

Abstract
Purpose
To describe the phenotypic characteristics and clinical course of a sporadic case of congenital fibrosis of the extraocular muscles (CFEOM) and Möbius syndrome with a de novo mutation in the KIF21A gene encoding a kinesin motor protein.
Methods
An individual with the rare combination of CFEOM and Möbius syndrome underwent comprehensive ophthalmologic and neurological evaluations. Magnetic resonance imaging (MRI) including diffusion tensor imaging (DTI) tractigraphy at 3T field strength was used to evaluate orbital, encephalic, and intracranial nerve integrity. The proband and her healthy parents underwent screening for mutations in the KIF21A, PHOX2A, and TUBB3 genes.
Results
The patient exhibited congenital, nonprogressive, bilateral external ophthalmoplegia, bilateral ptosis, bilateral facial palsy, and developmental delay. Her inability to blink resulted in severe exposure keratopathy and subsequent corneal perforation requiring a penetrating keratoplasty. MRI revealed an unremarkable configuration of the axial central nervous system and preservation of the intracranial portion of cranial nerves I, II, III, V, VI, VII, and VIII (cranial nerve IV is not normally visualized by MRI). A novel and de novo heterozygous KIF21A mutation (c.1056C>G, p.Asp352Glu) in a highly conserved region of the gene was present in the proband.
Conclusions
The reported KIF21A D352E mutation and associated phenotype further expand the clinical and mutational spectrum of CFEOM and Möbius syndrome.
Introduction
Congenital cranial dysinnervation disorders include congenital fibrosis of the extraocular muscles (CFEOM), Duane’s retraction syndrome, Möbius syndrome, congenital ptosis, and congenital facial palsy [1,2]. “Classic” autosomal dominant CFEOM (CFEOM1) is the most common form of this paralytic strabismus syndrome characterized by bilateral blepharoptosis and ophthalmoplegia with the eyes fixed in an infraducted (downward) primary position [1]. Patients often have a compensatory chin-up posture. Residual eye movements are notable for jerky horizontal divergent or convergent movements on attempted vertical gaze, suggesting aberrant innervation. Neuroimaging typically shows hypoplasia of the muscles and the oculomotor nerve. CFEOM1 results from heterozygous mutations in the KIF21A (gene ID: 300158; OMIM 608283) gene encoding a kinesin motor protein [3]. Autosomal recessive CFEOM (CFEOM2) is marked by bilateral blepharoptosis and a large-angle exotropia without significant vertical deviation caused by loss of function splice-site and missense mutations in the ARIX/PHOX2A gene (gene ID: 401; OMIM 602753) [4]. ARIX is a homeobox gene expressed in the central and peripheral nervous system [5]. CFEOM3 is another subset of the disorder that is inherited as an autosomal dominant trait with incomplete penetrance with unilateral or bilateral ptosis and/or restrictions in ocular motility. Heterozygous mutations in the TUBB3 gene (gene ID: 22152; OMIM 602661) encoding for the neuron-specific beta-tubulin isotype III have been found in CFEOM3 patients [6]. A subset of these individuals may also manifest facial paralysis, intellectual impairments, and structural cerebral abnormalities such as dysgenesis of the corpus callosum, basal ganglia, or corticospinal tracts.
Minimal criteria for Möbius syndrome include congenital facial diplegia and abduction deficits, but abnormalities of the extremities or mental retardation are present in some cases [1,7]. Although Möbius syndrome has been attributed to brainstem ischemia in some cases, cytogenetic abnormalities have been identified in some individuals. Two loci (one on chromosome 3 and another on chromosome 10), as well as other chromosomal abnormalities, have been implicated in familial cases of the disorder [1].
Total external ophthalmoplegia combined with Möbius syndrome has been previously reported in a few patients. In a review of congenital cranial dysinnervation disorders, Traboulsi described two patients with the rare combination Möbius syndrome and CFEOM reminiscent of CFEOM1, but underlying genetic deficits were not reported [8]. Verzijl reported that three of 37 patients with Möbius syndrome had congenital fibrosis of the extraocular muscles [7]. Furthermore, Chew et al. reported a “TUBB3 E410K phenotype” syndrome in which eight unrelated individuals sharing this de novo mutation exhibited features of CFEOM, Möbius syndrome, Kallmann syndrome (hypogonadotropic hypogonadism and anosmia), characteristic midface hypoplasia, intellectual impairment, and cyclic vomiting [9]. The present study describes the phenotype and clinical course of a patient with congenital fibrosis of the extraocular muscles, Möbius syndrome, and developmental delay with a novel, de novo mutation in KIF21A.
Methods
The study was approved by the institutional review board of the University of Texas Southwestern Medical Center, Dallas, TX. Informed consent was obtained from the parents. The study adhered to the Declaration of Helsinki and ARVO statement for human subject research.
The patient, a 10-year-old, right-handed girl, underwent ophthalmological examination, physical examination, and neurological evaluation in addition to cranial magnetic resonance imaging (MRI) and diffusion tensor imaging (DTI) at 3T field strength. The parents and three siblings also underwent neurological and ophthalmological examination. Additional sources of information included medical records and family photographs.
Comparative genomic hybridization microarray studies were performed in the patient via genomic DNA labeling with 9 Cy5-dCTP. Purification of labeling products, array hybridization, washing, scanning, and data analysis were conducted by Signature Genomics Laboratories (Spokane, WA) using commercial methods. Interpretation of cytogenomic microarray was performed using the Signature Genomics Genoglyphix software. Blood samples were obtained from the patient and unaffected parents for DNA sequencing. Genomic DNA was directly isolated from leukocytes following standard methods. Paternity was examined by Promega PowerPlex 1.2 System genotyping (Madison, WI) with the ABI PRISM 310 Genetic Analyzer (Carlsbad, CA). PCR amplification and direct sequencing of exons 2, 8, 20, and 21 of the KIF21A gene (in which all mutations in CFEOM1/CFEOM3 individuals have been detected to date) [3,10-15], as well as all of the exons of PHOX2A and TUBB3 genes, was performed. All splice sites flanking the exons of interest were also sequenced. PCR primers for PHOX2A and KIF21A are shown in Table 1. The primers for TUBB3 were published previously [6]. The targeted exons were amplified with Taq DNA polymerase from 5Prime (Gaithersburg, MD) and/or AccuPrime GC-Rich DNA Polymerase from Invitrogen (Carlsbad, CA). Products were analyzed using an Applied Biosystems 3730×l DNA Analyzer (Life Technologies, Carlsbad, CA). Genomic DNA from 288 Hispanic controls from the Dallas Heart Study, a population-based sample, were screened for the KIF21A mutation (c.1056C>G) using real-time allelic discrimination assay (TaqMan SNP Genoptyping Assay; Life Technologies) [16].
Table 1. Sequencing primers.
| Gene | Exon | Forward primer 5’-3’ | Reverse primer 5’-3’ |
|---|---|---|---|
|
PHOX2A |
1 |
GTCCTGCGCGTTAAAAGG |
ATTCACTTGGCGAGCGG |
|
2 |
GATCTCCCTCCGCCTTTATC |
CATTAAGCTCCCACACCTCC |
|
|
3 |
TTTCCGAACCAGGATCTCAC |
GGAGGAGGTCCCGGTATAAA |
|
|
KIF21A |
2 |
TCATGATTTTGGGGGATTGT |
CAGCTCGAGAAATAATACCCAGTT |
|
8 |
TTCCCCAAAGTAAACAAAAGC |
GTTAAAGACTGTCCACAAGGAAAA |
|
|
20 |
TAGATACGCTCCCCCTTAGC |
GAAAAGCAAGCAGGAAGTGG |
|
| 21 | AATATGTGAAACAATAGGCTGTTG | TTTCTTACTCTTCCTGACTCTAAAGAA |
Results
Clinical manifestations and therapeutic course
The patient presented for medical attention at 5 months of age with bilaterally restricted ocular movements, an inability to blink, and lower facial weakness more pronounced on the right side. Ethnicity was Hispanic and there was no family history of consanguinity or eye movement abnormalities. The patient was born at term in March 2001 via spontaneous vaginal delivery following an unremarkable pregnancy. Karyotype analysis performed in August 2001 was normal. Her medical history was significant for developmental delay: Ambulation was accomplished at 1.5 years (typical ambulation was achieved at 10 months of age in the family) and expressive language developed at 3 years of age. Head circumference had remained between the 10th and 50th percentiles throughout her life. The patient exhibited facial nerve palsy, which was first noticed on examination at 5 months and was a persistent feature of her syndrome. Ophthalmic examination in 2002 revealed both eyes fixed in infraduction and severe restriction of ocular movements (under positive forced duction testing).
The patient underwent bilateral inferior rectus recessions in May 2002. In October 2003, she underwent bilateral lower lid horizontal tightening with lateral tarsal strips and bilateral lower lid retraction repair using Alloderm grafts (Lifecell, Branchburg, NJ). However, because of her inability to blink, she remained at risk for exposure keratopathy and developed a descemetocele in the right eye (oculus dexter [OD]), requiring emergent penetrating keratoplasty in October 2006 and bilateral medial and lateral tarsal pillar tarsorrhaphies in November 2006. The patient’s postkeratoplasty course was additionally complicated by recurrent epithelial defects and episodes of bacterial keratitis despite intact tarsal pillars and aggressive topical lubrication.
Her most recent exam had revealed bilateral facial nerve territory weakness, more pronounced on the right side (Figure 1A,B), with mask facies, inability to fully close her eyelids spontaneously, severe impairment of partial closure of her eyelids against minimal manual resistance, and inability to smile or elevate the angle of the mouth bilaterally or to puff her cheeks and blow. Hearing in the office setting was normal, as illustrated by her capacity to carry out a conversation. Auditory function as assessed by the Weber and Rinne maneuvers was also normal. Her visual acuity was 20/400 in the OD and 20/200 in the left eye (oculus sinister [OS]). Pupils were 2.5 mm OD and 1.5 mm OS and were minimally reactive bilaterally, without afferent pupillary defect. Alignment testing performed using the modified Krimsky method revealed 20–30 prism diopters of exotropia at near and distance with OS fixing. Extraocular motility remained severely restricted in all directions of gaze (−4) with minimal rotary nystagmus with bilateral incyclotorsion, suggestive of partial superior oblique function (Figure 2). Stereopsis was absent.
Figure 1.

Subject with CFEOM combined with Möbius syndrome. A: External photograph. Ptosis is present due to poor levator function; lower facial nerve weakness is present bilaterally, and is greater on the right. B: Photograph of subject attempting to smile. Note the weak contraction of the risorius muscle with subtle creasing and dimpling of skin on her lower face (arrow). The angles of the mouth are minimally elevated under the maximal forced smile.
Figure 2.
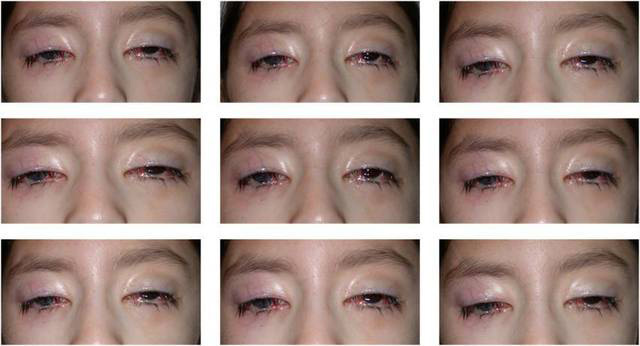
Subject in different fields of gaze. Motility examination demonstrates severe restriction in all fields of gaze. The only muscle movement was minimal rotary nystagmus with bilateral incyclotorsion suggestive of partial superior oblique muscle function.
The Worth four-dot test showed suppression of OD. Slit-lamp examination of the OD revealed normal tarsal pillars, an inferiorly decentered penetrating keratoplasty graft with stromal scarring, and a posterior chamber intraocular lens with posterior capsular opacification (Figure 3). The left eye exhibited mild anterior stromal scarring and thinning of the inferior cornea without an epithelial defect (Figure 3). MRI with DTI tractigraphy obtained at 3T field strength in June 2010 illustrated thinning of all extraocular muscles bilaterally with an unremarkable configuration of the axial central nervous system (CNS) and preservation of the intracranial portion of cranial nerves I, II, III, V, VI, VII, and VIII, although it was not possible to estimate oculomotor nerve thickness accurately by MRI (Figure 4). Ophthalmological and neurological examinations of the parents and siblings were normal.
Figure 3.
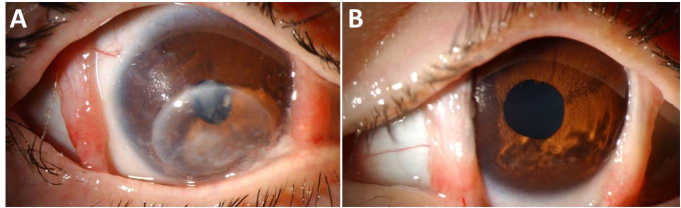
Slit lamp biomicroscopy. A: Right eye slit lamp photo. Note the medial and lateral tarsal pillars, inferiorly decentered corneal graft, and posterior chamber intraocular lens with posterior capsular opacification. B: Left eye slit-lamp photo.
Figure 4.
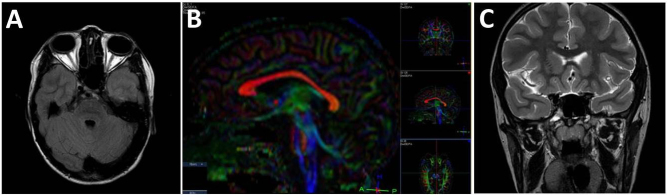
Magnetic resonance imaging. A: T1-weighted axial magnetic resonance imaging (MRI) shows diffuse thinning of the extraocular muscles. B: Sagittal diffusion tension imaging (DTI) demonstrates a normal corpus callosum configuration. C: T2-weighted coronal MRI illustrates a normal optic chiasm.
Molecular analysis
Paternity was confirmed. Genotyping identified a novel heterozygous 1056 C→G mutation at the third nucleotide position of codon 352 (D352E) on exon 8 of KIF21A, resulting in an aspartic acid to glutamic acid substitution. The unaffected parents and three siblings did not harbor the mutation (Figure 5). The missense variant occurred in a highly conserved region of the protein (Figure 6). Additionally, the variant was not found in public databases of dbSNP, 1000 Genomes, or the Exome Variant Server (EVS) [17-19]. The EVS alone had data from 6,503 exomes at the time of the query. The KIF21A mutation (c.1056C>G) was found to be absent in all 288 Hispanic controls using the allelic discrimination assay. DNA sequencing of the exons of PHOX2A and TUBB3 was normal. The E410K missense mutation was not present in TUBB3. Comparative genomic hybridization microarray results revealed no significant copy gains or losses at the KIF21A, PHOX2A, and TUBB3 loci or throughout the genome at any disease-associated loci, despite several copy number changes of unknown significance that included gains at 2q24.3, 14q32.33, 16p12.1, and losses at Xp22.33 ranging from 89.2 kb to 230.9 kb.
Figure 5.
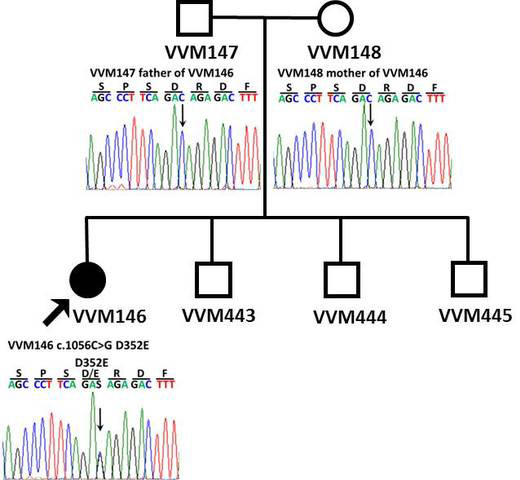
DNA sequencing. Sequence chromatographs of the family. The healthy parents harbor normal DNA sequences (top), whereas the patient with congenital fibrosis of the extraocular muscles (CFEOM) exhibits a de novo 1056 C 1 → G mutation at the third nucleotide position of codon 352 (D352E) on exon 8 of the KIF21A gene locus.
Figure 6.

Conservation of amino acid sequence. Aspartic acid residue at the position of the reported mutation is highly conserved across species; this region of the protein domain is highly conserved.
Discussion
The subject exhibited an extremely rare clinical presentation of CFEOM combined with Möbius syndrome. The patient’s phenotype is most consistent with CFEOM1 given her bilateral blepharoptosis and ophthalmoplegia with the eyes fixed in an infraducted position. Her amblyopia and loss of stereopsis are typical for her severe paralytic strabismus disorder. Although her developmental delay would be unusual for CFEOM in the absence of TUBB3 mutations, it is certainly compatible with her Möbius syndrome [6,7]. Her severe exposure keratopathy would be unusual in any type of CFEOM in the absence of ptosis surgery [20]. However, significant facial nerve weakness that results in disruption of the tear film and exposure keratopathy are more commonly reported with Möbius syndrome [21]. The patient’s inability to blink has resulted in a very challenging clinical course, despite surgical intervention with corneal transplantation followed by conjunctival pillar tarsorrhaphies and aggressive topical lubrication. The CNS, as well as the intracranial portions of the cranial nerves that are usually detectable by high-field 3T MRI and DTI, were unremarkable (in the case of her intraaxial CNS) or at least present (intracranial cranial nerves), although it is difficult to estimate nerve thickness by this method.
Using a candidate gene screening approach, we identified a novel, de novo 1056 C→G mutation in exon 8 of the KIF21A gene, which has not been previously identified via analysis of genomic databases (dbSNP, 1000 Genomes, and EVS) [17-19]. Additionally, this variant was absent in 288 Hispanic controls. The mutation occurs in a highly conserved region of the protein (Figure 6). Recent estimates place the per generation mutation rate at approximately one in 100 million positions in the haploid genome that corresponds to 0.86 de novo amino acid altering mutations per newborn [22,23]. This low de novo missense mutation rate and the fact that this reported mutation is in KIF21A, a previously mapped locus for CFEOM, are further evidence of its pathogenicity.
The KIF21A gene consists of 38 exons encoding a protein part of the kinesin superfamily, involved in the anterograde transportation of vesicles and organelles [24]. To date, 13 KIF21A mutations resulting in CFEOM have been reported (84C→G, 1,067T→C, 2,830G→C, 2,839A→G, 2,840T→G, 2,840T→C, 2,841G→A, 2,860C→T, 2,861G→A, 2,861G→T, 3,022G→C, 3,029T→C, 3000_3002delTGA; Figure 7) [3,10-15]. Of these, 11 have been found in families with CFEOM1; one was found in a patient with CFEOM3 (2841 G→A); and one has been associated with both CFEOM1 and CFEOM3 (2860 C→T).
Figure 7.
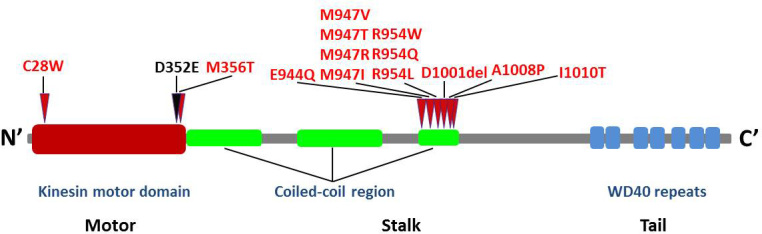
KIF21A mutations. Schematic of the kinesin protein structure and the relative locations of all KIF21A mutations identified to date. The novel mutation detected in the patient is shown in black at the junction between the N-terminal motor domain and the central coiled-coil stalk.
Kinesin, a molecular motor, contains an N-terminal motor domain that interacts with a microtubule track, a central coiled-coil stalk, and a C-terminus that interacts with transported cargo (Figure 7) [24]. Mutations in KIF21A may result in an inability to transfer cargo that is essential to the development of ocular motor axons, neuromuscular junctions, or extraocular muscles. It is noteworthy that 10 of the mutations identified to date result in the alteration of five amino acid residues clustered at the coiled-coil region of the protein. The previously described 1067 C→T mutation, and the novel mutation 1056 C→G described here, potentially disrupt the N-terminal kinesin motor domain, a region of high structural conservation [13]. It is noteworthy that the mutation identified here is conservative: The aspartate to glutamate substitution preserves the negative charge of the amino acid side chain but distorts the size of the residue by virtue of the elongated side chain of glutamate. This suggests that a relatively small volume change is not tolerated at this location, perhaps leading to abnormal interactions with microtubules or disrupting other close-range protein-protein interactions at a functionally critical region of the protein.
Typically, charge-preserving substitutions are of little import when amino acid residues are exposed in the bulk aqueous phase surrounding a protein by virtue of maintenance of the electrostatic field. In contrast, functionally relevant side chain size dependence indicates close interaction with neighboring residues during protein folding or catalysis [25,26]. Therefore, the simplest interpretation of the functional effect of the D352E mutation is that D352 potentially lies in an area critical for protein function.
Based on the functional impact of other KIF21A mutations, two pathogenic mechanisms have been hypothesized. The motor domain of the kinesin molecule interacts with the microtubule tracks and kinesin molecules typically work in pairs to move along microtubules efficiently. The coiled-coil region of the kinesin protein may interact with another kinesin protein, resulting in homo- or heterodimerization to facilitate movement in pair form. Mutation in either of these regions may thus result in the inability of dimer formation or the inability to interact effectively with microtubules, and thus the inability to deliver cargo [3]. A second mechanistic hypothesis involves the inability of mutated KIF21A to move in and out of an active state, resulting in the inability to deliver cargo [3,27]. It is noteworthy that in consanguineous families, there is evidence of CFEOM1 phenotype without mutations in KIF21A, PHOX2A, or TUBB3, giving rise to the possibility of a recessive form of the disease that may involve another locus or another mechanism [28].
We hypothesize that KIF21A D352E may result in abnormal kinesin/microtubule binding, perhaps in a dominant negative fashion, which can lead to a phenocopy state that exhibits the manifestations considered typical of TUBB3 mutation to date [6]. The precise identification of interacting proteins, kinesin/microtubule binding sites, and cargo will lead to further insights into the pathogenesis of CFEOM and the specific role of the kinesin in ocular development. The reported KIF21A D352E mutation helps to further expand the mutational spectrum of CFEOM and Möbius syndrome.
Acknowledgments
We thank the family for their participation in this study. We thank Dr. Nancy Rollins for performing MRI/DTI. We thank Dr. Helen H. Hobbs for providing access to Dallas Heart Study samples. Support: This study was supported by grants R01EY022161 and 2P30EY020799 from the National Eye Institute, National Institutes of Health, Bethesda, MD and an unrestricted grant from Research to Prevent Blindness, New York. JMP acknowledges the support of The Once Upon a Time Foundation. Presented in part at 2012 Annual Meeting of Association of Research in Vision and Ophthalmology, Fort Lauderdale, FL.
References
- 1.Heidary G, Traboulsi E, Engle E. The Genetics of Strabismus and Associated Disorders. Genetic Diseases of the Eye. Traboulsi, Elias (editor). New York: Oxford University Press, 2012. 657–686. [Google Scholar]
- 2.Oystreck DT, Engle EC, Bosley TM. Recent Progress in Understanding Congenital Cranial Dysinnervation Disorders. J Neuroophthalmol. 2011;31:69–77. doi: 10.1097/WNO.0b013e31820d0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamada K, Andrews C, Chan WM, McKeown CA, Magli A, De Berardinis T, Loewenstein A, Lazar M, O’Keefe M, Letson R, London A, Ruttum M, Matsumoto N, Saito N, Morris L, Del Monte M, Johnson RH, Uyama E, Houtman WA, De Vries B, Carlow TJ, Hart BL, Krawiecki N, Shoffner J, Vogel MC, Katowitz J, Goldstein SM, Levin AV, Sener EC, Ozturk BT, Akarsu AN, Brodsky MC, Hanisch F, Cruse RP, Zubcov AA, Robb RM, Roggenkaemper P, Gottlob I, Kowal L, Battu R, Traboulsi EI, Franceshini P, Newlin A, Demer JL, Engle EC. Heterozygous mutations of the kinesin KIF21A in congenital fibrosis of the extraocular muscles type 1 (CFEOM1). Nat Genet. 2003;35:318–21. doi: 10.1038/ng1261. [DOI] [PubMed] [Google Scholar]
- 4.Nakano M, Yamada K, Fain J, Sener EC, Selleck CJ, Awad AH, Zwaan J, Mullaney PB, Bosley TM, Engle EC. Homozygous mutations in ARIX (PHOX2A) result in congenital fibrosis of the extraocular muscles type 2. Nat Genet. 2001;29:315–20. doi: 10.1038/ng744. [DOI] [PubMed] [Google Scholar]
- 5.Pattyn A, Morin X, Cremer H, Goridis C, Brunet JF. Expression and interactions of the two closely related homeobox genes Phox2a and Phox2b during neurogenesis. Development. 1997;124:4065–75. doi: 10.1242/dev.124.20.4065. [DOI] [PubMed] [Google Scholar]
- 6.Tischfield MA, Baris HN, Wu C, Rudolph G, Maldergem LV, He W, Chan W, Andrews C, Demer JL, Robertson RL, Mackey DA, Ruddle JB, Bird TD, Gottlob I, Pieh C, Traboulsi EI, Pomeroy SL, Hunter DG, Soul JS, Newlin A, Sabol LJ, Doherty EJ, De Uzcategui CE, De Uzcategui N, Collins MLZC, Sener EC, Wabbels B, Hellebrand H, Meitinger T, de Berardinis T, Magli A, Schiavi C, Pastore-Trossello M, Koc F, Wong AM, Levin AV, Geraghty MT, Descartes M, Flaherty M, Jamieson RV, Moller HU, Meuthen I, Callen DF, Kerwin J, Lindsay S, Meindl A, Gupta ML, Pellman D, Engle EC. Human TUBB3 Mutations Perturb Microtubule Dynamics, Kinesin Interactions, and Axon Guidance. Cell. 2010;140:74–87. doi: 10.1016/j.cell.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verzijil HT, van der Zwaag B, Cruysberg JRM, Padberg GW. Möbius syndrome redefined: a syndrome of rhombencephalic maldevelopment. Neurology. 2003;61:327–33. doi: 10.1212/01.wnl.0000076484.91275.cd. [DOI] [PubMed] [Google Scholar]
- 8.Traboulsi EI. Congenital Abnormalities of Cranial Nerve Development: Overview, Molecular Mechanisms, and Further Evidence of Heterogeneity and Complexity of Syndromes with congenital Limitation of Eye Movements. Trans Am Ophthalmol Soc. 2004;102:373–89. [PMC free article] [PubMed] [Google Scholar]
- 9.Chew S, Balasubramanian R, Chan WM. A novel syndrome caused by the E410K amino acid substitution in the neuronal β-tubulin isotype 3. Brain. 2013;136:522–35. doi: 10.1093/brain/aws345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamada K, Hunter DG, Andrews C, Engle ECA. Novel KIF21A Mutation in a Patient with Congenital Fibrosis of the Extraocular Muscles and Marcus Gunn Jaw Winking Phenomenon. Arch Ophthalmol. 2005;123:1254–9. doi: 10.1001/archopht.123.9.1254. [DOI] [PubMed] [Google Scholar]
- 11.Yamada K, Chan WM, Andrews C, Bosley TM, Sener EC, Zwaan JT, Mullaney PB, Oztürk BT, Akarsu AN, Sabol LJ, Demer JL, Sullivan TJ, Gottlob I, Roggenkäemper P, Mackey DA, De Uzcategui CE, Uzcategui N, Ben-Zeev B, Traboulsi EI, Magli A, de Berardinis T, Gagliardi V, Awasthi-Patney S, Vogel MC, Rizzo JF, Engle EC. Identification of KIF21A mutations as a rare cause of congenital fibrosis of the extraocular muscles type 3 (CFEOM3). Invest Ophthalmol Vis Sci. 2004;45:2218–23. doi: 10.1167/iovs.03-1413. [DOI] [PubMed] [Google Scholar]
- 12.Lu S, Zhao C, Zhao K, Li N, Larsson C. Novel and Recurrent KIF21A mutations in Congenital Fibrosis of the Extraocular Muscles Type 1 and 3. Arch Ophthalmol. 2008;126:388–94. doi: 10.1001/archopht.126.3.388. [DOI] [PubMed] [Google Scholar]
- 13.Chan WM, Andrews C, Dragan L, Fredrick D, Armstrong L, Lyons C, Geraghty MT, Hunter DG, Yazdani A, Traboulsi EI, Pott JW, Gutowski NJ, Ellard S, Young E, Hanisch F, Koc F, Schnall B, Engle EC. Three novel mutations in KIF21A highlight the importance of the third coiled-coil stalk domain in the etiology of CFEOM1. BMC Genet. 2007;8:26. doi: 10.1186/1471-2156-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uyama E, Yamada K, Kawano H, Chan WM, Andrews B. A Japanese family with FEOM1-linked congenital fibrosis of the extraocular muscles type 1 associated with spinal canal stenosis and refinement of the FEOM1 critical region. Neuromuscul Disord. 2003;13:472–8. doi: 10.1016/s0960-8966(03)00065-8. [DOI] [PubMed] [Google Scholar]
- 15.Wang P, Li S, Xiao X, Guao X, Zhang Q. KIF21A novel deletion and recurrent mutation in patients with congenital fibrosis of the extraocular muscles. Int J Mol Med. 2011;28:973–5. doi: 10.3892/ijmm.2011.759. [DOI] [PubMed] [Google Scholar]
- 16.Victor RG, Haley RW, Willett DL, Peshock RM, Vaeth PC, Leonard D, Basit M, Cooper RS, Iannacchione VG, Visscher WA, Staab JM, Hobbs HH, Dallas Heart Study Investigators The Dallas Heart Study: a population-based probability sample for the multidisciplinary study of ethnic differences in cardiovascular health. Am J Cardiol. 2004;93:1473–80. doi: 10.1016/j.amjcard.2004.02.058. [DOI] [PubMed] [Google Scholar]
- 17.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–11. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The Thousand Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Exome Variant Server, NHLBI Exome Sequencing Project (ESP), Seattle, WA (URL:http://evs.gs.washington.edu/EVS/) [December15,2011
- 20.Yazdani A, Traboulsi E. Classification and surgical management of patients with familial and sporadic forms of congenital fibrosis of the extraocular muscles. Ophthalmology. 2004;111:1035–42. doi: 10.1016/j.ophtha.2003.08.030. [DOI] [PubMed] [Google Scholar]
- 21.Carta A, Mora P, Nerti A, Favilla S, Sadun AA. Ophthalmologic and systemic features in Möbius syndrome: An Italian Case Series. Ophthalmology. 2011;118:1518–23. doi: 10.1016/j.ophtha.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 22.Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci USA. 2010;107:961–8. doi: 10.1073/pnas.0912629107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roach JC, Glusman G, Smit AF, Huff CD, Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M, Shendure J, Drmanac R, Jorde LB, Hood L, Galas DJ. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–9. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marszalek JR, Weiner JA, Farlow SJ, Chun J, Goldstein LS. Novel dendritic kinesin sorting identified by different process targeting of two related kinesins: KIF21A and KIF21B. J Cell Biol. 1999;145:469–79. doi: 10.1083/jcb.145.3.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirsch GE, Pascual JM, Shieh CC. Functional role of a conserved aspartate in the external mouth of voltage-gated potassium channels. Biophys J. 1995;68:1804–13. doi: 10.1016/S0006-3495(95)80357-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pascual JM, Shieh CC, Kirsch GE, Brown AM. Multiple residues specify external tetraethylammonium blockade in voltate-gated potassium channels. Biophys J. 1995;69:428–34. doi: 10.1016/S0006-3495(95)79915-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Engle EC. The genetic basis of complex strabismus. Pediatr Res. 2006;59:343–8. doi: 10.1203/01.pdr.0000200797.91630.08. [DOI] [PubMed] [Google Scholar]
- 28.Khan AO, Shinwari J, Omar A, Al-Sharif L, Khalil DS, Alanazi M, Al-Amri A, Al Tassan N. Lack of KIF21A mutations in congenital fibrosis of the extraocular muscles type I patients from consanguineous Saudi Arabian families. Mol Vis. 2011;17:218–24. [PMC free article] [PubMed] [Google Scholar]