

Abstract
The endocannabinoids anandamide and 2-arachidonoylglycerol (2-AG) are lipid mediators that signal via CB1 and CB2 cannabinoid receptors and Gi/o-proteins to inhibit adenylyl cyclase and stimulate mitogen-activated protein kinase (MAPK). In the brain, CB1 receptors interact with opioid receptors in close proximity, and these receptors may share G-proteins and effector systems. In the striatum, CB1 receptors function in coordination with D1 and D2 receptors, and combined stimulation of CB1-D2 receptor heteromeric complexes promotes a unique interaction to stimulate cAMP production. CB1 receptors also trigger growth factor receptor signaling cascades in cells by engaging in cross-talk or inter-receptor signal transmission with the receptor tyrosine kinase (RTK) family. Mechanisms for CB1 receptor-RTK transactivation can include stimulation of signal transduction pathways regulated by second messengers such as phospholipase C (PLC), metalloprotease cleavage of membrane-bound precursor proteins such as epidermal growth factor which activate RTKs, RTK autophosphorylation, and recruitment of non-receptor tyrosine kinases. CB1 and CB2 receptors are expressed in peripheral tissues including liver and adipose tissue, and are induced in pathological conditions. Novel signal transduction resulting from endocannabinoid regulation of AMP-regulated kinase (AMPK) and peroxisome proliferator-activated receptors (PPARs) have been discovered from studies of hepatocytes and adipocytes. It can be predicted that drug discovery of the future will be based upon these novel signal transduction mechanisms for endocannabinoid mediators.
Keywords: anandamide, 2-arachidonoylglycerol, cannabinoid receptor heteromers, endocannabinoid, extracellular signal-regulated kinase (ERK), G-proteins, receptor tyrosine kinases, peroxisome proliferator-activated receptor (PPAR)
Overview and recent cannabinoid receptor signaling reviews
The endocannabinoids anandamide and 2-arachidonoylglycerol (2-AG) are the most well-characterized of lipid mediators that signal via CB1 and CB2 cannabinoid receptors, and the pharmacology has recently been reviewed [1,2]. Both established cannabinoid receptors are G-protein coupled receptors (GPCRs) that predominantly signal to effectors via Gi/o proteins. G-protein activation studies have shown that although 2-AG exhibits a high efficacy in stimulating the CB1 receptor to trigger these G-proteins, anandamide is only a weak partial agonist [3,4]. Compounds such as WIN55212-2, CP55940 and HU210 are high efficacy agonists, whereas Δ9-tetrahydrocannabinol (THC) is a partial agonist [3]. Antagonists that are selective for the CB1 receptor include rimonabant (also known as SR141716), taranabant, AM251, AM281 and LY320135, and antagonists for the CB2 receptor include SR144528 and AM630 [4,5].
Several recent reviews have provided an overview of the Gi/o-mediated inhibition of adenylyl cyclase and stimulation of mitogen-activated protein kinase (MAPK) by both CB1 and CB2 cannabinoid receptors [6–9]. CB1 receptors also regulate voltage-gated Ca2+ channels and certain K+ channels by direct interaction with the G-proteins released by agonist stimulation. This regulation of ion channel activity is the mechanism by which endocannabinoids can serve as retrograde neuromodulators in short-term synaptic plasticity including depolarization-induced suppression of inhibition and excitation, as well as the long-term depression of synaptic activity that suppresses neurotransmitter release for extended durations (see recent reviews [10–13]).
The CB2 receptor has been of particular interest because of its importance in immune surveillance in the brain as well as peripheral tissues (see recent reviews [14–17]). Novel lipid modulators that are analogous to the endocannabinoids have been discovered that play a role in neuropathic pain and other neuroinflammatory responses [18,19]. Pharmaceutical drug design is targeting selective CB2 receptor signaling, and compounds are becoming available that will be instrumental in promoting further investigation of signal transduction regulated by CB2 receptors [20,21].
The present review discusses recent investigations of cannabinoid receptor signaling that involve interactions with other GPCRs in close proximity that perhaps share G-proteins in novel ways. We also discuss cannabinoid receptor signaling that involves receptor and non-receptor tyrosine kinases, leading to divergence of signaling pathways beyond traditional G-protein effectors. Finally, the appreciation of cannabinoid receptors in peripheral tissues has opened the investigation of alternative signaling pathways used by these tissues. Greater understanding of these recent research findings may redirect development of therapeutic strategies to incorporate novel signal transduction enzymes and receptors as targets for future drug design.
CB1 cannabinoid receptor signaling: Coordination with other GPCRs in the brain
The CB1 receptor is found in relatively high densities throughout the CNS, where endocannabinoids act as retrograde neuromodulators that are synthesized by the postsynaptic neuron upon depolarization, transverse the extracellular space and decrease neurotransmitter release by activating CB1 receptors located on the presynaptic terminal [22]. The signal transduction machinery responsible includes Gi/o via direct Gβγ-mediated inhibition of Ca2+ influx through voltage gated calcium channels [23–25] as well as inhibition of cAMP/PKA-induced phosphorylation of ion channels proteins [26].
Interactions between the cannabinoid and opiate systems have been suspected for several decades, due largely to the fact that drugs from these classes share many pharmacological actions, such as antinociception, sedation, hypoactivity and hypothermia (for review, see [27,28]). Opioid and cannabinoid receptors share similar signal transduction pathways involving Gi/o-mediated inhibition of adenylyl cyclase and a decrease in cAMP concentrations in the striatum [29–31]. CB1 and μ-opioid receptors are colocalized on axons and dendrites and also share trans-synaptic interactions within striatal patch GABAergic neurons and within the shell of the nucleus accumbens [32,33]. CB1 receptors and μ-opioid receptors interact in signal transduction complexes that result in the functional endpoint of attenuating GABA and glutamate release [34,35]. Although Δ9-THC has been demonstrated to non-competitively decrease μ-opioid receptor binding to ligands [36], cannabidiol, which exhibits very poor affinity for CB1 receptors, has also been shown to influence opioid receptor binding [37]. Because these cannabinoid compounds were used at concentrations that exceeded their aqueous solubility, it is not clear whether the modulation observed using in vitro radioligand binding assays results from heteromerization of receptor proteins, allosteric modulation by these ligands at the μ-opioid receptor protein, or membrane fluidity changes imposed by lipophilic ligands altering signal transduction properties. In an oocyte model co-expressing exogenous CB1 or μ-opioid receptors plus K+ channels, cannabinoid agonists that regulated the CB1 receptor had no influence on μ-opioid receptor-mediated conductances, suggesting inability of the cannabinoid ligands to serve as allosteric ligands directly at the μ-opioid receptor protein [38].
Effects of Δ9-THC [36] but not the highly efficacious CB1 agonist desacetyllevonanatradol (DALN) [29] were reported for δ-opioid receptor binding. In a neuronal cell line endogenously expressing both CB1 and δ-opioid receptors, no pharmacological interactions were observed between receptors for the Gi-mediated inhibition of adenylyl cyclase [29]. However, the observation that a ceiling on the maximal inhibition could be obtained upon stimulation of both receptor types suggests the existence of shared components of the signal transduction module (such as the G-proteins or the effectors) limiting responses when both receptor types were stimulated [29]. Detailed investigation indicated that endogenously expressed CB1 and δ-opioid receptors in a neuronal cell do not acutely regulate the same G-proteins [39,40]. Chronic exposure to opioid ligands could cross-desensitize the cellular response to cannabinoid agonists, but not the converse, suggesting the occurrence of modulatory interactions between these receptors at the cellular level [39].
Partial cross-tolerance has been demonstrated between opioids and cannabinoids in catalepsy and hypoactivity [41,42], behaviors believed to be mediated by the striatum. For these cannabinoid responses, the MAPK pathway was modified in the development of tolerance [43,44] and the mechanism could involve src kinase [45]. Mechanisms for long-term cross-regulation might include the cannabinoid-mediated synthesis of enkephalin peptides [46,47] or the opioid-mediated regulation of CB1 receptor synthesis [48].
Cannabinoid and opioid-mediated antinociception may be predominantly regulated at the level of the thalamus, amygdala, and the descending noradrenergic pathways and spinal cord [49,50]. One mechanism by which this interaction occurs is the cannabinoid-regulated release of dynorphin peptides in the spinal cord [51,52]. Asymmetrical cross-tolerance has been observed such that morphine-tolerant animals are tolerant to the antinociceptive effects of Δ9-THC [53]. On the other hand, blocking CB1 receptors with daily injections of rimonabant had little effect on the development of analgesic tolerance to morphine [54]. PKA and PKC were involved in the maintenance of the morphine analgesic tolerance at the level of supraspinal structures, but not in the spinal cord [55,56]. Okadaic acid-sensitive PP1/PP2A phosphatase appeared to play a role in the sensitivity to morphine in tolerant mice, although calcineurin (also known as PP2B) was not involved [57]. In contrast, PKA, MAPK, as well as src kinases (but not PKC, PKG, phosphatidylinositol-3 kinase (PI-3K), or G-protein receptor kinases) were involved in the maintenance of tolerance to Δ9-THC [43,45,58]. An increase in PKA activity was observed in Δ9-THC analgesic-tolerant rats [59,60].
Evidence of GPCR cross-talk exists for the functional interaction of D1 and D2 dopamine and CB1 receptors. In rat striatal slice preparations, both receptors inhibited D1 receptor-stimulated cAMP production [30]. Similarly, in isolated striatal membranes from rat and monkey, both cannabinoid and D2 agonists inhibited forskolin-stimulated adenylyl cyclase activity, and the cannabinoid agonist inhibited D1-stimulated adenylyl cyclase [61]. However, concurrent stimulation of CB1 and D2 receptors in primary striatal cultures resulted in an increase in cAMP production attributed to a stimulation of Gs rather than Gi [62]. A similar increased cAMP production was observed by sub-maximal stimulation of both CB1 and D2 receptors exogenously expressed in host cells [63]. Cannabinoid-induced cAMP accumulation was seen with co-expression of receptors in HEK293 cells, in the absence of a D2 agonist, suggesting the expression of the D2 receptor was sufficient to shift the signaling of the CB1 receptor from Gi to Gs [64]. The shift in signaling was reversed by a CB1 antagonist, indicating this was a CB1 receptor specific mechanism [62,64]. It was postulated that D2 receptors might sequester Gi/o proteins away from the CB1 receptors, thereby promoting the interaction of CB1 with Gs [64]. However, co-stimulation by cannabinoid and D2 agonists in rat striatal membranes did not produce an increase in forskolin-stimulated adenylyl cyclase activity, even after treatment of the membranes with pertussis toxin A subunit [61]. This discrepancy observed between exogenously expressed receptors in intact cells versus striatal membranes could be attributed to differences between the composition and abundance of D2-CB1 receptor-G-protein modules available in the expression systems versus the striatal tissue. Another explanation is the diversity of adenylyl cyclase effectors: the increase in cAMP by CB1 and D2 receptor co-stimulation may be mediated by Gβγ dimers stimulating adenylyl cyclase isozymes II and IV [65]. In support of the latter, a study by Yao et. al. revealed brief D2 or CB1 agonist treatment of NG108-15 cells expressing exogenous D2 receptors increased cAMP and promoted PKA catalytic subunit α translocation into the nucleus [66]. Combinations of sub-threshold doses of CB1 and D2 agonists, which exerted no effects individually, produced a synergistic stimulation of PKA catalytic subunit α translocation when applied together. These effects were blocked by pertussis toxin, indicating a requirement for Gi/o proteins, and were also attenuated by overexpression of a Gβγ inhibitor peptide, indicating the requirement for Gβγ [66].
The first evidence of CB1 multimers came from the discovery of a high molecular weight band in CB1 Western blots, indicative of a multimeric CB1 complex, but devoid of G-proteins [67]. Later studies, using an antibody preferentially recognizing an epitope in the multimeric form of the CB1 receptor, demonstrated CB1 oligomers exist throughout the brain in a pattern similar to that seen using antibodies recognizing both monomeric and multimeric forms of the receptor, indicating the natural state of the CB1 receptor may be in a multimeric form [68]. The propensity of CB1 receptors to form homo-dimers has further been suggested by co-immunoprecipitation experiments using co-expressed epitope-tagged versions of the CB1 receptors [69]. Heteromers of CB1 receptors have been demonstrated by interactions with a variety of other GPCRs including D2-dopamine [63,70], A2A-adenosine [71], orexin [72], and μ-opioid receptors [34,73]. Most recent studies have utilized fluorescent and bioluminescence resonance energy transfer techniques (FRET and BRET respectively), which require a close physical proximity between individual components, in order to demonstrate CB1-GPCR multimeric relationships. Marcellino et al. [70] demonstrated a functional heteromer between CB1 and D2 receptors using FRET analysis. Behavioral experiments in this study indicated a sub-threshold dose of the CB1 agonist CP55940 effectively blocked D2 agonist quinpirole-mediated increases in locomotor activity. CB1-A2A heteromers have been demonstrated using co-immunoprecipitation and BRET experiments in striatal membranes [71]. Functional assays also revealed the motor depressant effects of a CB1 agonist administered directly into the striatum were blocked by pretreatment with an A2A antagonist [71]. These results have been followed up with an enhanced resonance energy transfer technique known as sequential BRET-FRET, which is designed to detect the close association of three intramolecular components [74]. Co-transfection and overexpression of D2, A2A and CB1 receptors in vitro resulted in a strong BRET-FRET signal implying the existence of a trimeric complex under such conditions. The functional significance of this heteromer has yet to be determined, and its existence in vivo has yet to be established. However, several technical hurdles exist for establishing the role of these complexes in vivo. The majority of evidence implicating a direct physical relationship between GPCRs depends heavily on overexpressing tagged forms of the receptors. However, most GPCRs are expressed at very low levels endogenously, and the non-physiological concentrations achieved in most in vitro studies may push the interactions between GPCRs to physiologically irrelevant pathways. The natural complexity of the striatum which possesses a multitude of cell types, the potential for interactions between specific GPCRs, and diversity in neuronal network connections, greatly limits the ability to study only one particular intra-molecular interaction between GPCRs in vivo. Therefore, although the CB1 receptor has the potential to interact with a variety of other GPCRs, both in terms of physical intramembrane reactions and through cross-talk between effector systems, combined in vitro and in vivo approaches are necessary to fully understand the functional nature of these interactions.
Cross-talk between CB1 receptors and Receptor Tyrosine Kinases (RTKs)
GPCRs have traditionally been recognized for their ability to transduce information provided by extracellular stimuli to the cell interior through their interaction with heterotrimeric G-proteins which positively or negatively regulate the activity of a variety of downstream intracellular effectors to influence second messenger levels and ultimately cellular responses. Many GPCRs also trigger growth factor receptor signaling cascades in cells by engaging in cross-talk or inter-receptor signal transmission with the RTK family [75]. RTK transactivation by GPCRs can involve multiple mechanisms, including (1) second messengers and/or signal transduction pathways regulated by second messengers, such as phospholipase C (PLC), (2) metalloprotease cleavage of membrane-bound precursor proteins (e.g., heparin-binding epidermal growth factor (EGF)) which binds to and activates RTKs, (3) RTK auto-phosphorylation, and (4) recruitment of non-receptor tyrosine kinases (e.g. Src family kinases) [75–77].
Cross-talk between CB1 receptors and RTKs was first described in non-neuronal Chinese hamster ovary (CHO) cells expressing endogenous RTKs for insulin and insulin-like growth factor 1 (IGF-1) and transfected with the human CB1 receptor [78]. RTK activation by their natural ligands produced MAPK activation, which was antagonized by the CB1 antagonist SR141716A (rimonabant). In contrast, rimonabant had no effect on MAPK activation by either ligand in untransfected CHO cells, suggesting the rimonabant-mediated effect in CB1-CHO cells involved cross-talk between CB1 receptors and RTKs for insulin or IGF-1. Cannabinoid agonists also induced MAPK activation in U373-MG glioblastoma cells and NCI-H292 lung carcinoma cells via EGF receptor transactivation [79]. CB1 receptor-mediated EGF receptor transactivation involved activation of tumor necrosis factor α-converting enzyme (TACE/ADAM17), which is a membrane-bound metalloprotease catalyzing the release of growth factor-like ligands to activate the EGF receptor [79]. In addition to cancer cells and non-neuronal cells, CB1 receptor-mediated RTK transactivation may play a role in a novel signaling pathway leading to intracellular Ca2+ influx in neurons. The CB1 agonist DALN could facilitate opening of L-type voltage-gated Ca2+ channels to increase intracellular Ca2+ levels in N18TG2 cells [80,81]. This effect appeared to be mediated by the MAPK cascade because Ca2+ uptake in these cells was diminished when DALN-mediated ERK1/2 phosphorylation was inhibited by the MAP-ERK kinase (MEK) inhibitor PD98059 [81]. DALN-mediated ERK1/2 phosphorylation and Ca2+ influx were both blocked by the PKC inhibitor chelerythrine, the calmodulin antagonist W-7, and the metalloprotease inhibitor o-phenanthroline [81]. The role of PKC in CB1 receptor-mediated ERK1/2 activation may involve a dual mechanism, because PKC can activate ERK1/2 by: (1) directly phosphorylating and activating Raf, the initial component of the MAPK cascade, or (2) activating metalloprotease enzymes, cleaving ligands that bind to and activate RTKs [76,82,83]. The finding that DALN-mediated Ca2+ uptake in N18TG2 cells was antagonized by the metalloprotease inhibitor o-phenanthroline suggested RTK transactivation plays a role in this process. This suspicion was supported by the observation that oxindole-1, a selective vascular endothelial growth factor (VEGF) receptor inhibitor, abolished the effect of DALN on ERK1/2 phosphorylation [81,84]. These findings indicate VEGF receptor transactivation in the CB1-MAPK-Ca2+ cascade in N18TG2 cells. The entire process begins with DALN stimulation of CB1 receptors, which promotes release of Gβγ, which activates PI-3K, thereby promoting Raf-mediated ERK1/2 activation and ERK modulates voltage-gated Ca2+ channels to cause Ca2+ influx [85,86]. A proposed model for CB1 receptor-mediated Ca2+ influx is shown in Fig. 1A. These pathways could potentially contribute to the neurotoxic effects of cannabinoids, because both cannabinoids and Ca2+ influx through L-type voltage gated Ca2+ channels have been shown to induce neurodegeneration [87–89].
Fig. 1. Putative mechanisms of cross-talk between the CB1 receptor and RTKs in neurons.
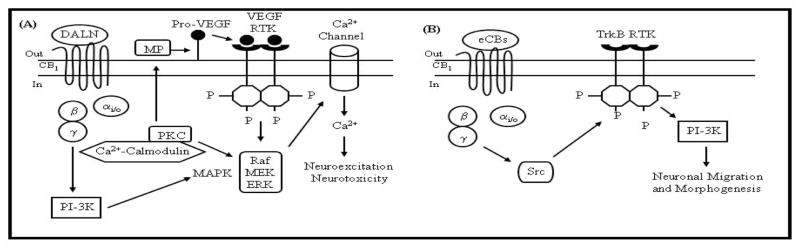
(A) The synthetic CB1 receptor agonist desacetyllevonantradol (DALN) potentiates Ca2+ influx in N18TG2 neuroblastoma cells via CB1 receptor-mediated transactivation of the VEGF receptor [80–89]. Agonist-stimulated CB1 receptors interact with Gi/o to release Gβγ, which activate PI-3K, thereby promoting Raf-mediated ERK1/2 activation. By this scenario, Ca2+ enters the cell following CB1 receptor-mediated ERK1/2 modulation of voltage-gated Ca2+ channel activity. Activated PKC could stimulate the MAPK cascade by phosphorylating Raf or by stimulating a metalloprotease that catalyzes production of VEGF (or other growth factor proteins) to activate RTKs. The transactivated VEGF receptor might also activate the MAPK cascade and further potentiate Ca2+ influx into neuronal cells. (B) The CB1 receptor mediates Src kinase-dependent TrkB RTK transactivation in neurons (see text).
CB1 receptor-mediated transactivation of RTKs could also play an important role in regulating neuronal migration and differentiation during embryonic development [90]. The effects of CB1 receptor activation on neuronal positioning and morphogenesis were described in cortical cholecystokinin-positive GABAergic interneurons expressing CB1 receptors [90]. These interneurons migrated towards anandamide (100 nM) in a Boyden chamber assay. CB1 receptor involvement in anandamide-induced chemotaxis was supported by evidence that (1) the CB1 receptor antagonist AM251 blocked the chemoattractive properties of anandamide and (2) parvalbumin-expressing interneurons lacking CB1 receptors failed to exhibit a migratory response towards anandamide [90]. The chemotaxic effects of anandamide were additive with brain-derived neurotrophic factor (BDNF), an endogenous ligand of the TrkB receptor. However, anandamide blocked neurite extension and also attenuated BDNF-induced neurite growth in a CB1 receptor-dependent manner, which prompted the investigators to determine if anandamide’s effects on developing interneurons were mediated through CB1 receptor transactivation of TrkB receptors (Fig. 1B). Immunocytochemical analysis revealed colocalization of CB1 receptors and TrkB receptors on terminal axon segments of developing interneurons [90]. PC12 pheochromocytoma cells co-expressing exogenous TrkB and CB1 were treated with anandamide and 2-AG, which promoted TrkB-CB1 receptor complex formation and TrkB receptor phosphorylation [90], suggesting cross-talk occurs between CB1 and TrkB receptors. The mechanism for transactivation could involve Src kinases, because the Src kinase inhibitor PP2 prevented CB1 receptor transactivation of TrkB receptors [90]. Cross-talk between CB1 receptors and TrkB receptors may have important physiological implications because anandamide-induced interneuron migration was blocked by the Trk receptor inhibitor K252a [90].
Cannabinoid Signal Transduction in Liver and Adipose Tissues
It is well established that the endocannabinoid system plays a role in the regulation of appetite, food intake and energy balance within the mesolimbic and hypothalamic areas of the brain [91–93]. In addition to their effects in the brain, cannabinoid receptors and endocannabinoids are present and contribute to the metabolic regulation of glucose homeostasis, lipid metabolism, and other metabolic functions in the gastrointestinal tract, liver, white adipose, skeletal muscle and pancreas [94].
Hepatic Cannabinoid Signaling
CB1 receptor expression has been reported in hepatocytes and stellate cells, as well as the vascular endothelial cells which line the portal vein [95–99] [95][96–99]. CB2 receptors are expressed in stellate cells [98,100] and Kupffer cells, which are the resident macrophages of the liver [101]. Hepatic CB1 receptor mRNA and anandamide levels were increased in mice on a high fat diet [96]. In these animals, an increase in fatty acid synthesis was promoted by the CB1 receptor via increases in de novo hepatic lipogenesis through activation of the fatty acid biosynthetic pathway [96], a response not observed in transgenic CB1-knockout or liver-specific CB1-knockout mice [97]. Mice on an alcohol (low-fat) diet exhibited increased stellate cell production of 2-AG and increased hepatocyte expression of CB1 receptors, associated with lipogenesis and hepatic steatosis [97].
Cannabinoid effects on fatty acid synthesis are a function of CB1 receptors on hepatocytes [96,102]. Agonist stimulation of CB1 receptors increased hepatic gene expression of the lipogenic transcription factor sterol regulatory element binding protein (SREBP-1c), leading to increased expression of the enzymes acetyl-CoA carboxylase-1 and fatty acid synthase [96]. CB1 receptor stimulation also decreased carnitine palmitoyltransferase-1 activity [97]. Phylogenetically, this mechanism has been conserved from at least as early as fish [103]. Agonist-stimulated CB1 receptors also promoted SREBP-1c and FAS expression in the hypothalamus, which regulates high-fat feeding behaviors [96].
Anandamide levels were increased due to cirrhosis in the liver, and this was associated with the pathological responses mediated by fibrin-producing stellate cells as well as the vascular endothelium [101,104]. The necrotic cell death associated with endocannabinoids in stellate cells appears to be associated with the accumulation of reactive oxygen species, rather than effects due to CB1- or CB2-mediated signal transduction [98,99,105]. Increased density of CB1 receptors was detected in hepatic endothelial cells cultured from a patient with cirrhosis compared with non-cirrhotic controls [95]. Anandamide promoted hepatic vascular relaxation, and this response was potentiated by chronic (7 day) bile duct ligation [104]. This enhancement was attributed to NO production via inducible nitric oxide synthesis (iNOS) [104]. Inasmuch as endogenous anandamide was uniquely able to regulate vascular reactivity via CB1 and VR1 receptors in cirrhosis, but not in normal tissue [106], it is possible that signaling via these receptors, when combined with the high concentrations of NO produced by iNOS, elicits an additive or synergistic response.
Discovery of the weight loss benefit of the CB1 antagonist, rimonabant, has elicited research in the area of metabolic energy balance regulation by the endocannabinoid system. Although rimonabant decreased appetite initially, its long-lasting effects may be due to its actions on peripheral tissues [107]. Kola and colleagues [108] explored the possible link between the fuel-sensor, AMP-activated protein kinase (AMPK), and the endocannabinoid system in the hypothalamus, liver and adipose tissue. In the rat hypothalamus, central or peripheral injection of 2-AG increased AMPK activity by 50% and 75%, respectively [108]. In contrast, Δ9-THC treatment inhibited rat liver AMPK activity by 62% and phosphorylation by 74%, whereas Δ9-THC inhibited AMPK activity by 25% in both subcutaneous and visceral rat adipose tissue [108]. AMPK activation inhibited fat deposition and enhanced fat breakdown in adipose tissue, thereby resulting in reduction of body weight, whereas activation of AMPK resulted in enhanced fatty acid oxidation in the liver [109]. Therefore, inhibition of AMPK activity may be a mechanism by which cannabinoid agonists promote fuel storage.
As predicted, CB1 antagonists elicit effects opposite to those of cannabinoid agonists on AMPK in the liver. Lee and colleagues [110] reported the potent CB1 antagonist, AM251, activated AMPK in HepG2 hepatoma cells. AMPK phosphorylation was dose-dependently and transiently increased by AM251 treatment, peaking at 30 min and then decreasing to basal levels [110]. Tedesco and colleagues [111] provided evidence that CB1 antagonism activates AMPK in adipose tissue. Rimonabant treatment of cultured mouse white adipocytes increased AMPK phosphorylation by 50% and activity by 75%. Additionally, in vivo studies revealed the effects of diet on AMPK. AMPK activity in fat tissue of CB1 knock-out mice was similar to wild-type mice and the activity level was not altered by high-fat diet. However, AMPK phosphorylation and activity were decreased by 60% in adipocyte lysates when wild-type mice were fed a high-fat diet [111]. Therefore, CB1 receptor blockade in vivo prevents inhibition of AMPK activity by a high-fat diet, providing further evidence in support of the endocannabinoid system playing a role in energy homeostasis in peripheral tissues.
Peroxisome proliferator-activated receptors (PPARs) are important modulators of cellular metabolism in metabolically active tissues including liver and adipose tissue [112,113]. PPARs are nuclear receptors which, in response to ligand binding, transactivate target genes involved in metabolic regulation, energy homeostasis and cell differentiation [112,113]. The three PPAR subtypes include: PPARα, expressed predominantly in liver, muscle, and heart tissue; PPARγ, expressed predominately in adipose tissue and macrophages; and PPARδ, expressed ubiquitously. PPAR ligands include fatty acids so it is not surprising that cannabinoids have been found to bind some PPAR isoforms. Cannabinoids able to bind PPARα include anandamide, noladin ether, virodhamine, WIN55212-2 and palmitoylethanolamide (PEA) [114–117]. Oleoylethanolamide (OEA), a non-CB1-binding analog of anandamide [118], has been shown to induce satiety and reduce body weight gain in animals [119–121]. In searching for the mechanism of action, OEA was found to bind and increase transcriptional activity of PPARα [122]. In isolated rat adipocytes and hepatocytes, OEA treatment increased fatty acid oxidation [123]. In addition, in vivo administration of OEA induced lipolysis in rats and wild type mice, but not genetically deleted PPARα (−/−) mice [123]. These data suggest an important role for OEA in hepatic lipid metabolism.
Adipose Signal Transduction
White adipocytes express CB1 and CB2 receptors in both preadipocyte and mature differentiation states [124–127], and anandamide and 2-AG were detected in mouse epididymal fat and human visceral fat [128]. Conditions of obesity in humans and diet-induced obesity in mice led to increased levels of 2-AG in visceral fat compared to lean controls [128]. The expression of CB1 receptor mRNA has also been confirmed in rat subcutaneous adipose tissue and in a mouse preadipocyte cell line, 3T3 F442A, by reverse transcription polymerase chain reaction. Additionally, CB1 was found to be up-regulated threefold in obese Zucker fa/fa rats compared with lean littermates and similarly up-regulated in differentiated (mature) 3T3 F442A adipocyte compared with preadipocyte cultures [125]. Expression of the CB1 receptor may respond to metabolic changes in adipose tissue induced by fat accumulation and/or by differentiation of cells to encompass a fat-storing function. Furthermore, functionality of the CB1 receptor was confirmed in 3T3 F442A cells by showing that rimonabant treatment was able to inhibit basal and serum-induced ERK1/2 activation [126].
The classical signal transduction pathways and second messengers associated with CB1 receptor function in the brain, such as cAMP, NO, and the MAPK pathways, have not been rigorously studied in peripheral tissues. However, several groups have examined these pathways while studying the role of the endocannabinoid system in adipose tissue. Roche and colleagues [129] reported the presence of CB1 and CB2 receptors in human subcutaneous and omental adipose tissue with 130- and 61-fold higher expression in mature adipocytes than in pre-adipocytes. The functionality of the receptors was demonstrated by treating mature or preadipocytes with CB1- and CB2- specific agonists and antagonists and measuring cAMP production. A dose-dependent increase in cAMP levels by treatment of cultured primary adipocytes with 2-AG was reversed by the CB1 specific antagonist, AM251 [129]. The CB2 specific antagonist, SR 144528, reversed the palmitoylethanolamide-induced inhibition of forskolin-stimulated cAMP levels, consistent with mediation by the CB2 receptor [129]. This study demonstrates (1) CB1 and CB2 receptor expression is induced by adipocyte differentiation and (2) CB1 and CB2 receptors can activate or inhibit the cAMP signal transduction pathway in adipocytes, respectively.
In their investigation into the role and underlying mechanism of anandamide-mediated glucose uptake in adipoctyes, Gasperi and colleagues [130] reported the presence of a functional AEA-related endocannabinoid system in 3T3-L1 cells. Both preadipocytes and mature adipocytes were found to express CB1 and CB2 receptors, as well as the enzymes necessary to synthesize (N-acylphosphatidylethanolamine-Phospholipase D), and degrade (fatty acid amide hydrolase) anandamide [130]. In addition, Western blot analysis detected inducible nitric oxide synthase (iNOS) expression in 3T3-L1 adipocytes treated with 10 μM anandamide, but not in untreated controls. These researchers concluded that anandamide induces a two-fold increase in insulin-stimulated glucose uptake in differentiated 3T3-L1 adipocytes in a CB1 receptor-dependent manner involving increased expression of iNOS [130].
Tedesco and colleagues [111] demonstrated cannabinoid receptor manipulation of NOS levels by using mouse primary white adipocytes, in which they tested the effects of CB1 receptor blockade on parameters of mitochondrial biogenesis and epithelial nitric oxide synthase (eNOS) expression. Rimonabant treatment of primary adipocytes resulted in increased mitochondrial mass, DNA levels, and gene expression. In addition, rimonabant treatment dose-dependently increased eNOS expression in cultured mouse primary white adipocytes [111]. The effects of rimonabant on mitochondrial biogenesis were counteracted by siRNA-mediated reduction of eNOS. In comparing these results to the Gasperi study [130], it is important to note that observations in rats have correlated iNOS upregulation with the down regulation of eNOS in fat and muscle tissues [131]. Interestingly, the results in these studies suggest opposite effects of cannabinoid receptor mediation on two different NOS enzymes in adipocytes.
PPARγ is an important receptor in adipose tissue where it is involved in adipocyte differentiation and inflammation. Adipogenesis is a prominent property of PPARγ ligands [132]. Anandamide, 2-AG, and the synthetic cannabinoid, ajulemic acid, bind PPARγ [130,133] [134] and induce adipocyte differentitation [132,133]. Similarly, the potent cannabinoid agonist, HU210, induced adipogenesis and increased PPARγ mRNA levels [128]. Other cannabinoids binding PPARγ include Δ9-THC, WIN55212-2, CP55940 and cannabidiol [135,136]. Unlike for PPARα, OEA and PEA do not bind or activate PPARγ [122] [133].
Although PPARδ has not been well studied, Yan and colleagues [127] provided evidence supporting participation in the endocannabinoid system. In 3T3-L1-preadipocyte cells subjected to RNA interference of PPARδ, CB1 expression was increased two-fold. Conversely, adenovirus-mediated overexpression of PPARδ significantly reduced CB1 expression by 50% and impeded adipocyte differentiation [127]. More research is necessary to elucidate the significance of PPARδ on CB1 expression and adipocyte metabolism. However, PPARα and PPARγ agonists are used in the management of type 2 diabetes and metabolic syndrome due to their lipid-lowering effects and their improvement of insulin sensitivity [112,113]. Therefore, it is possible that cannabinoids as ligands of PPARs may prove to be useful as therapeutic agents for metabolic diseases.
Summary and Future Directions
Endocannabinoid signaling in the brain has been investigated based upon prototypic cannabinoid compounds and synthetic analogs. Our recent appreciation that endocannabinoid mediators serve as retrograde messengers has increased interest in co-regulation of closely associated GPCRs and other receptors including RTKs. These novel interactions are likely sources of new drug development with the goal of narrowing selectivity such that unwanted side effects might be reduced. An understanding of how endocannabinoid drugs signal in peripheral tissues such as liver and adipose tissue is critical to development of novel treatments for disorders such as obesity and metabolic syndrome. The significance of novel signal transduction pathways in liver, adipose tissue, and other organs, is not yet fully elucidated and requires future studies. Nevertheless, the presence of novel signal transduction pathways in addition to classic CB1 and CB2 receptor signaling suggests that the endocannabinoid system may be fertile ground for future drug discovery.
Acknowledgments
The authors wish to thank the National Institute on Drug Abuse for funding for our own research and that of many of the research laboratories cited in this review.
Abbreviations
- 2-AG
2-arachidonoylglycerol
- AMPK
AMP-activated kinase
- BDNF
brain-derived neurotrophic factor
- CHO
Chinese hamster ovary
- DALN
desacetyllevonantradol
- EGF
epidermal growth factor
- eNOS
endothelial NO synthase
- ERK
extracellular signal-regulated kinase
- FRET, BRET
fluorescence or bioluminescence resonance energy transfer
- GPCR
G-protein coupled receptor
- IGF
insulin-like growth factor
- iNOS
inducible NO syn-thase
- MAPK
mitogen-activated protein kinase
- MEK
MAP-ERK kinase
- OEA
oleoylethanolamide
- PEA
pal-mitoylethanolamide
- PI-3K
phosphatidylinositol-3-kinase
- PLC
phospholipase C
- PKA
protein kinase A
- PKC
protein kinase C
- PPAR
peroxisome proliferator-activated receptors
- THC
Δ9-tetrahydrocannabinol
- RTK
receptor tyrosine kinase
- SREBP
sterol regulatory element binding protein
- VEGF
vascular endothelial growth factor
Footnotes
Conflict of Interest
The authors report no financial conflict of interest. The authors were supported by NIH grants R01-DA03690 (ACH), K01-DA024763 (CEB), and Reynolds Post-doctoral Fellowship (GDD).
References
- 1.Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG. Pharmacol Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- 2.Pertwee RG. Handb Exp Pharmacol. 2005:1–51. doi: 10.1007/3-540-26573-2_1. [DOI] [PubMed] [Google Scholar]
- 3.Childers SR. AAPS J. 2006;8:E112–E117. doi: 10.1208/aapsj080113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howlett AC, Padgett LW, Shim JY. Cannabinoid agonist and inverse agonist regulation of G-protein coupling. In: Reggio PH, editor. The Cannabinoid Receptors. Chapter 7. Humana Press Inc; 2009. pp. 173–202. [Google Scholar]
- 5.Pertwee RG. Life Sci. 2005;76:1307–1324. doi: 10.1016/j.lfs.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 6.Demuth DG, Molleman A. Life Sci. 2006;78:549–563. doi: 10.1016/j.lfs.2005.05.055. [DOI] [PubMed] [Google Scholar]
- 7.Diaz-Laviada I, Ruiz-Llorente L. Mini Rev Med Chem. 2005;5:619–630. doi: 10.2174/1389557054368808. [DOI] [PubMed] [Google Scholar]
- 8.Howlett AC. Handb Exp Pharmacol. 2005:53–79. doi: 10.1007/3-540-26573-2_2. [DOI] [PubMed] [Google Scholar]
- 9.McAllister SD, Glass M. Prostaglandins Leukot Essent Fatty Acids. 2002;66:161–171. doi: 10.1054/plef.2001.0344. [DOI] [PubMed] [Google Scholar]
- 10.Chevaleyre V, Takahashi KA, Castillo PE. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- 11.Freund TF, Katona I, Piomelli D. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- 12.Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- 13.Lovinger DM. Handb Exp Pharmacol. 2008:435–477. doi: 10.1007/978-3-540-74805-2_14. [DOI] [PubMed] [Google Scholar]
- 14.Cabral GA, Raborn ES, Griffin L, Dennis J, Marciano-Cabral F. Br J Pharmacol. 2008;153:240–251. doi: 10.1038/sj.bjp.0707584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Centonze D, Battistini L, Maccarrone M. Curr Pharm Des. 2008;14:2370–82. doi: 10.2174/138161208785740018. [DOI] [PubMed] [Google Scholar]
- 16.Miller AM, Stella N. Br J Pharmacol. 2008;153:299–308. doi: 10.1038/sj.bjp.0707523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wright KL, Duncan M, Sharkey KA. Br J Pharmacol. 2008;153:263–270. doi: 10.1038/sj.bjp.0707486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hohmann AG, Suplita RL. AAPS J. 2006;8:E693–E708. doi: 10.1208/aapsj080479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandez-Ruiz J, Pazos MR, Garcia-Arencibia M, Sagredo O, Ramos JA. Mol Cell Endocrinol. 2008;286:S91–S96. doi: 10.1016/j.mce.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 20.Anand P, Whiteside G, Fowler CJ, Hohmann AG. Brain Res Rev. 2008 doi: 10.1016/j.brainresrev.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marriott KS, Huffman JW. Curr Top Med Chem. 2008;8:187–204. doi: 10.2174/156802608783498014. [DOI] [PubMed] [Google Scholar]
- 22.Mackie K. J Neuroendocrinol. 2008;20(Suppl 1):10–14. doi: 10.1111/j.1365-2826.2008.01671.x. [DOI] [PubMed] [Google Scholar]
- 23.Guo J, Ikeda SR. Mol Pharmacol. 2004;65:665–674. doi: 10.1124/mol.65.3.665. [DOI] [PubMed] [Google Scholar]
- 24.Twitchell W, Brown S, Mackie K. J Neurophysiol. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- 25.Wilson RI, Kunos G, Nicoll RA. Neuron. 2001;31:453–462. doi: 10.1016/s0896-6273(01)00372-5. [DOI] [PubMed] [Google Scholar]
- 26.Deadwyler SA, Hampson RE, Mu J, Whyte A, Childers S. J Pharmacol Exp Ther. 1995;273:734–743. [PubMed] [Google Scholar]
- 27.Manzanares J, Corchero J, Romero J, Fernandez-Ruiz JJ, Ramos JA, Fuentes JA. Trends Pharmacol Sci. 1999;20:287–294. doi: 10.1016/s0165-6147(99)01339-5. [DOI] [PubMed] [Google Scholar]
- 28.Vigano D, Rubino T, Parolaro D. Pharmacol Biochem Behav. 2005;81:360–368. doi: 10.1016/j.pbb.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 29.Devane WA, Spain JW, Coscia CJ, Howlett AC. J Neurochem. 1986;46:1929–1935. doi: 10.1111/j.1471-4159.1986.tb08515.x. [DOI] [PubMed] [Google Scholar]
- 30.Bidaut-Russell M, Howlett AC. J Neurochem. 1991;57:1769–1773. doi: 10.1111/j.1471-4159.1991.tb06379.x. [DOI] [PubMed] [Google Scholar]
- 31.Childers SR, Fleming L, Konkoy C, Marckel D, Pacheco M, Sexton T, Ward S. Ann N Y Acad Sci. 1992;654:33–51. doi: 10.1111/j.1749-6632.1992.tb25954.x. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez JJ, Mackie K, Pickel VM. J Neurosci. 2001;21:823–833. doi: 10.1523/JNEUROSCI.21-03-00823.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pickel VM, Chan J, Kash TL, Rodriguez JJ, Mackie K. Neuroscience. 2004;127:101–112. doi: 10.1016/j.neuroscience.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 34.Rios C, Gomes I, Devi LA. Br J Pharmacol. 2006;148:387–395. doi: 10.1038/sj.bjp.0706757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schoffelmeer AN, Hogenboom F, Wardeh G, De Vries TJ. Neuropharmacology. 2006;51:773–781. doi: 10.1016/j.neuropharm.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 36.Vaysse PJ, Gardner EL, Zukin RS. J Pharmacol Exp Ther. 1987;241:534–539. [PubMed] [Google Scholar]
- 37.Kathmann M, Flau K, Redmer A, Trankle C, Schlicker E. Naunyn Schmiedebergs Arch Pharmacol. 2006;372:354–361. doi: 10.1007/s00210-006-0033-x. [DOI] [PubMed] [Google Scholar]
- 38.Kracke GR, Stoneking SP, Ball JM, Tilghman BM, Washington CC, Hotaling KA, Johnson JO, Tobias JD. Naunyn Schmiedebergs Arch Pharmacol. 2007;376:285–293. doi: 10.1007/s00210-007-0201-7. [DOI] [PubMed] [Google Scholar]
- 39.Shapira M, Gafni M, Sarne Y. Brain Res. 1998;806:26–35. doi: 10.1016/s0006-8993(98)00697-0. [DOI] [PubMed] [Google Scholar]
- 40.Shapira M, Vogel Z, Sarne Y. Cell Mol Neurobiol. 2000;20:291–304. doi: 10.1023/A:1007058008477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tulunay FC, Ayhan IH, Sparber SB. Psychopharmacology (Berl) 1982;78:358–360. doi: 10.1007/BF00433741. [DOI] [PubMed] [Google Scholar]
- 42.Narimatsu S, Yamamoto I, Watanabe K, Yoshimura H. Eur J Pharmacol. 1987;141:437–443. doi: 10.1016/0014-2999(87)90562-0. [DOI] [PubMed] [Google Scholar]
- 43.Rubino T, Forlani G, Vigano D, Zippel R, Parolaro D. Mol Cell Neurosci. 2004;25:355–362. doi: 10.1016/j.mcn.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 44.Rubino T, Forlani G, Vigano D, Zippel R, Parolaro D. J Neurochem. 2005;93:984–991. doi: 10.1111/j.1471-4159.2005.03101.x. [DOI] [PubMed] [Google Scholar]
- 45.Bass CE, Welch SP, Martin BR. Eur J Pharmacol. 2004;496:99–108. doi: 10.1016/j.ejphar.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 46.Valverde O, Noble F, Beslot F, Dauge V, Fournie-Zaluski MC, Roques BP. Eur J Neurosci. 2001;13:1816–1824. doi: 10.1046/j.0953-816x.2001.01558.x. [DOI] [PubMed] [Google Scholar]
- 47.Gerald TM, Howlett AC, Ward GR, Ho C, Franklin SO. Psychopharmacology (Berl) 2008 doi: 10.1007/s00213-008-1141-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Navarro M, Carrera MR, Fratta W, Valverde O, Cossu G, Fattore L, Chowen JA, Gomez R, Del AI, Villanua MA, Maldonado R, Koob GF, Rodriguez DF. J Neurosci. 2001;21:5344–5350. doi: 10.1523/JNEUROSCI.21-14-05344.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walker JM, Hohmann AG, Martin WJ, Strangman NM, Huang SM, Tsou K. Life Sci. 1999;65:665–673. doi: 10.1016/s0024-3205(99)00289-1. [DOI] [PubMed] [Google Scholar]
- 50.Walker JM, Hohmann AG. Handb Exp Pharmacol. 2005:509–554. doi: 10.1007/3-540-26573-2_17. [DOI] [PubMed] [Google Scholar]
- 51.Houser SJ, Eads M, Embrey JP, Welch SP. Brain Res. 2000;857:337–342. doi: 10.1016/s0006-8993(00)01981-8. [DOI] [PubMed] [Google Scholar]
- 52.Welch SP, Eads M. Brain Res. 1999;848:183–190. doi: 10.1016/s0006-8993(99)01908-3. [DOI] [PubMed] [Google Scholar]
- 53.Bloom AS, Dewey WL. Psychopharmacology (Berl) 1978;57:243–248. doi: 10.1007/BF00426745. [DOI] [PubMed] [Google Scholar]
- 54.Rubino T, Massi P, Vigano D, Fuzio D, Parolaro D. Life Sci. 2000;66:2213–2219. doi: 10.1016/s0024-3205(00)00547-6. [DOI] [PubMed] [Google Scholar]
- 55.Bernstein MA, Welch SP. Drug Alcohol Depend. 1997;44:41–46. doi: 10.1016/s0376-8716(96)01320-8. [DOI] [PubMed] [Google Scholar]
- 56.Smith FL, Javed R, Elzey MJ, Welch SP, Selley D, Sim-Selley L, Dewey WL. Brain Res. 2002;958:28–35. doi: 10.1016/s0006-8993(02)03394-2. [DOI] [PubMed] [Google Scholar]
- 57.Bernstein MA, Welch SP. Eur J Pharmacol. 1998;341:173–177. doi: 10.1016/s0014-2999(97)01502-1. [DOI] [PubMed] [Google Scholar]
- 58.Lee MC, Smith FL, Stevens DL, Welch SP. J Pharmacol Exp Ther. 2003;305:593–599. doi: 10.1124/jpet.102.044446. [DOI] [PubMed] [Google Scholar]
- 59.Rubino T, Vigano’ D, Massi P, Spinello M, Zagato E, Giagnoni G, Parolaro D. Neuropharmacology. 2000;39:1331–1336. doi: 10.1016/s0028-3908(99)00196-3. [DOI] [PubMed] [Google Scholar]
- 60.Vigano D, Rubino T, Vaccani A, Bianchessi S, Marmorato P, Castiglioni C, Parolaro D. Psychopharmacology (Berl) 2005;182:527–536. doi: 10.1007/s00213-005-0114-4. [DOI] [PubMed] [Google Scholar]
- 61.Meschler JP, Howlett AC. Neuropharmacology. 2001;40:918–926. doi: 10.1016/s0028-3908(01)00012-0. [DOI] [PubMed] [Google Scholar]
- 62.Glass M, Felder CC. J Neurosci. 1997;17:5327–5333. doi: 10.1523/JNEUROSCI.17-14-05327.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kearn CS, Blake-Palmer K, Daniel E, Mackie K, Glass M. Mol Pharmacol. 2005;67:1697–1704. doi: 10.1124/mol.104.006882. [DOI] [PubMed] [Google Scholar]
- 64.Jarrahian A, Watts VJ, Barker EL. J Pharmacol Exp Ther. 2004;308:880–886. doi: 10.1124/jpet.103.057620. [DOI] [PubMed] [Google Scholar]
- 65.Rhee MH, Nevo I, Avidor-Reiss T, Levy R, Vogel Z. Mol Pharmacol. 2000;57:746–752. doi: 10.1124/mol.57.4.746. [DOI] [PubMed] [Google Scholar]
- 66.Yao L, Fan P, Jiang Z, Mailliard WS, Gordon AS, Diamond I. Proc Natl Acad Sci USA. 2003;100:14379–14384. doi: 10.1073/pnas.2336093100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mukhopadhyay S, McIntosh HH, Houston DB, Howlett AC. Mol Pharmacol. 2000;57:162–170. [PubMed] [Google Scholar]
- 68.Wager-Miller J, Westenbroek R, Mackie K. Chem Phys Lipids. 2002;121:83–89. doi: 10.1016/s0009-3084(02)00151-2. [DOI] [PubMed] [Google Scholar]
- 69.Mackie K. Life Sci. 2005;77:1667–1673. doi: 10.1016/j.lfs.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 70.Marcellino D, Carriba P, Filip M, Borgkvist A, Frankowska M, Bellido I, Tanganelli S, Muller CE, Fisone G, Lluis C, Agnati LF, Franco R, Fuxe K. Neuropharmacology. 2008;54:815–823. doi: 10.1016/j.neuropharm.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 71.Carriba P, Ortiz O, Patkar K, Justinova Z, Stroik J, Themann A, Muller C, Woods AS, Hope BT, Ciruela F, Casado V, Canela EI, Lluis C, Goldberg SR, Moratalla R, Franco R, Ferre S. Neuropsychopharmacology. 2007;32:2249–2259. doi: 10.1038/sj.npp.1301375. [DOI] [PubMed] [Google Scholar]
- 72.Ellis J, Pediani JD, Canals M, Milasta S, Milligan G. J Biol Chem. 2006;281:38812–38824. doi: 10.1074/jbc.M602494200. [DOI] [PubMed] [Google Scholar]
- 73.Canals M, Milligan G. J Biol Chem. 2008;283:11424–11434. doi: 10.1074/jbc.M710300200. [DOI] [PubMed] [Google Scholar]
- 74.Carriba P, Navarro G, Ciruela F, Ferre S, Casado V, Agnati L, Cortes A, Mallol J, Fuxe K, Canela EI, Lluis C, Franco R. Nat Methods. 2008 doi: 10.1038/nmeth.1229. [DOI] [PubMed] [Google Scholar]
- 75.Lowes VL, Ip NY, Wong YH. Neurosignals. 2002;11:5–19. doi: 10.1159/000057317. [DOI] [PubMed] [Google Scholar]
- 76.Ferguson SS. Trends Neurosci. 2003;26:119–122. doi: 10.1016/S0166-2236(03)00022-5. [DOI] [PubMed] [Google Scholar]
- 77.Shah BH, Catt KJ. Trends Neurosci. 2004;27:48–53. doi: 10.1016/j.tins.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 78.Bouaboula M, Perrachon S, Milligan L, Canat X, Rinaldi-Carmona M, Portier M, Barth F, Calandra B, Pecceu F, Lupker J, Maffrand JP, Le Fur G, Casellas P. J Biol Chem. 1997;272:22330–22339. doi: 10.1074/jbc.272.35.22330. [DOI] [PubMed] [Google Scholar]
- 79.Hart S, Fischer OM, Ullrich A. Cancer Res. 2004;64:1943–1950. doi: 10.1158/0008-5472.can-03-3720. [DOI] [PubMed] [Google Scholar]
- 80.Rubovitch V, Gafni M, Sarne Y. Brain Res Mol Brain Res. 2002;101:93–102. doi: 10.1016/s0169-328x(02)00174-2. [DOI] [PubMed] [Google Scholar]
- 81.Rubovitch V, Gafni M, Sarne Y. Brain Res Mol Brain Res. 2004;120:138–144. doi: 10.1016/j.molbrainres.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 82.Cai H, Smola U, Wixler V, Eisenmann-Tappe I, Diaz-Meco MT, Moscat J, Rapp U, Cooper GM. Mol Cell Biol. 1997;17:732–741. doi: 10.1128/mcb.17.2.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Belcheva MM, Coscia CJ. Neurosignals. 2002;11:34–44. doi: 10.1159/000057320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Korzh A, Keren O, Gafni M, Bar-Josef H, Sarne Y. Brain Res. 2008;1189:23–32. doi: 10.1016/j.brainres.2007.10.070. [DOI] [PubMed] [Google Scholar]
- 85.Gomez del Pulgar T, Velasco G, Guzman M. Biochem J. 2000;347:369–373. doi: 10.1042/0264-6021:3470369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Galve-Roperh I, Rueda D, Gomez del Pulgar T, Velasco G, Guzman M. Mol Pharmacol. 2002;62:1385–1392. doi: 10.1124/mol.62.6.1385. [DOI] [PubMed] [Google Scholar]
- 87.Chan GC, Hinds TR, Impey S, Storm DR. J Neurosci. 1998;18:5322–5332. doi: 10.1523/JNEUROSCI.18-14-05322.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ho R, Ortiz D, Shea TB. J Alzheimers Dis. 2001;3:479–483. doi: 10.3233/jad-2001-3507. [DOI] [PubMed] [Google Scholar]
- 89.Pozzoli G, Tringali G, Vairano M, D’Amico M, Navarra P, Martire M. J Neurosci Res. 2006;83:1058–1065. doi: 10.1002/jnr.20794. [DOI] [PubMed] [Google Scholar]
- 90.Berghuis P, Dobszay MB, Wang X, Spano S, Ledda F, Sousa KM, Schulte G, Ernfors P, Mackie K, Paratcha G, Hurd YL, Harkany T. Proc Natl Acad Sci USA. 2005;102:19115–19120. doi: 10.1073/pnas.0509494102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pagotto U, Marsicano G, Cota D, Lutz B, Pasquali R. Endocr Rev. 2006;27:73–100. doi: 10.1210/er.2005-0009. [DOI] [PubMed] [Google Scholar]
- 92.Cota D, Tschop MH, Horvath TL, Levine AS. Brain Res Rev. 2006;51:85–107. doi: 10.1016/j.brainresrev.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 93.Kirkham TC. Behav Pharmacol. 2005;16:297–313. doi: 10.1097/00008877-200509000-00004. [DOI] [PubMed] [Google Scholar]
- 94.Cota D. Diabetes Metab Res Rev. 2007;23:507–517. doi: 10.1002/dmrr.764. [DOI] [PubMed] [Google Scholar]
- 95.Batkai S, Jarai Z, Wagner JA, Goparaju SK, Varga K, Liu J, Wang L, Mirshahi F, Khanolkar AD, Makriyannis A, Urbaschek R, Garcia N, Jr, Sanyal AJ, Kunos G. Nat Med. 2001;7:827–832. doi: 10.1038/89953. [DOI] [PubMed] [Google Scholar]
- 96.Osei-Hyiaman D, Depetrillo M, Pacher P, Liu J, Radaeva S, Batkai S, Harvey-White J, Mackie K, Offertaler L, Wang L, Kunos G. J Clin Invest. 2005;115:1298–1305. doi: 10.1172/JCI23057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Osei-Hyiaman D, Liu J, Zhou L, Godlewski G, Harvey-White J, Jeong WI, Batkai S, Marsicano G, Lutz B, Buettner C, Kunos G. J Clin Invest. 2008;118:3160–3169. doi: 10.1172/JCI34827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Siegmund SV, Uchinami H, Osawa Y, Brenner DA, Schwabe RF. Hepatology. 2005;41:1085–1095. doi: 10.1002/hep.20667. [DOI] [PubMed] [Google Scholar]
- 99.Siegmund SV, Qian T, de Minicis S, Harvey-White J, Kunos G, Vinod KY, Hungund B, Schwabe RF. FASEB J. 2007;21:2798–2806. doi: 10.1096/fj.06-7717com. [DOI] [PubMed] [Google Scholar]
- 100.Julien B, Grenard P, Teixeira-Clerc F, Van Nhieu JT, Li L, Karsak M, Zimmer A, Mallat A, Lotersztajn S. Gastroenterology. 2005;128:742–755. doi: 10.1053/j.gastro.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 101.Siegmund SV, Schwabe RF. Am J Physiol Gastrointest Liver Physiol. 2008;294:G357–G362. doi: 10.1152/ajpgi.00456.2007. [DOI] [PubMed] [Google Scholar]
- 102.Jeong WI, Osei-Hyiaman D, Park O, Liu J, Batkai S, Mukhopadhyay P, Horiguchi N, Harvey-White J, Marsicano G, Lutz B, Gao B, Kunos G. Cell Metab. 2008;7:227–235. doi: 10.1016/j.cmet.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 103.Migliarini B, Carnevali O. Mol Cell Endocrinol. 2008;286:S12–S16. doi: 10.1016/j.mce.2008.01.021. [DOI] [PubMed] [Google Scholar]
- 104.Moezi L, Rezayat M, Samini M, Shafaroodi H, Mehr SE, Ebrahimkhani MR, Dehpour AR. Eur J Pharmacol. 2004;486:53–59. doi: 10.1016/j.ejphar.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 105.Siegmund SV, Seki E, Osawa Y, Uchinami H, Cravatt BF, Schwabe RF. J Biol Chem. 2006;281:10431–10438. doi: 10.1074/jbc.M509706200. [DOI] [PubMed] [Google Scholar]
- 106.Moezi L, Gaskari SA, Liu H, Baik SK, Dehpour AR, Lee SS. Br J Pharmacol. 2006;149:898–908. doi: 10.1038/sj.bjp.0706928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sandoval D, Cota D, Seeley RJ. Annu Rev Physiol. 2008;70:513–535. doi: 10.1146/annurev.physiol.70.120806.095256. [DOI] [PubMed] [Google Scholar]
- 108.Kola B, Hubina E, Tucci SA, Kirkham TC, Garcia EA, Mitchell SE, Williams LM, Hawley SA, Hardie DG, Grossman AB, Korbonits M. J Biol Chem. 2005;280:25196–25201. doi: 10.1074/jbc.C500175200. [DOI] [PubMed] [Google Scholar]
- 109.Misra P. Expert Opin Ther Targets. 2008;12:91–100. doi: 10.1517/14728222.12.1.91. [DOI] [PubMed] [Google Scholar]
- 110.Lee YM, Uhm KO, Lee ES, Kwon J, Park SH, Kim HS. Biochem Biophys Res Commun. 2008;370:641–645. doi: 10.1016/j.bbrc.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 111.Tedesco L, Valerio A, Cervino C, Cardile A, Pagano C, Vettor R, Pasquali R, Carruba MO, Marsicano G, Lutz B, Pagotto U, Nisoli E. Diabetes. 2008;57:2028–2036. doi: 10.2337/db07-1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gross B, Staels B. Best Pract Res Clin Endocrinol Metab. 2007;21:687–710. doi: 10.1016/j.beem.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 113.Lee CH, Olson P, Evans RM. Endocrinology. 2003;144:2201–2207. doi: 10.1210/en.2003-0288. [DOI] [PubMed] [Google Scholar]
- 114.O’Sullivan SE. Br J Pharmacol. 2007;152:576–582. doi: 10.1038/sj.bjp.0707423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.LoVerme J, La Rana G, Russo R, Calignano A, Piomelli D. Life Sci. 2005;77:1685–1698. doi: 10.1016/j.lfs.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 116.Sun Y, Alexander SP, Garle MJ, Gibson CL, Hewitt K, Murphy SP, Kendall DA, Bennett AJ. Br J Pharmacol. 2007;152:734–743. doi: 10.1038/sj.bjp.0707478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jhaveri MD, Richardson D, Robinson I, Garle MJ, Patel A, Sun Y, Sagar DR, Bennett AJ, Alexander SP, Kendall DA, Barrett DA, Chapman V. Neuropharmacology. 2008;55:85–93. doi: 10.1016/j.neuropharm.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 118.Boring DL, Berglund BA, Howlett AC. Prostaglandins Leukot Essent Fatty Acids. 1996;55:207–210. doi: 10.1016/s0952-3278(96)90100-3. [DOI] [PubMed] [Google Scholar]
- 119.Rodriguez DF, Navarro M, Gomez R, Escuredo L, Nava F, Fu J, Murillo-Rodriguez E, Giuffrida A, LoVerme J, Gaetani S, Kathuria S, Gall C, Piomelli D. Nature. 2001;414:209–212. doi: 10.1038/35102582. [DOI] [PubMed] [Google Scholar]
- 120.Gaetani S, Oveisi F, Piomelli D. Neuropsychopharmacology. 2003;28:1311–1316. doi: 10.1038/sj.npp.1300166. [DOI] [PubMed] [Google Scholar]
- 121.Proulx K, Cota D, Castaneda TR, Tschop MH, D’Alessio DA, Tso P, Woods SC, Seeley RJ. Am J Physiol Regul Integr Comp Physiol. 2005;289:R729–R737. doi: 10.1152/ajpregu.00029.2005. [DOI] [PubMed] [Google Scholar]
- 122.Fu J, Gaetani S, Oveisi F, Lo VJ, Serrano A, Rodriguez DF, Rosengarth A, Luecke H, Di Giacomo B, Tarzia G, Piomelli D. Nature. 2003;425:90–93. doi: 10.1038/nature01921. [DOI] [PubMed] [Google Scholar]
- 123.Guzman M, Lo VJ, Fu J, Oveisi F, Blazquez C, Piomelli D. J Biol Chem. 2004;279:27849–27854. doi: 10.1074/jbc.M404087200. [DOI] [PubMed] [Google Scholar]
- 124.Cota D, Marsicano G, Tschop M, Grubler Y, Flachskamm C, Schubert M, Auer D, Yassouridis A, Thone-Reineke C, Ortmann S, Tomassoni F, Cervino C, Nisoli E, Linthorst AC, Pasquali R, Lutz B, Stalla GK, Pagotto U. J Clin Invest. 2003;112:423–431. doi: 10.1172/JCI17725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bensaid M, Gary-Bobo M, Esclangon A, Maffrand JP, Le Fur G, Oury-Donat F, Soubrie P. Mol Pharmacol. 2003;63:908–914. doi: 10.1124/mol.63.4.908. [DOI] [PubMed] [Google Scholar]
- 126.Gary-Bobo M, Elachouri G, Scatton B, Le Fur G, Oury-Donat F, Bensaid M. Mol Pharmacol. 2006;69:471–478. doi: 10.1124/mol.105.015040. [DOI] [PubMed] [Google Scholar]
- 127.Yan ZC, Liu DY, Zhang LL, Shen CY, Ma QL, Cao TB, Wang LJ, Nie H, Zidek W, Tepel M, Zhu ZM. Biochem Biophys Res Commun. 2007;354:427–433. doi: 10.1016/j.bbrc.2006.12.213. [DOI] [PubMed] [Google Scholar]
- 128.Matias I, Gonthier MP, Orlando P, Martiadis V, De Petrocellis L, Cervino C, Petrosino S, Hoareau L, Festy F, Pasquali R, Roche R, Maj M, Pagotto U, Monteleone P, Di M., V J Clin Endocrinol Metab. 2006;91:3171–3180. doi: 10.1210/jc.2005-2679. [DOI] [PubMed] [Google Scholar]
- 129.Roche R, Hoareau L, Bes-Houtmann S, Gonthier MP, Laborde C, Baron JF, Haffaf Y, Cesari M, Festy F. Histochem Cell Biol. 2006;126:177–187. doi: 10.1007/s00418-005-0127-4. [DOI] [PubMed] [Google Scholar]
- 130.Gasperi V, Fezza F, Pasquariello N, Bari M, Oddi S, Agro AF, Maccarrone M. Cell Mol Life Sci. 2007;64:219–229. doi: 10.1007/s00018-006-6445-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Valerio A, Cardile A, Cozzi V, Bracale R, Tedesco L, Pisconti A, Palomba L, Cantoni O, Clementi E, Moncada S, Carruba MO, Nisoli E. J Clin Invest. 2006;116:2791–2798. doi: 10.1172/JCI28570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mueller E, Drori S, Aiyer A, Yie J, Sarraf P, Chen H, Hauser S, Rosen ED, Ge K, Roeder RG, Spiegelman BM. J Biol Chem. 2002;277:41925–41930. doi: 10.1074/jbc.M206950200. [DOI] [PubMed] [Google Scholar]
- 133.Bouaboula M, Hilairet S, Marchand J, Fajas L, Le Fur G, Casellas P. Eur J Pharmacol. 2005;517:174–181. doi: 10.1016/j.ejphar.2005.05.032. [DOI] [PubMed] [Google Scholar]
- 134.Liu J, Li H, Burstein SH, Zurier RB, Chen JD. Mol Pharmacol. 2003;63:983–992. doi: 10.1124/mol.63.5.983. [DOI] [PubMed] [Google Scholar]
- 135.O’Sullivan SE, Kendall DA, Randall MD. Br J Pharmacol. 2004;142:435–442. doi: 10.1038/sj.bjp.0705810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.O’Sullivan SE, Tarling EJ, Bennett AJ, Kendall DA, Randall MD. Biochem Biophys Res Commun. 2005;337:824–831. doi: 10.1016/j.bbrc.2005.09.121. [DOI] [PubMed] [Google Scholar]