

Abstract
Inflammation plays a central role in neonatal brain injury. During brain inflammation the resident macrophages of the brain, the microglia cells, are rapidly activated. In the periphery, α7 nicotinic acetylcholine receptors (α7R) present on macrophages can regulate inflammation by suppressing cytokine release. In the current study we investigated α7R expression in neonatal mice after hypoxia-ischemia (HI). We further examined possible anti-inflammatory role of α7R stimulation in vitro and microglia polarization after α7R agonist treatment. Real-time PCR analysis showed a 33% reduction in α7R expression 72 h after HI. Stimulation of primary microglial cells with LPS in combination with increasing doses of the selective α7R agonist AR-R 17779 significantly attenuated TNFα release and increased α7R transcript in microglial cells. Gene expression of M1 markers CD86 and iNOS, as well as M2 marker CD206 was not influenced by LPS and/or α7R agonist treatment. Further, Mox markers heme oxygenase (Hmox1) and sulforedoxin-1 (Srx1) were significantly increased, suggesting a polarization towards the Mox phenotype after α7R stimulation. Thus, our data suggest a role for the α7R also in the neonatal brain and support the anti-inflammatory role of α7R in microglia, suggesting that α7R stimulation could enhance the polarization towards a reparative Mox phenotype.
1. Introduction
Perinatal hypoxia-ischemia (HI) is a major cause of brain injury in newborns, resulting in an increased risk of developmental impairment and permanent neurological deficits such as cerebral palsy and mental retardation [1]. Inflammation plays a central role in the development of brain injury in newborns [2]. Both neonatal hypoxia-ischemia and stroke trigger an inflammatory response [3, 4] and experimental studies show that inhibition of proinflammatory mediators is neuroprotective [5, 6].
Microglia, the resident macrophage of the brain, are central in this process being the main cell providing immunosurveillance in the brain. During pathological conditions, such as hypoxia-ischemia, microglia are rapidly activated with antigen presentation and secretion of cytokines and other inflammatory mediators as a consequence [7]. In the periphery, macrophages are highly dynamic cells that can be polarized into different macrophage phenotypes depending on the microenvironment, that is, the classical proinflammatory M1 macrophage and the wound-healing M2 macrophage being the most discussed phenotypes [8, 9]. Recently, also other macrophage phenotypes have been described, the Mox macrophage that develops in response to oxidative stress [10] and the M4 macrophage, induced by the platelet-derived cytokine CXCL4 [11]. Interestingly, also primary microglial cells can be polarized in vitro into different microglial phenotypes [12].
In the brain, nicotinic acetylcholine receptors (nAChRs) contribute to regulation of neuronal plasticity [13] and neuroprotection [14, 15]. These ion channels, forming homo- or heteropentamers, have been suggested to play important roles in neurodegenerative diseases such as Alzheimer's disease [16], Parkinson's disease [17], and schizophrenia [18, 19]. The most abundant nicotinic receptors in the brain are the α4β2 (α4β2R) and the α7 receptors (α7R). In the periphery, the α7 nicotinic receptor (α7R) can modulate inflammation [20], that is, signaling via the α7R inhibits cytokine release, thereby suppressing inflammation and providing protection against tissue damage in inflammatory states [21]. The α7R is expressed by leucocytes; however, macrophages have been identified as the primary effector cell [22].
Recently, stimulation of the α7R was described to have a neuroprotective role in adult brain injury [23], and to be expressed by microglial cells [24, 25], however, little is known about the α7R expression in the neonate and its role in perinatal brain injury. In the present study we hypothesized that the expression of α7R is decreased after perinatal hypoxia-ischemic brain injury and that stimulation of α7R with a selective α7R agonist, AR-R 17779, has an anti-inflammatory effect on microglia. Further, we investigated the microglial phenotype after stimulation with α7R agonist.
2. Material and Methods
2.1. Animals
C57BL/6J mice obtained from Charles River were housed and bred in a room with 12 h light/dark cycle. Water and standard laboratory food were available ad libitum. All procedures involving animals were approved by the regional ethics committee of Gothenburg and performed according to the Swedish guidelines for the Care and Use of Laboratory animals.
2.2. Hypoxia-Ischemia Model
Hypoxia-ischemia (HI) in neonatal mice was performed as previously described [26]. In brief, at postnatal day (P) 9-10, the left common carotid artery was ligated under isoflurane anesthesia. After ligation, the wound was closed, anesthesia discontinued, and the mice were allowed to recover for one hour. After recovery, the mice were placed in an incubator circulated with firstly normal air for 10 minutes, secondly with a humidified gas mixture (10.00 ± 0.01% oxygen in nitrogen) for 45 minutes, and thirdly with normal air again for 10 minutes. Incubator temperature was kept at 36°C throughout the experiment. Thereafter, the pups were returned to their dam until sacrifice. The method induces hypoxic-ischemic injury to the left cerebral hemisphere [26]. At 24 and 72 h after HI the animals were terminally anesthetized and intracardially perfused with saline to remove blood from the brain. Brains were collected, snap frozen in N (l) and stored at −80°C until further analysis. Control mice underwent sham surgery. Mice at P9-10 were used given that their brains are approximately at a developmental stage equivalent to the near-term human infant [27].
2.3. Microglial Cell Culture
Mixed glial cell cultures were prepared from whole brains of P2-3 mice. Brains were homogenized by pipetting in Dulbecco's modified Eagle's medium (DMEM) with 20% fetal bovine serum (FBS) and 1% Penicillin-Streptomycin (Sigma Aldrich, Stockholm, Sweden) followed by filtration through 70 μm cell strainer (BD Biosciences, Stockholm, Sweden). Cells were seeded in DMEM 20% FBS and 1% Penicillin-Streptomycin in 75 cm2 flasks (Sarstedt AB, Helsingborg, Sweden) and cultured in 5% CO2/95% air at 37°C. After 7 days in vitro the medium was replaced with DMEM with 10% FBS/1% Penicillin-Streptomycin. Mixed glial cells reached confluency after 14 days in vitro. Primary microglia were mechanically isolated by using a reciprocating shaker at 250 rpm for 3 hours at 36°C. Microglia cells were pelleted via centrifugation at 250 g for 10 min, resuspended in DMEM with 2% FBS/1% Penicillin-Streptomycin and 200 000–250 000 cells were plated/well in 12-well plates (BD Biosciences, Stockholm, Sweden). Following incubation for 24 h, cells were stimulated with LPS (10 ng/mL in PBS, List Biological Laboratories Inc., Campbell, CA) with or without the α7R agonist AR-R 17779 (Tocris Bioscience, Bristol, UK) with the indicated doses. AR-R 17779 was dissolved in Dimethyl Sulfoxide (DMSO) and then diluted in culture media to a maximal final concentration of 3.3% DMSO. After 4 h incubation, supernatant and cells were harvested and stored at −80°C for further analysis.
2.4. RNA Extraction, cDNA Synthesis, and Gene Expression Analysis
Brains collected after HI and microglia samples obtained from cell cultures were homogenized by pipetting in RNase free PBS and using 30G insulin syringes (BD Biosciences, Stockholm Sweden) with RLT buffer (Oiagen GmbH, Hilden, Germany), respectively. RNA was extracted by using the RNAeasy Lipid Tissue Mini/Micro Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer's protocol. RNA concentration was determined with NanoDrop analysis (NanoDrop Products, DE, USA). QuantiTect Reverse Transcription kit (Qiagen GmbH, Hilden, Germany) was used to synthesize first strand cDNA according to the manufacturer's protocol.
Real-time PCR analysis was run on a LightCycler 480 (Roche Diagnostics GmbH, Mannheim, Germany) using the following cycling program: denaturation at 95°C for 10 minutes followed by 45 cycles of denaturation at 95°C for 15 seconds and annealing/extension at 60°C for 4 seconds and 72°C for 8–12 seconds, respectively. Melting-curve analysis was performed to ensure that only one PCR product was obtained. PCR products were further validated on agarose gel. All samples were run in duplicate. Intersample differences were limited to 0.5 cycles and samples with >0.5 cycles difference being excluded from the analysis. The following primers were used: α7R (chrna7, QT00143626), α4R (chrna4, QT00144662), β2R (chrnb2, QT00127708), CD86 (QT01055250), iNOS (QT01547980), CD206 (mrc1, QT00103012), Arginase 1 (Arg1, QT00134288), heme oxygenase (Hmox1, QT00159915), and sulforedoxin-1 (Srx1/npn3, QT00289443, all from Qiagen). The expression level of each target gene was normalized against the reference gene YWHAZ (tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, QT00105350), calculated as 2−ΔΔCT, where ΔCT was the CT of the target gene after subtracting the CT value of the reference gene and ΔΔCT was the CT value corrected by the average CT of each group.
2.5. TNFα and IL-6 Analysis in Cell Culture Supernatants
Microglial cell supernatants were obtained by collecting the media of stimulated cells followed by centrifugation at 8000 g for 3 min. The supernatants were transferred to new tubes and analysis of TNFα and IL-6 levels were performed by ELISA (BioLegend Inc., San Diego, USA) according to the manufacturer's protocol.
2.6. Statistical Analysis
All data are presented as mean ± SEM. Normality was tested using the Shapiro-Wilk normality test and parametric or nonparametric tests were used accordingly. Normally distributed data were analyzed with ANOVA followed by Dunnett's or Tukey's multiple comparison test. Data that did not fulfill the test for normality was analyzed by Kruskall-Wallis one-way analysis of variance followed by Dunn's multiple comparison test. All statistical analyses were performed by SPSS (IBM SPSS Statistics 20, IBM Corporation, CHI, USA) or Prism (GraphPad Prism 5, GraphPad Software, Inc., CA, USA). The significance level was set to P ≤ 0.05.
3. Results and Discussion
3.1. HI Decrease α7R Expression at 72 Hours in the Neonatal Brain
The expression of nicotine receptors are decreased in patients with neurodegenerative disorders such as schizophrenia [28] and Alzheimer's disease [16]. Also the expression of α7R is decreased in experimental models of adult brain injury [29]. Little is known of the expression of α7R in the neonatal brain or following brain injury in neonates. Hence, we investigated the expression levels of the α7R in a well-documented HI model in neonatal mice. Mice at P9-10 were chosen, as this can be approximated to a term human infant in terms of brain development [27]. At 24 h after HI there was no difference in α7R expression (Figure 1(a)); however, at 72 h after HI α7R gene expression was decreased by 33% in the injured versus noninjured hemisphere (Figure 1(a)). The gene expression of α7R was not altered in the noninjured hemisphere compared to mice undergoing sham surgery (Figure 1(a)). Thus, similar to traumatic brain injury in adult [30], the α7R gene expression is diminished after neonatal brain injury.
Figure 1.

Decreased α7R expression 72 h after HI. Gene expression analysis of α7R, α4, and β2 receptor subunit levels in brains 24 and 72 h after hypoxia-ischemia (HI) in neonatal mice at age P9-10 ((a)–(c)). (a) At 24 h after HI there was no difference in gene expression of α7R; however, at 72 h α7R gene expression was significantly reduced in the injured (ipsilateral, Ipsi) hemisphere compared to the noninjured (contralateral, Contra) hemisphere. (b) Gene expression of α4R subunit was not influenced at 24 or at 72 h after HI. (c) β2R subunit expression was significantly increased in the injured (Ipsi) hemisphere compared to the noninjured (Contra) hemisphere at 24 h after HI. There was no difference in gene expression of β2R subunit at 72 h after HI. There was no difference between the sham animals (ipsilateral hemisphere) compared to the noninjured contralateral hemisphere in HI mice. Gene expression was normalized to YWHAZ and analyzed using ΔΔCT method. Data are expressed as mean ± SEM, *P < 0.05 for contralateral versus ipsilateral hemisphere in HI mice, n = 4-5 mice/group.
In the brain, the most abundant nicotinic receptors are the α7R and the receptor consisting of α4 and β2 subunits, respectively (α4β2R) [31]. To explore whether HI influence a general change in nicotinic receptor expression or if this was specific for α7R we analyzed the expression of the receptor subunits of the α4β2R. The gene expression of the α4 subunit was not altered by HI (Figure 1(b)); however, β2 gene expression was significantly increased 24 h after HI (Figure 1(c)). This is particularly interesting since a recent study suggests that lack of, or, antagonists to β2-containing nicotinic receptors decrease brain injury in adult stroke [32]. Whether the increased β2 gene expression after HI contributes to brain injury in neonatal mice remains to be explored.
3.2. α7 Receptor Agonist Increase α7R Gene Expression in Microglial Cultures
After establishing that α7R is regulated in HI we sought to investigate its expression in microglial cultures and its possible anti-inflammatory effect. Primary rat and mouse microglial cultures express α7R [24, 25]. We could confirm these findings in the present study. We further explored the expression of α7R after proinflammatory stimulation, using LPS, with or without the selective α7R agonist AR-R 17779. Interestingly, α7R expression was not altered by LPS stimulation per se; however, α7R expression increased by 86% in microglial cells treated with LPS and α7R agonist AR-R 17779 (Figure 2(a)). Similar to the expression in the brain tissue, gene expression of the α4 subunit was not altered by the different treatments (Figure 2(b)). However, β2 gene expression was significantly increased by agonist treatment (Figure 2(c)). From our results we cannot determine whether this increase have functional implications [33].
Figure 2.

α7R agonist increase α7R gene expression in microglia cultures. Primary microglia cultures were stimulated with LPS (10 ng/mL) with or without α7R agonist AR-R 17779 (10 µM) for 4 h. Cells were collected and gene expression of (a) α7, (b) α4, and (c) β2 receptor subunits was investigated using real-time PCR. α7R agonist AR-R 17779 significantly increased α7R expression (a), whereas the α4 receptor subunit was not influenced by the treatment (b). Further, gene expression of β2 receptor subunits was increased by α7R agonist treatment (c). Graph represents pooled data from 4 independent experiments and n = 4/group. Gene expression was normalized to YWHAZ and analyzed using ΔΔCT method. Data are expressed as mean ± SEM, *P < 0.05.
Long-term treatment with nicotine, selective α7R agonists [34, 35], as well as, treatment with acetylcholine esterase inhibitors (AChE) [36], the enzyme responsible for acetylcholine degradation, increase α7R on protein level. Several mechanisms are suggested to be involved in agonist-induced upregulation of nicotinic receptors, for example, increased receptor trafficking to the surface, decreased cell surface turnover, increased subunit maturation, and decreased subunit degradation [35]. When comparing α7R expression between different mouse strains, α7R gene expression does not correlate with protein levels [37]. However, the mice in the previous study were not treated with α7R agonists, it is possible that α7R agonists treatment could influence α7R expression on both gene and protein level. Thus, further studies are needed to explore whether the increase in microglial α7R gene expression after agonist treatment is translated into protein.
3.3. α7 Receptor Agonist AR-R 17779 Decrease TNFα in a Dose-Dependent Manner
Previous evaluation of the anti-inflammatory role of α7R in microglial cultures has mostly been based on nicotine or acetylcholine (ACh) stimulations in combination with proinflammatory stimuli such as LPS [24, 25] and few studies have evaluated other α7R ligands. In the current study we investigate the effect of the selective α7R agonist AR-R 17779 in microglial cultures. After 4 hours stimulation with LPS, in combination with increasing doses of α7R agonist AR-R 17779, we detected a significant decrease in TNFα levels in the cell culture supernatant (Figure 3(a)). Further, we also investigated the effect of α7R agonist AR-R 17779 on the pleiotropic cytokine IL-6. Interestingly, there was a numerical dose-dependent decrease in IL-6 levels, similar to the TNF response; however, this did not reach significance (Figure 3(b)). Hence, our results support the earlier studies in microglial cultures using nicotine and ACh [24, 25] as well as in hippocampal cultures [23]; that is, α7R agonist have an anti-inflammatory effect on microglia.
Figure 3.

α7R stimulation decrease TNFα levels in a dose-dependent manner. Primary microglial cultures were stimulated with LPS (10 ng/mL) in combination with 1, 5, 10, or 15 µM of α7R agonist AR-R 17779 for 4 h and levels of TNFα (a) and IL-6 (b) were determined in cell culture supernatants by ELISA. 1 µM α7R agonist AR-R 17779 did not influence the LPS induced TNFα response, however, 5, 10, and 15 µM significantly decreased the TNFα levels (a). Treatment with LPS and α7R agonist AR-R 17779 did not influence the level of IL-6 between groups (b). Graph represents pooled data from 3-4 independent experiments, n = 3-4/group. Data are expressed as mean ± SEM, *P < 0.05, ***P < 0.001.
The mechanism behind the suppressed cytokine response is intriguing. In microglia, the properties of the α7R differ from the neuronal α7R [25]. Rather than functioning as a conventional ligand-gated ion channel causing Ca2+ influx, the α7R activates intracellular pathways including phospholipase C (PLC) and release of Ca2+ from intracellular stores [25]. In the periphery, several different intracellular signaling pathways have been suggested to be involved in the anti-inflammatory effect mediated via the α7R, including the JAK2/STAT3 pathway [38, 39], MAPK [24, 40], and NFκB [41]. At present, only MAPK have been demonstrated to participate in the anti-inflammatory effect after α7R agonist treatment in microglia [24], whether other signaling pathways also are engaged remains to be investigated.
3.4. α7 Receptor Agonist AR-R 17779 Polarize Microglia into Mox Phenotype
It was recently shown that in the periphery, oxidative stress drives macrophages towards a novel macrophage phenotype (Mox) mediated via activation of nuclear factor erythroid 2-like factor 2 (Nrf2) [10]. Interestingly, the α7R agonist PNU282987 is neuroprotective and decreases inflammation in adult brain injury, an effect mediated via Nrf2 [23]. Based on these finding we sought to determine the microglial phenotype in the current experiments. By real-time PCR we analyzed M1 markers CD86 and iNOS, M2 markers CD206 and Arginase 1 (Arg1), and Mox markers heme oxygenase (Hmox1) and sulfiredoxin-1 (Srx1) [10]. None of the M1 markers, CD86 and iNOS, or M2 marker, CD206 was regulated by LPS or the combination of LPS and α7R agonist AR-R 17779 (Figures 4(a)-4(b)). Interestingly, M2 marker Arg1 was downregulated by LPS and gene expression was then normalized with the combination of LPS and α7R agonist (Figure 4(b)). Further, Mox markers Hmox1 and Srxn1 were both significantly upregulated by LPS and AR-R 17779 treatment (Figure 4(c)), suggesting that α7R stimulation drives microglial cells towards the Mox phenotype. The exact role of the Mox phenotype in vivo remains to be investigated although Mox macrophages have been proposed to exert anti-inflammatory and anti-oxidizing effects in vivo [42]. Possibly, the decreased TNFα levels after α7R agonist could partly be due to the Mox phenotype.
Figure 4.
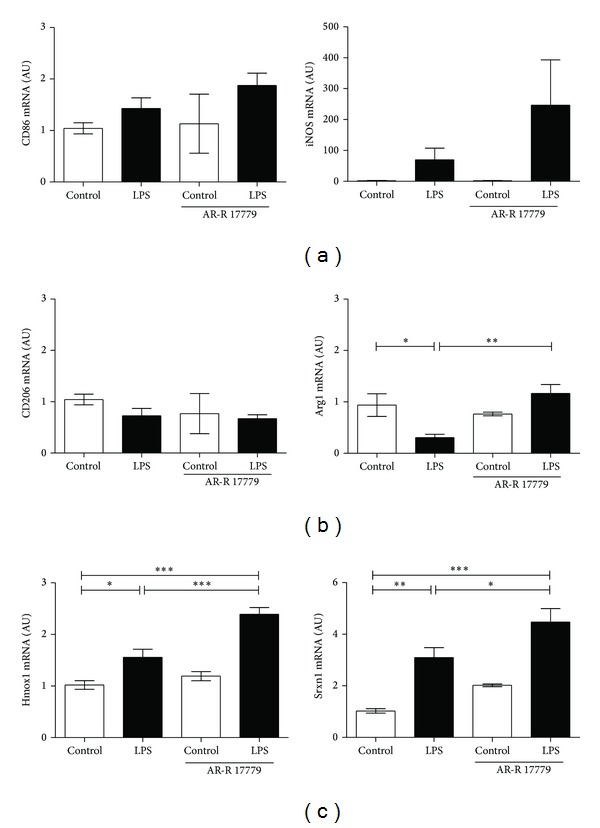
α7 receptor agonist AR-R 17779 polarize microglia towards Mox phenotype. Primary microglia cultures were stimulated with LPS (10 ng/mL) with or without α7R agonist AR-R 17779 (10 µM) for 4 h. Cells were collected and gene expression of (a) M1 markers CD86 and iNOS, (b) M2 markers CD206 and Arginase 1 (Arg1) and (c) Mox markers heme oxygenase (Hmox1) and sulfiredoxin-1 (Srxn1) was investigated. Treatment with LPS and α7R agonist AR-R 17779 did not influence microglial expression of M1 markers and M2 marker CD206; however, M2 marker Arg1 was downregulated by LPS, and upregulated by the combination of LPS and α7R agonist AR-R 17779 ((a)-(b)). Mox markers Hmox1 and Srxn1 were both upregulated by LPS and α7R agonist AR-R 17779 treatment (c). Graph represents pooled data from 4-5 independent experiments, n = 6–8/group for all except Control AR-R 17779; n = 2. Gene expression was normalized to YWHAZ and analyzed using ΔΔCT method. Data are expressed as mean ± SEM, *P < 0.05, **P < 0.01,***P < 0.001.
4. Conclusions
In line with what is seen in patients with neurodegenerative disorders and in experimental models of adult brain injury, HI in neonatal mice decrease the expression of the α7R. This regulation proposes an important role for α7R also in the developing brain. Further, we demonstrated an anti-inflammatory effect of the α7R agonist AR-R 17779 on microglial cells, possibly, partly due to upregulation of the α7R transcript after stimulation with α7R agonist, but potentially also partly due to microglial polarization towards the Mox phenotype. Thus, our data suggest a role for the α7R in neonatal brain injury and support the anti-inflammatory role of the α7R in microglial cultures, suggesting that α7R stimulation could enhance the polarization towards a reparative Mox phenotype.
Acknowledgments
The authors thank Ana Baburamani and Anna-Lena Leverin for helpful assistance. This study was supported by The Swedish Research Council (VR2010-2627, VR2012-2992), a Government grant to a researcher in Public Health Service at the Sahlgrenska University Hospital (ALFGBG-142881), the Leducq Foundation (DSRR_P34404), Åhlén-stiftelsen, Wilhelm and Martina Lundgren Foundation, The Sigurd and Elsa Golje Foundation, The Swedish Heart-Lung Foundation, The Swedish Society of Medicine, Magnus Bergvall Foundation, and The Swedish Stroke Association.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Perez A, Ritter S, Brotschi B, et al. Long-term neurodevelopmental outcome with hypoxic-ischemic encephalopathy. Journal of Pediatrics. 2013;163:454–459. doi: 10.1016/j.jpeds.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 2.Dammann O, Leviton A. Inflammatory brain damage in preterm newborns—dry numbers, wet lab, and causal inferences. Early Human Development. 2004;79(1):1–15. doi: 10.1016/j.earlhumdev.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 3.Hedtjärn M, Mallard C, Hagberg H. Inflammatory gene profiling in the developing mouse brain after hypoxia-ischemia. Journal of Cerebral Blood Flow and Metabolism. 2004;24(12):1333–1351. doi: 10.1097/01.WCB.0000141559.17620.36. [DOI] [PubMed] [Google Scholar]
- 4.Vexler ZS, Tang XN, Yenari MA. Inflammation in adult and neonatal stroke. Clinical Neuroscience Research. 2006;6(5):293–313. doi: 10.1016/j.cnr.2006.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hedtjärn M, Leverin A-L, Eriksson K, Blomgren K, Mallard C, Hagberg H. Interleukin-18 involvement in hypoxic-ischemic brain injury. The Journal of Neuroscience. 2002;22(14):5910–5919. doi: 10.1523/JNEUROSCI.22-14-05910.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Svedin P, Hagberg H, Sävman K, Zhu C, Mallard C. Matrix metalloproteinase-9 gene knock-out protects the immature brain after cerebral hypoxia-ischemia. The Journal of Neuroscience. 2007;27(7):1511–1518. doi: 10.1523/JNEUROSCI.4391-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vexler ZS, Yenari MA. Does inflammation after stroke affect the developing brain differently than adult brain? Developmental Neuroscience. 2009;31(5):378–393. doi: 10.1159/000232556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leitinger N, Schulman IG. Phenotypic polarization of macrophages in atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013;33:1120–1126. doi: 10.1161/ATVBAHA.112.300173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. Journal of Clinical Investigation. 2012;122(3):787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kadl A, Meher AK, Sharma PR, et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circulation Research. 2010;107(6):737–746. doi: 10.1161/CIRCRESAHA.109.215715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gleissner CA, Shaked I, Little KM, Ley K. CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. Journal of Immunology. 2010;184(9):4810–4818. doi: 10.4049/jimmunol.0901368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chhor V, Le Charpentier T, Lebon S, et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain, Behavior, and Immunity. 2013;32:70–85. doi: 10.1016/j.bbi.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albuquerque EX, Alkondon M, Pereira EFR, et al. Properties of neuronal nicotinic acetylcholine receptors: pharmacological characterization and modulation of synaptic function. Journal of Pharmacology and Experimental Therapeutics. 1997;280(3):1117–1136. [PubMed] [Google Scholar]
- 14.Kihara T, Shimohama S, Sawada H, et al. α7 Nicotinic Receptor Transduces Signals to Phosphatidylinositol 3-Kinase to Block A β-Amyloid-induced Neurotoxicity. The Journal of Biological Chemistry. 2001;276(17):13541–13546. doi: 10.1074/jbc.M008035200. [DOI] [PubMed] [Google Scholar]
- 15.Zoli M, Picciotto MR, Ferrari R, Cocchi D, Changeux J-P. Increased neurodegeneration during ageing in mice lacking high-affinity nicotine receptors. EMBO Journal. 1999;18(5):1235–1244. doi: 10.1093/emboj/18.5.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burghaus L, Schütz U, Krempel U, et al. Quantitative assessment of nicotinic acetylcholine receptor proteins in the cerebral cortex of Alzheimer patients. Molecular Brain Research. 2000;76(2):385–388. doi: 10.1016/s0169-328x(00)00031-0. [DOI] [PubMed] [Google Scholar]
- 17.Guan Z-Z, Nordberg A, Mousavi M, Rinne JO, Hellström-Lindahl E. Selective changes in the levels of nicotinic acetylcholine receptor protein and of corresponding mRNA species in the brains of patients with Parkinson’s disease. Brain Research. 2002;956(2):358–366. doi: 10.1016/s0006-8993(02)03571-0. [DOI] [PubMed] [Google Scholar]
- 18.Freedman R, Olincy A, Buchanan RW, et al. Initial phase 2 trial of a nicotinic agonist in schizophrenia. American Journal of Psychiatry. 2008;165(8):1040–1047. doi: 10.1176/appi.ajp.2008.07071135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olincy A, Harris JG, Johnson LL, et al. Proof-of-concept trial of an α7 nicotinic agonist in schizophrenia. Archives of General Psychiatry. 2006;63(6):630–638. doi: 10.1001/archpsyc.63.6.630. [DOI] [PubMed] [Google Scholar]
- 20.Wang H, Yu M, Ochani M, et al. Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation. Nature. 2003;421(6921):384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 21.Andersson U, Tracey KJ. Neural reflexes in inflammation and immunity. The Journal of Experimental Medicine. 2012;209:1057–1068. doi: 10.1084/jem.20120571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H, Liao H, Ochani M, et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nature Medicine. 2004;10(11):1216–1221. doi: 10.1038/nm1124. [DOI] [PubMed] [Google Scholar]
- 23.Parada E, Egea J, Buendia I, et al. The microglial alpha7-acetylcholine nicotinic receptor Is a key element in promoting neuroprotection by Inducing heme oxygenase-1 via nuclear factor erythroid-2-related factor 2. Antioxidants & Redox Signaling. 2013;19:1135–1148. doi: 10.1089/ars.2012.4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shytle RD, Mori T, Townsend K, et al. Cholinergic modulation of microglial activation by α7 nicotinic receptors. Journal of Neurochemistry. 2004;89(2):337–343. doi: 10.1046/j.1471-4159.2004.02347.x. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki T, Hide I, Matsubara A, et al. Microglial α7 nicotinic acetylcholine receptors drive a phospholipase C/IP3 pathway and modulate the cell activation toward a neuroprotective role. Journal of Neuroscience Research. 2006;83(8):1461–1470. doi: 10.1002/jnr.20850. [DOI] [PubMed] [Google Scholar]
- 26.Stridh L, Mottahedin A, Johansson ME, et al. Toll-like receptor-3 activation increases the vulnerability of the neonatal brain to hypoxia-ischemia. The Journal of Neuroscience. 2013;33:12041–12051. doi: 10.1523/JNEUROSCI.0673-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Craig A, Luo NL, Beardsley DJ, et al. Quantitative analysis of perinatal rodent oligodendrocyte lineage progression and its correlation with human. Experimental Neurology. 2003;181(2):231–240. doi: 10.1016/s0014-4886(03)00032-3. [DOI] [PubMed] [Google Scholar]
- 28.Leonard S, Breese C, Adams C, et al. Smoking and schizophrenia: abnormal nicotinic receptor expression. European Journal of Pharmacology. 2000;393(1–3):237–242. doi: 10.1016/s0014-2999(00)00035-2. [DOI] [PubMed] [Google Scholar]
- 29.Verbois SL, Scheff SW, Pauly JR. Time-dependent changes in rat brain cholinergic receptor expression after experimental brain injury. Journal of Neurotrauma. 2002;19(12):1569–1585. doi: 10.1089/089771502762300238. [DOI] [PubMed] [Google Scholar]
- 30.Verbois SL, Scheff SW, Pauly JR. Chronic nicotine treatment attenuates α7 nicotinic receptor deficits following traumatic brain injury. Neuropharmacology. 2003;44(2):224–233. doi: 10.1016/s0028-3908(02)00366-0. [DOI] [PubMed] [Google Scholar]
- 31.Buisson B, Bertrand D. Nicotine addiction: the possible role of functional upregulation. Trends in Pharmacological Sciences. 2002;23(3):130–136. doi: 10.1016/S0165-6147(00)01979-9. [DOI] [PubMed] [Google Scholar]
- 32.Liu Q, Tang Z, Gan Y, et al. Genetic deficiency of beta2-containing nicotinic receptors attenuates brain injury in ischemic stroke. Neuroscience. 2014;256:170–177. doi: 10.1016/j.neuroscience.2013.10.049. [DOI] [PubMed] [Google Scholar]
- 33.Timmermann DB, Sandager-Nielsen K, Dyhring T, et al. Augmentation of cognitive function by NS9283, a stoichiometry-dependent positive allosteric modulator of alpha2- and alpha4-containing nicotinic acetylcholine receptors. British Journal of Pharmacology. 2012;167:164–182. doi: 10.1111/j.1476-5381.2012.01989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Christensen DZ, Mikkelsen JD, Hansen HH, Thomsen MS. Repeated administration of 7 nicotinic acetylcholine receptor (nAChR) agonists, but not positive allosteric modulators, increases 7 nAChR levels in the brain. Journal of Neurochemistry. 2010;114(4):1205–1216. doi: 10.1111/j.1471-4159.2010.06845.x. [DOI] [PubMed] [Google Scholar]
- 35.Thomsen MS, Mikkelsen JD. Type I and II positive allosteric modulators differentially modulate agonist-induced up-regulation of alpha7 nicotinic acetylcholine receptors. Journal of Neurochemistry. 2012;123:73–83. doi: 10.1111/j.1471-4159.2012.07876.x. [DOI] [PubMed] [Google Scholar]
- 36.Kume T, Sugimoto M, Takada Y, et al. Up-regulation of nicotinic acetylcholine receptors by central-type acetylcholinesterase inhibitors in rat cortical neurons. European Journal of Pharmacology. 2005;527(1–3):77–85. doi: 10.1016/j.ejphar.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 37.Brooks NP, Mexal S, Stitzel JA. Chrna7 genotype is linked with alpha7 nicotinic receptor expression but not alpha7 RNA levels. Brain Research. 2009;1263:1–9. doi: 10.1016/j.brainres.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chatterjee PK, Al-Abed Y, Sherry B, Metz CN. Cholinergic agonists regulate JAK2/STAT3 signaling to suppress endothelial cell activation. American Journal of Physiology: Cell Physiology. 2009;297(5):C1294–C1306. doi: 10.1152/ajpcell.00160.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Jonge WJ, van der Zanden EP, The FO, et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nature Immunology. 2005;6(8):844–851. doi: 10.1038/ni1229. [DOI] [PubMed] [Google Scholar]
- 40.Toborek M, Son KW, Pudelko A, King-Pospisil K, Wylegala E, Malecki A. ERK 1/2 signaling pathway is involved in nicotine-mediated neuroprotection in spinal cord neurons. Journal of Cellular Biochemistry. 2007;100(2):279–292. doi: 10.1002/jcb.21013. [DOI] [PubMed] [Google Scholar]
- 41.Saeed RW, Varma S, Peng-Nemeroff T, et al. Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. Journal of Experimental Medicine. 2005;201(7):1113–1123. doi: 10.1084/jem.20040463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Butcher MJ, Galkina EV. Phenotypic and functional heterogeneity of macrophages and dendritic cell subsets in the healthy and atherosclerosis-prone aorta. Frontiers in Physiology. 2012;3, article 44 doi: 10.3389/fphys.2012.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]