

Abstract
Hereditary sensory neuropathy type 1 (HSN-1) is an autosomal dominant neurodegenerative disease caused by missense mutations in the SPTLC1 gene. The SPTLC1 protein is part of the SPT enzyme which is a ubiquitously expressed, critical and thus highly regulated endoplasmic reticulum bound membrane enzyme that maintains sphingolipid concentrations and thus contributes to lipid metabolism, signalling, and membrane structural functions. Lipid droplets are dynamic organelles containing sphingolipids and membrane bound proteins surrounding a core of neutral lipids, and thus mediate the intracellular transport of these specific molecules. Current literature suggests that there are increased numbers of lipid droplets and alterations of lipid metabolism in a variety of other autosomal dominant neurodegenerative diseases, including Alzheimer’s and Parkinson’s disease. This study establishes for the first time, a significant increase in the presence of lipid droplets in HSN-1 patient-derived lymphoblasts, indicating a potential connection between lipid droplets and the pathomechanism of HSN-1. However, the expression of adipophilin (ADFP), which has been implicated in the regulation of lipid metabolism, was not altered in lipid droplets from the HSN-1 patient-derived lymphoblasts. This appears to be the first report of increased lipid body accumulation in a peripheral neuropathy, suggesting a fundamental molecular linkage between a number of neurodegenerative diseases.
Keywords: Hereditary sensory neuropathy type 1, Serine palmitoyltransferase, Serine palmitoyltransferase long chain subunit 1, Lipid droplets, Nile red, ADFP
Introduction
Serine palmitoyltransferase (SPT) is a critical, ubiquitously expressed, and highly regulated endoplasmic reticulum bound membrane enzyme that maintains cellular sphingolipid concentrations [1–3]. SPT is a pyridoxal-5′-sphosphate (PLP)-dependent multimeric enzyme that catalyses the first and rate limiting step in the de novo synthesis of sphingolipids [1, 2].
SPT is a multimeric enzyme composed of three similar subunits: serine palmitoyltransferase long chain subunit 1 (SPTLC1), SPTLC2, and SPTLC3. SPTLC2 and SPTLC3 both contain a lysine residue at the active site required for PLP binding; therefore, these two subunits are essential for activating the SPT enzyme [2, 4]. SPTLC1, however, lacks this PLP binding site and other key catalytic residues, suggesting that SPTLC1 plays a more regulatory role in the SPT complex [2, 3].
The SPTLC1 gene is located on chromosome 9p22, and positional cloning has identified three missense mutations associated with Hereditary sensory neuropathy type 1 (HSN-1), an autosomal dominant sensory neuropathy affecting peripheral sensory neurons [1, 5]. These mutations result in a single amino acid substitution of cysteine to tryptophan at position 133 (C133W), cysteine to tyrosine at position 133 (C133Y), and valine to aspartic acid at position 144 (V144D) [6]. HSN-1 is the most common HSN subtype resulting in the progressive degeneration and dying back of neurons in the dorsal root ganglia. Despite its initial characterisation over 50 years ago [7] and the identification of critical mutations in the SPTLC1 gene, the molecular mechanisms underlying disease development and progression still remain poorly understood [8, 9].
There are currently two main hypotheses as to the pathomechanism(s) in HSN-1, suggesting either a ‘gain of toxic function’ of the SPT enzyme or a dominant negative effect [1, 10, 11]. Peripheral neurons may be sensitive to the perturbation of sphingolipid metabolism (i.e. decrease in functional levels) caused by a mutation-induced reduction in SPT enzyme activity [11]. This hypothesis has been shown to be consistent with recent studies on C133W and V144D, demonstrating that both mutations reduce normal SPT activity in various cell types, including cultured patient lymphoblasts [11]. A concomitant change in the membrane lipid composition would be expected to be seen but recent data has been contradictory. Initially, an increase in glucosylceramide synthesis was reported; however, a decrease in ceramide levels and sphingomyelin synthesis yielded no change in the overall sphingolipid composition [11].
Studies of SPT activity using patient lymphoblasts that endogenously expressed the SPTLC1 mutation reported greater than 50 % reduction of SPT activity [8, 10]. While the mechanism by which SPT activity is reduced is yet to be confirmed, Bejaoui et al. [8] observed that the mutation did not directly affect the stability of the protein as translated but may interfere with the function of the enzyme. As SPTLC1 mutations have a direct effect on the activity of SPT, this supports the dominant negative effect theory. Thus, competition possibly arising between mutated and wild type SPTLC1 for interaction with SPTLC2 may represent an underlying disease mechanism, with the mutated SPTLC1 possessing a higher affinity then wild type [8]. Nonetheless, the SPTLC1 mutation does not reduce SL levels despite SPT activity being reduced by more than half [11]. Therefore, the remaining 50 % of SPT activity may be sufficient to maintain normal sphingolipid homeostasis in these cells, presumably because the total SPT activity is normally than sufficient; indeed, this is suggested by in vivo SPT downregulation [10].
The alternative theory is that mutations in the SPTLC1 gene cause a gain of toxic function. Mutations in the SPTLC1 gene are thought to induce a shift in the substrate specificity of the SPT enzyme resulting in the production of one or more toxic lipid species [1]. The production of two atypical deoxysphingoid bases (DSB) has been linked to the mutant SPT enzyme [1]. These DSB metabolites can neither be converted to sphingolipids nor degraded and therefore accumulate in the cell, producing a neurotoxic affect. This gain of toxic function occurs in many other autosomal dominant, inherited neurodegenerative disorders, including Alzheimer’s disease and Parkinson’s disease [12, 13].
Increases in the number of lipid droplets and changes in lipid metabolism have also been identified in a variety of autosomal dominant neurodegenerative diseases, including Alzheimer’s and Parkinson’s [12–14]. Lipid droplets are organelles which contribute to cellular homeostasis by regulating lipid metabolism and the transport of proteins and lipids (including sphingolipids) throughout the cell [15–19]. Current research into Alzheimer’s disease emphasises that accumulation of lipid droplets and abnormalities in lipid metabolism may cause or exacerbate the disease phenotype [13]. Similarly, in Parkinson’s disease, the dysregulation of intracellular lipid droplet interactions and expressions, as well as changes in lipid metabolism, may also contribute to the disease phenotype [12, 14].
Considering the potential linkages between central and peripheral neurodegenerative conditions [13], including the accumulation of lipid droplets and alterations of lipid metabolism in other autosomal dominant neurodegenerative diseases [12], we have tested the hypothesis that an accumulation of lipid droplets would also be associated with HSN-1 disease. The data confirm a significantly increased number of lipid droplets in lymphoblasts from HSN-1 patients expressing the C133W and V144D mutant SPTLC1 proteins. This appears to be the first report of increased lipid body accumulation in an autosomal dominant sensory neuropathy. We discuss these findings in terms of probable molecular mechanisms underlying the development and progression of HSN-1.
Results
SPTLC1 mutations do not alter cellular morphology despite increases in lipid droplet accumulation
EBV transformed, patient-derived lymphoblasts endogenously express the mutant SPTLC1 enzymes associated with HSN-1 [10]. Detailed analysis indicated no gross morphological changes in lymphoblasts derived from healthy controls and HSN-1 patients expressing the C133W and V144D mutant SPTLC1 proteins (Fig. 1a). To establish that an increase could be effectively detected, control lymphoblasts were also treated with oleic acid, which is known to result in the accumulation of lipid droplets [20]. Confocal analysis of patient-derived lymphoblasts, stained with DAPI (4′,6-diamidino-2-phenylindolenucleus; nuclear stain) and Nile red (for lipid droplets), confirmed an accumulation of lipid droplets within the cytoplasm of oleic acid-treated lymphoblasts compared to cells from healthy untreated controls (Fig. 1b). Notably, an obvious increase in the number of lipid droplets was also seen in the HSN-1 patient-derived lymphoblasts expressing the C133W and V144D mutant SPTLC1 proteins compared to the healthy control lymphoblasts (Fig. 1b). Interestingly, punctate Nile red (i.e. lipid droplet) staining appears largely localised to the ER which is also where the SPTLC1 protein is bound.
Fig. 1.

SPTLC1 mutations cause no change to gross morphology but yield increased lipid droplets. a Representative bright field micrographs showing gross morphology of health control and patient-derived lymphoblasts. b Representative confocal micrographs showing Nile red-stained lipid droplets (red) and DAPI nuclear stain (blue). Scale bar = 20 μm. c Fluorescence spectroscopy of the Nile red-stained lipid droplets in patient-derived lymphoblasts (n = 5 separate experiments). Plus symbol = oleic acid treatment; minus symbol = no oleic acid treatment. Asterisk indicates p < 0.05, relative to control (−)
To quantitatively assess the increase in lipid droplet accumulation caused by the HSN-1 mutant SPTLC1 genes, Nile red-stained patient-derived lymphoblasts were analysed using fluorescence spectroscopy (Fig. 1c). The oleic acid-treated positive control lymphoblasts showed a statistically significant increase in lipid droplets compared to untreated cells derived from healthy control subjects. Although proportionally lower, the HSN-1 patient-derived lymphoblasts also showed statistically significant increases in lipid droplet accumulation compared to the healthy control lymphoblasts; relative fluorescence increases amounted to 0.0353 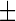 0.0018 OD (a 2.3-fold increase) and 0.0420
0.0018 OD (a 2.3-fold increase) and 0.0420 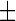 0.0057 OD (a 2.7-fold increase) for the V144D and C133W mutations, respectively.
0.0057 OD (a 2.7-fold increase) for the V144D and C133W mutations, respectively.
Quantification of lipid droplets in HSN-1 patient-derived lymphoblasts
In order to best characterise the relationship between increased lipid droplet accumulation and HSN-1, Nile red-stained patient-derived lymphoblasts were analysed using flow cytometry to determine the lipid droplet fluorescence per cell (Fig. 2). Labelling with Nile red resulted in an overall rightward shift of 102 units in the fluorescence histograms relative to unlabelled cells, indicating that no endogenous fluorescence affected the analysis. Control cells previously treated with oleic acid showed a further rightward shift relative to those that were untreated, and Nile red fluorescence in the HSN-1 patient-derived cells yielded peak midpoints and distributions comparable to the oleic acid-treated controls. Overall, the oleic acid-treated control lymphoblasts, as well as the HSN-1 patient-derived lymphoblasts showed statistically significant increases in total lipid droplet staining per cell compared to the untreated healthy controls; for the C133W and V144D, mutants this amounted to increases in relative fluorescence of 46.004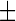 1.563 OD (a 1.6-fold increase) and 40.89
1.563 OD (a 1.6-fold increase) and 40.89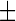 1.099 OD (a 1.8-fold increase), respectively, relative to the stained controls.
1.099 OD (a 1.8-fold increase), respectively, relative to the stained controls.
Fig. 2.

Relative quantification of lipid droplets in HSN-1 patient-derived lymphoblasts expressing the C133W and V144D mutant SPTLC1 genes. a Representative flow cytometry scatter plots of Nile red-stained lipid droplets in control and patient-derived lymphoblasts. Number symbol = unstained, non-treated; plus symbol = oleic acid treated, stained; and minus symbol = non-treated, stained lymphoblasts. b Flow cytometry analysis of the relative fluoresence intensity of Nile red-stained lipid droplets in patient-derived lymphoblasts. Plus symbol = oleic acid treatment. Minus symbol = no oleic acid treatment (n = 5 separate experiments). Asterisk indicates p < 0.05, relative to control (−)
Expression of lipid droplet marker protein ADFP in HSN-1 patient-derived lymphoblasts
Adipophilin (ADFP) is a membrane-associated protein present in mature lipid droplets, and is thus widely used as a marker for lipid droplets. Western blot analysis was performed for ADFP on total cell lysates from oleic acid-treated control and on untreated control and HSN-1 patient-derived lymphoblasts (Fig. 3a), revealing ADFP expression is below the level of detection in the untreated samples. To confirm this, western blot analysis was performed on total cell lysates of oleic acid-treated control and HSN-1 patient-derived lymphoblasts (Fig. 3c). This western blot analyses show that ADFP protein expression is relatively abundant in the oleic acid-treated healthy control and patient lymphoblasts, thus highlighting the need for oleic acid treatment for total cell lysate analysis.
Fig. 3.

Expression of lipid droplet marker protein ADFP in HSN-1 patient-derived lymphoblasts. a Immunoblot detection of ADFP in oleic acid-treated and oleic acid-untreated total cell lysates; 1 represents treated controls; 2, untreated controls; 3, untreated V144D mutant; and 4, untreated C133W mutant. Immunoblots of total protein lysates from oleic acid-treated cells probed for SPTLC1 (b), ADFP (c), and GAPDH (d); 1–3 represents treated controls; 4–6, treated V144D mutants; and 7–9, treated C133W mutants. Histograms of the immunoblotting results of oleic acid-treated controls and of SPTLC1 (e) and ADFP (f), normalised to GAPDH (n = 3)
Further western blot analyses of SPTLC1 (Fig. 3b) and ADFP (Fig. 3c) were carried out, normalising to the housekeeping protein glyceraldehyde-3-phosphate dehydrogenase (GAPDH) to establish relative protein expression levels of SPTLC1 and ADFP from cells treated with oleic acid. Despite some variability in expression levels, there were no statistically significant changes in the expression of SPTLC1 (Fig. 3e) or ADFP (Fig. 3f) in the HSN-1 patient-derived lymphoblasts compared to those derived from healthy control subjects.
Lipid droplets show no co-localisation with mitochondria
Previous ultrastructural analysis has shown lipid droplet presence in close proximity to the ER and mitochondria membranes in the patient lymphoblasts (Myers, S.J, unpublished observations). In order to determine if the lipid droplets co-localise with mitochondria, oleic acid treated-healthy control and HSN-1 patient-derived lymphoblasts were fractionated into mitochondrial and cytosolic fractions and immunoblotted for ADFP and MTCO2; the latter is part of Cytochrome c oxidase subunit 2 of complex IV, located in the inner membrane of mitochondria. Fluorescence micrographs of patient-derived lymphoblasts, stained with DAPI (nucleus) and anti-MTCO2 antibody, confirmed normal healthy mitochondrial structures within the cytoplasm (Fig. 4a). The mitochondrial marker MTCO2 was detected solely in the mitochondrial fractions, and the lipid droplet marker, ADFP, only in the cytosolic fractions, suggesting that the mitochondria and lipid droplets do not co-localise and are thus less likely to interact.
Fig. 4.
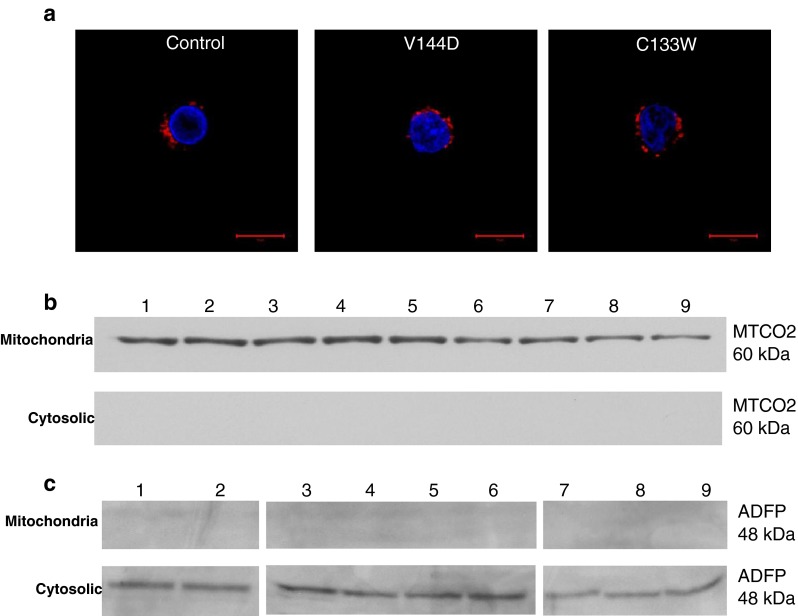
Mitochondria show no co-localisation with lipid droplets. a Representative confocal micrographs showing MTCO2 stained mitochondria (red) and DAPI nuclear stain (blue). Scale bar = 20 μm. b Immunoblot detection of MTCO2 at 60 kDa from mitochondrial and cytosolic fractions, 1–3 represents oleic acid-treated controls; 4–6, treated V144D mutants; and 7–i, treated C133W mutants. c Immunoblot detection of ADFP at 48 kDa from oleic acid-treated mitochondrial and cytosolic fractions
Protein expression of ADFP, enriched from a lipid droplet fractionation
In order to identify whether an isolated untreated lipid droplet fraction expressed SPTLC1 and ADFP, lipid droplets were isolated using a well-established protocol [20] whereby, from an equivalent number of lymphoblasts lipid droplets were isolated and an equal concentration of protein from each lipid droplet fraction (7 μg) was loaded onto an SDS-PAGE and assessed by western blot analysis. SPTLC1 was never detected in the isolated lipid droplet fraction; however, ADFP was consistently detected in the lipid droplet fractions from both the healthy control and the patient lymphoblasts. This highlighted the successful isolation of an enriched lipid droplet fraction from untreated lymphoblasts, indicating that oleic acid treatment was not required for this level of analysis (Fig. 5). Most importantly, there was no change in the amount of ADFP in control vs. patient lipid droplets.
Fig. 5.

Expression of ADFP in an enriched lipid droplet fraction. Immunoblot detection of ADFP and SPTLC1 proteins from untreated lipid droplet enriched fractions, 1–3, untreated controls; 4–6, untreated V144D mutants; and 7–9, untreated C133W mutants (n = 3)
Discussion
HSN-1 is an autosomal dominant neuropathy resulting in the progressive degeneration and dying back of the peripheral sensory neurons in the dorsal root ganglia [5]. HSN-1 is caused by missense mutations in the SPTLC1 gene; however, the actual cellular and molecular mechanisms underlying the disease still remain poorly understood [10]. This study quantified increases in lipid droplets in HSN-1 patient-derived lymphoblasts, and also determined that there was no association between these inclusions and mitochondria, the latter being often also effected in neurodegenerative diseases [12, 13].
Increased numbers of lipid droplets and changes in lipid metabolism have been seen in a variety of autosomal dominant neurodegenerative disorders such as Alzheimer’s disease and Parkinson’s disease [12, 13]. Current research into Alzheimer’s disease emphasises that accumulation of lipid droplets and abnormalities in lipid metabolism may cause or exacerbate the disease phenotype [13]. Parkinson’s disease research has identified a deregulation of intracellular lipid droplet interactions and expressions, and also changes in lipid metabolism, which may contribute to the disease phenotype. The data presented here establish a significant increase in lipid droplets in lymphoblasts from patients with HSN-1, a peripheral neurodegenerative disorder. With a breadth of research identifying accumulation of lipid droplets and changes in lipid metabolism in other autosomal dominant neurodegenerative diseases, it is now reasonable to suggest that the increased presence of lipid droplets may cause or exacerbate the HSN-1 disease phenotype. Indeed, such alterations suggest a more central connection with the pathomechanism(s) underlying a host of neurodegenerative disease, both central and peripheral.
Bright field micrographs indicated no gross morphological changes between healthy control and HSN-1 patient-derived lymphoblasts; however, confocal micrographs of Nile red stained lipid droplets showed an increase of these inclusions within the oleic acid-treated positive controls compared to the healthy controls and patient-derived lymphoblasts (Fig. 1). Therefore, oleic acid-induced lipid droplet formation [21] was used as a positive control. More importantly the results indicated a significant quantitative increase of lipid droplets within the patient-derived lymphoblasts compared to the healthy controls. This increase in lipid droplet abundance in HSN-1 patient-derived lymphoblasts was confirmed using fluorescence spectroscopy. The confocal and fluorescence spectroscopy analyses thus indicated a possible connection between increased lipid droplet numbers and the cellular mechanism of HSN-1. The data confirmed using Nile red staining and flow cytometric analysis indicating a significant increase in lipid droplet numbers within individual patient-derived lymphoblasts (Fig. 2).
ADFP is a lipid droplet-associated membrane protein, and has thus been used as a marker for these inclusions [19]. Immunoblotting revealed that only the oleic acid-treated samples showed detectable ADFP protein from total cell lysates (Fig. 3a). Analysis of SPTLC1 from oleic acid-treated healthy control and patient-derived lymphoblasts revealed no significant difference in expression (Fig. 3f), comparable to the results of another study that found no change in the levels of SPTLC1 in HSN-1 patient lymphoblasts [10]. By both confocal and immunoblotting analyses, the mitochondrial membrane marker MTCO2 was detected only in the mitochondrial fraction, and not in the cytoplasmic fraction isolated from oleic acid-treated healthy control patient-derived lymphoblasts (Fig. 4b). In contrast, immunoblotting of the lipid droplet marker ADFP identified it only in the cytosolic fraction, and not in the mitochondrial fraction, suggesting no co-localisation between the mitochondrial and lipid droplet membranes.
Immunoblotting for ADFP in lipid droplet fractions isolated from healthy control and HSN-1 patient-derived lymphoblasts expressing the C133W and V144D mutant SPTLC1 proteins indicated no detectable changes in protein expression. ADFP is currently thought to be involved in lipid homeostasis and lipolysis by protecting triacylglycerol, within the lipid droplets, from cytosolic lipases [17, 19]. These results suggest that the increase in lipid droplets within the HSN-1 patient lymphoblasts, without a change in ADFP, may cause abnormalities in lipid metabolism or be indicative of a protein trafficking defect that involve elements of the cytoskeleton [17]. Previously, abnormalities in lipid metabolism have been linked to a variety of other autosomal dominant neurodegenerative diseases, including Alzheimer’s disease and Parkinson’s disease, suggesting that changes in lipid metabolism may either cause or exacerbated the disease phenotype. Therefore, comparable changes in lipid metabolism, along with a trafficking defect, may also cause or exacerbate the HSN-1 disease phenotype, a peripheral neurodegenerative disorder. Further analyses are warranted to determine if there is a deregulation of lipid metabolism within the HSN-1 patient lymphoblasts and to identify its link with HSN-1 disease and other autosomal dominant neurodegenerative diseases.
This is the first study to investigate lipid droplet formation in an autosomal dominant sensory neuropathy. These data indicate a possible connection between increased lipid droplet abundance and the cellular mechanism underlying HSN-1. The increase in lipid droplets without a parallel increase in the expression of ADFP in the HSN-1 patient-derived lymphoblasts may well indicate a deregulation of lipid metabolism which may exacerbate the HSN-1 phenotype. The data thus also suggest a more common and/or central role for these molecular alterations in specific types of central and peripheral neurodegeneration.
Materials
All cell culture stock solutions, including RPMI-1640, fetal bovine serum (FBS), penicillin (100 U/mL), streptomycin (100 μg/mL), l-glutamine (2 mM), HEPES (1 M), and phosphate buffered saline (PBS) were purchased from Gibco Invitrogen (Australia). Cell culture consumables were purchased from BD Falcon (Greiner, USA). MTCO2 and GAPDH primary antibodies were purchased from Abcam (USA); SPTLC1 primary antibody was purchased from Santa Cruz Biotechnology (USA). ADFP primary antibody, secondary HRP Mouse and Rabbit antibodies, oleic acid, Nile red, and DAPI stains were purchased from Sigma-Aldrich (Australia).
Methods
EBV-transformed lymphoblasts
Epstein-Barr Virus (EBV)-transformed control and HSN-1 patient lymphoblasts were graciously provided by Prof. Garth Nicholson (Molecular Medicine Laboratory, Anzac Institute, Sydney) [10].
Lymphoblast cultures
Lymphoblasts were cultured in RPMI-1640 media (Gibco), supplemented with FBS (10 % v/v), penicillin (1 U/mL), streptomycin (1 μg/mL), l-glutamine (2 mM), and HEPES (1 M) at 37 °C in a humidified atmosphere of 5 % CO2, using T75 cm2 culture flasks (Greiner, Interpath). Prior to use in biochemical assays, lymphoblasts were collected by centrifugation at 1,000×g (3 min at RT) and washed in PBS. Normalisation of cell count and viability was obtained using the Countess Automated Cell Counter (Invitrogen, Australia).
Oleic acid treatment
For positive controls, lymphoblasts from normal healthy donors were treated (in culture) with a 400 μM solution of oleic acid (Sigma, Australia) for 24 h prior to isolation. This is a standardised and routine protocol to induce lipid droplet formation [21].
Fluorescence microscopy
For fluorescence microscopy analyses, glass cover slips were prepared in advance (i.e. overnight). The cover slips were dipped 20 times into a 1:50 dilution of HistoGrip (Invitrogen, Australia) in pure acetone. The cover slips were then dipped 10 times in dH2O to wash off excess HistoGrip and dried overnight. Lymphoblasts were collected by centrifugation at 1,000×g for 5 min at RT, and resuspended in PBS. Cell suspensions (containing 1 × 106 to 2 × 106 cells) were centrifuged at 1,000×g for 5 min at RT, resuspended in 1 mL of 4 % paraformaldehyde in PBS and incubated for 15 min at RT.
For lipid droplet analysis, the cell suspension was centrifuged at 1,000×g for 5 min at RT, resuspended in 1 mL of 320 nM Nile red (Sigma, Australia) in Hank’s Buffered Salt Solution (HBSS; prewarmed to 37 °C) and incubated in the dark for 15 min at RT. For mitochondrial analysis the cell suspension was permiabilised with a 0.3 % Triton X-100 for 10 min at 37 °C. The cells were centrifuged at 1,000×g for 5 min, resuspended in 1 mL of 1 % BSA in PBS, and incubated for 30 min at 37 °C. The cells were subsequently centrifuged at 1,000×g for 5 min, resuspended in 200 μL of a solution of 1 % BSA in PBS that also contained primary antibody (MTCO2 at 1:50 dilution), and incubated for 1 h at RT. The cells were then centrifuged at 1,000×g for 5 min, washed in 1 mL of PBS, centrifuged again, and resuspended in 200 uL of a solution of 1 % BSA in PBS that also contained secondary antibody (1:200 dilution, anti-mouse, rhodamine conjugated) for 1 h at RT. Thereafter, the cell suspension was centrifuged at 1,000×g for 5 min at RT, resuspended in 1 mL of DAPI (10 mg/mL stock) in PBS, and incubated for 2 min at RT. The cell suspension was then washed twice in 1 mL of PBS then suspended in 2 mL of PBS, aliquoted into individual wells on 6-well plates, and centrifuged at 500×g (10 min, at RT) to pellet the cells onto the cover slips at the bottom of each well. The PBS and non-adherent cells were then aspirated from the wells and the cover slips allowed to dry for 20 min at RT. Thereafter, the cover slips were mounted using 20 μL of DAKO solution (Dako, Australia) and sealed with nail polish prior to assessment using a LSM-5 Exciter Confocal Microscope (Carl Zeiss, Australia).
Fluorescence detection
For fluorescence detection, lymphoblasts were isolated as above; the cells were then suspended in 1 mL of 4 % paraformaldehyde in PBS, and incubated for 15 min at RT. Thereafter, the cell suspension was centrifuged at 1,000×g for 5 min at RT, and the resulting pellet suspended in 1 mL of 320 nM Nile red (Sigma, Australia), (10 mM stock) in Hank’s Buffered Salt Solution (HBSS), and incubated in the dark for 15 min at RT. After incubation, the cell suspension was centrifuged at 1,000×g for 5 min and resuspended in 1 mL of PBS at RT. The cell suspension was then analysed using a BMG Polar Star Omega Fluorescence Plate Reader (BMG Labtech, Germany) and a MACS Quant Flow Cytometry (Miltenyi Biotech, Australia).
Isolation of mitochondrial and cytosolic proteins
Briefly, mitochondria were isolated using a Mitochondrial Protein Isolation kit (Amresco Scientific, USA). Lymphoblasts were first centrifuged at 1,000×g for 5 min, and the cells were then washed in 10 ml of ice cold 1X PBS prior to suspension in 1 ml ice cold 1X PBS. Cells were transferred to a 1.5 ml microcentrifuge tube, centrifuged at 1,000×g (4 °C for 5 min) and then suspended in the Mitochondrial Protein Isolation Buffer (including 1X Protease Inhibitor cocktail). Cells were then homogenized on ice by 20 passages through a 26 ½ G needle attached to a 1 cm3 syringe prior to centrifugation at 1,000×g (4 °C for 10 min). The supernatant was collected, transferred to a fresh 1.5-ml tube, and centrifuged at 14,000×g (15 min at 4 °C). The supernatant was collected and transferred into a new tube; this fraction contained the cytosolic proteins. The pellet, containing mitochondrial proteins, was suspended in 1 ml Mitochondrial Protein Isolation Buffer and centrifuged at 14,000×g (1 min at 4 °C). The supernatant was discarded and the pellet suspended in 100 μl of Mitochondrial Protein Isolation Buffer.
Lipid droplet isolation
Lipid droplets were isolated essentially as previously described [20]. Briefly, lymphoblasts were collected by centrifugation at 1,000×g (5 min at 4 °C) to yield an equivalent number of lymphoblasts, suspended in 30 mL of dissociation buffer (25 mM Tris-HCl pH 7.4, 1 mM EGTA, 1 mM EDTA, and 100 mM KCl). This cell suspension was then centrifuged at 1,000×g (5 min at 4 °C); the pellet was then suspended in 10 mL of dissociation buffer and transferred to 15-mL tubes. The cell suspension was then centrifuged at 1,000×g (5 min at 4 °C), the cells suspended in 750 μL of dissociation buffer containing 1× protease inhibitors, and transferred to 1.5 mL microcentrifuge tubes. The cell suspension was then placed on ice and homogenised using a 26 ½ G needle, as described above. The resulting cell lysate was then centrifuged at 15,000×g (10 min at 4 °C) to remove whole cells, nuclei, and large organelles. The supernatant was collected and transferred into new 1.5 mL microcentrifuge tubes. An equal volume of 1.08 M sucrose solution (approximately 300 μL) was added to the supernatant and the total volume then transferred to an ultracentrifuge tube. The supernatant/sucrose mixture was overlayed sequentially with 700 μL of 0.27 M sucrose, 500 μL 0.135 M sucrose, and 500 μL of top solution (25 mM Tris-HCl pH 7.4, 1 mM EGTA, and 1 mM EDTA). The gradient was then centrifuged at 149,711×g (60 min at 4 °C). The middle and bottom layers were subsequently removed using a fine glass pipette, leaving only the white top layer containing lipid droplets.
Total protein preparation
For a positive control, normal healthy donor lymphoblasts were treated with a 400 μM solution of oleic acid (Sigma-Aldrich, Australia) in standard media, 24 h prior to isolation. Lymphoblasts were collected by centrifugation at 1,000×g for 3 min at RT and resuspended in 30 mL of PBS. The cell suspension was then centrifuged at 500×g for 3 min at RT, resuspended in 1 mL of PBS, and 10 μL was aliquoted for cell counts. The cell suspension was then centrifuged at 500×g for 3 min at RT, resuspended in 300 μL of NDRM (non-detergent resistant membrane lysis buffer; 10 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 % Triton X-100, and 1× protease inhibitors in PBS), and lysed for 20 min on ice. Following lysis, the cell lysate was centrifuged at 18,000×g for 15 min at 4 °C. The supernatant containing total cell proteins were collected and transferred to fresh 1.5 mL microcentrifuge tubes ready for protein concentration and analysis. The pellet containing whole cells and detergent resistant membranes were resuspended in 200 μL PBS containing 1× protease inhibitors.
Protein concentration
Determination of total cellular protein was performed using the bicinchoninic acid (BCA) protein assay (Sigma-Aldrich, Australia).
SDS-PAGE and immunoblotting
Cell lysates and lipid droplet fractions (50 μg total protein) were subjected to SDS-PAGE on 15 % resolving gels and transferred to PVDF membrane. The membranes were blocked with 5 % skim milk in TBS buffer containing 0.1 % Tween-20. The blocked membranes were incubated with anti-ADFP, anti-SPTLC1, and anti-MTCO2 at 1:1,000 and anti-GAPDH at 1:5,000, for 16 h. The membrane was then incubated with secondary horse radish peroxidase antibody (1:2,000 dilution) for 1 h at RT. Blots were developed using an enhanced chemiluminescence (ECL) detection kit (WEST-ZOL, Biotech, Korea).
Acknowledgments
We are grateful to Prof Garth Nicholson (Molecular Medicine Laboratory and Northcott Neuroscience Laboratory Anzac Research Institute, Sydney) for providing all EBV-transformed lymphoblast lines [10] used in this study. LLM was supported by a UWS Honours Scholarships, the School of Science and Health Honours support and an anonymous private foundation; SES was supported by APA Research Scholarship, the UWS School of Science and Health Postgraduate research fund; RH was supported by a UWS Postgraduate Research Award and anonymous private foundation. SJM notes the support of an anonymous Private Foundation. JRC acknowledges the support of the UWS School of Medicine.
Footnotes
Lee L. Marshall and Scott E. Stimpson contributed equally to this work.
Contributor Information
Jens R. Coorssen, Phone: +61-2-46203802, FAX: +61-2-46203890, Email: j.coorssen@uws.edu.au
Simon J. Myers, Phone: +61-2-46203383, FAX: +61-2-46203025, Email: s.myers@uws.edu.au
References
- 1.Penno A, Reilly M, Houlden H, Laura M, Rentsch K, Niederkofler V, Stoeckli E, Nicholson G, Eichler F, Brown R, Von-Eckardstein A, Hornemann T. Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J Biol Chem. 2010;285(15):11178–11187. doi: 10.1074/jbc.M109.092973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hornemann T, Richard S, Rutti M, Wei Y, Von-Eckardstein A. Cloning and initial characterization of a new subunit for mammalian serine-palmitoyltransferase. J Biol Chem. 2006;281(49):37275–37281. doi: 10.1074/jbc.M608066200. [DOI] [PubMed] [Google Scholar]
- 3.Yard B, Carter L, Johnson K, Overton I, Dorward M, Liu H, McMahon S, Oke M, Puech D, Barton G, Naismith J, Campopiano D. The structure of serine palmitoyltransferase; gateway to sphingolipid biosynthesis. J Mol Biol. 2007;370(5):870–886. doi: 10.1016/j.jmb.2007.04.086. [DOI] [PubMed] [Google Scholar]
- 4.Han G, Gupta S, Gable K, Niranjanakumari S, Moitra P, Eichler F, Brown R, Harmon J, Dunn T. Identification of small subunits of mammalian serine palmitoyltransferase that confer distinct acyl-CoA substrate specificities. Proc Natl Acad Sci U S A. 2009;106(20):8186–8191. doi: 10.1073/pnas.0811269106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCampbell A, Broom D, Truong D, Allchorne A, Gable K, Cutler RG, Mattson M, Woolf C, Frosch M, Harmon J, Dunn T, Brown R. Mutant SPTLC1 dominantly inhibits serine palmitoyltransferase activity in vivo and confers an age-dependent neuropathy. Hum Mol Genet. 2005;14(22):3507–3521. doi: 10.1093/hmg/ddi380. [DOI] [PubMed] [Google Scholar]
- 6.Hornemann T, Penno A, Richard S, Nicholson G, Van-Dijk F, Rotthier A, Timmerman V, Von-Eckardstein A. A systematic comparison of all mutations in hereditary sensory neuropathy type I (HSAN I) reveals that the G387A mutation is not disease associated. Neurogenetics. 2009;10(2):135–143. doi: 10.1007/s10048-008-0168-7. [DOI] [PubMed] [Google Scholar]
- 7.Houlden H, King R, Blake J, Groves M, Love S, Woodward C, Hammans S, Nicoll J, Lennox G, O'Donovan DG, Gabriel C, Thomas PK, Reilly MM. Clinical, pathological and genetic characterization of hereditary sensory and autonomic neuropathy type 1 (HSAN I) Brain. 2006;129:411–425. doi: 10.1093/brain/awh712. [DOI] [PubMed] [Google Scholar]
- 8.Bejaoui Y, Uchida Y, Yasuda S, Ho M, Nishijima M, Brown RH, Jr, Holleran WM, Hanada K. Hereditary sensory neuropathy type 1 mutations confer dominant negative effects on serine palmitoyltransferase, critical for sphingolipid synthesis. J Clin Invest. 2002;110:1301–1308. doi: 10.1172/JCI0216450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dawkins JL, Hulme DJ, Brahmbhatt SB, Auer-Grumbach M, Nicholson GA. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat Genet. 2001;27:309–312. doi: 10.1038/85879. [DOI] [PubMed] [Google Scholar]
- 10.Dedov V, Dedova I, Merrill A, Nicholson G. Activity of partially inhibited serine palmitoyltransferase is sufficient for normal sphingolipid metabolism and viability of HSN1 patient cells. Biochim Biophys Acta. 2004;1688(2):168–175. doi: 10.1016/j.bbadis.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 11.Verhoeven K, Timmerman V, Mauko B, Pieber TR, De Jonghe P, Auer-Grumbach M. Recent advances in hereditary sensory and autonomic neuropathies. Curr Opin Neurol. 2006;19:474–480. doi: 10.1097/01.wco.0000245370.82317.f6. [DOI] [PubMed] [Google Scholar]
- 12.Cole N, Murphy D, Grider T, Rueter S, Brasaemle D, Nussbaum R. Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein alpha-synuclein. J Biol Chem. 2002;277(8):6344–6352. doi: 10.1074/jbc.M108414200. [DOI] [PubMed] [Google Scholar]
- 13.Lane R, Farlow M. Lipid homeostasis and apolipoprotein E in the development and progression of Alzheimer’s disease. J Lipid Res. 2005;46(5):949–968. doi: 10.1194/jlr.M400486-JLR200. [DOI] [PubMed] [Google Scholar]
- 14.Gitler A, Chesi A, Geddie M, Strathearn K, Hamamichi S, Hill K, Caldwell K, Caldwell G, Cooper A, Rochet J, Lindquist S. α-Synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet. 2008;41(3):308–315. doi: 10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beller M, Thiel K, Thul P, Jackle H. Lipid droplets: a dynamic organelle moves into focus. FEBS Lett. 2010;584(11):2176–2182. doi: 10.1016/j.febslet.2010.03.022. [DOI] [PubMed] [Google Scholar]
- 16.Farese R, Walther T. Lipid droplets finally get a little R-E-S-P-E-C-T. Cell. 2009;139(5):855–860. doi: 10.1016/j.cell.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ducharme N, Bickel P. Minireview: lipid droplets in lipogenesis and lipolysis. Endocrinology. 2008;149(3):942–949. doi: 10.1210/en.2007-1713. [DOI] [PubMed] [Google Scholar]
- 18.Zehmer J, Huang Y, Peng G, Pu J, Anderson R, Liu P. A role for lipid droplets in inter-membrane lipid traffic. Proteomics. 2009;9(4):914–921. doi: 10.1002/pmic.200800584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hodges B, Wu C. Proteomic insights into an expanded cellular role for cytoplasmic lipid droplets. J Lipid Res. 2010;51(2):262–273. doi: 10.1194/jlr.R003582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lay, S.L, Hajduch, E, Linday, M.R, Liepvre, X.L, Thiele, C, Ferre, P, Parton, R.G, Kurzchalia, T, Simons, K & Dugail, I. 2006. Cholesterol-Induced Caveolin Targeting to Lipid Droplets in Adipocytes: A Role for Caveolar Endocytosis. Traffic, vol. 7, pp. 549–561. Journal of Biological Chemistry. Vol. 280, No. 52, pp. 42841–42847 [DOI] [PubMed]
- 21.Xu G, Sztalryd C, Lu X, Tansey JT, Gan J, Dorward H, Kimmel AR, Londos C (2005) Post-translational regulation of adipose differentiation related protein by the ubiquitin/proteasome pathway [DOI] [PubMed]