

Abstract
Trypanosoma evansi and Trypanosoma vivax, which are the major causative agents of animal trypanosomosis in Venezuela, have shown a very high immunological cross-reactivity. Since the production of T. vivax antigens is a limiting factor as this parasite is difficult to propagate in experimental animal models, our goal has been to identify and isolate antigens from T. evansi that cross-react with T. vivax. Here, we used the Venezuelan T. evansi TEVA1 isolate to prepare the total parasite lysate and its corresponding cytosolic and membranous fractions. In order to extract the T. evansi integral membrane proteins, the particulate portion was further extracted first with Triton X-100, and then with sodium dodecyl sulfate. After discarding the cytosolic and Triton X-100 solubilized proteins, we employed sedimentation by centrifugation on linear sucrose gradients to partially purify the sodium dodecyl sulfate-solubilized proteins from the Triton X-100 resistant particulate fraction of T. evansi. We obtained enriched pools containing polypeptide bands with apparent molecular masses of 27 kDa, 31 kDa, and 53 kDa, which were recognized by anti-T. vivax antibodies from experimentally and naturally infected bovines.
1. Introduction
Mammal-infecting Trypanosoma species are grouped into the sections Stercoraria and Salivaria. Trypanosomes classified as Stercoraria develop in the posterior part of the vector digestive tract and are transmitted through feces. In contrast, the biological transmission of trypanosomes classified as Salivaria occurs by insects that harbor the parasites in their salivary glands, although mechanical transmission also occurs. Salivarian trypanosomes include the subgenera Tejeraia (Trypanosoma rangeli), Duttonella (Trypanosoma vivax), Nannomonas (Trypanosoma congolense), Trypanozoon (Trypanosoma brucei, Trypansoma evansi, and Trypanosoma equiperdum), and Pycnomonas (Trypanosoma suis).
Cross-reactions among evolutionarily close parasites are generally explored either to prevent false positive interpretation of the tests [1, 2] or to take advantage of them through an interspecific detection assay [3]. Antigenic similarities among salivarian trypanosomes have been known for a long time; indeed cross-reactions between T. congolense, T. vivax, T. evansi, and T. brucei spp. have been repeatedly reported [4–6]. Even monoclonal antibodies developed for the diagnosis of specific salivarian trypanosome species have been shown to cross-react all together [7, 8]. Recently, similarities between salivarian and stercorarian trypanosomes have also been recorded [9], showing an important cross-reactivity between T. evansi and T. cruzi.
T. vivax and T. evansi are salivarian parasite species that cause animal trypanosomosis predominantly in bovines and equines, respectively. They are the major causative agents of animal trypanosomosis in Venezuela. Given that it is more convenient to produce T. evansi which grows easily in rodents, than T. vivax which does not, the use of T. evansi crude antigen or purified cross-reacting antigenic proteins for the detection of T. vivax infections in cattle has been under investigation for the last two decades [3, 10–12]. Moreover, the use of T. evansi antigens in the diagnosis of T. vivax infections is an interesting alternative for laboratories which do not have the facilities to produce T. vivax antigens. Since the parasite cell surface is an intuitive place to explore for antigenic potential proteins, we partially purified sodium dodecyl sulfate- (SDS-) solubilized membrane-associated integral proteins from the Venezuelan T. evansi TEVA1 isolate, aka TeAp-N/D1 [13], by using sedimentation by centrifugation on linear sucrose gradients. We obtained fractions containing T. evansi polypeptides with apparent molecular weights of 27,000, 31,000, and 53,000, which were recognized by anti-T. vivax antibodies from infected cows.
2. Materials and Methods
2.1. Materials
Reagents were purchased from the following sources: anti-mouse IgG horseradish peroxidase conjugate, middle range molecular weight protein markers, Promega; anti-rabbit IgG horseradish peroxidase conjugate, anti-bovine IgG horseradish peroxidase conjugate, anti-bovine IgG alkaline phosphatase conjugate, anti-equine IgG alkaline phosphatase conjugate, diaminobenzidine (DAB), fibrous DEAE-cellulose, benzamidine, iodoacetamide, L-trans-epoxysuccinyl-leucylamido(4-guanidino)butane (E-64), leupeptin, phenyl methyl sulfonyl fluoride (PMSF), Sigma; prestained high molecular weight protein markers, Gibco BRL; 5-bromo-4-chloro-3 indolyl phosphate (BCIP), nitro blue tetrazolium (NBT), protein assay dye reagent concentrate, bovine serum albumin (BSA), broad range molecular weight standards, and nitrocellulose membranes (0,45 μm pore size), BioRad. All other chemicals were of the highest quality grade available.
2.2. Source of Antigens
We used a Venezuelan field isolate of T. evansi named TEVA1 [13]. Cryopreserved T. evansi-infected blood was inoculated intraperitoneally into adult albino rats (Sprague-Dawley). When the number of parasites reached ≥106 trypanosomes/mL, the blood was extracted from the rats by cardiac puncture using 0.5 M EDTA as anticoagulant. Parasites were purified by anion exchange chromatography using a fibrous DEAE-cellulose column [14] and were kept frozen at −80°C until further use.
2.3. Isolation of the T. evansi Particulate Fraction
Purified T. evansi parasites (~5 × 109) were extracted on ice, by sonication (4 cycles of 7 watts, 30 sec each, with a 2 min resting period in between) using 16 mL of lysis buffer (5 mM Tris-HCl buffer (pH 8.0) containing 1 mM benzamidine, 1 mM PMSF, 10 mM EDTA, 10 mM EGTA, 1 mM iodoacetamide, 20 μM E-64, and 20 μM leupeptin). The parasite homogenate (H) was centrifuged at 100,000 ×g for 30 min, at 4°C, to separate the pellet containing the particulate fraction (P) from the supernatant holding the clarified soluble fraction (S). This procedure was repeated three times to completely wash all soluble antigens from the remaining parasite membranous fraction. In an attempt to solubilize this fraction, P was subjected to three successive extractions with lysis buffer containing 2% Triton X-100. A centrifugation step at 100,000 ×g for 30 min, at 4°C, was employed to separate the supernatant (STX-100) from the neutral detergent-washed pellet (PTX-100).
2.4. Solubilization with SDS of the Triton X-100 Washed Parasite Particulate Fraction
The Triton X-100 washed pellet was resuspended in lysis buffer containing 4% SDS and homogenized by passage through a number 23 needle. A centrifugation step at 100,000 ×g for 30 min, at room temperature, was employed to separate the supernatant (SSDS) from the anionic detergent washed pellet (PSDS). The concentration of SDS was reduced to 2% in the resulting supernatant sample, before assaying its polypeptide and antigenic composition by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot, respectively.
2.5. Partial Purification of Antigens from the T. evansi SDS-Solubilized Particulate Fraction Using Zonal Sedimentation on Sucrose Gradients
Linear 10–30% and 5–20% sucrose analytical gradients (4 mL) were prepared in 100 mM Tris-HCl (pH 8.0), 0,2 mM EDTA, 0,4 mM dithiothreitol, 10 mM MgCl2, 0.3 M NH4Cl, and 2% SDS. After loading the parasite SDS-solubilized particulate fraction sample onto the sucrose gradients, the tubes were centrifuged at 200,000 ×g, at room temperature, for 18 h. Fractions were collected through the bottom of the tube, and an aliquot of each fraction was analyzed by SDS-PAGE and Western blot using sera from T. evansi-infected horses and T. vivax-infected cows.
2.6. Animal Sera
Two healthy horses were experimentally infected with cryopreserved blood samples containing ~106 parasites of different T. evansi isolates. The first horse was inoculated with the TEVA1 T. evansi isolate (H-TEVA1), and the second horse was inoculated with the TeAp-ElFrio01 isolate [13] (H-TeApEF). Three healthy bovines were infected with a cryopreserved blood sample containing ~106 parasites of the LIEM-176 T. vivax isolate [15] (B-103, B-173, and B-303). Blood samples from the experimentally infected animals were taken every day, for a two-month period, and sera were stored frozen at −20°C and used as positive controls. Table 1 summarizes the samples of sera obtained from experimentally infected animals.
Table 1.
Identification of sera from experimentally infected animals.
| Sample number | Sera name | Host | Infection agent | Isolate |
|---|---|---|---|---|
| 1 | B-103 | Bovine | T. vivax | LIEM-176 |
| 2 | B-173 | Bovine | T. vivax | LIEM-176 |
| 3 | B-303 | Bovine | T. vivax | LIEM-176 |
| 4 | H-TEVA1 | Horse | T. evansi | TEVA1 |
| 5 | H-TeApEF | Horse | T. evansi | TeAp-ElFrio01 |
In addition, blood samples were collected from the jugular vein of parasitologically negative and trypanosome-infected field animals. Horses and cows were examined for the presence of trypanosomes in the circulation by the microhematocrit technique [16] and were diagnosed as positive or negative for trypanosomosis by indirect ELISA, using the clarified antigenic fraction from T. evansi as the source of antigen [10, 17]. Following blood clotting at room temperature and centrifugation, each serum was stored frozen at −20°C and employed for immunodetection. Table 2 summarizes the samples of sera obtained from field animals.
Table 2.
Identification of sera from field animals.
| Sample number | Sera name | Natural host | Infection state | Infection agent | Locality |
|---|---|---|---|---|---|
| 1 | B-LC12 | Bovine | Infected | T. vivax | La Candelaria Farm, Guárico State, Venezuela |
| 2 | B-LC29 | Bovine | Not infected | — | La Candelaria Farm, Guárico State, Venezuela |
| 3 | B-LC31 | Bovine | Infected | T. vivax | La Candelaria Farm, Guárico State, Venezuela |
| 4 | B-LC43 | Bovine | Infected | T. vivax | La Candelaria Farm, Guárico State, Venezuela |
| 5 | B-LC54 | Bovine | Infected | T. vivax | La Candelaria Farm, Guárico State, Venezuela |
| 6 | B-LC69 | Bovine | Not infected | — | La Candelaria Farm, Guárico State, Venezuela |
| 7 | B-LL19 | Bovine | Infected | T. vivax | La Loma Farm, Guárico State, Venezuela |
| 8 | B-LE14 | Bovine | Infected | T. vivax | La Esperanza Farm, Guárico State, Venezuela |
| 9 | B-P13 | Bovine | Infected | T. vivax | Paradero Farm, Guárico State, Venezuela |
| 10 | B-F5315 | Bovine | Not infected | — | France |
| 11 | B-F5683 | Bovine | Not infected | — | France |
| 12 | H-LR | Horse | Not infected | — | La Rinconada Racetrack, Caracas, Venezuela |
| 13 | H-N/D | Horse | Infected | T. evansi | N/D*, Apure State, Venezuela |
*N/D: not determined.
2.7. Other Procedures
Protein concentration was measured according to Bradford [18], using bovine serum albumin as protein standard. The p64 antigen, which is the soluble form of a T. evansi variant surface glycoprotein (VSG) that displays cross-reactivity between T. evansi and T. vivax, was purified as described by Uzcanga et al. [11]. The purified p64 was used to produce polyclonal antibodies in mice ascitic fluid [12]. Polyclonal anti-VSG antibodies were also generated in rabbit serum, following the procedure described by Harlow and Lane [19]. SDS-PAGE was carried out on 1.5 mm thick slab gels containing 12 or 15% polyacrylamide [20]. Coomassie blue R-250 was used for protein staining. For Western blot analyses; the proteins were electrotransferred from the gels to nitrocellulose filters [21]. For immunodetection, the filters were incubated with bovine sera (dilution 1 : 100) or specific anti-VSG polyclonal antibodies generated in mice (dilution 1 : 5,000) or rabbits (dilution 1 : 150). The sheets were then incubated with the appropriate dilution of alkaline phosphatase-conjugated or horseradish peroxidase-conjugated secondary antibodies against bovine, mouse, or rabbit IgG, depending on the case, following the instructions of the supplier. Finally, the polypeptide bands were visualized by the addition of NBT and BCIP when alkaline phosphatase-conjugated antibodies were used, or DAB and hydrogen peroxide when horseradish peroxidase-conjugated antibodies were employed, according to the provider. A lane containing a mixture of molecular weight protein markers was included in the blot to determine the apparent size of the polypeptide bands.
3. Results
Figure 1(a) illustrates the Coomassie blue stained polypeptide profiles, which were acquired following SDS-PAGE separation of various T. evansi fractions (H, S, P, STX-100 and PTX-100). As seen in the figure, a variety of polypeptide bands possessing a broad range of sizes were obtained in the different fractions. Then, serum from a bovine experimentally infected with T. vivax was used to identify the TEVA1 T. evansi antigenic polypeptides that exhibited cross-reactivity with T. vivax by Western blotting. As previously reported [10], a series of cross-reacting antigens with apparent molecular masses ranging from approximately 10,000 to 110,000 daltons were evident in the T. evansi homogenate (Figure 1(b)). Both T. evansi fractions, S and P, also contained antigens that display cross-reactivity with T. vivax (Figure 1(b)). Moreover, a 64 kDa polypeptide band (p64) was the major polypeptide species of T. evansi, which was present in the H, S, and P fractions, and was recognized by bovine anti-T. vivax antibodies. Interestingly, the STX-100 sample was highly enriched with the 64 kDa polypeptide band that exhibited cross-reactivity with T. vivax, but no p64 was observed in the PTX-100 sample (Figure 1(b)). Additionally, a series of cross-reacting antigenic polypeptide bands were resistant to the neutral detergent treatment and remained in the PTX-100 sample. The resulting pattern showed that the most intensive antigenic bands corresponded to polypeptide bands migrating at about 68–70 kDa, 52–55 kDa, and 27–31 kDa, respectively (Figure 1(b)).
Figure 1.
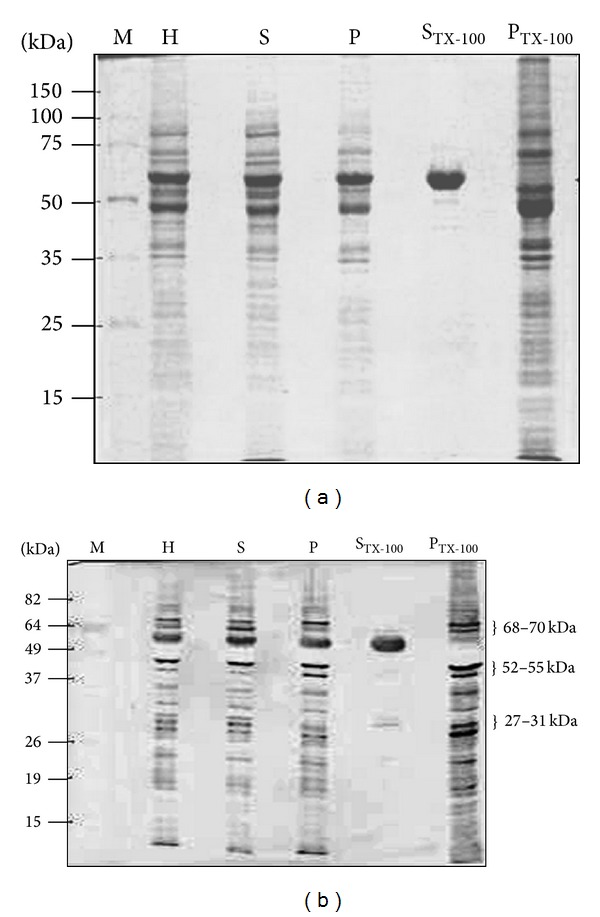
Analysis of T. evansi antigenic polypeptide bands that display cross-reactivity with T. vivax. Purified T. evansi parasites were homogenized to generate the whole-cell extract (H), which was centrifuged to separate the soluble fraction (S) from the particulate fraction (P). Then, P was extracted with 2% Triton X-100, and the Triton X-100 solubilized fraction (STX-100) was separated from the remaining pellet (PTX-100) by centrifugation. (a) Coomassie blue staining. (b) Immunoblot developed with serum B-303 (Table 1). M = protein molecular weight markers.
Previously, Uzcanga et al. [11] demonstrated that the predominant 64 kDa cross-reacting antigen between T. evansi and T. vivax, which was purified from the TEVA1 T. evansi Venezuelan isolate, represented the soluble form of a VSG. The 64 kDa band obtained in the H, S, P, and STX-100 parasite fractions was identified here as the soluble form of the same VSG (p64), by using specific anti-VSG polyclonal antibodies that were raised in rabbits and mice (Figure 2). As expected from the results shown in Figure 1(b), no VSG remained in the PTX-100 fraction following treatment with Triton X-100 (Figure 2). Interestingly, several of the polypeptide bands that remained in the PTX-100 fraction, including the polypeptides of approximately 68–70 kDa, 52–55 kDa, and 27–31 kDa, were immunorecognized by sera from bovines experimentally and naturally infected with T. vivax (Figure 3). These results demonstrated that these cross-reacting antigenic bands are parasite integral proteins that are probably localized in Triton X-100 resistant membrane microdomains. As also seen in the figure (lanes 7–9), sera from horses experimentally and naturally infected with T. evansi recognized the same antigenic polypeptide bands than anti-T. vivax antibodies.
Figure 2.
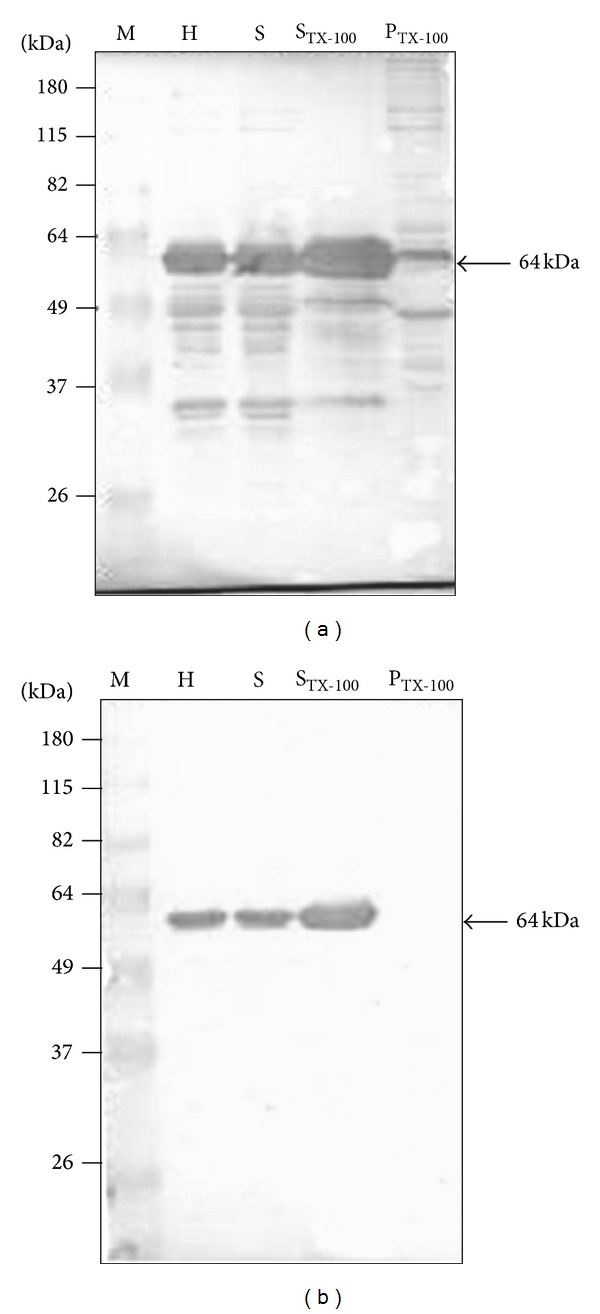
The 64 kDa band that is enriched in the STX-100 fraction corresponds to the previously reported p64 antigen [10, 11], that is partially responsible for the cross-reaction between T. evansi and T. vivax. The p64 antigen was observed in the parasite homogenate (H), soluble (S), and Triton X-100 solubilized (STX-100) fractions. No p64 was obtained in the resulting pellet after treatment with Triton X-100 (PTX-100). (a) Western blot using polyclonal anti-p64 antibodies prepared in rabbit sera. (b) Western blot using polyclonal anti-p64 antibodies prepared in mice ascitic fluid. M = protein markers.
Figure 3.

Identification of T. vivax-cross-reacting antigens from the PTX-100 fraction of T. evansi. An aliquot of the PTX-100 fraction (350 μg of total protein) was separated by electrophoresis on a preparative 15% polyacrylamide slab gel. Following SDS-PAGE, the proteins were electrotransferred to nitrocellulose, and the blot was cut into 3 mm strips. Strips containing the PTX-100 fraction were developed using sera B-303 (lane 1), B-LC31 (lane 2), B-LE14 (lane 3), B-P13 (lane 4), B-LC43 (lane 5), B-LL19 (lane 6), H-TEVA1 (lane 7), H-TeApEF (lane 8), and H-N/D (lane 9). Sera are described in Tables 1 and 2.
Since SDS is a strong anionic detergent, which is capable of disintegrating membrane structures and denaturing most integral membrane proteins, the PTX-100 fraction was reextracted with lysis buffer containing 4% SDS, in order to solubilize its antigenic proteins. Figure 4 shows a separation by SDS-PAGE of the polypeptide bands that were released with the SDS treatment (SSDS), and those that remained in the final pellet (SSDS), following centrifugation. In the presence of 4% SDS, most proteins were solubilized (see the SSDS fraction), and a clear decrease in the amount of polypeptide bands was evident in the PSDS sample (Figure 4). Polypeptides with apparent molecular masses ranging from <25,000 to >150,000 Da were observed in the T. evansi SSDS fraction (Figure 4). The sizes of the major polypeptide bands contained in the parasite SSDS fraction are indicated in the figure.
Figure 4.
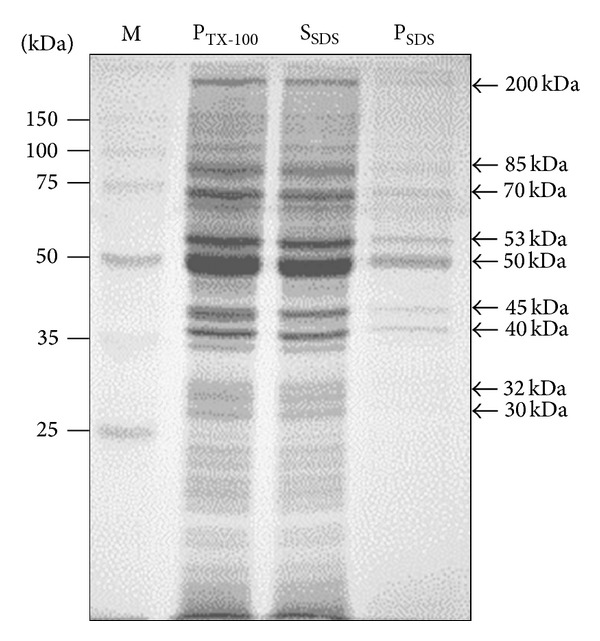
Solubilization of the proteins contained in the PTX-100 fraction using SDS. The T. evansi PTX-100 fraction was homogenized using 4% SDS. Following centrifugation, the supernatant (SSDS) was separated from the SDS-washed pellet (PSDS). The concentration of SDS was reduced to 2% and the polypeptide composition of the SSDS and PSDS fractions was evaluated by SDS-PAGE. Shown is the Coomassie blue staining of the gel. M = protein molecular weight standards.
In order to separate the T. vivax-cross-reacting antigens, the T. evansi SDS-solubilized particulate fraction was subjected to ultracentrifugation by zonal sedimentation on sucrose gradients. A 200 μL aliquot of the SSDS sample was loaded on a 4 mL linear 10–30% sucrose gradient. Following centrifugation, 47 fractions of 80 μL each were collected through the bottom of the tube. An aliquot of each fraction was analyzed by SDS-PAGE and Western blot using serum from a naturally T. vivax-infected bovine. The bulk of the proteins were separated between fractions 24 and 47 (Figure 5), and almost no proteins were seen in the first 23 fractions (data not shown). As illustrated in the figure by SDS-PAGE, the majority of the sedimented polypeptide bands were of low and middle apparent molecular weights. Western blot analysis showed a major cross-reacting polypeptide band of 53 kDa that was present in all the protein containing fractions (Figure 5). The cross-reacting antigen of 53 kDa showed a peak between fractions 37 and 39. Although fractions 30 to 36 were contaminated with other nonantigenic polypeptide bands, the only cross-reacting antigen contained in these fractions was the 53 kDa polypeptide band. Particularly, fractions 35 and 36 were enriched in the 53 kDa cross-reacting antigenic band (Figure 5). In addition, cross-reacting antigenic bands of 31 and 27 kDa were also obtained between fractions 37 and 43, showing a peak in fractions 39 and 40 (Figure 5).
Figure 5.
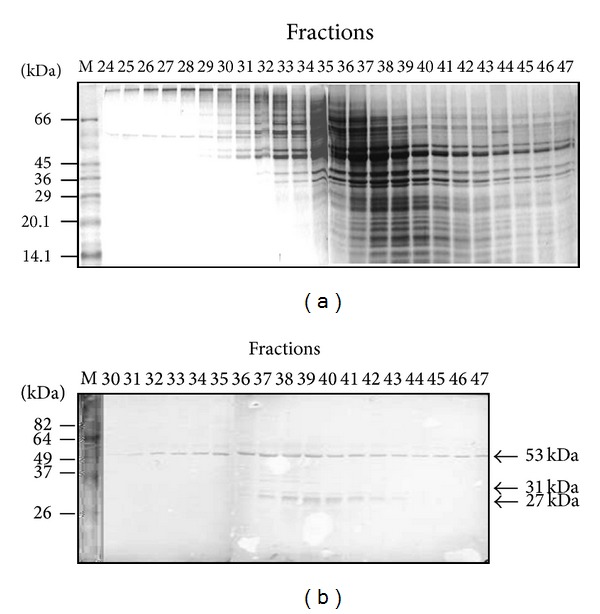
Separation of the SSDS fraction using sedimentation on a linear 10–30% sucrose gradient. A 200 μL aliquot of the T. evansi SSDS sample was loaded on a 4 mL linear 10–30% sucrose gradient. Fractions (80 μL) were collected through the bottom of the tube following ultracentrifugation. (a) SDS-PAGE analysis of the resulting fractions; shown is the Coomassie blue staining. (b) Western blot developed with serum B-173 (Table 1). M = protein molecular weight markers.
Given that no proteins were obtained in the first 23 fractions of the 10–30% sucrose gradient, a 4 mL linear 5–20% sucrose gradient was also employed in order to improve the antigenic separation. Following centrifugation, 41 fractions of 110 μL each were collected through the bottom of the tube and were analyzed as above. Western blot analysis showed that the major 53 kDa cross-reacting antigen eluted between fractions 13 and 31 and showed a peak around fraction 28 (Figure 6). The cross-reacting antigenic bands of 27 and 31 kDa were observed between fractions 25 and 32, showing a peak around fractions 29-30 (Figure 6). In conclusion, fractions enriched in three predominant antigens with apparent molecular masses of 27, 31, and 53 kDa, which are partially responsible of the cross-reactivity between T. vivax and T. evansi, were obtained from the Triton X-100 resistant particulate fraction of T. evansi. Moreover, an improvement on the separation of these cross-reacting antigens was obtained when the sedimentation was carried out on a linear 5–20% sucrose analytical gradient (Figure 6).
Figure 6.

Separation of the SSDS fraction using sedimentation on a linear 5–20% sucrose gradient. A 200 μL aliquot of the T. evansi SSDS sample was loaded on a 4 mL linear 5–20% sucrose gradient. Fractions (110 μL) were collected through the bottom of the tube following ultracentrifugation. (a) SDS-PAGE analysis of the resulting fractions; shown is the Coomassie blue staining. (b) Western blot developed with serum B-173 (Table 1). M = protein molecular weight markers.
We pooled fractions 13–19 from the 5–20% sucrose gradient since they only contained the 53-kDa cross-reacting antigenic band (Pool I). Additionally, we also pooled fractions 25 to 33 (Pool II), which contained a mixture of all major cross-reacting bands (27, 31, and 53 kDa polypeptides). The cross-reactivity of the partially purified antigens from each pool was analyzed by Western blot employing different sera from bovines experimentally or naturally infected with T. vivax. Aliquots of 400 μL of Pool I and Pool II were loaded on 12% polyacrylamide preparative gels. Once, the separation was finished, the gels were electrotransferred to nitrocellulose membranes, which were cut into strips and analyzed using various sera from T. vivax-infected bovines. As shown in Figures 7(a) and 7(b) (Pools I and II, resp.), the partially purified antigens of 27, 31, and 53 kDa were recognized by sera from horses infected with the TEVA1 and the TeAp-ElFrio01 T. evansi isolates and from cows experimentally and naturally infected with T. vivax. Hence, the polypeptides of 27, 31, and 53 kDa correspond to T. evansi antigens that exhibit cross-reactivity with T. vivax.
Figure 7.
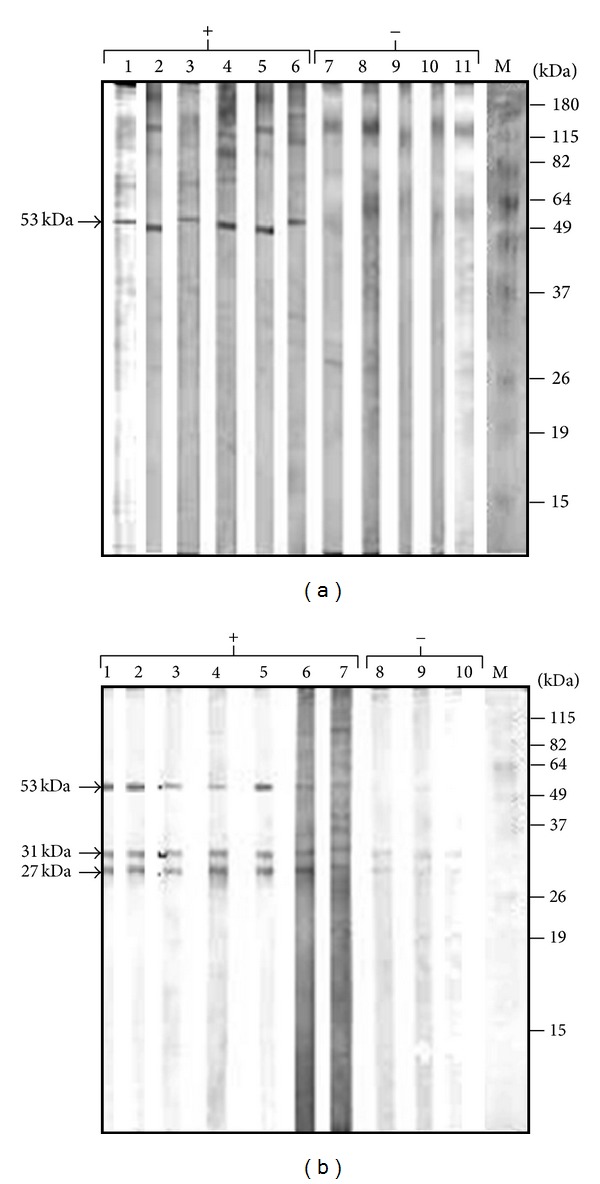
Reactivity of the T. evansi polypeptide bands contained in Pools I and II against sera from bovines experimentally and naturally infected with T. vivax. Aliquots (400 μL) of Pools I and II (fractions 13–19 and 25–33 from Figure 6, resp.) were separated by electrophoresis on a preparative 12% polyacrylamide slab gel. Following SDS-PAGE, the proteins were electrotransferred to nitrocellulose filters, and the blots were cut into 3 mm strips. Strips containing Pool I (a) or Pool II (b) were developed using positive sera (+) from horses experimentally infected with T. evansi or from bovines experimentally or naturally infected with T. vivax. Parasitologically and serologically negative sera (−) were also employed as controls. In (a), lanes 1–11, sera H-TEVA1, H-TeApEF, B-303, B-103, B-173, B-LC12, B-LC29, B-LC69, B-F5315, B-F5683, and H-LR, respectively. In (b), lanes 1–10, sera H-TEVA1, H-TeApEF, B-303, B-103, B-173, B-LC12, B-LC54, B-F5315, B-F5683, and H-LR, respectively. Sera are described in Tables 1 and 2.
4. Discussion
Absolute control of bovine trypanosomosis cannot be achieved with the methods that are currently available, which are inadequate to prevent the enormous economic impact caused by this disease. Early and accurate diagnosis of the trypanosomosis caused by T. vivax is of paramount importance for the strategic use of the available antiparasitic drugs. However, the diagnosis of this disease may be difficult as there are no pathognomonic clinical signs of infection, and standard trypanosome detection methods are not sensitive enough. Bovine trypanosomosis is detected using parasitological (microhematocrit centrifugation procedure) [16], immunological (indirect immunofluorescence, indirect ELISA, Western blot, etc.), and molecular (polymerase chain reaction or PCR) diagnostic techniques. In particular, the immunological tests are valuable methods for T. vivax diagnosis because of their high sensitivity in detecting antibodies to trypanosome antigens. Despite advances in the diagnosis of animal trypanosomosis, many acute infections go unnoticed and a chronic form of disease, frequently with no parasitemia, is more prevalent [22].
As in other infectious diseases, the early diagnosis of animal trypanosomosis is essential before local outbreaks become an epidemic of substantial proportions. This is particularly fundamental in bovine farms, where the disease can be transmitted mechanically by the vector flies (Tabanids) from one host to another, with the possibility of reaching the whole herd in a very short time. Woo [23] has shown that the magnitude of enzootic trypanosomosis caused by non-tsetse borne T. evansi is about three times greater than due to tsetse borne trypanosomes. Thus, cross-reactions among evolutionarily close parasites can be capitalized by using an interspecific detection assay. In particular, cross-reaction has been extensively reportedamong the immunogenic components of T. vivax and T. evansi. A predominant 64 kDa glycosylated cross-reacting antigen, p64, was previously purified from the TEVA1 T. evansi isolate and identified as the soluble form of a VSG [10, 11]. We have also purified two additional proteins with native molecular masses of approximately 51 and 68 kDa from the cytosolic fraction of the same T. evansi isolate, which were proven to be recognized by anti-T. vivax bovine antibodies and were not related to the purified p64 [12]. In the present work, we have continued with the isolation and characterization of the antigenic proteins from T. evansi, which are partially responsible for the immunological cross-reaction with T. vivax.
Similar to the bloodstream forms of T. brucei, T. evansi contains a glycosylphosphatidylinositol-specific phospholipase C (GPI-PLC). This enzyme cleaves the GPI-anchor of the VSG, forming free diacylglycerol in the membrane and, probably, a 1,2-cyclic phosphate on the inositol ring, which remains attached to the released VSG [24, 25]. This cleavage converts the membrane-bound form of the VSG (mVSG) to the soluble released form of the VSG (sVSG) [26]. As demonstrated in Figure 2 for p64, the GPI-PLC from the TEVA1 T. evansi isolate is active and functional. Then, other parasite GPI-anchored cell surface proteins must also be cleaved by the parasite GPI-PLC enzyme throughout the various extraction steps performed here, yielding their corresponding soluble released forms. Our results revealed that three integral membrane-associated polypeptide bands from T. evansi, which do not contain GPI anchors and with apparent molecular masses of 27, 31, and 53 kDa, contained common epitopes with T. vivax proteins. Consequently, these common or highly conserved antigenic polypeptides from T. evansi are good candidates to be considered as tools for immunodiagnosis of the trypanosomosis caused by T. vivax.
The subgenus Trypanozoon is the most homogeneous group of salivarian trypanosomes, which contains three recognized species that are morphologically indistinguishable, T. brucei, T. evansi, and T. equiperdum. The typical classification of T. brucei, T. equiperdum, and T. evansi as separate species is based on differences in the mode of transmission, host range and pathogenicity, and longstanding understanding that T. brucei contains interlocked maxicircle and minicircle kinetoplast DNA molecules (kDNA), T. equiperdum retains at least a part of maxicircle kDNA (dyskinetoplastidy), and T. evansi completely loses it (akinetoplastidy) [27–29]. There is a widely accepted paradigm that holds that T. evansi evolved, via T. equiperdum, when camels infected with T. brucei moved to tsetse-free areas [30], and a recent study has even suggested that T. equiperdum and T. evansi can be classified as subspecies of T. brucei [31]. Consequently, T. brucei, T. evansi, and T. equiperdum are phylogenetically very close. Although the genome of T. evansi has not been solved yet, the genome of a taxonomically relative parasite, T. brucei brucei (strain 927), which causes African sleeping sickness in humans, is now complete [32], providing both a milestone for trypanosome biology and an opportunity to consider a multitude of questions at the genome level. More recently, Jackson et al. [33] have produced high-quality draft genome sequences for two related African trypanosomes, T. congolense (subgenera Nannomonas) and T. vivax (subgenera Duttonella), specifically T. congolense IL3000 and T. vivax Y486. All these genome sequences are accessible through GeneDB (http://www.genedb.org/) or TriTrypDB (http://tritrypdb.org/). In an attempt to identify plausible candidates for the three T. vivax-cross-reacting antigens with apparent molecular masses of 27, 31, and 53 kDa, which were found in the Triton X-100 resistant membrane fraction of T. evansi, we analyzed the T. brucei brucei genome searching for gene products that (i) were putative integral proteins, (ii) were identified as immunodiagnostic antigens, and (iii) possessed similar sizes.
Sullivan et al. [34] took a nonbiased proteomic approach to identify potential diagnostic antigens for human African trypanosomosis, by asking which T. brucei proteins bind to the antibodies in sera of T. brucei gambiense-infected patients and not to the antibodies of uninfected individuals. This approach provided a list of twenty-four trypanosome proteins that selectively bound to the antibodies of infected patients and that might, therefore, be considered as immunodiagnostic antigens. In the same year, Jackson et al. generated a cell surface phylome for African trypanosomes, by comparing genes predicted to encode cell surface proteins of T. brucei with those from T. congolense and T. vivax (http://www.genedb.org/Page/trypanosoma_surface_ phylome). This cell surface phylome provided a detailed analysis of species-specific gene families and of gene gain and loss in shared families, aiding in the identification of surface proteins that may mediate specific aspects of pathogenesis and disease progression. In order to further discuss our results, we evaluated which proteins from the T. brucei cell surface phylome [35] were common to the list of twenty-four T. brucei immunodiagnostic antigens that were found by Sullivan et al. [34]. African trypanosome genomes contain large VSG gene families [32, 33], but monoallelic expression of a single gene is ensured because transcription is restricted to telomeric VSG expression sites (ES) [36–38]. Several other expression site-associated Genes (ESAG1-12) [39–41] are located in the ES and are cotranscribed with the active VSG [32, 42]. The product for the T. brucei ESAG3 gene (Tb927.2.2020) has been reported as a protein of 44.2 kDa that contains two putative transmembrane helices. As ESAG3 is an integral membrane protein that was selectively recognized by T. brucei gambiense infection IgG, and most membrane-embedded proteins have posttranslational modifications (e.g., glycosylations) that will increase their apparent size by SDS-PAGE, we suggest that the T. evansi orthologous gene product for ESAG3 might correspond to the 53 kDa cross-reacting antigen that was recognized here by anti-T. vivax antibodies from infected bovines. In fact, two potential N-glycosylation sites were detected in the T. brucei ESAG3 protein using the NetNGlyc 1.0 server (http://www.cbs.dtu.dk/services/NetNGlyc/, R. Gupta, E. Jung, S. Brunak, 2004. Prediction of N-glycosylation sites in human proteins, manuscript in preparation). Yadav et al. [43] also identified a 52–55 kDa cluster of polypeptides from T. evansi as immunodominant antigens by Western blot using serum from an experimentally infected equine. Interestingly, two ESAG3 orthologous genes have been reported in the T. vivax genome (TvY486 0042500 and Tvy486 0043380). A similar analysis yielded the gene for the hypothetical protein 4180 (Tb927.6.4180), which corresponds to a transmembrane protein of 16,317 Da and was also identified as an immunodiagnostic antigen since it bound to the infection IgG fraction. A potential N-glycosylation site was also detected in the Tb927.6.4180 protein. Thus, we proposed that the T. evansi orthologous gene product for the hypothetical protein 4180 may correspond to the polypeptide band of 27 kDa that exhibited cross-reactivity with T. vivax. An orthologous gene for the hypothetical protein 4180 has also been reported in the T. vivax genome (TvY486 0603610).
By only using the T. brucei cell surface phylome generated by Jackson et al. [35], three additional genes that encode for hypothetical proteins of similar sizes, and contain transmembrane regions, were also found (Tb927.8.7720 of 24.9 kDa; Tb927.6.380 of 33.9 kDa; and Tb927.4.5070 of 51.3 kDa). Since the molecular masses of these hypothetical proteins coincide approximately with those of the T. evansi antigens that displayed cross-reactivity with T. vivax (27 kDa, 31 kDa, and 53 kDa), they might correspond to the T. evansi orthologous genes that encode for these hypothetical proteins. Yet, none of these genes were identified within the group of the T. brucei immunodiagnostic antigens reported by Sullivan et al. [34]. Genes Tb927.8.7720 and Tb927.4.5070 appeared to be specific for T. brucei; however, an ortholog for gene Tb927.6.380 has also been reported in the T. vivax genome (TvY486 0600040). T. brucei contains a number of other cell-surface immunodiagnostic antigens that were proteomically selected by Sullivan et al. [34], such as the gene related to ESAG (GRESAG) 4, which encodes for an adenylyl cyclase enzyme [44], and invariant surface glycoproteins (ISG) with molecular masses of 75 kDa, 65 kDa, and 64 kDa. However, the sizes of these proteins did not match with any of the integral membrane antigenic proteins from T. evansi that were identified here by their immunological cross-reactivity with T. vivax. Various members of the twenty-four T. brucei immunodiagnostic antigens reported by Sullivan et al. [34], which must also exist in T. evansi, possess comparable molecular masses to polypeptide bands identified in this work, for example, ESAG6 and ESAG7, which encoded for transferrin receptor subunits of 44,221 Da and 38,433 Da, respectively. However, ESAG6 and ESAG7 contain GPI anchor motifs that must be cleaved by the parasite GPI-PLC during the extraction steps, producing their corresponding soluble released forms. Consequently, ESAG6 and ESAG7 cannot be found in the SDS-solubilized particulate fraction of T. evansi.
Since previous results have demonstrated that one antigen may not be sufficient for diagnostic purposes, we strongly feel that a pool of antigens should be evaluated to develop a good immunodiagnosis assay. The identification and characterization of these antigens could serve not only for diagnosis, but also for prophylaxis and chemotherapy against bovine trypanosomosis. Moreover, the identification of the antigenic proteins that lead to the cross-reactivity between these two trypanosomes could help to better understand the evolution of these parasites. The specificity and sensitivity of all these cross-reacting antigens, either individually or in groups, are parameters being evaluated at the present time, in order to establish the possibility of utilizing them as tools for the serological immunodiagnosis of bovine trypanosomosis. However, differences in antigen expression among parasite isolates or immunogenetic variations among hosts could determine the ability for antigen recognition by the various animal species.
5. Conclusions
Based on the cross-reaction between T. evansi and T. vivax, the use of T. evansi antigens for the diagnosis of T. vivax infections represents an excellent alternative for laboratories lacking the facilities to produce T. vivax antigens. Three membrane-associated integral polypeptide bands from T. evansi, possessing apparent molecular masses of 27, 31, and 53 kDa, were partially purified here by centrifugation on linear sucrose gradients and were proven to be antigens that display cross-reactivity with T. vivax. These T. evansi polypeptides are attractive candidates to be considered as tools for immunodiagnosis of the trypanosomosis caused by T. vivax.
Acknowledgments
This research was supported by Grants from FONACIT (no. G-2000001152 and no. LAB-2000001639) and from Decanato de Investigación y Desarrollo, Universidad Simón Bolívar (no. S1-IC-CB-017-06). We would like to thank A. Reyna-Bello (Universidad Nacional Experimental Simón Rodríguez) for providing sera from the French bovines that were used as negative controls and the personnel from Fondo Nacional de Investigaciones Veterinarias for contributing with the bovines and their maintenance.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Monzon CM, Colman OLR. Estudio seroepidemiológico de la tripanosomiasis equina (o Mal de Caderas) mediante la prueba de immunofluorescencia indirecta en la Provincia de Formosa (Argentina). Años 1983 a 1987. Arquivo Brasileiro de Medicina Veterinária e Zootecnia. 1988;40:279–285. [Google Scholar]
- 2.Desquesnes M, Bengaly Z, Millogo L, Meme Y, Sakande H. The analysis of the cross-reactions occurring in antibody-ELISA for the detection of trypanosomes can improve identification of the parasite species involved. Annals of Tropical Medicine and Parasitology. 2001;95(2):141–155. doi: 10.1080/00034980120050251. [DOI] [PubMed] [Google Scholar]
- 3.Desquesnes M, Tresse L. Utilization of T. evansi antigens in indirect-ELISA for the diagnosis of Trypanosoma sp. in livestock. In: Vokaty S, Desquesnes M, editors. Proceedings of First Symposium on New World Trypanosomes. Georgetown, Guyana, November 20–22, 1996. 1999. pp. 105–110. [Google Scholar]
- 4.Silayo RS, Gray AR, Luckins AG. Use of antigens of cultured Trypanosoma brucei in tests for bovine trypanosomiasis. Tropical Animal Health and Production. 1980;12(3):127–131. doi: 10.1007/BF02242642. [DOI] [PubMed] [Google Scholar]
- 5.Ijagbone IF, Staak C, Reinhard R. Fractionation of trypanosome antigens for species-specific sero-diagnosis. Veterinary Parasitology. 1989;32(4):293–299. doi: 10.1016/0304-4017(89)90040-x. [DOI] [PubMed] [Google Scholar]
- 6.Ferenc SA, Stopinski V, Courtney CH. The development of an enzyme-linked immunosorbent assay for Trypanosoma vivax and its use in a seroepidemiological survey of the Eastern Caribbean Basin. International Journal for Parasitology. 1990;20(1):51–56. doi: 10.1016/0020-7519(90)90172-j. [DOI] [PubMed] [Google Scholar]
- 7.Desquesnes M. Evaluation of three antigen detection tests (monoclonal trapping ELISA) for African trypanosomes, with an isolate of Trypaaosoma vivax from French Guyana. Annals of the New York Academy of Sciences. 1996;791:172–184. doi: 10.1111/j.1749-6632.1996.tb53524.x. [DOI] [PubMed] [Google Scholar]
- 8.Giardina S, Giansante D, Mercante T, Paganico G. Monoclonal antibodies produced against Trypanosoma equiperdum and Trypanosoma evansi recognize both species. Revista Cientifica de la Facultad de Ciencias Veterinarias de la Universidad del Zulia. 2002;12(2):82–93. [Google Scholar]
- 9.Desquesnes M, Bosseno M-F, Brenière SF. Detection of Chagas infections using Trypanosoma evansi crude antigen demonstrates high cross-reactions with Trypanosoma cruzi . Infection, Genetics and Evolution. 2007;7(4):457–462. doi: 10.1016/j.meegid.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 10.Uzcanga G, Mendoza M, Aso PM, Bubis J. Purification of a 64 kDa antigen from Trypanosoma evansi that exhibits cross-reactivity with Trypanosoma vivax . Parasitology. 2002;124(3):287–299. doi: 10.1017/s0031182001001214. [DOI] [PubMed] [Google Scholar]
- 11.Uzcanga GL, Perrone T, Noda JA, et al. Variant surface glycoprotein from Trypanosoma evansi is partially responsible for the cross-reaction between Trypanosoma evansi and Trypanosoma vivax . Biochemistry. 2004;43(3):595–606. doi: 10.1021/bi0301946. [DOI] [PubMed] [Google Scholar]
- 12.Camargo RE, Uzcanga GL, Bubis J. Isolation of two antigens from Trypanosoma evansi that are partially responsible for its cross-reactivity with Trypanosoma vivax . Veterinary Parasitology. 2004;123(1-2):67–81. doi: 10.1016/j.vetpar.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 13.Perrone TM, Gonzatti MI, Villamizar G, Escalante A, Aso PM. Molecular profiles of Venezuelan isolates of Trypanosoma sp. by random amplified polymorphic DNA method. Veterinary Parasitology. 2009;161(3-4):194–200. doi: 10.1016/j.vetpar.2009.01.034. [DOI] [PubMed] [Google Scholar]
- 14.Lanham SM, Godfrey DG. Isolation of salivarian trypanosomes from man and other mammals using DEAE-cellulose. Experimental Parasitology. 1970;28(3):521–534. doi: 10.1016/0014-4894(70)90120-7. [DOI] [PubMed] [Google Scholar]
- 15.González LE, García JA, Núñez C, et al. Trypanosoma vivax: a novel method for purification from experimentally infected sheep blood. Experimental Parasitology. 2005;111(2):126–129. doi: 10.1016/j.exppara.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 16.Woo PT. The haematocrit centrifuge technique for the diagnosis of African trypanosomiasis. Acta Tropica. 1970;27(4):384–386. [PubMed] [Google Scholar]
- 17.Aray C, Uzcanga G, Soto H, Mendoza M. Ensayo immunoenzimático para el diagnóstico de la tripanosomiasis bovina causada por Trypanosoma sp. Seroprevalencia en el municipio Monagas del Estado Guárico-Venezuela. Revista Científica Facultad de Ciencias Veterinarias de La Universidad del Zulia. 1998;8(supplement 1):114–116. [Google Scholar]
- 18.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Analytical Biochemistry. 1976;72(1-2):248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 19.Harlow E, Lane D. Antibodies. A Laboratory Manual. Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory; 1988. [Google Scholar]
- 20.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 21.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proceedings of the National Academy of Sciences of the United States of America. 1979;76(9):4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nantulya VM. Trypanosomiasis in domestic animals: the problems of diagnosis. Revue Scientifique et Technique. 1990;9(2):357–367. doi: 10.20506/rst.9.2.507. [DOI] [PubMed] [Google Scholar]
- 23.Woo PTK. Salivarian trypanosomes producing disease in livestock outside of sub-saharan Africa. In: Kreier JP, editor. Parasitic Protozoa. Vol. 1. New York, NY, USA: Academic Press; 1977. pp. 269–296. [Google Scholar]
- 24.Ferguson MAJ, Haldar K, Cross GAM. Trypanosoma brucei variant surface glycoprotein has a sn-1,2-dimyristyl glycerol membrane anchor at its COOH terminus. Journal of Biological Chemistry. 1985;260(8):4963–4968. [PubMed] [Google Scholar]
- 25.Ferguson MA, Homans SW, Dwek RA, Rademacher TW. The glycosylphosphatidylinositol membrane anchor of Trypanosoma brucei variant surface glycoprotein. Biochemical Society Transactions. 1988;16(3):265–268. doi: 10.1042/bst0160265. [DOI] [PubMed] [Google Scholar]
- 26.Cardoso De Almeida ML, Turner MJ. The membrane form of variant surface glycoproteins of Trypanosoma brucei . Nature. 1983;302(5906):349–352. doi: 10.1038/302349a0. [DOI] [PubMed] [Google Scholar]
- 27.Schnaufer A, Domingo GJ, Stuart K. Natural and induced dyskinetoplastic trypanosomatids: how to live without mitochondrial DNA. International Journal for Parasitology. 2002;32(9):1071–1084. doi: 10.1016/s0020-7519(02)00020-6. [DOI] [PubMed] [Google Scholar]
- 28.Brun R, Hecker H, Lun Z-R. Trypanosoma evansi and T. equiperdum: distribution, biology, treatment and phylogenetic relationship (a review) Veterinary Parasitology. 1998;79(2):95–107. doi: 10.1016/s0304-4017(98)00146-0. [DOI] [PubMed] [Google Scholar]
- 29.Gibson W. Resolution of the species problem in African trypanosomes. International Journal for Parasitology. 2007;37(8-9):829–838. doi: 10.1016/j.ijpara.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 30.Hoare CA. The Trypanosomes of Mammals. A Zoological Monograph. Oxford, UK: Blackwell Scientific; 1972. [Google Scholar]
- 31.Lai D-H, Hashimi H, Lun Z-R, Ayala FJ, Lukes J. Adaptations of Trypanosoma brucei to gradual loss of kinetoplast DNA: Trypanosoma equiperdum and Trypanosoma evansi are petite mutants of T. brucei . Proceedings of the National Academy of Sciences of the United States of America. 2008;105(6):1999–2004. doi: 10.1073/pnas.0711799105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berriman M, Hall N, Sheader K, et al. The architecture of variant surface glycoprotein gene expression sites in Trypanosoma brucei . Molecular and Biochemical Parasitology. 2002;122(2):131–140. doi: 10.1016/s0166-6851(02)00092-0. [DOI] [PubMed] [Google Scholar]
- 33.Jackson AP, Berry A, Aslett M, et al. Antigenic diversity is generated by distinct evolutionary mechanisms in African trypanosome species. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(9):3416–3421. doi: 10.1073/pnas.1117313109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sullivan L, Wall SJ, Carrington M, Ferguson MA. Proteomic selection of immunodiagnostic antigens for human African trypanosomiasis and generation of a prototype lateral flow immunodiagnostic device. PLoS Neglected Tropical Diseases. 2013;7(2) doi: 10.1371/journal.pntd.0002087.e2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jackson AP, Allison HC, Barry JD, Field MC, Hertz-Fowler C, Berriman M. A cell-surface phylome for African trypanosomes. PLoS Neglected Tropical Diseases. 2013;7(3) doi: 10.1371/journal.pntd.0002121.e2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horn D. The molecular control of antigenic variation in Trypanosoma brucei . Current Molecular Medicine. 2004;4(6):563–576. doi: 10.2174/1566524043360078. [DOI] [PubMed] [Google Scholar]
- 37.Pays E. The variant surface glycoprotein as a tool for adaptation in African trypanosomes. Microbes and Infection. 2006;8(3):930–937. doi: 10.1016/j.micinf.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 38.Horn D, McCulloch R. Molecular mechanisms underlying the control of antigenic variation in African trypanosomes. Current Opinion in Microbiology. 2010;13(6):700–705. doi: 10.1016/j.mib.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alexandre S, Guyaux M, Murphy NB, et al. Putative genes of a variant-specific antigen gene transcription unit in Trypanosoma brucei . Molecular and Cellular Biology. 1988;8(6):2367–2378. doi: 10.1128/mcb.8.6.2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Son HJ, Cook GA, Hall T, Donelson JE. Expression site associated genes of Trypanosoma brucei rhodesiense. Molecular and Biochemical Parasitology. 1989;33(1):59–66. doi: 10.1016/0166-6851(89)90042-x. [DOI] [PubMed] [Google Scholar]
- 41.Pays E, Tebabi D, Pays A, et al. The genes and transcripts of an antigen gene expression site from T. brucei . Cell. 1989;57(5):835–845. doi: 10.1016/0092-8674(89)90798-8. [DOI] [PubMed] [Google Scholar]
- 42.Hertz-Fowler C, Figueiredo LM, Quail MA, et al. Telomeric expression sites are highly conserved in Trypanosoma brucei . PLoS ONE. 2008;3(10) doi: 10.1371/journal.pone.0003527.e3527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yadav SC, Kumar R, Kumar V, et al. Identification of immuno-dominant antigens of Trypanosoma evansi for detection of chronic trypanosomosis using experimentally infected equines. Research in Veterinary Science. 2013;95(2):522–528. doi: 10.1016/j.rvsc.2013.04.030. [DOI] [PubMed] [Google Scholar]
- 44.Paindavoine P, Rolin S, Van Assel S, et al. A gene from the variant surface glycoprotein expression site encodes one of several transmembrane adenylate cyclases located on the flagellum of Trypanosoma brucei . Molecular and Cellular Biology. 1992;12(3):1218–1225. doi: 10.1128/mcb.12.3.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]