

Abstract
Mitochondrial disorders are nowadays recognized as impinging on most areas of medicine. They include specific and widespread organ involvement, including both tissue degeneration and tumour formation. Despite the spectacular progresses made in the identification of their underlying molecular basis, effective therapy remains a distant goal. Our still rudimentary understanding of the pathophysiological mechanisms by which these diseases arise constitutes an obstacle to developing any rational treatments. In this context, the idea of using a heterologous gene, encoding a supplemental oxidase otherwise absent from mammals, potentially bypassing the defective portion of the respiratory chain, was proposed more than 10 years ago. The recent progress made in the expression of the alternative oxidase in a wide range of biological systems and disease conditions reveals great potential benefit, considering the broad impact of mitochondrial diseases. This review addresses the state of the art and the perspectives that can be now envisaged by using this strategy.
Linked Articles
This article is part of a themed issue on Mitochondrial Pharmacology: Energy, Injury & Beyond. To view the other articles in this issue visit http://dx.doi.org/10.1111/bph.2014.171.issue-8
Keywords: allospecific gene expression, mitochondrial diseases, alternative oxidase
Mitochondrial defects
Respiratory chain (RC) subunits only represent a subset of the more than a thousand components required for the biosynthesis and function of the mitochondria. Nevertheless, RC dysfunction, as narrowly defined, leads to a striking number of different diseases in humans (Munnich and Rustin, 2001). Primary genetic defects are presumably only the tip of the iceberg of diseases involving functional deficiency of the mitochondrial RC (Nunnari and Suomalainen, 2012). In addition, they almost certainly include major age-related cancers, neurodegenerative and cardiac diseases (Wallace, 2005). Depending on their nature and expression, RC defects can impair, to a variable extent, the many intra-mitochondrial catabolic and biosynthetic pathways, the handling of oxidative stress, signals governing cell death and the structural organization of the mitochondrial network. It is therefore not surprising that they can affect virtually any organ of the human body, either alone or in combination, and at any age. So far, while loss of mitochondrial function is increasingly recognized as an important underlying factor of human disease, with a significant number of genetic causes identified (Chinnery and Hudson, 2013), our current understanding of major features of even typical mitochondrial diseases remains poor. The reasons for their progressive nature, for their often unpredictable course and their tissue specificity are still largely mysterious (Briere et al., 2004). As a result, except for a tiny minority of cases, we still lack any effective treatments to cure or even stabilize such conditions (Dimauro and Rustin, 2008). So far, strategies envisaged to combat mitochondrial dysfunction have mainly focused on negating the effects of mutant material (nucleic acids or proteins) or on alleviating a supposed increased oxidative stress. However, these strategies have never been demonstrated to be successful, except in the case of a few specific animal models (Suomalainen, 2011). This was our major motivation for investigating systems used in nature to cope with mitochondrial blockade.
What has evolved in nature to cope with a mitochondrial blockade?
It remains the case that, in most situations, nature copes only poorly with mitochondrial blockade. Poisoning of mitochondrial function(s), especially the RC, is lethal under most conditions. Nevertheless, one situation of blockade is encountered every day in plants around us. Each morning, when solar energy radiates upon plant leaves, it causes photosynthesis to be turned on. This mobilizes most of the adenine nucleotide pool of the cell to the detriment of the mitochondria (Hampp et al., 1982). The presence of high levels of cellular ATP, produced by photosynthesis, combined with the corresponding lack of ADP to drive ATP production in mitochondria, prevents the dissipation of the proton gradient (ΔμH+) across the inner mitochondrial membrane (IMM). In turn, respiratory electron flow and thus substrate oxidization are disabled, a necessary consequence of the tight coupling of respiration and oxidative phosphorylation (OXPHOS) dictated by the chemiosmotic mechanism (Figure 1). Under such conditions, intra-mitochondrial metabolism would be stopped almost in its entirety, due to the centrality of the tricarboxylic acid cycle, which is directly dependent upon respiration. Any reducing power entering the RC can escape only as potentially deleterious superoxides. However, mitochondrial metabolism must somehow proceed during the day because many vital biochemical reactions are carried out uniquely in the mitochondria of the plant cell. In fact, this happens without plants suffering any catastrophe due to excessive superoxide overproduction. This is due to the activation of a set of bioenergetic safety valves provided by two specific classes of electron-transporting protein (Rustin et al., 1980). These safety valves allow electrons to be diverted from the standard RC to oxygen, despite the impedance of the proton gradient across the IMM. In plants, the 40 subunits of the rotenone-sensitive respiratory chain complex I can be bypassed by a single-subunit NADH dehydrogenase (e.g. nda1 of Arabidopsis, Michalecka et al., 2003), which is resistant to rotenone, while the rest of the RC (from quinones to oxygen) can be bypassed by the activity of one other single protein, the so-called alternative oxidase (AOX, (Palmer, 1976). This entire non-proton motive electron transfer circuit (nda1 plus AOX), bypassing all proton-translocation sites of the RC, permits the necessary operation of mitochondrial metabolism during the day (Rustin and Queiroz, 1985), simultaneously avoiding mitochondrial overproduction of superoxides (Cvetkovska and Vanlerberghe, 2012). For several decades, the occurrence and operation of such a non-proton motive circuit (also detected in various fungi and microorganisms) was considered to be one of the major differences between the plant and animal kingdoms (Henry and Nyns, 1975). This belief was challenged by the observation, by Dr A MacDonald, of the occurrence of a gene encoding a protein similar to AOX in the genomes of a subset of lower animals (McDonald and Vanlerberghe, 2007; McDonald et al., 2009).
Figure 1.
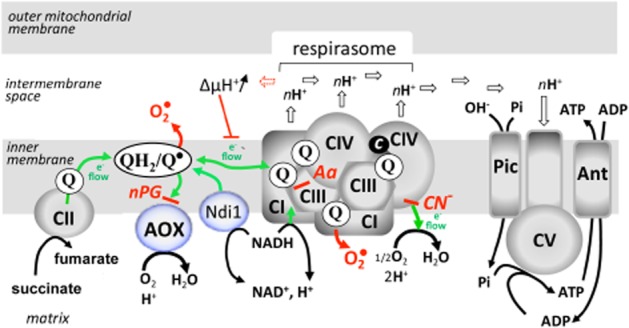
A schematized view of the mitochondrial respiratory chain of the animal kingdom. Electron flow (green line) through the chain is coupled to proton extrusion (open arrows) by the respirasome, which associates various proportions of respiratory complexes I, III and IV (CI, CIII, CIV). Protons released in the intermembrane space are subsequently used by complex V (CV; ATP synthase) to phosphorylate ADP (imported by the adenylate carrier; Ant) to ATP, making use of the inorganic phosphate imported by the phosphate carrier (Pic). Build-up of the proton gradient (ΔμH+) in case of impairment of ATP synthase function exerts a negative feedback control on electron flow in the respirasome. Under these conditions, quinones (Q) are essentially converted to reduced forms (QH2), with formation of highly unstable semi-quinones (Q°) prone to react with oxygen to produce superoxides (O2°). The scheme additionally features the two allotopic bypasses that have been successfully introduced in human cultured cells, Drosophila, and rodents, namely the internal NADH dehydrogenase from yeast (Ndi1; see Yagi et al., 2006 for a review) and the AOX. Site of action (red dash) of inhibitors is indicated for nPG, Aa (antimycin A) and cyanide (CN−).
Given the biological roles of their homologues in plants, this discovery provided an incentive to determine the functional properties of metazoan AOX-like proteins and test their potential to rescue RC impairment in different biological contexts (Rustin and Jacobs, 2009).
Nature and function of AOX
The nature of the AOX has been a matter of debate since the 1980s. At one point, it was even proposed that the oxidase did not really exist and that the corresponding cyanide-insensitive oxygen consumption resulted from underlying lipoperoxidation in the absence of any specific protein (Rustin et al., 1983). The situation was clarified by the biochemical isolation of the AOX (Elthon and McIntosh, 1987), whose structure is now resolved (Moore et al., 2013). The AOX is an integral membrane protein that is bound to the matrix side of the IMM. The enzyme was suggested to contain a coupled di-iron centre and it was shown by electron spin resonance that the Arabidopsis thaliana AOX AOX1a contains a hydroxo-bridged mixed-valent Fe(II)/Fe(III) binuclear iron centre. The catalytic cycle would make use of this di-iron centre and a tyrosine-residue free radical. Two major functions have been attributed to AOX in plants. First, under light conditions in the green parts of plants, AOX allows the mitochondria to work in parallel to photosynthesis (see above). Accordingly, the expression of the AOX was shown to be triggered by the greening of A. thaliana (Zhang et al., 2010). Second, under numerous stress conditions, especially those resulting in decreased functioning of the standard cytochrome pathway, the AOX would avoid the conversion of the excess reducing power accumulated in the ubiquinone pool into deleterious superoxides (Figure 1) (Vanlerberghe, 2013). Because the AOX, similarly to nda1, is a non-proton motive oxidase, its permanent functioning might be problematic, as it would decrease the yield of ADP phosphorylation associated with substrate oxidation. However, due to a very low affinity for its substrate, ubiquinol, as compared with the first complex of the cytochrome pathway (complex III, also known as the bc1 complex), AOX does not efficiently compete with the latter (Bahr and Bonner, 1973a,1973b). In other words, the AOX essentially works only when the cytochrome pathway does not. This unique property was predicted to make it innocuous when expressed in any cell with a functional cytochrome pathway (Rustin and Jacobs, 2009).
Allospecific expression
Based on the above considerations, Hakkaart et al. (2006) successfully expressed an AOX in cultured human cells. In order to maximize the chance of successful expression of an active protein, the AOX gene from an animal species endowed with AOX, the chordate Ciona intestinalis was chosen rather than the gene from plants. The gene was placed under the control of a tetracycline-inducible promoter so as to allow the cells to grow in case the AOX had an unsuspected deleterious effect (Hakkaart et al., 2006). Subsequently, this was revealed to have been an unnecessary precaution because human cells expressing the AOX grew normally, without any sign of AOX-related toxicity. Cell growth rate, cell and mitochondrial morphology, cell respiration and mitochondrial substrate oxidation, RC activities and OXPHOS process were all found to be unaffected by the presence of the AOX protein. Indeed, just as predicted, none of the parameters tested suggested that the AOX was working in these cells as long as the cytochrome pathway operated freely. However, in the case of a chemical blockade of the cytochrome pathway triggered by cyanide (targeting complex IV, cytochrome c oxidase) or by antimycin (targeting complex III), the AOX was activated, allowing significant cell respiration to proceed in the presence of these inhibitors, which fully inhibited the respiration of the parental cells devoid of AOX. Accordingly, the AOX-endowed cells were able to grow in the presence of cyanide or antimycin, conditions under which control cells rapidly died. A few years subsequently, a plant AOX from Arum concinnatum was successfully expressed in HeLa cells, indicating that the plant enzyme and the gene that encodes it can also be functional in human cells (Kakizaki et al., 2010; Kakizaki and Ito, 2013).
The C. intestinalis AOX was next engineered for conditional expression in Drosophila melanogaster (Fernandez-Ayala et al., 2009). Ubiquitous AOX expression in Drosophila at the level of a typical abundant mRNA appeared to be benign. The protein was stable and correctly targeted to mitochondria, conferring substantial cyanide resistance to mitochondrial substrate oxidation in vitro. As in human cells, it appeared to be enzymatically inert in the absence of a cytochrome pathway inhibitor (Fernandez-Ayala et al., 2009). Consistent with this, it had only a minimal effect on development with a slightly increased weight loss experienced by young adult flies, suggestive of only a minimal drop in the overall efficiency of catabolism. AOX-expressing flies were also fertile, indicative that AOX expression clearly does not compromise any somatic functions required for fertility in this model organism. A slight impairment in energetic efficiency might nevertheless be sufficient to account for the evolutionary loss of AOX in both vertebrates and arthropods, organisms where maximal muscle-force generation, dependent on aerobic ATP supply, may be needed to escape predators or catch prey. Among metazoans, AOX genes have so far been found only in phyla composed of slow-moving or sessile organisms (McDonald et al., 2009).
The successful allotopic expression of the AOX was an incentive to revisit conditions in which superoxide overproduction has been suspected to be instrumental. In keeping with this, AOX expression in HEK cells was used to demonstrate that superoxides produced by the RC do not play an instrumental role in hypoxia-dependent HIF-1α stabilization triggered by RC inhibitors (Chua et al., 2010). HIF-1α plays an essential role in cellular and systemic responses to hypoxia. In addition to transcriptional regulation, HIF-1α abundance depends on the coordinated activity of the oxygen-dependent superoxide-sensitive prolyl hydroxylases (PHDs), which maintain the appropriate balance of HIF-1α protein by targeting it for ubiquitination and proteasome-dependent degradation. Accordingly, it has been hypothesized that superoxides are instrumental in controlling the activity of the PHDs. However, using AOX as a tool to decrease mitochondrial superoxide production, it was observed that this decrease did not affect the HIF-1α balance, suggesting that superoxides did not play a major role in this balance (Chua et al., 2010). It was rather found that an increase in cellular oxygen resulting from inhibition of the RC leads to activation of the PHDs. According to the free radical theory of ageing, organisms age because cells accumulate oxidative damage over time (Harman, 1956; 1972). In keeping with this, we used the AOX to investigate the role of mitochondrial superoxide overproduction in the aging of the flies, and established that mitochondrial superoxide production correlates with but does not directly regulate lifespan in Drosophila. According to this study (Sanz et al., 2010), mtDNA influences longevity in (female) flies, but does so independently of superoxide production which was maintained at a low level by the AOX.
More recently, the C. intestinalis AOX gene was successfully expressed in the mouse (El-Khoury et al., 2013). The gene was recoded to maximize its expression and introduced into early mouse embryos by germ line lentiviral transduction. It was placed under the control of the ubiquitously active, chimeric cytomegalovirus/β-actin/β-globin promoter, together with the Woodchuck hepatitis virus post-transcriptional regulatory element, in order to further enhance AOX transgene expression. This was the first demonstration that a functional AOX can be expressed in a mammal and transmitted between generations, conferring significant cyanide resistance to mitochondrial substrate oxidation and tissue respiration as well as the whole organism (El-Khoury et al., 2013). The enzyme was targeted to the mitochondria but did not interfere or compete significantly with the cytochrome pathway. The AOX in this mouse (MitAOX mouse) was enzymatically functional only upon blockade of the latter, when the pool of ubiquinone is expected to become highly reduced. Altogether, the expression of the AOX did not result in any deleterious consequence, while spectacularly increasing the survival of the mice in the presence of a lethal concentration of gaseous cyanide. Thus, the proposed protective mechanism provided by the AOX to organisms naturally harbouring the enzyme was preserved when the oxidase was expressed in a mammal (El-Khoury et al., 2013).
AOX expression in disease contexts
Human cultured cells
Based on the observation that AOX expression provided an efficient bypass of a chemically-induced blockade of the RC, the expression of AOX gene was next attempted in cultured skin fibroblasts derived from a patient presenting an early onset fatal hypertrophic cardiomyopathy due to a deleterious cytochrome oxidase (COX) 15 gene mutation (Dassa et al., 2009b). COX15 encodes an enzyme required for synthesis of haem a, an essential prosthetic group of complex IV required for assembly of the functional complex (Petruzzella et al., 1998). The respiration of the COX15-mutant cells decreased by 30%, compared with control cells, and was fully cyanide sensitive. The cells were transduced by a lentiviral construct containing the C. intestinalis AOX coding sequence under the control of the EF1a promoter. The respiration of the cells was increased by 25–30% compared with parental COX15-mutant cells, and was >50% cyanide resistant. When cultured under respiratory restrictive conditions (low glucose), the growth of untransduced COX15-mutant cells was significantly impaired, but was restored to levels observed in high glucose by the expression of AOX. The study next focused on the ability of the AOX to protect RC-deficient cells from oxidative insults, taking advantage of the observation that oxidant sensitivity is greatly increased in cultured human fibroblasts rendered COX deficient by mutations in SURF1, COX10 or COX15 (Dassa et al., 2009b). Cells grown in non-restrictive medium were treated with antimycin, which causes endogenous oxidative insult, or with exogenously supplied H2O2. Measurements of cell viability confirmed that COX-deficient cells were significantly more sensitive to both types of oxidative insult than control cells. In particular, antimycin caused massive cell death under respiratory restrictive conditions, which was fully counteracted by AOX. The protection of cell growth by AOX expression was abolished if antimycin-treated cells were further treated with n-propyl gallate (nPG), a specific inhibitor of AOX (Ki of about 2 μM) (Siedow and Girvin, 1980). The protection afforded by AOX expression was attributable to avoidance of the excess superoxide production caused by antimycin inhibition of complex III. Next, HEK293-derived cells harbouring tetracycline-inducible AOX were made COX-deficient using shRNA targeted against COX10, another gene in the haem a biosynthetic pathway required for assembly of complex IV. The decreased respiration of these cells was largely compensated when AOX expression was boosted by tetracycline (Dassa et al., 2009b). The inhibitory effect of nPG on respiration further confirmed the involvement of AOX in the recovery of respiration. Finally, in a restrictive growth medium requiring competent respiration, the COX10-knockdown cells showed a significant growth defect that was fully eliminated upon AOX induction.
Drosophila
The innocuousness of AOX expression in otherwise wild-type Drosophila (Fernandez-Ayala et al., 2009) and the successful rescue of phenotypes resulting from COX deficiency in cultured cells (Dassa et al., 2009b) were an incentive to study the effect of AOX expression on mutant flies. The expression of AOX using the ubiquitous da-GAL4 driver was shown to complement the semi-lethality of a partial RNAi knockdown of either cyclope (encoding the COXVIc structural subunit of complex IV) or the complex IV assembly factor Surf1. AOX expression allowed hatching of flies knocked down for cyclope at 18°C, conditions that normally prohibit the completion of development. At a higher temperature (25°C), AOX prevented the developmental delay and small size of the eclosing progeny knocked down for cyclope (Fernandez-Ayala et al., 2009). Surf1 was next targeted by RNAi technology using a mifepristone (RU486) inducible tub-GS driver. At a dose where Surf1 knockdown was lethal or semi-lethal, concomitant AOX expression under the control of the same driver prevented the lethality (Fernandez-Ayala et al., 2009). In subsequent applications of this approach (K.K. Kemppainen et al., unpublished observations), AOX was shown to be capable of preventing neurodegeneration resulting from a point mutation in the gene levy, encoding subunit COXVIa of complex IV (Liu et al., 2007) or from the neuron-specific knockdown of essential complex IV subunits. It also corrected the late developmental lethality produced by knockdown of complex IV subunits in muscle.
In human cultured cells, AOX expression inhibits superoxide overproduction because it prevents over-reduction of the RC (Rustin and Jacobs, 2009). This effect was further investigated in the Drosophila mutant dj-1β, a model of Parkinson's disease associated with defects in ROS handling (Park et al., 2005). The human homologue (DJ1) has been identified as the causative gene in some cases of familial Parkinson's disease (Tan and Skipper, 2007). The fly mutant exhibits progressive locomotor decline. The locomotor ability of dj-1b mutant flies was studied over a period of 4 weeks post-eclosion, with or without the concomitant expression of AOX (Fernandez-Ayala et al., 2009). The presence of the AOX transgene conferred a partial rescue of locomotor decline, which was more obvious in the case of ubiquitous or nervous system-directed AOX expression. In AOX-expressing males, the dj-1β locomotor defect was almost completely abolished. Along with the improved phenotype, the increased production of mitochondrial ROS observed in the dj-1b mutant fly was decreased to wild-type levels by ubiquitous AOX expression. This work demonstrates that, at least in flies, phenotypes associated with partial COX deficiency, whatever its genetic cause, or with defects in ROS handling, can be successfully rescued by AOX.
Prospects
From data that have accumulated over the past few years, it appears that the predictions made on the potential use of AOX (Dassa et al., 2009a; Rustin and Jacobs, 2009) have been verified. Firstly, the expression of AOX in human cultured cells, in the fly and in the mouse is essentially innocuous. Secondly, it renders human cells, flies and mice resistant to inhibitors of the cytochrome pathway, such as antimycin or cyanide. Thirdly, it prevents superoxide overproduction triggered by an excessive reduction of the RC. As a result, in accordance with our fourth prediction, it offers an efficient way to prevent the consequences of a wide set of genetic or environmentally determined lesions targeting the cytochrome pathway in model systems.
Based on this body of data, it is now possible to define additional perspectives, taking advantage of AOX expression (Figure 2). Firstly, it is tempting to use the MitAOX mouse to investigate the long list of pathological conditions where overproduction of superoxide by mitochondria has been postulated to be instrumental (Fernandez-Checa et al., 2010), especially when already modelled in the mouse. This can be achieved by crossing selected mouse lines with transgenic mice either expressing AOX ubiquitously (El-Khoury et al., 2013) or in a tissue-restricted fashion (under construction). As already done in Drosophila, its effects should be determined in physiological conditions where mitochondrial superoxide overproduction has thus far merely been suggested to be of aetiological or pathological significance (Brand et al., 2013). This includes, for example, ageing, but also many types of physiological insult affecting mitochondrial function in diverse tissues. AOX-expressing mice should also be crossed with mouse lines in which a deficiency of complex IV or other cytochrome pathway components has been observed or engineered (Fukui et al., 2007). AOX-expressing mice can also be used directly in other types of models, such as surgical or toxicological paradigms of human disease. In such cases, the mechanism of any eventual rescue brought about by AOX expression (restoration of metabolic homeostasis, energetic compensation, and/or alleviation of oxidative stress) will be of major interest: the consequences of RC deficiency are far from fully understood. Finally, as an alternative to the tissue-restricted AOX-expressing mouse, the use of injectable AOX, including its mitochondrial targeting sequence, delivered by a viral or similar vector, can also be considered. Recent progress made in ensuring the safety and longevity of viral vectors (Bouaita et al., 2012) makes this approach particularly attractive, as it might allow the therapeutic use of the AOX gene.
Figure 2.
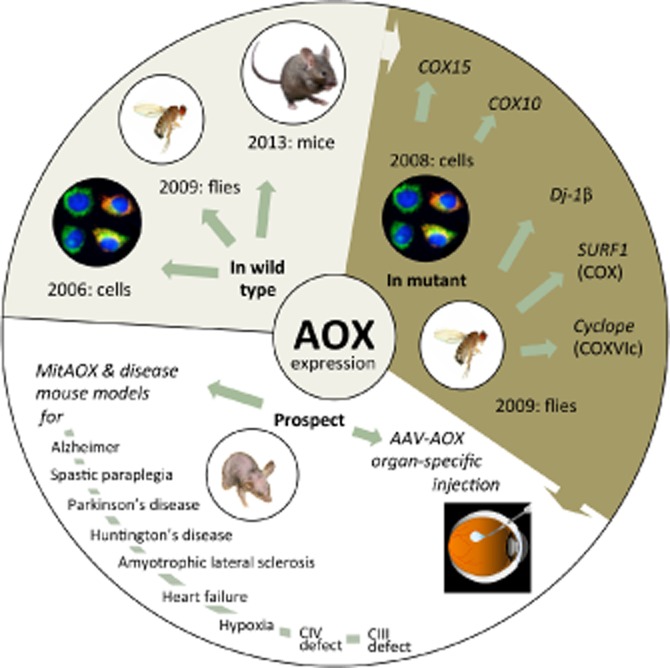
State of the art and prospect for allotopic AOX expression. Upper left sector: first successful expression of the AOX gene in human cultured cells (cells), Drosophila melanogaster (flies) and Mus musculus (CD-1/B6 MitAOX mice). Upper right sector: first complementation of respiratory chain defect in human COX15-mutant fibroblasts, with siRNA-down-regulated COX10 HEK cells, in cyclope, Surf1 and dj-1β mutant D. melanogaster. Lower sector (left): a partial list of mouse models which should be informative to cross with the MitAOX mouse, either to establish/rule out the pathological involvement of mitochondrial superoxides, and/or of components from the mitochondrial cytochrome pathway, or to demonstrate the ability of AOX to complement genetic defects of the mitochondrial cytochrome pathway (CIV, CIII). One on the right-hand side: potential use of adeno-associated virus (AAV) constructs containing the AOX gene as a therapeutic strategy to target affected organs, for example, the eyes in cases of mutations affecting the cytochrome pathway.
The findings with Drosophila already suggest that AOX expression could be of benefit in a wide spectrum of OXPHOS disorders. In keeping with this, it is worth emphasizing that the single AOX gene can compensate for the impairment of any of at least 30 genes encoding structural components of RC complexes III or IV, as well as assembly factors required specifically for their biosynthesis, mutations that cause a wide spectrum of currently intractable diseases. However, even in the mouse, a number of questions remain to be answered before it is possible to conclude that AOX expression is a feasible therapeutic strategy. In particular, as the AOX gene we used so far derives from C. intestinalis which lives in highly oxygenated (intertidal zone) and cold water (<20°C), we have to consider the possibility that different temperature and oxygen tension in mammalian organs might affect the functionality of the AOX, and thus its capacity to counteract RC deficiencies, although the first data with regard to the brain are highly encouraging (El-Khoury et al., 2013).
Acknowledgments
Our research is supported by Ammi (Association contre les Maladies Mitochondriales), AFAF (Association Française contre l'Ataxie de Friedreich), ANR AifInter (R. E-K., P. R.), Academy of Finland, the European Research Council (MITO BY-PASS project), Sigrid Juselius Foundation and Tampere University Medical Research Fund.
Glossary
- AOX
alternative oxidase
- COX
cytochrome oxidase
- IMM
inner mitochondrial membrane
- OXPHOS
oxidative phosphorylation
- PHD
prolyl hydroxylase
- RC
respiratory chain
Conflict of interest
None.
References
- Bahr JT, Bonner WD., Jr Cyanide-insensitive respiration. I. The steady states of skunk cabbage spadix and bean hypocotyl mitochondria. J Biol Chem. 1973a;248:3441–3445. [PubMed] [Google Scholar]
- Bahr JT, Bonner WD., Jr Cyanide-insensitive respiration. II. Control of the alternate pathway. J Biol Chem. 1973b;248:3446–3450. [PubMed] [Google Scholar]
- Bouaita A, Augustin S, Lechauve C, Cwerman-Thibault H, Benit P, Simonutti M, et al. Downregulation of apoptosis-inducing factor in Harlequin mice induces progressive and severe optic atrophy which is durably prevented by AAV2-AIF1 gene therapy. Brain. 2012;135:35–52. doi: 10.1093/brain/awr290. (Pt 1) [DOI] [PubMed] [Google Scholar]
- Brand MD, Orr AL, Perevoshchikova IV, Quinlan CL. The role of mitochondrial function and cellular bioenergetics in ageing and disease. Br J Dermatol. 2013;169(Suppl. 2):1–8. doi: 10.1111/bjd.12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briere JJ, Chretien D, Benit P, Rustin P. Respiratory chain defects: what do we know for sure about their consequences in vivo. Biochim Biophys Acta. 2004;1659:172–177. doi: 10.1016/j.bbabio.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Chinnery PF, Hudson G. Mitochondrial genetics. Br Med Bull. 2013;106:135–159. doi: 10.1093/bmb/ldt017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua YL, Dufour E, Dassa EP, Rustin P, Jacobs HT, Taylor CT, et al. Stabilization of hypoxia-inducible factor-1alpha protein in hypoxia occurs independently of mitochondrial reactive oxygen species production. J Biol Chem. 2010;285:31277–31284. doi: 10.1074/jbc.M110.158485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvetkovska M, Vanlerberghe GC. Alternative oxidase modulates leaf mitochondrial concentrations of superoxide and nitric oxide. New Phytol. 2012;195:32–39. doi: 10.1111/j.1469-8137.2012.04166.x. [DOI] [PubMed] [Google Scholar]
- Dassa EP, Dufour E, Goncalves S, Jacobs HT, Rustin P. The alternative oxidase, a tool for compensating cytochrome c oxidase deficiency in human cells. Physiol Plant. 2009a;137:427–434. doi: 10.1111/j.1399-3054.2009.01248.x. [DOI] [PubMed] [Google Scholar]
- Dassa EP, Dufour E, Goncalves S, Paupe V, Hakkaart GA, Jacobs HT, et al. Expression of the alternative oxidase complements cytochrome c oxidase deficiency in human cells. EMBO Mol Med. 2009b;1:30–36. doi: 10.1002/emmm.200900001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimauro S, Rustin P. A critical approach to the therapy of mitochondrial respiratory chain and oxidative phosphorylation diseases. Biochim Biophys Acta. 2008;1792:1159–1167. doi: 10.1016/j.bbadis.2008.10.015. [DOI] [PubMed] [Google Scholar]
- El-Khoury R, Dufour E, Rak M, Ramanantsoa N, Grandchamp N, Csaba Z, et al. Alternative oxidase expression in the mouse enables bypassing cytochrome c oxidase blockade and limits mitochondrial ROS overproduction. PLoS Genet. 2013;9:e1003182. doi: 10.1371/journal.pgen.1003182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elthon TE, McIntosh L. Identification of the alternative terminal oxidase of higher plant mitochondria. Proc Natl Acad Sci U S A. 1987;84:8399–8403. doi: 10.1073/pnas.84.23.8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Ayala DJ, Sanz A, Vartiainen S, Kemppainen KK, Babusiak M, Mustalahti E, et al. Expression of the Ciona intestinalis alternative oxidase (AOX) in Drosophila complements defects in mitochondrial oxidative phosphorylation. Cell Metab. 2009;9:449–460. doi: 10.1016/j.cmet.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Fernandez-Checa JC, Fernandez A, Morales A, Mari M, Garcia-Ruiz C, Colell A. Oxidative stress and altered mitochondrial function in neurodegenerative diseases: lessons from mouse models. CNS Neurol Disord Drug Targets. 2010;9:439–454. doi: 10.2174/187152710791556113. [DOI] [PubMed] [Google Scholar]
- Fukui H, Diaz F, Garcia S, Moraes CT. Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2007;104:14163–14168. doi: 10.1073/pnas.0705738104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakkaart GA, Dassa EP, Jacobs HT, Rustin P. Allotopic expression of a mitochondrial alternative oxidase confers cyanide resistance to human cell respiration. EMBO Rep. 2006;7:341–345. doi: 10.1038/sj.embor.7400601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampp R, Goller M, Ziegler H. Adenylate levels, energy charge, and phosphorylation potential during dark-light and light-dark transition in chloroplasts, mitochondria, and cytosol of mesophyll protoplasts from Avena sativa L. Plant Physiol. 1982;69:448–455. doi: 10.1104/pp.69.2.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- Henry MF, Nyns ED. Cyanide-insensitive respiration. An alternative mitochondrial pathway. Subcell Biochem. 1975;4:1–65. [PubMed] [Google Scholar]
- Kakizaki Y, Ito K. Engineering plant alternative oxidase function in mammalian cells: substitution of the motif-like sequence ENV for QDT diminishes catalytic activity of Arum concinnatum AOX1a expressed in HeLa cells. Appl Biochem Biotechnol. 2013;170:1229–1240. doi: 10.1007/s12010-013-0235-x. [DOI] [PubMed] [Google Scholar]
- Kakizaki Y, Seymour RS, Ito K. A novel functional element in the N-terminal region of Arum concinnatum alternative oxidase is indispensable for catalytic activity of the enzyme in HeLa cells. Biochim Biophys Acta. 2010;1797:20–28. doi: 10.1016/j.bbabio.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Liu W, Gnanasambandam R, Benjamin J, Kaur G, Getman PB, Siegel AJ, et al. Mutations in cytochrome c oxidase subunit VIa cause neurodegeneration and motor dysfunction in Drosophila. Genetics. 2007;176:937–946. doi: 10.1534/genetics.107.071688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald A, Vanlerberghe G. Branched mitochondrial electron transport in the Animalia: presence of alternative oxidase in several animal phyla. IUBMB Life. 2004;56:333–341. doi: 10.1080/1521-6540400000876. [DOI] [PubMed] [Google Scholar]
- McDonald AE, Vanlerberghe GC, Staples JF. Alternative oxidase in animals: unique characteristics and taxonomic distribution. J Exp Biol. 2009;212:2627–2634. doi: 10.1242/jeb.032151. (Pt 16) [DOI] [PubMed] [Google Scholar]
- Michalecka AM, Svensson AS, Johansson FI, Agius SC, Johanson U, Brennicke A, et al. Arabidopsis genes encoding mitochondrial type II NAD(P)H dehydrogenases have different evolutionary origin and show distinct responses to light. Plant Physiol. 2003;133:642–652. doi: 10.1104/pp.103.024208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AL, Shiba T, Young L, Harada S, Kita K, Ito K. Unraveling the heater: new insights into the structure of the alternative oxidase. Annu Rev Plant Biol. 2013;64:637–663. doi: 10.1146/annurev-arplant-042811-105432. [DOI] [PubMed] [Google Scholar]
- Munnich A, Rustin P. Clinical spectrum and diagnosis of mitochondrial disorders. Am J Med Genet. 2001;106:4–17. doi: 10.1002/ajmg.1391. [DOI] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer JM. The organization and regulation of electron transport in plant mitochondria. Annu Rev Plant Physiol. 1976;27:133–157. [Google Scholar]
- Park J, Kim SY, Cha GH, Lee SB, Kim S, Chung J. Drosophila DJ-1 mutants show oxidative stress-sensitive locomotive dysfunction. Gene. 2005;361:133–139. doi: 10.1016/j.gene.2005.06.040. [DOI] [PubMed] [Google Scholar]
- Petruzzella V, Tiranti V, Fernandez P, Ianna P, Carrozzo R, Zeviani M. Identification and characterization of human cDNAs specific to BCS1, PET112, SCO1, COX15, and COX11, five genes involved in the formation and function of the mitochondrial respiratory chain. Genomics. 1998;54:494–504. doi: 10.1006/geno.1998.5580. [DOI] [PubMed] [Google Scholar]
- Rustin P, Dupont J, Lance C. Oxidative interactions between fatty acid peroxy radicals and quinones: possible involvement in cyanide-resistant electron transport in plant mitochondria. Arch Biochem Biophys. 1983;225:630–639. doi: 10.1016/0003-9861(83)90074-7. [DOI] [PubMed] [Google Scholar]
- Rustin P, Jacobs HT. Respiratory chain alternative enzymes as tools to better understand and counteract respiratory chain deficiencies in human cells and animals. Physiol Plant. 2009;137:362–370. doi: 10.1111/j.1399-3054.2009.01249.x. [DOI] [PubMed] [Google Scholar]
- Rustin P, Moreau F, Lance C. Malate oxidation in plant mitochondria via malic enzyme and the cyanide-insensitive electron transport pathway. Plant Physiol. 1980;66:457–462. doi: 10.1104/pp.66.3.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustin P, Queiroz C. Changes in oxidative properties of Kalanchoe blossfeldiana leaf mitochondria during development of Crassulacean acid metabolism. Planta. 1985;164:415–422. doi: 10.1007/BF00402955. [DOI] [PubMed] [Google Scholar]
- Sanz A, Soikkeli M, Portero-Otin M, Wilson A, Kemppainen E, McIlroy G, et al. Expression of the yeast NADH dehydrogenase Ndi1 in Drosophila confers increased lifespan independently of dietary restriction. Proc Natl Acad Sci U S A. 2010;107:9105–9110. doi: 10.1073/pnas.0911539107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siedow JN, Girvin ME. Alternative respiratory pathway: its role in seed respiration and its inhibition by propyl gallate. Plant Physiol. 1980;65:669–674. doi: 10.1104/pp.65.4.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomalainen A. Therapy for mitochondrial disorders: little proof, high research activity, some promise. Semin Fetal Neonatal Med. 2011;16:236–240. doi: 10.1016/j.siny.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Tan EK, Skipper LM. Pathogenic mutations in Parkinson disease. Hum Mutat. 2007;28:641–653. doi: 10.1002/humu.20507. [DOI] [PubMed] [Google Scholar]
- Vanlerberghe GC. Alternative oxidase: a mitochondrial respiratory pathway to maintain metabolic and signaling homeostasis during abiotic and biotic stress in plants. Int J Mol Sci. 2013;14:6805–6847. doi: 10.3390/ijms14046805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi T, Seo BB, Nakamaru-Osigo E, Marella M, Barber-Singh J, Yamashita T, et al. Can a single subunit yeast NADH dehydrogenase (Ndi1) remedy diseases caused by respiratory complex I defects? Rejuvenation Res. 2006;9:191–197. doi: 10.1089/rej.2006.9.191. [DOI] [PubMed] [Google Scholar]
- Zhang DW, Xu F, Zhang ZW, Chen YE, Du JB, Jia SD, et al. Effects of light on cyanide-resistant respiration and alternative oxidase function in Arabidopsis seedlings. Plant Cell Environ. 2010;33:2121–2131. doi: 10.1111/j.1365-3040.2010.02211.x. [DOI] [PubMed] [Google Scholar]