

Abstract
A mixture of structural isomers was separated and identified at nanomolar concentrations (~100,000 molecules) by incorporating capillary zone electrophoresis (CZE) with a sheath flow surface-enhanced Raman scattering (SERS) detector. Baseline resolution was obtained from three structural isomers of rhodamine using a planar silver SERS substrate, demonstrating the utility of this approach for trace chemical analysis.
The ability to identify and characterize molecules purified through separation lies at the heart of chemical analysis. For column-based separations, common methods of detection include UV-visible absorption, laser-induced fluorescence (LIF), and mass spectrometry. Despite its low cost and flexibility, on-column UV-visible absorption suffers from poor molecular specificity and a lack of sensitivity.1, 2 On the other hand, LIF offers a high degree of sensitivity but requires fluorescent labels.3–5 Since structure determination by migration times alone requires extensive knowledge of the samples beforehand, the use of these two methods is limited for explicit analyte characterization. Mass spectrometry provides exquisite analyte identification for many samples. However, many classes of molecules, such as structural isomers and other molecules with the same mass (isobars) are still challenging to characterize. The cost of high-resolution mass spectrometers necessary for characterizing similar compounds limits the utility of this technique for routine characterization.6, 7 As a result, there is a need for new detection techniques capable of providing structural information with high sensitivity and selectivity for chemical analysis.
Here we demonstrate surface-enhanced Raman scattering (SERS) for characterization of three rhodamine isomers separated by capillary zone electrophoresis (CZE). CZE is a powerful analytical technique for separation of charged analytes8, 9 and has been incorporated into microfluidic devices for high efficiency separations10–12. SERS provides extensive structural and quantitative information about a variety of molecules based on their vibrational transitions13 and can be readily performed in solution to facilitate detection in-line with chemical separations.14 Given these attributes, SERS has the potential to provide chemical identity of solutes following CZE separation.
There have been previous attempts to couple SERS to CZE. In these studies, CZE-SERS was accomplished by interfacing detection directly on-column or at-line. Direct on-column SERS detection has been achieved using running buffers containing silver colloidal solutions and by laser-induced growth of silver particles at the end of the capillary.15, 16 The use of colloidal particles has shown detection limits in the nM or pM range; however, memory effects commonly prevent the regeneration of the detection window and limit these configurations to a one-time-use only. Planar SERS substrates in CZE suffer an additional challenge; specifically, a metal in an electric field will form a bipolar electrode and cause electrochemical formation of bubbles and degradation of the sample.17 In-line CZE-SERS with planar substrates has been limited to μM limits of detection.16 An at-line approach to CZE-SERS deposits the effluent onto a moving substrate.18 Drying the sample adsorbs molecules to the surface and avoids challenges associated with mass transport. This approach also avoids challenges associated with the formation of a bipolar electrode across the SERS substrate; however, designing an interface that guarantees maintenance of the electrical current during the deposition onto the substrate is not trivial.
By incorporating our recently demonstrated sheath flow SERS detector,19 we are able to circumvent the challenges noted above and achieve online detection in CZE separations. In particular, the potential drop (bipolar electrode formation) across the SERS substrate is minimized by the increased volume of the sheath flow and confined sample near the electrical ground. Changes observed in the silver oxide background signals suggest a small electrochemical potential is still present. However, we have successfully used the same SERS substrate in CE applications for up to three days without significant signal degradation. The sheath flow SERS detector enables sequential and high throughput detection of the separated dyes at nanomolar concentrations (attomole - femtomole injections) using a 50 ms acquisition without significant “memory effect” or fouling of the SERS substrate.
The sheath flow SERS detector was coupled online to a CZE system. The CZE system is similar to the one previously reported except for the detection module.20, 21 CZE separation was performed in positive mode on a 50 cm bare fused silica capillary (Polymicro Technologies, Phoenix, AZ) with 72 μm i.d. and 143 μm o.d.. A constant potential of 300 V/cm was supplied by a Spellman, CZE 1000R power supply (Spellman High Voltage Electronics Corp., Hauppauge, NY).21 The sample, containing 10−8 M rhodamine 6G (R6G), 10−10 M rhodamine B (RB), and10−7 M 5-carboxytetramethylrhodamine (5-TAMRA), was prepared in 15 mM sodium tetraborate buffer (pH 9.4). The CZE separation was performed using a 2 s pressure injection, which injects 34 nL of sample. After injection, the capillary was placed in 15 mM sodium tetraborate buffer solution (pH 9.4) and 15kV (~40μA) was applied to the Pt electrode at the sample end of the capillary. SERS measurements were performed in kinetic series with 50 ms acquisition times and by using a sheath flow rate of 10 μL/min (a sheath flow to capillary flow rate ratio of 100:1). The Raman spectrometer used in this study has been previously described.22 Raman scattering was detected from a 633 nm laser, away from the absorption band of the rhodamine dyes and thus without the benefit of resonance enhancement. Full details on the instrument setups and experimental procedures are provided in the ESI. Figure S1 presents the schematic of the experimental setup used for the CZE-SERS experiments.
Figure 1A shows the heatmap of the SERS intensity as a function of Raman shift and migration time following the electrophoretic separation of three rhodamine isomers (R6G, RB, and 5-TAMRA). The Raman spectrum observed indicates that R6G migrates at tm=180 ± 13 s, RB at tm=220 ± 19 s, and finally 5-TAMRA at tm=290 ± 15 s. The SERS signal for each peak persists for about 1–2 s or less at these low concentrations. The short duration of the SERS signal is more clearly observed in the 2 s zooms shown in Figure 1B, which illustrate the difference in width of each migration peak.
Figure 1.
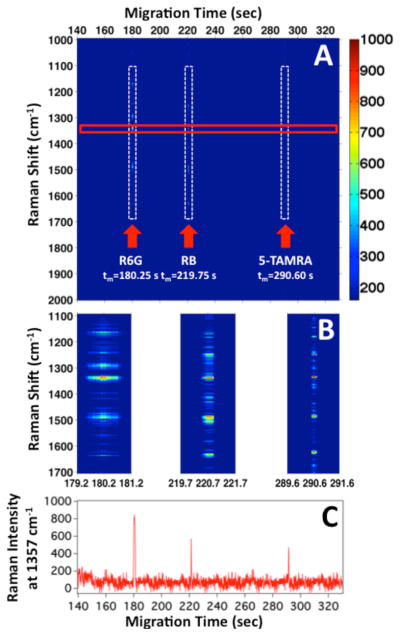
(A) Heatmap of the observed SERS intensity at each Raman shift as a function of migration time for the electrophoretic separation of R6G, RB, and 5-TAMRA. (B) Zoom in on the dashed vertical rectangles in (A) show 2 s windows corresponding to the detected analytes. (C) SERS intensity profile of the Raman band at 1357 cm−1 is plotted against migration time, extracted from the red rectangle shown in (A). This band is attributed to the combined aromatic C-C and C=N stretching modes of rhodamine compounds. The dashed vertical rectangles in (A) highlight the detection of each analyte.
Figure 1C shows the SERS electropherogram constructed from the SERS intensity at 1357 cm−1 as a function of migration time. This band is attributed to the combined aromatic C-C and C=N stretching modes of rhodamine compounds.23–27 The intensity profile at 1357 cm−1 provides a convenient signal to characterize the separation efficiency with SERS detection. The spectrally resolved SERS electropherogram of the three rhodamine dyes is characterized by a low and constant background.
Analysis of the SERS electropherogram (Figure 1C) shows a peak for R6G at t=180.25 s with a full width at half max (FWHM) of 1.25 s, which suggests a separation efficiency of N = 115,000 ± 35,000 theoretical plates. The SERS electropherogram peak for RB at t=219.75 s shows a more symmetric peak with a FWHM of 0.55 s. This corresponds to N = 898,000 ± 115,000 theoretical plates. The electropherogram peak for 5-TAMRA at t= 290.60 s has a FWHM of 0.40 s, which corresponds to a separation efficiency of N = 2,900,000 ± 620,000 theoretical plates. Because our analytes fluoresce when excited at shorter wavelengths, we performed laser-induced fluorescence (LIF) to compare the migration times and separation efficiency. Figure S-2 shows the electropherogram of the same three analyte mixture using LIF detection. The analyte concentrations and separation conditions were kept identical to those used in the optimized SERS experiments to provide a direct comparison. The LIF electropherogram shows three bands associated with the elution of R6G, RB, and 5-TAMRA with a separation efficiency N=1,000 – 6,000 theoretical plates (analyte dependent), which is low for a CZE separation with LIF detection. The poor separation efficiencies are the result of the large injection volume and the high concentration of analytes used for the CZE-LIF experiments. However, CZE-SERS and CZE-LIF generated identical elution order and equivalent migration times under identical separation conditions.
The difference in observed number of theoretical plates provides insight into the mechanism of SERS detection. Only molecules located within a close proximity to the SERS substrate surface can be detected. Our previous work suggests the observed signal arises from adsorbed molecules. However, it is known that Langmuir behavior inhibits analyte adsorption at low concentrations, typically below 1 nM.28 We have successfully detected RB at a concentration below this in Figure 1. This suggests that hydrodynamic confinement may provide a transiently increased concentration at the surface, such that the SERS detection is only obtained from the highest concentration portion of the migrating analyte band. This is in contrast to LIF, where the greater sensitivity enables detection of the width of the entire eluting sample. In Figures S-3 and S-4, we show the SERS results from a longer injection and a higher concentration of analytes. The apparent efficiency with SERS detection decreases to a level comparable to LIF due to molecules remaining on the surface for longer periods.
The role of adsorption is further evident in the width of the R6G peak relative to the widths observed for RB and 5-TAMRA. Increased adsorption of R6G to the surface results in a longer observed peak width in the SERS electropherogram (Figure 1C), suggesting R6G has a stronger binding affinity for silver surfaces than RB and 5-TAMRA. The CZE-SERS efficiency appears to correlate to the sample desorption rate from the substrate. In these results, the longer desorption rate observed for R6G can be directly attributed to the difference in molecular structure of the three rhodamine dyes (Figure S-5). R6G is the only dye out of the three containing a secondary amine group. The pKa of this amine group has a value of 6.13. When dissolved in borate buffer (pH 9.4), the basic form of R6G predominates (pH>pKa). As a result, the secondary amine group is deprotonated and more electron rich. Under these conditions, the nitrogen atom on the R6G molecules is more likely to adsorb to the silver SERS substrate than the other amine groups in RB and 5-TAMRA. These properties explain the higher affinity of R6G for the silver SERS substrate and the resulting slower desorption mechanism observed for this rhodamine dye. Despite these variations, it is worth noting that baseline resolution is achieved between each analyte, demonstrating no memory effects.
In our earlier publication,19 our SERS detector demonstrated a linear response from nM to μM concentrations of R6G, indicating that quantitation is possible for trace analyte detection. Our previous work further showed that chemical effects alter the desorption rate, which we are investigating to understand their impact on the observed separation.
The main advantage of using SERS over conventional detection techniques (UV and LIF) is that it can provide chemical information to identify and characterize analytes beyond migration times. Figure 2a shows a single 50 ms SERS spectrum of R6G (10−8 M) from the electrophoretic separation of the three dye mixture extracted from Figure 1A at tm=180.25 s. The main features of the R6G spectrum are the bands at 1175, 1306, 1357, 1506, and 1648 cm−1. These bands are associated with the characteristic stretching modes of the C-H band, C=N, and aromatic C-C stretching vibrations of R6G.23–27 Figure 2b shows a single 50 ms SERS spectrum of RB (10−10 M) extracted from Figure 1A at tm=219.75 s. The RB bands are assigned to the aromatic C-H bending (1197 cm−1), the C-C bridge-bands stretching (1276 cm−1), and the aromatic C-H bending vibrations (1357 cm−1, 1506 cm−1, and 1645 cm−1).29 Finally, Figure 2c shows a single 50 ms SERS spectrum of 5-TAMRA (10−7 M) extracted from Figure 1A at tm=290.60 s. The main features of the 5-TAMRA spectrum are the bands at 1197, 1276, 1354, 1506, and 1643 cm−1. These bands are assigned to the aromatic C-H bending, C-C bridge-band stretching, and aromatic C-C stretching modes of 5-TAMRA.30
Figure 2.
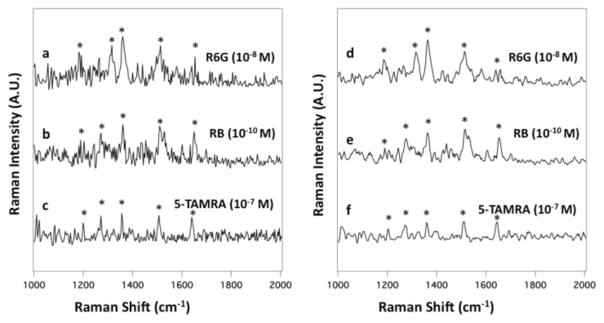
Single 50 ms SERS spectrum of (a) R6G (10−8 M) extracted from Figure 1A at t=180.25 s, (b) RB (10−10 M) extracted at t=219.75 s, and (c) 5-TAMRA (10−7 M) extracted at t=290.60 s. The average SERS spectrum of (d) R6G (10−8 M) extracted from Figure 1A between t=179.65 and 180.05 s, (e) RB (10−10 M) extracted between t=220.45 and 221.90 s, and (f) 5-TAMRA (10−7 M) extracted between t=290.45 and 290.95 s are shown. Asterisks indicate the bands intrinsic to each analyte.
Averaging the SERS signal over the duration of the electropherogram peak yields spectra with a S/N ratio ≥ 25 for all three analytes. Figure 2d shows the average SERS spectrum of R6G extracted from Figure 1A between t=179.65 and 180.05 s. The averaged SERS spectrum of RB extracted between t=220.45 and 220.90 s is shown in Figure 2e. Of note, the SERS spectrum of RB was acquired from the injection of a few attomoles (~100,000 molecules). Figure 2f shows the averaged spectrum of 5-TAMRA extracted from Figure 1A between t=290.45 and 290.80 s. While all three dyes show similar spectra, as expected based on their structures, the differences observed enable identification of the analytes.
In conclusion, we have demonstrated highly sensitive and ultrafast online SERS detection of structural isomers of rhodamine separated by CZE. SERS spectra of the analytes provided direct spectral signatures associated with the subtle structural differences of the three rhodamine dyes. The limit of detection for SERS reported here is more than 1000x better when compared to the best previously reported LOD using a planar substrate.16 The observed Raman scattering allowed differentiation of two isobaric compounds (R6B and RB, M.W=479.02 g/mol) at nanomolar concentrations, which is not achievable by mass spectrometry. The SERS flow detector should be readily incorporated into any liquid separation, such as liquid chromatography. The implementation of this robust and sensitive online SERS flow detector suggests an alternative for the characterization of pharmaceuticals, metabolites, and other analytes.
Supplementary Material
Acknowledgments
The University of Notre Dame, NIH Award R21 GM107893, and the Cottrell Scholar Award from the Research Corporation for Science Advancement supported this work. The authors thank Norman Dovichi for helpful comments and access to CE equipment.
Footnotes
Electronic Supplementary Information (ESI) available: Detailed experimental and supporting figures S1–S5. See DOI: 10.1039/c000000x/
Notes and references
- 1.Beck W, Engelhardt H. Chromatographia. 1992;33:313–316. [Google Scholar]
- 2.Song JZ, Huang DP, Tian SJ, Sun ZP. Electrophoresis. 1999;20:1850–1855. doi: 10.1002/(SICI)1522-2683(19990701)20:9<1850::AID-ELPS1850>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 3.Chen DY, Dovichi NJ. J Chromatogr B. 1994;657:265–269. doi: 10.1016/0378-4347(94)00014-x. [DOI] [PubMed] [Google Scholar]
- 4.MacTaylor CE, Ewing AG. Electrophoresis. 1997;18:2279–2290. doi: 10.1002/elps.1150181218. [DOI] [PubMed] [Google Scholar]
- 5.Santesson S, Andersson M, Degerman E, Johansson T, Nilsson J, Nilsson S. Anal Chem. 2000;72:3412–3418. doi: 10.1021/ac000140r. [DOI] [PubMed] [Google Scholar]
- 6.Banks JF. Electrophoresis. 1997;18:2255–2266. doi: 10.1002/elps.1150181216. [DOI] [PubMed] [Google Scholar]
- 7.Figeys D, Aebersold R. Electrophoresis. 1998;19:885–892. doi: 10.1002/elps.1150190603. [DOI] [PubMed] [Google Scholar]
- 8.Dovichi NJ, Hu S. Curr Opin Chem Biol. 2003;7:603–608. doi: 10.1016/j.cbpa.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 9.Jorgenson JW, Lukacs KD. Science. 1983;222:266–272. doi: 10.1126/science.6623076. [DOI] [PubMed] [Google Scholar]
- 10.Ohno K, Tachikawa K, Manz A. Electrophoresis. 2008;29:4443–4453. doi: 10.1002/elps.200800121. [DOI] [PubMed] [Google Scholar]
- 11.Squires TM, Quake SR. Rev Mod Phys. 2005;77:977–1026. [Google Scholar]
- 12.Fiorini GS, Chiu DT. Biotechniques. 2005;38:429–446. doi: 10.2144/05383RV02. [DOI] [PubMed] [Google Scholar]
- 13.Stiles PL, Dieringer JA, Shah NC, Van Duyne RR. Annu Rev Anal Chem. 2008;1:601–626. doi: 10.1146/annurev.anchem.1.031207.112814. [DOI] [PubMed] [Google Scholar]
- 14.Chen CY, Morris MD. J Chromatogr. 1991;540:355–363. doi: 10.1016/s0021-9673(01)88822-1. [DOI] [PubMed] [Google Scholar]
- 15.Nirode WF, Devault GL, Sepaniak MJ. Anal Chem. 2000;72:1866–1871. doi: 10.1021/ac991248d. [DOI] [PubMed] [Google Scholar]
- 16.Leopold N, Lendl B. Anal Bioanal Chem. 2010;396:2341–2348. doi: 10.1007/s00216-010-3468-3. [DOI] [PubMed] [Google Scholar]
- 17.Mavre F, Anand RK, Laws DR, Chow KF, Chang BY, Crooks JA, Crooks RM. Anal Chem. 2010;82:8766–8774. doi: 10.1021/ac101262v. [DOI] [PubMed] [Google Scholar]
- 18.He L, Natan MJ, Keating CD. Anal Chem. 2000;72:5348–5355. doi: 10.1021/ac000583v. [DOI] [PubMed] [Google Scholar]
- 19.Negri P, Jacobs KT, Dada OO, Schultz ZD. Anal Chem. 2013;85:10159–10166. doi: 10.1021/ac401537k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dada OO, Huge BJ, Dovichi NJ. Analyst. 2012;137:3099–3101. doi: 10.1039/c2an35321k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krylov SN, Starke DA, Arriaga EA, Zhang Z, Chan NW, Palcic MM, Dovichi NJ. Anal Chem. 2000;72:872–877. doi: 10.1021/ac991096m. [DOI] [PubMed] [Google Scholar]
- 22.Asiala SM, Schultz ZD. Analyst. 2011;136:4472–4479. doi: 10.1039/c1an15432j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kneipp K, Wang Y, Kneipp H, Perelman LT, Itzkan I, Dasari R, Feld MS. Phys Rev Lett. 1997;78:1667–1670. [Google Scholar]
- 24.Hildebrandt P, Stockburger M. J Phys Chem. 1984;88:5935–5944. [Google Scholar]
- 25.Liu GL, Lee LP. Appl Phys Lett. 2005:87. [Google Scholar]
- 26.Pristinski D, Tan SL, Erol M, Du H, Sukhishvili S. J Raman Spectrosc. 2006;37:762–770. [Google Scholar]
- 27.Tao A, Kim F, Hess C, Goldberger J, He RR, Sun YG, Xia YN, Yang PD. Nano Lett. 2003;3:1229–1233. [Google Scholar]
- 28.Lee HJ, Li Y, Wark AW, Corn RM. Anal Chem. 2005;77:5096–5100. doi: 10.1021/ac050815w. [DOI] [PubMed] [Google Scholar]
- 29.Zhang JT, Li XL, Sun XM, Li YD. J Phys Chem B. 2005;109:12544–12548. doi: 10.1021/jp050471d. [DOI] [PubMed] [Google Scholar]
- 30.Smith E, Dent G. Modern Raman Spectroscopy: A Practical Approach. 2005 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.