depletion of this antioxidant pool is directly associated with dysfunction and loss of viability in a range of cells challenged with oxidative stress (Sims et al., 2004; Muyderman et al., 2007).
Mitochondrial glutathione in ALS
Although the role of mitochondrial antioxidant defences involving glutathione has been rigorously investigated in other neurological conditions and in other cell types (Sims et al., 2004) the evidence for a role in ALS is mostly indirect. Experimentally decreased glutathione levels speed up disease progression in the mutant SOD1G93A transgenic (Vargas et al., 2011) and loss of total glutathione content in motor neurons has been reported in cell lines carrying the same mutation (Rizzardini et al., 2003). Moreover, reduced levels of oxidized glutathione have been reported in cerebrospinal fluid from ALS patients (Tohgi et al., 1999). In addition, we have recently shown that the total glutathione content in the mutant SOD1G93A expressing NSC-34 cell is no different from the parent NSC-34 cell. However, these cells had significantly lower levels of mitochondrial glutathione when this pool was measured separately, and this phenomenon was associated with an increased susceptibility to oxidative stress with subsequent apoptotic cell death (Muyderman et al., 2009). Therefore it is plausible to suggest that at the very least, the SOD1G93A mutation induces a low grade of mitochondrial oxidative stress resulting in the consumption of glutathione or the export of oxidised glutathione from the mitochondria thereby making them more susceptible to mitochondrial oxidative stress than their normal counterparts. This hypothesis is consistent with studies in which treatments protecting mitochondria from oxidative stress have shown a highly beneficial effect on the survival of both CNS astrocytes and motor neurons (Cassina et al., 2008). Moreover, overexpression of the mitochondrial antioxidant genes, MnSOD and GPX4, protect against mutant SOD1 in the NSC-34 cell line (Liu et al., 2002). These findings represent some of the most direct evidence to demonstrate the key role of the mitochondrial glutathione pool in preserving viability under pathological conditions. Interestingly, studies involving intraventricular glutathione delivery have been shown to restore mitochondrial glutathione levels after ischaemia and protect against ischaemic damage (Anderson et al., 2004). However, to our knowledge, no attempts have been made to explore this potentially effective therapeutic approach in ALS.
Electron transport chain deficiency in ALS
Mitochondria are responsible for oxidative metabolism. ATP production is dependent on the ETC where complex I (NADH dehydrogenase) is believed to be the rate limiting step (Pathak and Davey, 2008). Although evidence is sparse and many times contradictory, deficiencies in the activities of complex I and II-III (Browne et al., 1998) and complex IV (Menzies et al., 2002; Kirkinezos et al., 2005) have been reported from human tissue as well as from animal and cell culture models. In contrast, however a marked increase in complex I activity has been demonstrated in post mortem material (Bowling et al., 1993). Moreover, platelet mitochondria from ALS patients are able to restore fully ETC activity in Rho-zero cells who lack these organelles (Gajewski et al., 2003). On the other hand, spinal cord mitochondria isolated from transgenic mutant SOD1 mice show decrease in the activity of several complexes which is associated with decreased oxygen consumption and ATP production (Mattiazzi et al., 2002). Consistent with this observation, expression of mutant SOD1 in neuroblastoma-derived SH-SY5Y leads to loss of mitochondrial membrane potential (Carri et al., 1997). Moreover decreases in complex IV activity associated with a loss of cytochrome c have been reported in mitochondria isolated from presymptomatic transgenic mutant SOD1 mice (Kirkinezos et al., 2005).
Mitochondrial permeability transition pore in ALS
Involvement of the mitochondrial permeability transition pore has been implicated in mitochondrial dysfunction in several studies of ALS. Proteins responsible for pore formation are highly expressed in motor neurons and cyclophilin D (a matrix protein with peptidyl-prolyl cis-trans isomerase activity with a key role in the development of the transition pore) is found in high concentrations in swollen mitochondria in the SOD1 animal model. The role of permeability transition pore formation is further supported from studies in which silencing of cyclophilin D expression resulted in delayed disease onset and increased survival in the mutant SOD1 mouse. Interestingly, in an attempt to investigate the role of calcium overload, Parone et al. (2013) found that eliminating cyclophilin D in the mutant SOD1 mouse resulted in a significantly increased calcium mitochondrial buffering capacity and improved oxidative metabolism associated with normalised mitochondrial morphology and reduced mitochondrial swelling (Table 1). However, despite significantly reduced motor neuron death, both disease progression and lifespan were unaltered suggesting that other non-mitochondrial processes are the primary contributors to motor neuron cell death in this model (Parone et al., 2013). In contrast, earlier studies demonstrated a beneficial effect of interfering with the permeability transition in similar mutant SOD1 mouse models. For example, cyclosporin A, which inhibits transition pore opening by binding to cyclophilin D, slowed disease progression and increased lifespan when administered to SOD1 transgenic mice (Karlsson et al., 2004). In another study, it was shown that FK506 that mimics some effects of cyclosporin but lack effects on the permeability transition, did not affect survival in the same transgenic mouse model (Anneser et al., 2001). Together these studies strongly indicate a role for the permeability transition pore in ALS; however, it should be noted that the effects on lifespan were modest and that motor neuron death eventually occurred in spite of the absence of transition pore formation. Hence, other mechanisms leading to mitochondrial dysfunction and the development of cell death must occur in parallel.
It has been suggested that oxidative stress or calcium overload are triggers for transition pore opening in ALS (Martin et al., 2009). As pore formation is sometimes associated with necrosis-like cell death, it is possible that the permeability transition is secondary to, or results from, a decline in energy metabolism, or alternatively that transient induction of the pore initiates changes leading to a more slowly developing programmed form of necrosis. A likely explanation for this observation could be that the final development of caspase-dependent apoptosis is overwhelmed by other molecular changes that result in these alternative forms of cell death, a view that would be consistent with findings of low ATP/ADP ratio and impairment of the ETC activity reported in cells from ALS patients.
New ALS mutations – new views
The rapid advances in the understanding of ALS pathology over the last decade have considerably changed our view of the pathogenesis of this disease. It is now believed that impaired RNA metabolism produce similar, if not identical, disease phenotypes as mutations in the SOD1 gene. This raises the question whether as mitochondrial dysfunction is exclusive to mutant SOD1 toxicity or if it can be applied to newly discovered forms of fALS. Even though the evidence is still indirect, several findings point in the direction of impaired mitochondrial function, at least in TDP-43 pathology, playing a central role to the overall pathogenesis of ALS (see Figure 2: Table 1).
TDP-43 is a highly conserved 43-kDa DNA-and RNA-binding protein regulating transcription and splicing (Bose et al., 2008). The protein is abundantly expressed in neurons and glia. TDP-43 predominantly localizes in nuclei, but is also found in cytosolic stress granules where it is believed to participate in local post-transcriptional modifications of mRNA and translational control (Buratti and Baralle, 2008). TDP-43 has a promiscuous protein interaction pattern with more than 200 targets reported, suggesting an involvement in a vast array of intracellular events (Freibaum et al., 2010). The physiological role of TDP-43 is, to a large extent, unknown although it has been suggested to act as a neuronal response factor and to play a prominent role in the stability of neurofilament mRNA and in microRNA biogenesis (Wang et al., 2008; Volkening et al., 2009). Abnormal processing and aggregation of TDP-43 is a characteristic of so called TDP-43 proteinopathies (Kwong et al., 2007). In these disorders, TDP-43 is relocated from the nuclei to the cytoplasm and sequestered into inclusions mainly composed of ubiquitinated and phosphorylated C-terminally truncated fragments (Neumann et al., 2006; Geser et al., 2008). Abnormal molecular weight TDP-43 fragments have been observed in neurons and astrocytes in patients from a spectrum of neurodegenerative diseases including 95% of familial and sporadic ALS (Mackenzie et al., 2007; Liscic et al., 2008) making it an interesting candidate for all forms of the disease.
It is currently not known if mutations in TDP-43 affect post-transcriptional modifications of gene products that control or modify mitochondrial function or if mitochondrial dysfunction in TDP-43 proteinopathies result from direct toxic effects by truncated and aggregated TDP-43. Nevertheless, transgenic animals display mitochondrial changes that resemble some of those found in human ALS. In non-vertebrate models, mutated TDP-43 aggregates around mitochondria and has been shown to cause negative effects on oxidative metabolism (Braun et al., 2011). Moreover, transgenic animals expressing mutant TDP-43 exhibit changes in mitochondrial trafficking, mitochondrial clustering, vacuolization and the expression of proteins responsible for mitochondrial fusion and fission (Figure 2: Table 1) (Shan et al., 2010; Xu et al., 2010; Wang et al., 2013). Interestingly, it seems that the presence of mutant TDP-43 may not be the only mechanism by which mitochondrial damage is induced in TDP-43 pathology. In a recent study, Hong et al. (2012) demonstrated that overexpression of wild-type TDP-43 results in mitochondrial damage and that this is associated with activation of mitophagy. In this study, both truncated and full-length TDP-43 was localized to the mitochondria thereby supporting the view of a direct toxic effect on these organelles, possibly mediated by impaired autoregulation of TDP-43 protein expression. These findings are important, as impaired TDP-43 homeostasis is likely to occur in sporadic occurring cases of ALS in which no genetic cause of the pathology can be found.
Mutations in the RNA binding protein FUS have also recently been linked to the pathogenesis of ALS although FUS is involved in different aspects of RNA metabolism than that of TDP-43. In FUS proteinopathies, the protein is believed to translocate to the cytoplasm causing ER stress with possible downstream effects on mitochondrial function (Farg et al., 2013). Little is known about how mutations in FUS affect mitochondrial function, however, transgenic rats overexpressing mutant FUS display motor neuron death with features of mitochondrial dysfunction (Huang et al., 2011). The introduction of a FUS mutation in murine spinal cord motor neuron cultures resulted in cell death and changes in mitochondrial morphology similar to those reported in the SOD1 animal model (Tradewell et al., 2012). Moreover, indirect evidence of mitochondrial dysfunction has been reported in post mortem tissue from one patient suffering from juvenile ALS with mutations in FUS as the only pathogenic factor (Huang et al., 2010). The recent advances in identifying new genes responsible for the pathogenesis in ALS suggest that genes encoding for proteins linked to rare cases of ALS such as UBQLN2, SQTM1/p62, VAPB and VCP, may all result in a common pathogenic pathway involving mitochondrial dysfunction (see Figure 2). Interactions between these proteins and those of the more common proteinopathies in ALS need to be investigated further.
Together these data are all indicative of mitochondrial dysfunction in non-SOD1 ALS but further studies are required to provide enough data to make pharmacological interventions possible. Thus it seems plausible to conclude that altered RNA metabolism has the potential to disrupt normal mitochondrial function.
Targeting mitochondria in the treatment of ALS
Rilazole is currently the only approved drug for the treatment of ALS but the compound only exerts modest effects on survival and is without effect on motor function (Bensimon et al., 1994; Hugon, 1996). Overall, the mean increase in survival is approximately 3 months (Miller et al., 2012). Rilazole has many potential targets but is believed to exert its main function by reducing presynaptic glutamate release and thereby reducing excitotoxicity. There have been numerous clinical trials over the last decades targeting mechanisms predominantly identified in SOD1 transgenic animals including protein clearing agents such as lithium (Miller et al., 2011); analeptic drugs such as topiramate which reduces extracellular glutamate (Cudkowicz et al., 2003); anti-inflammatory drugs primarily targeting glial activation such as minocycline (Gordon et al., 2007) and the broader 5-HT1A receptor agonist, xaliproden, a drug with proven neurotrophic and neuroprotective activities (Meininger et al., 2004). Unfortunately, these single drug approaches have proven ineffective.
There are many problems associated with identifying a single pharmacological target in ALS. Firstly, the ethology of the disease is still, to a large extent, unknown and the disease is relatively rare making large clinical trials difficult. The complex pathogenesis of ALS raises two possible explanations on why previous pharmacological approaches have failed when translated to humans: (i) findings from the SOD1 models may not apply to all cases of ALS, especially considering that 90% of patients present with TDP-43 pathology which is absent in patients with SOD1 mutations. This observation highlights the need for studies involving newly discovered ALS-causing mutations such as those of TDP-43, FUS and the C9orf72 hexanucleotide repeat expansion. (ii) The multiple pathogenic processes demonstrated in ALS support the view that multiple pathways are converging to a common endpoint resulting in motor neuron loss. This may also explain the disappointing results of targeting a single pathological process such as caspase inhibitors. As neuronal loss in ALS is most likely to reflect combinations of multiple pathogenic mechanisms including glutamate excitoxicity, oxidative stress, mitochondrial dysfunction and disruption of axonal transport processes, a multidrug approach is most likely to be needed. In addition, activation of glial cells results in secretion of pro-inflammatory cytokines resulting in further toxicity and glial cells expressing ALS-linked mutations are known to have detrimental effects on surrounding motor neurons highlighting the need for glia-directed pharmacological interventions.
In developing a multidrug approach, individual targets must be identified. Considering the vast amount of evidence of mitochondrial dysfunction in both the SOD1 model and in models of the later discovered genetic mutations, maintaining mitochondrial function is an appealing part of such an approach. There have been several studies addressing the role of pharmacological interventions targeted at mitochondrial function although the outcomes have been limited. One such drug is pramipexole (PPX). PPX is a dopamine analogue believed to exert neuroprotective effects through enhancing mitochondrial function and inhibiting activation of mitochondrial linked apoptotic pathways at the same time counteracting glutamate-mediated excitotoxicity (Alavian et al., 2012). PPX also scavenges reactive oxygen species and lowers free radical levels in ALS patients (Pattee et al., 2003). Dexpramipexole is the R(+) enantiomer of PPX and is tolerated in higher doses than PPX and both have shown promise in early clinical studies (Cudkowicz et al., 2011). Dexpramipexole has similar neuroprotective properties to that of PPX but lacks dopaminergic effects. Most likely, both PPX and Dexpramipexole, exert part of their neuroprotective effects though direct effects on mitochondria by stabilizing the proton gradients needed for ATP production. Dexpramipexole was recently shown to decrease mortality in a Phase 2 study in subjects with ALS (Rudnicki et al., 2013) making this an interesting candidate in a multidrug approach for the treatment of ALS.
Melatonin is another compound that recently gained interest by its mitochondria stabilizing effects in ALS. In the mutant SOD1G93A model, melatonin decreases cytochrome c release, caspase-3 activation and delays disease progression (Zhang et al., 2013). Moreover, treatment with melatonin also resulted in a reduction in the activation of astrocytes and microglia indicating an attenuated inflammatory response. Moreover, treatment with SOD1-like peptides has been shown to restore mitochondrial function in mutant SOD1G93A mice (Tan et al., 2013).
A possible drug target associated with mitochondrial function is the transcriptional co-activator PPARγ co-activator-1α (PGC-1α). PCG-1α is a regulator of mitochondrial oxidative metabolism and is activated by several converging pathways including cAMP and cytokines (Puigserver and Spiegelman, 2003) and reduced PCG-1α mRNA levels have been described in ALS patients (Thau et al., 2012). Overexpressing PCG-1α in the SOD1G93A model improves motor function and prolongs survival possibly by restoring close to normal ETC activity (Zhao et al., 2011). Interestingly, PCG-1α/G93A animals also show an increased expression of astrocytic GLT-1 glutamate transporters that may have been the ultimate cause of neuroprotection seen in this model. Whether this is a primary or secondary event is not known and the relation to enhanced mitochondrial function is unclear. In the context of ETC activity, treatment with creatine has been shown to enhance mitochondrial activity and slow disease progression in the G93A SOD1 model (Klivenyi et al., 2004); however, clinical trials have so far been disappointing (Shefner et al., 2004). Although many of these studies have shown potential to increase lifespan and delay disease onset, the effects are in general modest and highlights the need for larger multidrug studies in non-SOD1 models of the disease.
Concluding remarks
Evidence of mitochondrial dysfunction in ALS has been reported for more than 50 years and is today recognized as central in the disease process. However, it is still unclear if changes in mitochondrial biology is a primary event in the pathology or if it occurs secondary to other cellular processes resulting in oxidative stress and activation of apoptotic pathways. Nevertheless, the extent of preservation of key mitochondrial properties and the ability of these organelles to mount appropriate defensive responses must still be regarded as important determinants of tissue viability in ALS and hence is a strong indicator for mitochondrial-directed pharmacological interventions. Based on the current evidence of multifaceted mitochondrial dysfunction reported in human ALS and in most rodent animal models, it seems obvious that pharmacological interventions targeting mitochondrial function are essential for future treatment strategies in ALS Interventions that restore, or promote mitochondrial function seem a plausible approach, however, based on the data reviewed above it is unlikely that ALS is solely a mitochondrial disease and that any pharmacological intervention may have to have multiple targets, possibly including those in non-neuronal cells types as astrocytes and microglia. The time of a single drug approach for treatment of ALS is gone but mitochondria are still a valid target.
Conflict of interests
None.
Glossary
- AIF
apoptosis-inducing factor
- ALS
amyotrophic lateral sclerosis
- ETC
electron transport chain
- fALS
familial ALS
- PGC-1α
PPARγ co-activator 1α
- PPX
pramipexole
- SOD1
superoxide dismutase
References
- Ahtoniemi T, Jaronen M, Keksa-Goldsteine V, Goldsteins G, Koistinaho J. Mutant SOD1 from spinal cord of G93A rats is destabilized and binds to inner mitochondrial membrane. Neurobiol Dis. 2008;32:479–485. doi: 10.1016/j.nbd.2008.08.010. [DOI] [PubMed] [Google Scholar]
- Alavian KN, Dworetzky SI, Bonanni L, Zhang P, Sacchetti S, Mariggio MA, et al. Effects of dexpramipexole on brain mitochondrial conductances and cellular bioenergetic efficiency. Brain Res. 2012;1446:1–11. doi: 10.1016/j.brainres.2012.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MF, Nilsson M, Eriksson PS, Sims NR. Glutathione monoethyl ester provides neuroprotection in a rat model of stroke. Neurosci Lett. 2004;354:163–165. doi: 10.1016/j.neulet.2003.09.067. [DOI] [PubMed] [Google Scholar]
- Anneser JM, Gmerek A, Gerkrath J Borasio GD, Heumann R. Immunosuppressant FK506 does not exert beneficial effects in symptomatic G93A superoxide dismutase-1 transgenic mice. Neuroreport. 2001;12:2663–2665. doi: 10.1097/00001756-200108280-00015. [DOI] [PubMed] [Google Scholar]
- Bartolome F, Wu HC, Burchell VS, Preza E, Wray S, Mahoney CJ, et al. Pathogenic VCP mutations induce mitochondrial uncoupling and reduced ATP levels. Neuron. 2013;78:57–64. doi: 10.1016/j.neuron.2013.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF. Oxidatively modified proteins in aging and disease. Free Radic Biol Med. 2002;32:797–803. doi: 10.1016/s0891-5849(02)00780-3. [DOI] [PubMed] [Google Scholar]
- Beal MF, Ferrante RJ, Browne SE, Matthews RT, Kowall NW, Brown RH., Jr Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann Neurol. 1997;42:644–654. doi: 10.1002/ana.410420416. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Estevez AG, Crow JP, Barbeito L. Superoxide dismutase and the death of motoneurons in ALS. Trends Neurosci. 2001;24:S15–S20. doi: 10.1016/s0166-2236(00)01981-0. [DOI] [PubMed] [Google Scholar]
- Bendotti C, Calvaresi N, Chiveri L, Prelle A, Moggio M, Braga M, et al. Early vacuolization and mitochondrial damage in motor neurons of FALS mice are not associated with apoptosis or with changes in cytochrome oxidase histochemical reactivity. J Neurol Sci. 2001;191:25–33. doi: 10.1016/s0022-510x(01)00627-x. [DOI] [PubMed] [Google Scholar]
- Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330:585–591. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- Bose JK, Wang IF, Hung L, Tarn WY, Shen CK. TDP-43 overexpression enhances exon 7 inclusion during the survival of motor neuron pre-mRNA splicing. J Biol Chem. 2008;283:28852–28859. doi: 10.1074/jbc.M805376200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowling AC, Schulz JB, Brown RH, Jr, Beal MF. Superoxide dismutase activity, oxidative damage, and mitochondrial energy metabolism in familial and sporadic amyotrophic lateral sclerosis. J Neurochem. 1993;61:2322–2325. doi: 10.1111/j.1471-4159.1993.tb07478.x. [DOI] [PubMed] [Google Scholar]
- Braun RJ, Sommer C, Carmona-Gutierrez D, Khoury CM, Ring J, Buttner S, et al. Neurotoxic 43-kDa TAR DNA-binding protein (TDP-43) triggers mitochondrion-dependent programmed cell death in yeast. J Biol Chem. 2011;286:19958–19972. doi: 10.1074/jbc.M110.194852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner C, Moulin M. Physiological roles of the permeability transition pore. Circ Res. 2012;111:1237–1247. doi: 10.1161/CIRCRESAHA.112.265942. [DOI] [PubMed] [Google Scholar]
- Browne SE, Bowling AC, Baik MJ, Gurney M, Brown RH, Jr, Beal MF. Metabolic dysfunction in familial, but not sporadic, amyotrophic lateral sclerosis. J Neurochem. 1998;71:281–287. doi: 10.1046/j.1471-4159.1998.71010281.x. [DOI] [PubMed] [Google Scholar]
- Buratti E, Baralle FE. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci. 2008;13:867–878. doi: 10.2741/2727. [DOI] [PubMed] [Google Scholar]
- Carri MT, Ferri A, Battistoni A, Famhy L, Gabbianelli R, Poccia F, et al. Expression of a Cu,Zn superoxide dismutase typical of familial amyotrophic lateral sclerosis induces mitochondrial alteration and increase of cytosolic Ca2+ concentration in transfected neuroblastoma SH-SY5Y cells. FEBS Lett. 1997;414:365–368. doi: 10.1016/s0014-5793(97)01051-x. [DOI] [PubMed] [Google Scholar]
- Carriedo SG, Sensi SL, Yin HZ, Weiss JH. AMPA exposures induce mitochondrial Ca(2+) overload and ROS generation in spinal motor neurons in vitro. J Neurosci. 2000;20:240–250. doi: 10.1523/JNEUROSCI.20-01-00240.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassina P, Cassina A, Pehar M, Castellanos R, Gandelman M, de Leon A, et al. Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J Neurosci. 2008;28:4115–4122. doi: 10.1523/JNEUROSCI.5308-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu AY, Zhai P, Dal Canto MC, Peters TM, Kwon YW, Prattis SM, et al. Age-dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis. Mol Cell Neurosci. 1995;6:349–362. doi: 10.1006/mcne.1995.1027. [DOI] [PubMed] [Google Scholar]
- Chung MJ, Suh YL. Ultrastructural changes of mitochondria in the skeletal muscle of patients with amyotrophic lateral sclerosis. Ultrastruct Pathol. 2002;26:3–7. doi: 10.1080/01913120252934260. [DOI] [PubMed] [Google Scholar]
- Crow JP, Sampson JB, Zhuang Y, Thompson JA, Beckman JS. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J Neurochem. 1997;69:1936–1944. doi: 10.1046/j.1471-4159.1997.69051936.x. [DOI] [PubMed] [Google Scholar]
- Crugnola V, Lamperti C, Lucchini V, Ronchi D, Peverelli L, Prelle A, et al. Mitochondrial respiratory chain dysfunction in muscle from patients with amyotrophic lateral sclerosis. Arch Neurol. 2010;67:849–854. doi: 10.1001/archneurol.2010.128. [DOI] [PubMed] [Google Scholar]
- Cudkowicz M, Bozik ME, Ingersoll EW, Miller R, Mitsumoto H, Shefner J, et al. The effects of dexpramipexole (KNS-760704) in individuals with amyotrophic lateral sclerosis. Nat Med. 2011;17:1652–1656. doi: 10.1038/nm.2579. [DOI] [PubMed] [Google Scholar]
- Cudkowicz ME, Shefner JM, Schoenfeld DA, Brown RH, Jr, Johnson H, Qureshi M, et al. A randomized, placebo-controlled trial of topiramate in amyotrophic lateral sclerosis. Neurology. 2003;61:456–464. doi: 10.1212/wnl.61.4.456. [DOI] [PubMed] [Google Scholar]
- Damiano M, Starkov AA, Petri S, Kipiani K, Kiaei M, Mattiazzi M, et al. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J Neurochem. 2006;96:1349–1361. doi: 10.1111/j.1471-4159.2006.03619.x. [DOI] [PubMed] [Google Scholar]
- De Vos KJ, Morotz GM, Stoica R, Tudor EL, Lau KF, Ackerley S, et al. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet. 2012;21:1299–1311. doi: 10.1093/hmg/ddr559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–215. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- Echaniz-Laguna A, Zoll J, Ribera F, Tranchant C, Warter JM, Lonsdorfer J, et al. Mitochondrial respiratory chain function in skeletal muscle of ALS patients. Ann Neurol. 2002;52:623–627. doi: 10.1002/ana.10357. [DOI] [PubMed] [Google Scholar]
- Ekegren T, Grundstrom E, Lindholm D, Aquilonius SM. Upregulation of Bax protein and increased DNA degradation in ALS spinal cord motor neurons. Acta Neurol Scand. 1999;100:317–321. doi: 10.1111/j.1600-0404.1999.tb00403.x. [DOI] [PubMed] [Google Scholar]
- Estevez AG, Crow JP, Sampson JB, Reiter C, Zhuang Y, Richardson GJ, et al. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science. 1999;286:2498–2500. doi: 10.1126/science.286.5449.2498. [DOI] [PubMed] [Google Scholar]
- Farg MA, Soo KY, Warraich ST, Sundaramoorthy V, Blair IP, Atkin JD. Ataxin-2 interacts with FUS and intermediate-length polyglutamine expansions enhance FUS-related pathology in amyotrophic lateral sclerosis. Hum Mol Genet. 2013;22:717–728. doi: 10.1093/hmg/dds479. [DOI] [PubMed] [Google Scholar]
- Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68:1440–1446. doi: 10.1001/archneurol.2011.250. [DOI] [PubMed] [Google Scholar]
- Fernandez-Checa JC, Kaplowitz N, Garcia-Ruiz C, Colell A. Mitochondrial glutathione: importance and transport. Semin Liver Dis. 1998;18:389–401. doi: 10.1055/s-2007-1007172. [DOI] [PubMed] [Google Scholar]
- Ferri A, Cozzolino M, Crosio C, Nencini M, Casciati A, Gralla EB, et al. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc Natl Acad Sci U S A. 2006;103:13860–13865. doi: 10.1073/pnas.0605814103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri A, Fiorenzo P, Nencini M, Cozzolino M, Pesaresi MG, Valle C, et al. Glutaredoxin 2 prevents aggregation of mutant SOD1 in mitochondria and abolishes its toxicity. Hum Mol Genet. 2010;19:4529–4542. doi: 10.1093/hmg/ddq383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibaum BD, Chitta RK, High AA, Taylor JP. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res. 2010;9:1104–1120. doi: 10.1021/pr901076y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski CD, Lin MT, Cudkowicz ME, Beal MF, Manfredi G. Mitochondrial DNA from platelets of sporadic ALS patients restores normal respiratory functions in rho(0) cells. Exp Neurol. 2003;179:229–235. doi: 10.1016/s0014-4886(02)00022-5. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Morselli E, Kepp O, Kroemer G. Targeting post-mitochondrial effectors of apoptosis for neuroprotection. Biochim Biophys Acta. 2009;1787:402–413. doi: 10.1016/j.bbabio.2008.09.006. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Kroemer G. Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol. 2012;13:780–788. doi: 10.1038/nrm3479. [DOI] [PubMed] [Google Scholar]
- Geser F, Winton MJ, Kwong LK, Xu Y, Xie SX, Igaz LM, et al. Pathological TDP-43 in parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam. Acta Neuropathol. 2008;115:133–145. doi: 10.1007/s00401-007-0257-y. [DOI] [PubMed] [Google Scholar]
- Giustarini D, Rossi R, Milzani A, Colombo R, Dalle-Donne I. S-glutathionylation: from redox regulation of protein functions to human diseases. J Cell Mol Med. 2004;8:201–212. doi: 10.1111/j.1582-4934.2004.tb00275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsteins G, Keksa-Goldsteine V, Ahtoniemi T, Jaronen M, Arens E, Akerman K, et al. Deleterious role of superoxide dismutase in the mitochondrial intermembrane space. J Biol Chem. 2008;283:8446–8452. doi: 10.1074/jbc.M706111200. [DOI] [PubMed] [Google Scholar]
- Gordon PH, Moore DH, Miller RG, Florence JM, Verheijde JL, Doorish C, et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol. 2007;6:1045–1053. doi: 10.1016/S1474-4422(07)70270-3. [DOI] [PubMed] [Google Scholar]
- Griffith OW, Meister A. Origin and turnover of mitochondrial glutathione. Proc Natl Acad Sci U S A. 1985;82:4668–4672. doi: 10.1073/pnas.82.14.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guegan C, Vila M, Teismann P, Chen C, Onteniente B, Li M, et al. Instrumental activation of bid by caspase-1 in a transgenic mouse model of ALS. Mol Cell Neurosci. 2002;20:553–562. doi: 10.1006/mcne.2002.1136. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Harraz MM, Marden JJ, Zhou W, Zhang Y, Williams A, Sharov VS, et al. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest. 2008;118:659–670. doi: 10.1172/JCI34060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Thielen P, Fisher J, Pasinelli P, Brown RH, Korsmeyer S, et al. The proapoptotic BCL-2 family member BIM mediates motoneuron loss in a model of amyotrophic lateral sclerosis. Cell Death Differ. 2007;14:1386–1389. doi: 10.1038/sj.cdd.4402166. [DOI] [PubMed] [Google Scholar]
- Higgins CM, Jung C, Xu Z. ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci. 2003;4:16. doi: 10.1186/1471-2202-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong K, Li Y, Duan W, Guo Y, Jiang H, Li W, et al. Full-length TDP-43 and its C-terminal fragments activate mitophagy in NSC34 cell line. Neurosci Lett. 2012;530:144–149. doi: 10.1016/j.neulet.2012.10.003. [DOI] [PubMed] [Google Scholar]
- Huang C, Zhou H, Tong J, Chen H, Liu YJ, Wang D, et al. FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet. 2011;7:e1002011. doi: 10.1371/journal.pgen.1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Zhang J, Geser F, Trojanowski JQ, Strober JB, Dickson DW, et al. Extensive FUS-immunoreactive pathology in juvenile amyotrophic lateral sclerosis with basophilic inclusions. Brain Pathol. 2010;20:1069–1076. doi: 10.1111/j.1750-3639.2010.00413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugon J. Riluzole and ALS therapy. Wien Med Wochenschr. 1996;146:185–187. [PubMed] [Google Scholar]
- Iaccarino C, Mura ME, Esposito S, Carta F, Sanna G, Turrini F, et al. Bcl2-A1 interacts with pro-caspase-3: implications for amyotrophic lateral sclerosis. Neurobiol Dis. 2011;43:642–650. doi: 10.1016/j.nbd.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Israelson A, Arbel N, Da CS, Ilieva H, Yamanaka K, Shoshan-Barmatz V, et al. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron. 2010;67:575–587. doi: 10.1016/j.neuron.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68:857–864. doi: 10.1016/j.neuron.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan J, Cena V, Prehn JH. Mitochondrial control of neuron death and its role in neurodegenerative disorders. J Physiol Biochem. 2003;59:129–141. doi: 10.1007/BF03179878. [DOI] [PubMed] [Google Scholar]
- Julien JP, Kriz J. Transgenic mouse models of amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762:1013–1024. doi: 10.1016/j.bbadis.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Jung C, Higgins CM, Xu Z. Mitochondrial electron transport chain complex dysfunction in a transgenic mouse model for amyotrophic lateral sclerosis. J Neurochem. 2002;83:535–545. doi: 10.1046/j.1471-4159.2002.01112.x. [DOI] [PubMed] [Google Scholar]
- Kantari C, Walczak H. Caspase-8 and bid: caught in the act between death receptors and mitochondria. Biochim Biophys Acta. 2011;1813:558–563. doi: 10.1016/j.bbamcr.2011.01.026. [DOI] [PubMed] [Google Scholar]
- Karlsson H, Fong KS, Hansson MJ, Elmer E, Csiszar K, Keep MF. Life span extension and reduced neuronal death after weekly intraventricular cyclosporin injections in the G93A transgenic mouse model of amyotrophic lateral sclerosis. J Neurosurg. 2004;101:128–137. doi: 10.3171/jns.2004.101.1.0128. [DOI] [PubMed] [Google Scholar]
- Keep M, Elmer E, Fong KS, Csiszar K. Intrathecal cyclosporin prolongs survival of late-stage ALS mice. Brain Res. 2001;894:327–331. doi: 10.1016/s0006-8993(01)02012-1. [DOI] [PubMed] [Google Scholar]
- Kirkinezos IG, Bacman SR, Hernandez D, Oca-Cossio J, Arias LJ, Perez-Pinzon MA, et al. Cytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A-SOD1 mice. J Neurosci. 2005;25:164–172. doi: 10.1523/JNEUROSCI.3829-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klivenyi P, Kiaei M, Gardian G, Calingasan NY, Beal MF. Additive neuroprotective effects of creatine and cyclooxygenase 2 inhibitors in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurochem. 2004;88:576–582. doi: 10.1046/j.1471-4159.2003.02160.x. [DOI] [PubMed] [Google Scholar]
- Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G. Pathophysiological implications of mithochondrial cell death control. Bull Mem Acad R Med Belg. 2010;165:205–210. [PubMed] [Google Scholar]
- Kroemer G, Zamzami N, Susin SA. Mitochondrial control of apoptosis. Immunol Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- Kruman II, Pedersen WA, Springer JE, Mattson MP. ALS-linked Cu/Zn-SOD mutation increases vulnerability of motor neurons to excitotoxicity by a mechanism involving increased oxidative stress and perturbed calcium homeostasis. Exp Neurol. 1999;160:28–39. doi: 10.1006/exnr.1999.7190. [DOI] [PubMed] [Google Scholar]
- Kwong LK, Neumann M, Sampathu DM, Lee VM, Trojanowski JQ. TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol. 2007;114:63–70. doi: 10.1007/s00401-007-0226-5. [DOI] [PubMed] [Google Scholar]
- Lansbury PT, Lashuel HA. A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature. 2006;443:774–779. doi: 10.1038/nature05290. [DOI] [PubMed] [Google Scholar]
- Li M, Ona VO, Guegan C, Chen M, Jackson-Lewis V, Andrews LJ, et al. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science. 2000;288:335–339. doi: 10.1126/science.288.5464.335. [DOI] [PubMed] [Google Scholar]
- Li Q, Vande VC, Israelson A, Xie J, Bailey AO, Dong MQ, et al. ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc Natl Acad Sci U S A. 2010;107:21146–21151. doi: 10.1073/pnas.1014862107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liscic RM, Grinberg LT, Zidar J, Gitcho MA, Cairns NJ. ALS and FTLD: two faces of TDP-43 proteinopathy. Eur J Neurol. 2008;15:772–780. doi: 10.1111/j.1468-1331.2008.02195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Li B, Flanagan SW, Oberley LW, Gozal D, Qiu M. Increased mitochondrial antioxidative activity or decreased oxygen free radical propagation prevent mutant SOD1-mediated motor neuron cell death and increase amyotrophic lateral sclerosis-like transgenic mouse survival. J Neurochem. 2002;80:488–500. doi: 10.1046/j.0022-3042.2001.00720.x. [DOI] [PubMed] [Google Scholar]
- Liu W, Tian F, Morimoto N, Yamashita T, Ikeda Y, Deguchi K, et al. Dynamic changes of mitochondrial fusion and fission proteins in a murine model of amyotrophic lateral sclerosis. Curr Neurovasc Res. 2013;1:5–17. doi: 10.2174/15672026113109990060. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61:427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- Marinkovic P, Reuter MS, Brill MS, Godinho L, Kerschensteiner M, Misgeld T. Axonal transport deficits and degeneration can evolve independently in mouse models of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2012;109:4296–4301. doi: 10.1073/pnas.1200658109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ. Mitochondrial pathobiology in ALS. J Bioenerg Biomembr. 2011;43:569–579. doi: 10.1007/s10863-011-9395-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Gertz B, Pan Y, Price AC, Molkentin JD, Chang Q. The mitochondrial permeability transition pore in motor neurons: involvement in the pathobiology of ALS mice. Exp Neurol. 2009;218:333–346. doi: 10.1016/j.expneurol.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiazzi M, D'Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, et al. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277:29626–29633. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- Meininger V, Bensimon G, Bradley WR, Brooks B, Douillet P, Eisen AA, et al. Efficacy and safety of xaliproden in amyotrophic lateral sclerosis: results of two phase III trials. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5:107–117. doi: 10.1080/14660820410019602. [DOI] [PubMed] [Google Scholar]
- Meister A. Mitochondrial changes associated with glutathione deficiency. Biochim Biophys Acta. 1995;1271:35–42. doi: 10.1016/0925-4439(95)00007-q. [DOI] [PubMed] [Google Scholar]
- Menzies FM, Cookson MR, Taylor RW, Turnbull DM, Chrzanowska-Lightowlers ZM, Dong L, et al. Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain. 2002;125:1522–1533. doi: 10.1093/brain/awf167. [DOI] [PubMed] [Google Scholar]
- Meyerowitz J, Parker SJ, Vella LJ, Ng DC, Price KA, Liddell JR, et al. C-Jun N-terminal kinase controls TDP-43 accumulation in stress granules induced by oxidative stress. Mol Neurodegener. 2011;6:57. doi: 10.1186/1750-1326-6-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels J, Kepp O, Senovilla L, Lissa D, Castedo M, Kroemer G, et al. Functions of BCL-X L at the Interface between Cell Death and Metabolism. Int J Cell Biol. 2013;2013:705294. doi: 10.1155/2013/705294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RG, Moore DH, Forshew DA, Katz JS, Barohn RJ, Valan M, et al. Phase II screening trial of lithium carbonate in amyotrophic lateral sclerosis: examining a more efficient trial design. Neurology. 2011;77:973–979. doi: 10.1212/WNL.0b013e31822dc7a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND) Cochrane Database Syst Rev. 2012;(3) doi: 10.1002/14651858.CD001447.pub3. CD001447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morotz GM, De Vos KJ, Vagnoni A, Ackerley S, Shaw CE, Miller CC. Amyotrophic lateral sclerosis-associated mutant VAPBP56S perturbs calcium homeostasis to disrupt axonal transport of mitochondria. Hum Mol Genet. 2012;21:1979–1988. doi: 10.1093/hmg/dds011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muyderman H, Wadey AL, Nilsson M, Sims NR. Mitochondrial glutathione protects against cell death induced by oxidative and nitrative stress in astrocytes. J Neurochem. 2007;102:1369–1382. doi: 10.1111/j.1471-4159.2007.04641.x. [DOI] [PubMed] [Google Scholar]
- Muyderman H, Hutson PG, Matusica D, Rogers ML, Rush RA. The human G93A-superoxide dismutase-1 mutation, mitochondrial glutathione and apoptotic cell death. Neurochem Res. 2009;34:1847–1856. doi: 10.1007/s11064-009-9974-z. [DOI] [PubMed] [Google Scholar]
- Nagai M, Aoki M, Miyoshi I, Kato M, Pasinelli P, Kasai N, et al. Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. J Neurosci. 2001;21:9246–9254. doi: 10.1523/JNEUROSCI.21-23-09246.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalbandian A, Llewellyn KJ, Badadani M, Yin HZ, Nguyen C, Katheria V, et al. A progressive translational mouse model of human valosin-containing protein disease: the VCP(R155H/+) mouse. Muscle Nerve. 2013;47:260–270. doi: 10.1002/mus.23522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Nishimura AL, Mitne-Neto M, Silva HC, Richieri-Costa A, Middleton S, Cascio D, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75:822–831. doi: 10.1086/425287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parone PA, Da CS, Han JS, McAlonis-Downes M, Vetto AP, Lee SK, et al. Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J Neurosci. 2013;33:4657–4671. doi: 10.1523/JNEUROSCI.1119-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasinelli P, Houseweart MK, Brown RH, Jr, Cleveland DW. Caspase-1 and-3 are sequentially activated in motor neuron death in Cu,Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2000;97:13901–13906. doi: 10.1073/pnas.240305897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, et al. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43:19–30. doi: 10.1016/j.neuron.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Pathak RU, Davey GP. Complex I and energy thresholds in the brain. Biochim Biophys Acta. 2008;1777:777–782. doi: 10.1016/j.bbabio.2008.05.443. [DOI] [PubMed] [Google Scholar]
- Pattee GL, Post GR, Gerber RE, Bennett JP., Jr Reduction of oxidative stress in amyotrophic lateral sclerosis following pramipexole treatment. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:90–95. doi: 10.1080/14660820310012736. [DOI] [PubMed] [Google Scholar]
- Pedrini S, Sau D, Guareschi S, Bogush M, Brown RH, Jr, Naniche N, et al. ALS-linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl-2. Hum Mol Genet. 2010;19:2974–2986. doi: 10.1093/hmg/ddq202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesaresi MG, Amori I, Giorgi C, Ferri A, Fiorenzo P, Gabanella F, et al. Mitochondrial redox signalling by p66Shc mediates ALS-like disease through Rac1 inactivation. Hum Mol Genet. 2011;20:4196–4208. doi: 10.1093/hmg/ddr347. [DOI] [PubMed] [Google Scholar]
- Pickles S, Destroismaisons L, Peyrard SL, Cadot S, Rouleau GA, Brown RH, Jr, et al. Mitochondrial damage revealed by immunoselection for ALS-linked misfolded SOD1. Hum Mol Genet. 2013;22:3947–3959. doi: 10.1093/hmg/ddt249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polster BM. AIF, reactive oxygen species, and neurodegeneration: a ‘complex’ problem. Neurochem Int. 2013;62:695–702. doi: 10.1016/j.neuint.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, Cleveland DW. Motor neuron disease: the curious ways of ALS. Nature. 2008;454:284–285. doi: 10.1038/454284a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon HF, Hensley K, Thongboonkerd V, Merchant ML, Lynn BC, Pierce WM, et al. Redox proteomics analysis of oxidatively modified proteins in G93A-SOD1 transgenic mice – a model of familial amyotrophic lateral sclerosis. Free Radic Biol Med. 2005;39:453–462. doi: 10.1016/j.freeradbiomed.2005.03.030. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- Redler RL, Dokholyan NV. The complex molecular biology of amyotrophic lateral sclerosis (ALS) Prog Mol Biol Transl Sci. 2012;107:215–262. doi: 10.1016/B978-0-12-385883-2.00002-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes NA, Fisher JK, Austgen K, VandenBerg S, Huang EJ, Oakes SA. Blocking the mitochondrial apoptotic pathway preserves motor neuron viability and function in a mouse model of amyotrophic lateral sclerosis. J Clin Invest. 2010;120:3673–3679. doi: 10.1172/JCI42986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzardini M, Lupi M, Bernasconi S, Mangolini A, Cantoni L. Mitochondrial dysfunction and death in motor neurons exposed to the glutathione-depleting agent ethacrynic acid. J Neurol Sci. 2003;207:51–58. doi: 10.1016/s0022-510x(02)00357-x. [DOI] [PubMed] [Google Scholar]
- Rizzardini M, Mangolini A, Lupi M, Ubezio P, Bendotti C, Cantoni L. Low levels of ALS-linked Cu/Zn superoxide dismutase increase the production of reactive oxygen species and cause mitochondrial damage and death in motor neuron-like cells. J Neurol Sci. 2005;232:95–103. doi: 10.1016/j.jns.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14:248–264. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- Rudnicki SA, Berry JD, Ingersoll E, Archibald D, Cudkowicz ME, Kerr DA, et al. Dexpramipexole effects on functional decline and survival in subjects with amyotrophic lateral sclerosis in a Phase II study: subgroup analysis of demographic and clinical characteristics. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:44–51. doi: 10.3109/17482968.2012.723723. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Iwata M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2007;66:10–16. doi: 10.1097/nen.0b013e31802c396b. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Warita H, Murakami T, Abe K, Iwata M. Ultrastructural study of mitochondria in the spinal cord of transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol. 2004;107:461–474. doi: 10.1007/s00401-004-0837-z. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Horie Y, Iwata M. Mitochondrial alterations in dorsal root ganglion cells in sporadic amyotrophic lateral sclerosis. Acta Neuropathol. 2007;114:633–639. doi: 10.1007/s00401-007-0299-1. [DOI] [PubMed] [Google Scholar]
- Seibenhener ML, Du Y, Diaz-Meco MT, Moscat J, Wooten MC, Wooten MW. A role for sequestosome 1/p62 in mitochondrial dynamics, import and genome integrity. Biochim Biophys Acta. 2013;1833:452–459. doi: 10.1016/j.bbamcr.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan X, Chiang PM, Price DL, Wong PC. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc Natl Acad Sci U S A. 2010;107:16325–16330. doi: 10.1073/pnas.1003459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan D, Meade G, Foley VM, Dowd CA. Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem J. 2001;360:1–16. doi: 10.1042/0264-6021:3600001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shefner JM, Cudkowicz ME, Schoenfeld D, Conrad T, Taft J, Chilton M, et al. A clinical trial of creatine in ALS. Neurology. 2004;63:1656–1661. doi: 10.1212/01.wnl.0000142992.81995.f0. [DOI] [PubMed] [Google Scholar]
- Shelton MD, Chock PB, Mieyal JJ. Glutaredoxin: role in reversible protein s-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid Redox Signal. 2005;7:348–366. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- Sims NR, Muyderman H. Mitochondria, oxidative metabolism and cell death in stroke. Biochim Biophys Acta. 2010;1802:80–91. doi: 10.1016/j.bbadis.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Sims NR, Nilsson M, Muyderman H. Mitochondrial glutathione: a modulator of brain cell death. J Bioenerg Biomembr. 2004;36:329–333. doi: 10.1023/B:JOBB.0000041763.63958.e7. [DOI] [PubMed] [Google Scholar]
- Song W, Song Y, Kincaid B, Bossy B, Bossy-Wetzel E. Mutant SOD1G93A triggers mitochondrial fragmentation in spinal cord motor neurons: neuroprotection by SIRT3 and PGC-1alpha. Neurobiol Dis. 2013;51:72–81. doi: 10.1016/j.nbd.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soo KY, Atkin JD, Horne MK, Nagley P. Recruitment of mitochondria into apoptotic signalling correlates with the presence of inclusions formed by amyotrophic lateral sclerosis-associated SOD1 mutations. J Neurochem. 2009;108:578–590. doi: 10.1111/j.1471-4159.2008.05799.x. [DOI] [PubMed] [Google Scholar]
- Soo KY, Atkin JD, Farg M, Walker AK, Horne MK, Nagley P. Bim links ER stress and apoptosis in cells expressing mutant SOD1 associated with amyotrophic lateral sclerosis. PLoS ONE. 2012;7:e35413. doi: 10.1371/journal.pone.0035413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalloni A, Albo F, Ferrari F, Mercuri N, Bernardi G, Zona C, et al. Cu/Zn-superoxide dismutase (GLY93-->ALA) mutation alters AMPA receptor subunit expression and function and potentiates kainate-mediated toxicity in motor neurons in culture. Neurobiol Dis. 2004;15:340–350. doi: 10.1016/j.nbd.2003.11.012. [DOI] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan W, Naniche N, Bogush A, Pedrini S, Trotti D, Pasinelli P. Small peptides against the mutant SOD1/Bcl-2 toxic mitochondrial complex restore mitochondrial function and cell viability in mutant SOD1-mediated ALS. J Neurosci. 2013;33:11588–11598. doi: 10.1523/JNEUROSCI.5385-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateno M, Sadakata H, Tanaka M, Itohara S, Shin RM, Miura M, et al. Calcium-permeable AMPA receptors promote misfolding of mutant SOD1 protein and development of amyotrophic lateral sclerosis in a transgenic mouse model. Hum Mol Genet. 2004;13:2183–2196. doi: 10.1093/hmg/ddh246. [DOI] [PubMed] [Google Scholar]
- Thau N, Knippenberg S, Korner S, Rath KJ, Dengler R, Petri S. Decreased mRNA expression of PGC-1alpha and PGC-1alpha-regulated factors in the SOD1G93A ALS mouse model and in human sporadic ALS. J Neuropathol Exp Neurol. 2012;71:1064–1074. doi: 10.1097/NEN.0b013e318275df4b. [DOI] [PubMed] [Google Scholar]
- Tohgi H, Abe T, Yamazaki K, Murata T, Ishizaki E, Isobe C. Increase in oxidized NO products and reduction in oxidized glutathione in cerebrospinal fluid from patients with sporadic form of amyotrophic lateral sclerosis. Neurosci Lett. 1999;260:204–206. doi: 10.1016/s0304-3940(98)00986-0. [DOI] [PubMed] [Google Scholar]
- Tradewell ML, Yu Z, Tibshirani M, Boulanger MC, Durham HD, Richard S. Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum Mol Genet. 2012;21:136–149. doi: 10.1093/hmg/ddr448. [DOI] [PubMed] [Google Scholar]
- Tsai KJ, Yang CH, Fang YH, Cho KH, Chien WL, Wang WT, et al. Elevated expression of TDP-43 in the forebrain of mice is sufficient to cause neurological and pathological phenotypes mimicking FTLD-U. J Exp Med. 2010;207:1661–1673. doi: 10.1084/jem.20092164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner MR, Hardiman O, Benatar M, Brooks BR, Chio A, Ince PG, et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013;12:310–322. doi: 10.1016/S1474-4422(13)70036-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande VC, Miller TM, Cashman NR, Cleveland DW. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc Natl Acad Sci U S A. 2008;105:4022–4027. doi: 10.1073/pnas.0712209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas MR, Johnson DA, Johnson JA. Decreased glutathione accelerates neurological deficit and mitochondrial pathology in familial ALS-linked hSOD1(G93A) mice model. Neurobiol Dis. 2011;43:543–551. doi: 10.1016/j.nbd.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkening K, Leystra-Lantz C, Yang W, Jaffee H, Strong MJ. Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS) Brain Res. 2009;1305:168–182. doi: 10.1016/j.brainres.2009.09.105. [DOI] [PubMed] [Google Scholar]
- Vukosavic S, Dubois-Dauphin M, Romero N, Przedborski S. Bax and Bcl-2 interaction in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem. 1999;73:2460–2468. doi: 10.1046/j.1471-4159.1999.0732460.x. [DOI] [PubMed] [Google Scholar]
- Vukosavic S, Stefanis L, Jackson-Lewis V, Guegan C, Romero N, Chen C, et al. Delaying caspase activation by Bcl-2: a clue to disease retardation in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2000;20:9119–9125. doi: 10.1523/JNEUROSCI.20-24-09119.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IF, Wu LS, Chang HY, Shen CK. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem. 2008;105:797–806. doi: 10.1111/j.1471-4159.2007.05190.x. [DOI] [PubMed] [Google Scholar]
- Wang W, Li L, Lin WL, Dickson DW, Petrucelli L, Zhang T, et al. The ALS disease associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum Mol Genet. 2013;22:4706–4719. doi: 10.1093/hmg/ddt319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YF, Gendron TF, Zhang YJ, Lin WL, D'Alton S, Sheng H, et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci. 2010;30:10851–10859. doi: 10.1523/JNEUROSCI.1630-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YF, Zhang YJ, Lin WL, Cao X, Stetler C, Dickson DW, et al. Expression of mutant TDP-43 induces neuronal dysfunction in transgenic mice. Mol Neurodegener. 2011;6:73. doi: 10.1186/1750-1326-6-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Cook A, Kim J, Baranov SV, Jiang J, Smith K, et al. Melatonin inhibits the caspase-1/cytochrome c/caspase-3 cell death pathway, inhibits MT1 receptor loss and delays disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2013;55:26–35. doi: 10.1016/j.nbd.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Varghese M, Yemul S, Pan Y, Cheng A, Marano P, et al. Peroxisome proliferator activator receptor gamma coactivator-1alpha (PGC-1alpha) improves motor performance and survival in a mouse model of amyotrophic lateral sclerosis. Mol Neurodegener. 2011;6:51. doi: 10.1186/1750-1326-6-51. [DOI] [PMC free article] [PubMed] [Google Scholar]