

Abstract
NAGS catalyzes the conversion of glutamate and acetyl-CoA to N-acetylglutamate (NAG) the essential allosteric activator of carbamyl phosphate synthetase I, the first urea cycle enzyme in mammals. A 17-year-old female with recurrent hyperammonemia attacks, the cause of which remained undiagnosed for 8 years in spite of multiple molecular and biochemical investigations, showed markedly enhanced ureagenesis (measured by isotope incorporation) in response to N-carbamylglutamate (NCG). This led to sequencing of the regulatory regions of the NAG synthase (NAGS) gene and identification of a deleterious single-base substitution in the upstream enhancer.
The homozygous mutation (-3063C>A), affecting a highly conserved nucleotide within the Hepatic Nuclear Factor 1 (HNF-1) binding site, was not found in SNP databases and in a screen of 1086 alleles from a diverse population. Functional assays demonstrated that this mutation decreases transcription and binding of HNF-1 to the NAGS gene, while a consensus HNF-1 binding sequence enhances binding to HNF-1 and increases transcription. Oral daily NCG therapy appears to have restored ureagenesis in this patient, normalizing her biochemical markers, and allowing discontinuation of alternate pathway therapy and normalization of her diet with no recurrence of hyperammonemia.
Keywords: N-acetyl-L-glutamate, acetylglutamate, hyperammonemia, bio-informatics, urea cycle, stable isotope, gene expression regulation
Introduction
The reactions which permit the elimination of waste nitrogen are essential to human life (Caldovic and Tuchman, 2003). One of these essential biochemical pathways - the urea cycle – requires the coordinated function of 6 enzymes and 2 mitochondrial membrane transporters to convert ammonia into urea (Batshaw, 1984). An interruption in any of these steps can result in hyperammonemia. In the absence of adequate treatment, unchecked hyperammonemia progressively leads to brain damage, coma and death (Batshaw, 1984).
The initial step of the urea cycle is catalyzed by carbamyl phosphate synthetase I (CPSI) (EC 6.3.4.16), generating carbamyl phosphate from ammonia, bicarbonate and ATP. However, CPSI is only active in the presence of its allosteric activator, N-acetylglutamate (NAG), biosynthesized by N-acetylglutamate synthase (NAGS; EC 2.3.1.1) from glutamate and acetyl-coenzyme A.
Biochemically, a deficiency of NAG, as observed in inherited NAGS deficiency (Bachmann, et al., 1981) (MIM #237310), is indistinguishable from CPSI deficiency (MIM #237300), as both conditions result in hyperammonemia due to reduced flux through the CPSI enzyme. Initial diagnoses of NAGS deficiency were based on measurements of hepatic NAGS activity (Bachmann, et al., 1988; Bachmann, et al., 1981), though these assays may not be entirely reliable (Heckmann, et al., 2005; Hwu, et al., 2007; Vockley, et al., 1992). The cloning of the human NAGS gene in 2002 (Caldovic, et al., 2002) has allowed molecular testing to become the primary method of diagnosis. Mutations in the coding region of the NAGS gene have been identified in all reported cases of NAGS deficiency since 2002 (Belanger-Quintana, et al., 2003; Caldovic, et al., 2005; Caldovic and Tuchman, 2003; Elpeleg, et al., 2002; Haberle, et al., 2003; Nordenstrom, et al., 2007; Schmidt, et al., 2005; Tuchman, et al., 2008a).
Patients with NAGS deficiency respond remarkably well when administered N-carbamylglutamate (NCG), a stable structural analog of NAG, which has been shown to activate CPSI in vitro (Hall, et al., 1958), normalize blood ammonia (Caldovic, et al., 2004; Tuchman, et al., 2008a), and restore ureagenesis (Caldovic, et al., 2004; Tuchman, et al., 2008a). In fact, NAGS deficiency is the only urea cycle disorder (UCD) in which urea cycle function may be restored through a pharmacological agent. In other urea cycle disorders, medical treatment is suboptimal, and consists of dietary protein restriction and ammonia scavenging agents (Summar, 2001), in which nitrogen elimination by alternate pathways is dependent on a stoichiometric reaction (Brusilow, et al., 1979).
The regulatory regions of the NAGS gene were recently identified [Heibel, 2011, submitted]. NAGS transcription is regulated by a promoter, which contains multiple Sp1 (Specificity protein 1) and one CREB (cAMP response element binding) binding sites, and an enhancer 3kb upstream of the coding sequence to which HNF-1 (Hepatic Nuclear Factor 1) and NF-Y (Nuclear factor Y) bind.
Mutations within the regulatory regions of genes currently represent a very small proportion of known disease causing mutations (Stenson, et al., 2009), and are likely underdiagnosed. Indeed, there has been only one report of a disease-causing mutation in a regulatory region of a urea cycle gene (Luksan, et al.).
In this report, we describe the first NAGS enhancer mutation, identified in a patient with an unspecified proximal urea cycle disorder, who despite the absence of any identifiable NAGS mutations by conventional mutation analysis (Caldovic, et al., 2007; Haberle, et al., 2003), demonstrated a marked response to a short-term trial of NCG. We demonstrate that the mutation ascertained in our patient is indeed deleterious, and results in decreased transcription factor binding and expression of the NAGS gene. We also document the patient’s favorable subsequent response to chronic treatment with NCG, leading to apparent normalization of her health.
Case Report
The patient was a 17-year-old girl of Eastern European origin, with an unspecified proximal urea cycle disorder. At 10 years of age, she presented to the Emergency Department with a 3-day history of nausea, ataxia, and somnolence. Initial plasma ammonia was 203 μM, anion gap 13, glutamine 868 μM, citrulline 12 μM, BUN 3.6 mM. She was tentatively diagnosed with partial OTC deficiency, however, a subsequent allopurinol challenge revealed normal orotic acid excretion in response to the drug, making this diagnosis improbable. In addition, sequencing of exons and intron/exon boundaries for NAGS, CPSI and OTC did not reveal any pathogenic mutation. In a further attempt to reach a diagnosis, a liver biopsy was performed, but enzymatic analysis of hepatocyte CPSI and OTC demonstrated normal activity. Analysis of hepatic NAGS enzyme activity was not performed. Because of the succession of non-diagnostic investigations, we considered enrolling her into a short-term clinical trial of N-carbamylglutamate (NCG) (Caldovic, et al., 2004; Tuchman, et al., 2008a). We deemed that a diagnosis of partial NAGS deficiency was still plausible, and that a short-term treatment with NCG might not only be diagnostic, but would also correct the metabolic defect in this condition, as observed in previous studies (Caldovic, et al., 2004; Stenson, et al., 2009; Tuchman, et al., 2008a). In addition, a short-term trial was not likely to be harmful in the other proximal urea cycle disorders. Indeed, a partial CPSI deficiency might in fact respond to supraphysiological levels of a CPSI activator (Kuchler, et al., 1996; O’Connor, et al., 1985). We discussed an NCG trial with the patient and her parents, and they consented to the study.
Materials and Methods
N-Carbamylglutamate study
The study design is shown in Figure 1, and is identical to that used in our prior NCG studies (Ah Mew, et al.,; Tuchman, et al., 2008a). As shown, the patient underwent an identical study procedure before and immediately following treatment for 3 days with oral NCG (Carbaglu, Orphan Europe, Paris, 2.2g/m2/d in four divided doses). The study was approved by the Children’s National Medical Center Institutional Review Board and informed consent was obtained before enrollment.
Figure 1.
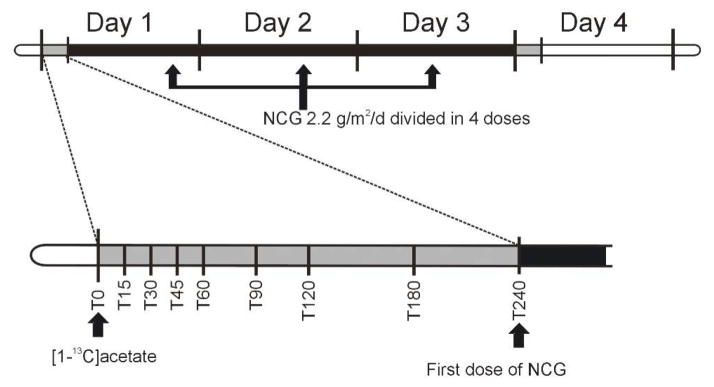
Experimental time line. Identical studies were performed before and immediately following a 3-day course of NCG at a dose of 2.2 g/m2/d. Plasma samples for ammonia, urea, amino acids, and [13C]urea were obtained at 0, 15, 30, 45, 60, 90, 120, 180, and 240 minutes prior (time 0) and following the ingestion of isotopic acetate.
On day 1 of the clinical study, an indwelling venous catheter was used for blood drawing. However, on day 4, peripheral intravenous access could not be obtained, and thus, a peripherally inserted central catheter (PICC) was introduced by an experienced interventional radiologist. The PICC remained in place for the remainder of the study, and was removed upon obtaining the last blood sample.
Following an overnight fast, a baseline sample (5 mL) of heparinized blood was obtained (time 0). Then, the subject ingested 0.33 mmol/kg, (27.5 mg/kg) of [1-13C] sodium acetate (98 atom % excess) dissolved in 60 mL of water. Blood samples (4 mL) were subsequently obtained at 15, 30, 45, 60, 75, 90, 120, 180, and 240 minutes, and drawn into a pre-cooled, heparinized tube. The sample was immediately centrifuged at 4°C to separate the plasma, which was directly transferred to cryogenic tubes, placed in dry ice, and kept frozen at −70°C until analysis, performed within 2 days after collection. Each sample was analyzed for plasma ammonia and urea (RXL Dade Behring, Siemens Healthcare Diagnostics, Deerfield, IL), quantitative amino acids (Biochrom, Cambridge, UK), and [13C]urea as previously described (Ah Mew, et al., 2009; Tuchman, et al., 2008a).
Mutation Detection
The patient’s NAGS gene was amplified using primers listed in Table 1 and previously described reagents (Caldovic, et al., 2003). Polymerase Chain Reaction (PCR) conditions were: 3 min initial denaturation at 98°C, followed by 30 cycles of 30 sec denaturation at 98°C, 30 sec of annealing at 65°C, 50 sec extension at 72°C, and final extension of 5 minutes at 72°C. The entire NAGS gene, from 3.5kb upstream of the start codon to 250bp downstream of the termination codon and including introns, was amplified and sequenced. Two small regions from −1414 to −1346 (68bp) and +2301 to 2575 (274bp), relative to the ATG codon of the NAGS gene, could not be sequenced likely due to the GC-rich nature of these segments. These two segments fall within a nonconserved 5′-untranscribed location, and within intron 4, respectively.
Table 1.
Primers used to amplify the human NAGS gene for mutation screening
| Name | Sequence |
|---|---|
| Tile1-1f | 5- GTG ACT GTG GCT GGT GTG GGT CTC C -3 |
| Tile1-1r | 5- GCC AGC TCT TCA TCA CAG CAG GCC ACC -3 |
| Tile 2-1f | 5- GAT ACC AGT CCT GCC CTC AGG GAG CC -3 |
| Tile2-1r | 5- CAG GGT CAC TCA GCT CAC AAG CCG TGG -3 |
| Tile3-1f | 5- GGGTCT GGC CAT GCC ACT GAC GAG C -3 |
| Tile3-1r | 5- GTG AGG AGG CGG GTG GGT CAT TTG AGG -3 |
| Tile4-2f | 5- AGG TTA AAG AGG GCC GGG CGT GGT GG -3 |
| Tile4-2r | 5- GGG TGG TGC TGG CCT TGT CTG TCC TG -3 |
| Tile5-2f | 5- GTC TGG GCA ACA TGG TGA ACA CCC GTC -3 |
| Tile5-2r | 5- CAG AAC CAC AGC CAT CAG CGC CGT C -3 |
| Tile6-2f | 5- CGA CGG CGC TGA TGG CTG TGG TTC TG -3 |
| Tile6-2r | 5- CCG TCT CGG TGA CCC TAC CCT TCC AG -3 |
| Tile7-2f | 5- CCA TCA CTC CGC GGA CAA GCC CTT CG -3 |
| Tile7-2r | 5- CGG GCA GAG TAG CAC GCT CTG AGG AC -3 |
| Tile8-2f | 5- TTT GGC GGC GGG TCT GTG CTA CG -3 |
| Tile8-2r | 5- TCC TGG ACA GGA GGA GAA GCC GC -3 |
| Tile9-1f | 5- CCC ACC ACT CCT CGG CCG TCA TCA CC -3 |
| Tile9-1r | 5- CCT TGG TCC GAA GCT GCC CGA CTG CCC -3 |
| Tile10-1f | 5- GGG ACG ACT ACC TGG CCT CGC TG -3 |
| Tile10-1r | 5- GAG GAC CTA CTG CAT GCT GGG CAC C -3 |
| Tile11-1f | 5- CCC ACG CAG CCC ACC TCT CAC AG -3 |
| Tile11-1r | 5- CCT GAG TAG AGC CCT GGA AGG CAC CC -3 |
f-forward and r-reverse
The patient’s parents’ NAGS enhancers were also sequenced using forward primer hNAGS-Enh 5′-CTGAACCTGGGCACAATGACAACA-3′, and reverse primer hNAGS-Enh 5′-TATGAGGTGGAAATGGGCTGGGAA-3′. The genomic DNA from a healthy individual was used as a control. The NM_153006.1 (www.ncbi.nlm.nih.gov/nuccore) sequence of the human NAGS gene was used as reference for sequence comparisons.
SNP Genotyping
Genotyping was completed using TaqMan allele discrimination assays from Applied Biosystems Inc. according to the manufacturer’s instructions (See Table 2 for the PCR primers and allele specific probes). The end point fluorescent readings were performed on an ABI 7900HT Sequence Detection System (SDS V 2.3 software; Applied Biosystems). For quality control, negative controls and duplicate samples (100% agreement) were included in the analysis.
Table 2.
TaqMan primers and probes used for SNP genotyping
| Mutation | Forward Primer | Reverse Primer | Common allele probe (5′ VIC) | Rare allele probe (5′ FAM) |
|---|---|---|---|---|
| -3063C>A | 5′GGTCAATGGGCCTGGAGTTAAT-3′ | 5′TCCCCAGGCAGCCAATG-3′ | CAGAGATACCAGAGTAGATG | CAGAGATACCAGATTAGATG |
Consensus sequence of HNF-1 Binding Motif
A consensus binding site for transcription factor HNF-1 was determined based on similarity matrices M01011, M00790, M00206, and M00132 in the TRANSFAC database (Figure 2) (Courtois, et al., 1988; Olsen, et al., 1994; Toniatti, et al., 1990; Wu, et al., 1994a; Wu, et al., 1994b). At sites with ambiguous nucleotide identify, selection of nucleotide deferred to human NAGS. The consensus binding site for HNF-1 at −3079 to −3059 was determined to be 5′-AGTTAATAATTAACTAT-3′.
Figure 2.

Consensus HNF-1 binding site. The human NAGS HNF-1 binding site and matrices for HNF-1 binding sites from the TRANSFAC database. The -3063C>A mutation is indicated. Standard nucleotide codes are used.
Site-Directed Mutagenesis
The –3063C>A (patient mutation) and -3069C>A, -3066C>T, -3065T>A, -3061C>A consensus HNF-1 binding site mutations were engineered into the NAGS gene using QuickChange Lightening Site-Directed Mutagenesis Kit (Agilent) according to manufacturer’s instructions. Primers NAGS–3063C>A Fw and Rv (Table 3) amplified 100ng of template plasmid 4.10Enh containing the wild-type NAGS enhancer and firefly (Photinus pyralis) luciferase luc2 [Heibel, 2011, submitted] to create 4.10EnhPat. Amplification of 100ng of 4.10WT by primers NAGS HNF-1con Fw and Rv (Table 3) created 4.10EnhCon plasmid. The presence of each mutation was confirmed by sequencing.
Table 3.
Primers used to generate -3063C>A and consensus HNF-1 binding site mutants via site-directed mutagenesis
| Primer Name | Primer Sequence |
|---|---|
| NAGS-3063C>A FW | 5′-GGCCTGGAGTTAATCATCTAATCTGGTATCTCTGG-3′ |
| NAGS-3063C>A RV | 5′-CCAGAGATACCAGATTAGATGATTAACTCCAGGCC-3′ |
| NAGS HNF-1con Fw | 5′-CCAGGGTCAATGGGCCTGGAGTTAATAATTAACTATGGTATCTCTGGGCC-3′ |
| NAGS HNF-1con Rv | 5′-GGCCCAGAGATACCATAGTTAATTATTAACTCCAGGCCCATTGACCCTGG-3′ |
Cloning of Wild-Type and Mutated DNA Sequence
The enhancer region of human NAGS was initially cloned into pGL4.10 vector (Promega) to create plasmid 4.10Enh as described by Heibel, et. al [2011, submitted]. The mutated plasmids, 4.10EnhPat and 4.10EnhCon, and pGL4.23 vector, containing a minimal TATA-promoter with luc2, were cut with XhoI and HindII and the vectors were treated with Antarctic Alkaline Phosphatase (AAP) (New England Biolabs). The mutated enhancers were then ligated into the pGL4.23 vector and named 4.23Pat and 4.23Con. 4.10EnhPat and 4.10EnhCon was also amplified with forward primer hEnhBS 5′-GGATCCAGGACCCTTCTGGGTGGAAGTTAT-3′ and reverse primer hEnhBS 5′-GTCGACTTCCTAGGGATCCACCCAATTCAGTC-3′, to introduce BamHI and SalI restriction enzyme cut sites at 5′ and 3′ of the enhancer respectively. The enhancers were amplified with Platinum Taq PCRx DNA Polymerase (Invitrogen) using the following conditions: initial denaturation at 95°C for 2 min, followed by 35 cycles of denaturation at 95°C for 30 sec, annealing at 60°C for 30 sec and extension at 68°C for 1 min, and final extension at 68°C for 6 min. This enhancer PCR product and the plasmid 4.10Prom containing the wild-type NAGS promoter [Heibel, 2011, submitted] were cut with BamHI and SalI, the vector was treated with AAP, and the enhancers were ligated into the vector. The resulting plasmids, expressing luc2, were named 4.10Pat and 4.10Con. Correct sequences were confirmed using sequencing primers specified by Promega.
Tissue culture and Reporter Assays
Cell culture and transfections
Human hepatoma cells (HepG2) (donated by Dr. Marshall Summar, Children’s National Medical Center, Washington, DC) were cultured in complete media containing RPMI 1640 media supplemented with 10% FBS (fetal bovine serum) and 5% Penicillin/Streptomycin under 5% CO2 at 37°C. Cells were plated at a density of 5 × 105 cells/well on 24-well culture plates 24 hours prior to transfection. The cells (90% confluent) were then transfected using Lipofectamine 2000 (Invitrogen) and cultured in transfection media containing RPMI 1640 media supplemented with 10% FBS. Cells were transfected with reporter plasmids described in the previous section or control plasmids pGL4.10 lacking a promoter, pGL4.13 with a SV40 promoter, or pGL4.23 with a TATA-box promoter for regulation of the firefly (Photinus pyralis) luciferase gene luc2. A total of 0.25 ug of DNA was co-transfected with 0.225 ug of luc2 containing expression vector and 0.025ug of Renilla reniformis luciferase gene hRluc containing expression vector (Promega) (as internal control).
Reporter assays
24 hours following transfection, cells were assayed for both firefly and Renilla luciferase activity using Dual-Luciferase Reporter Assay System (Promega) and Berthold Centro 960 luminometer (Berthold) according to the manufacturer’s protocol. All reporter assay values were corrected for transfection efficiency by normalizing the firefly luciferase signal to the Renilla luciferase values. Mean values are expressed relative to wild-type and calculated error was from three trials and reported as ± standard error of the means (SEM). To evaluate the effects of enhancer and promoter from repeated measurements over three series of experiments, mixed-effects linear regression (Stata 10) was employed.
Avidin-Agarose DNA-Protein Pull-Down Assay
Biotinylated DNA probes
Biotinylated and non-biotinylated probes for the enhancer region of human NAGS was amplified from genomic DNA isolated from donated mouse tail using Pure Gene DNA Purification Kit (Gentra) or 4.10EnhPat and 4.10EnhCon plasmids with primers NAGSEnh 5′-CTGAACCTGGGCACAATGACAACA-3′ and 5′-TATGAGGTGGAAATGGGCTGGGAA-3′ and conditions as described previously [Heibel, 2011, submitted]. Resulting probes were referred to as pWT, pPat, or pCon respectively.
Preparation of nuclear extracts
Nuclear extract was isolated from donated adult mouse livers of C57BL/6 mice using Nuclear Extraction Kit (Origene) according to manufacturer’s instructions. The protein concentration of the nuclear extract was determined using bovine serum albumin as the protein standard with Bradford Assay dye concentrate reagents (Bio-Rad, Hercules, CA). On average 10mg of nuclear protein was obtained from one mouse liver.
Binding Protocol
The binding assay was previously described in Heibel, et al (2011, submitted). Briefly, nuclear extract prepared from whole mouse liver was added to PBS buffer containing protease and phosphatase inhibitors (Roche) for a final protein concentration of 1mg in 500 ul of solution. The nuclear extract solution was pre-cleared for 1 hour and then samples were centrifuged at 600g for 1 min and the supernatant fluid was retained. 15 ug of the DNA probe was added to the pre-cleared nuclear extract solution with 100 ul of beads and incubated for 16 hrs on a rotating shaker at 4°. The biotinylated probe and bead concentrations were added in excess to ensure a complete pull-down of DNA–protein complexes. Following incubation, the beads were washed and resuspended in denaturing buffer before boiling to dissociate bound proteins. The proteins were resolved by electrophoresis, transferred electrophoretically to a nitrocellulose membrane, and identified by immunoblotting using specific antibodies against HNF-1α/β (Santa Cruz Biotech) at 1:500 dilution and against NF-Yα (Santa Cruz Biotech) at 1:1000 dilution. The membrane was than incubated with 1:20,000 dilution of donkey anti-rabbit secondary antibody conjugated to horseradish peroxidase (Pierce) and bands were visualized using SuperSignal West Pico Kit (Pierce) according to manufacturer’s instructions. Band volume (OD*mm2) was determined using a GS-800 Calibrated Densitometer (Bio-Rad) and values were corrected for loading error by normalizing the HNF-1 band volume to the NF-Y band volume. Mean values are expressed relative to wild-type and calculated error was from three trials and reported as ± SEM. Unpaired Student’s t test was performed to determine statistical significance.
Results
N-carbamylglutamate trial
The stable isotope method for measuring in vivo ureagenesis has been previously applied to assess ureagenesis in normal unaffected individuals (Tuchman, et al., 2008a), as well as to evaluate response to a 3-day trial with NCG in patients with NAGS deficiency and propionic acidemia (Ah Mew, et al.,; Tuchman, et al., 2008a). Figure 3 summarizes the results of the tracer study before and at the end of the 3-day treatment with NCG. In the described patient, the pre-treatment production of [13C]urea attained a peak of only 0.017 atom % excess (Fig 3A). Therapy with NCG for 3 days augmented this parameter over 7-fold to 0.12 atom % excess. Correspondingly, peak blood [13C]urea concentration before administration of NCG was 0.074μM, and 2.0μM, following treatment, a remarkable 27-fold increase above baseline (Fig 3B). In comparison, the peak concentration of this parameter in 38 untreated adult control studies was 4.01±0.59 μM (mean±SEM) (Tuchman, et al., 2008a).
Figure 3.
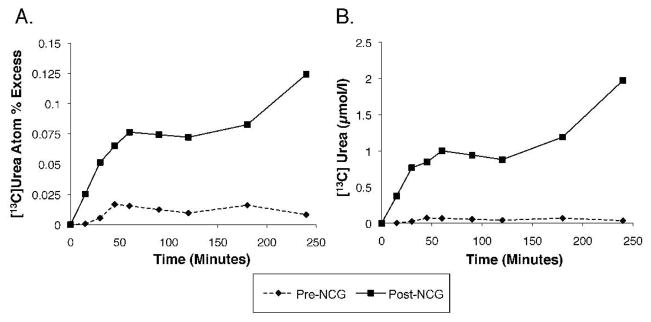
Isotopic enrichment of [13C]urea. Increase over time in isotopic enrichment of [13C]urea (A) and plasma concentration of [13C]urea (B) in the patient, who was administered 27.5 mg/kg of [1-13C]sodium acetate at time 0 of each study day.
This marked increase in nitrogen disposal was corroborated by changes in other biomarkers, as illustrated in Table 4. Ammonia at baseline was normal at 21 μM, and remained essentially unchanged at 18 μM following therapy. However, blood Urea Nitrogen (BUN) increased over three-fold (Table 4) and mean glutamine and alanine levels markedly decreased from 553 and 616 μM to 309 and 363 μM, respectively.
Table 4.
Plasma ammonia, urea, glutamine and alanine (mean ± SD) in the patient before and after 3 days treatment with NCG
| Biomarker | Pre-NCG | Post-NCG |
|---|---|---|
| Ammonia (μmol/l) | 21 ± 6 | 18 ± 3 |
| Urea (mmol/l) | 0.4 ± 0.1 | 1.4 ± 0.1 |
| Glutamine (μmol/l) | 553 ± 61 | 309 ± 29 |
| Alanine (μmol/l) | 616 ± 76 | 363 ± 31 |
Mutation detection and SNP Genotyping
Sequencing of the patient’s NAGS gene, from 3.5kb upstream of the translation start codon to 250bp downsteam of the termination codon, revealed 16 homozygous and 1 heterozygous sequence alteration compared to the reference NAGS sequence from the NCBI database. All but one were identified as commonly-occurring polymorphisms in dbSNP (build 132: http://www.ncbi.nlm.nih.gov/SNP/) (Sherry, et al., 2001), or were also present in the wild-type control sequence. The remaining change, a homozygous C to A transversion 3063bp upstream of the NAGS translation start codon (-3063C>A) (Figure 4), was determined to be in the recently identified hepatic nuclear factor 1 (HNF-1) binding site of a conserved NAGS regulatory region (Figure 5) (Heibel et al, 2011 submitted). The patient’s parents were both heterozygous for this change.
Figure 4.

DNA sequence of the HNF-1 binding site within the enhancer of the NAGS gene. The patient is homozygous for a single base substitution at position -3063. The patient’s parents are heterozygous for this allele at the same position. Asterisk (*) indicates the location of the mutation.
Figure 5.
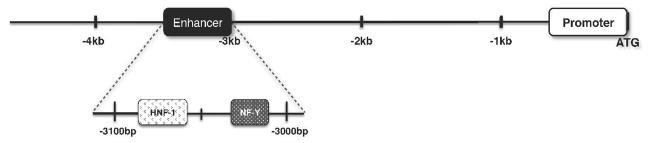
Transcription factor binding sites in the NAGS enhancer. Transcription factors HNF-1 and NF-Y bind within the human NAGS enhancer.
While, the mutated cytosine nucleotide is completely conserved among 7 mammalian species (Figure 6), single nucleotide polymorphism (SNP) genotyping was conducted to determine whether the -3063C>A change could be an uncommon change found in the general population. The mutation was not detected in 1086 control alleles (Table 5), with representation of the patient’s ethnic origin. The diverse cohort of subjects obtained through the Human Genome Diversity Panel (HGDP) (Cann, et al., 2002) included 36 individuals from Russia, 150 subjects from Asia including Japan, China Cambodia, and Papua New Guinea, 56 subjects from South America including native Mexicans and Columbians, and 36 subjects from Western Africa including Nigeria and Senegal, and 265 subjects of Northern European Caucasian decent, obtained from the FAMUSS population (Thompson, et al., 2004).
Figure 6.
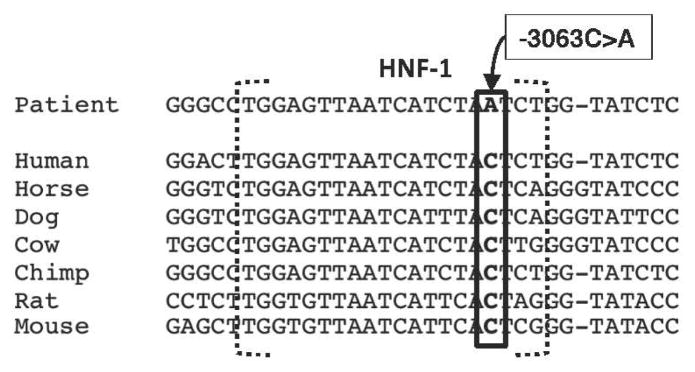
Alignment of the -3063C>A mutation with the HNF-1 binding site in mammalian species. Reference mammalian DNA sequence of the HNF-1 binding site was aligned with the patient’s HNF-1 binding site. The -3063C>A mutation (box) is indicated.
Table 5.
Allele frequencies in populations analyzed by SNP genotyping
| Genotype | |||
|---|---|---|---|
| CC | CA/AA | ||
| Number of Screened Individuals | N=543 | N=543 | |
| Ethnicity | Russian | 36 | 0 |
| Northern European | 265 | 0 | |
| South American* | 56 | 0 | |
| Asian+ | 150 | 0 | |
| Western African† | 36 | 0 | |
Subjects were native Mexican and native Columbian
Subjects were Japanese, Cambodian, Papau New Guinean, and Chinese
Subjects were Senegalese and Nigerian
Reporter Assays
The functional effect of the mutation on NAGS transcription was examined using a luciferase reporter assay, driven by the NAGS enhancer with either the wild-type, mutated, or consensus HNF-1 binding sites. Reporter plasmids included the NAGS wild-type promoter with the wild-type enhancer, 4.10WT, the enhancer containing the patient’s mutation -3063C>A, 4.10Pat, or the enhancer containing the consensus HNF-1 binding site, 4.10Con (Figure 7A). Additional reporter plasmids included a minimum TATA–promoter and the wild-type enhancer, 4.23WT, the enhancer containing the patient’s mutation, 4.23Pat, or the consensus enhancer, 4.23Con (Figure 7B) to isolate the role of the enhancers. HepG2 cells were transfected with the reporter plasmids or pGL4.10, pGL4.13, and pGL4.23 as negative, positive, and reference controls respectively. Cells were co-transfected with vector pGL4.74, containing Renilla luciferase hRluc, as a transfection efficiency control.
Figure 7.

Effects of the -3063C>A mutation and consensus HNF-1 binding site on gene expression. Luciferase expression is significantly different between wild-type, -3063C>A, and consensus enhancers when expressed in conjunction with the human wild-type NAGS promoter (A) or the minimal TATA-box promoter (B). All results are an average of three independent experiments that were each carried out in triplicate. Calculated error was from three trials and reported as ±SEM. One asterisk (*) indicates p<0.05 and two asterisks (**) indicate p<0.005.
Transcription of luciferase was decreased by 25% in cells transfected with plasmid harboring the -3063C>A mutation, compared to the wild-type enhancer, and either the NAGS or TATA-containing promoter (4.10Pat p<0.001, 4.23Pat p<0.001) (Figure 7A & B). Transcription of luciferase increased in cells transfected with plasmids containing the consensus enhancer, compared to wild-type, and either promoter (4.10Con, 35% greater, p<0.001, 4.23Con, 75% greater, p=0.006) (Figure 7A & B). No luciferase expression was detected for each enhancer alone, in the absence of a promoter (data not shown). Control vectors pGL4.10 and pGL4.23 did not stimulate luciferase expression while positive control vector pGL4.13 activated transcription in this cell culture system as previously shown (Heibel, 2011, submitted). Measured luciferase activity was normalized to hRluc expression to adjust for transfection efficiencies. Statistical analysis was performed for normalized values while graphical representation is displayed as normalized values relative to the selected promoter with the wild-type enhancer. The reporter assay results compared to wild-type, showed an increase in transcription of luciferase when the consensus HNF-1 binding site was present and a decrease when the wild-type HNF-1 binding site contained the -3063C>A mutation.
Pull Down assays
To examine the direct effect of the mutation, a DNA-protein pull-down assay was devised in which biotin-labeled DNA probes containing the wild-type (pWT), -3063C>A mutation (pPat), and consensus (pCon) HNF-1 binding site were incubated with nuclear proteins, precipitated using avidin-agarose beads, and analyzed by western blot.
The biotinylated enhancer probes pWT, pPat, and pCon contained 333bp of the NAGS sequence from −2834 to −3167 (Figure 8B, lanes 1, 3, and 5). Non-biotinylated enhancer probes were used as negative controls (Figure 8B, lanes 2, 4, and 6). Normalized results showed a 30% increase in band intensity for pCon (p=0.015) and a 40% decrease in band intensity for pPat (p=0.001) compared to pWT (Figure 8A). Negative control DNA probes did not bind HNF-1 (Figure 8B, lanes 2, 4, and 6). These results indicate that HNF-1 binding is reduced in the presence of the -3063C>A mutation and increased in the presence of the consensus site sequence.
Figure 8.

Effects of the -3063C>A mutation and consensus HNF-1 binding site on “pull-down” of HNF-1 protein. Amounts of HFN-1 protein pulled down were significantly different between enhancers containing the wild-type, -3063C>A mutated, or consensus HNF-1 binding site (A). Immunoblots (B) show HNF-1 and NF-Y protein following overnight incubation with whole nuclear extract. Values are normalized means from three replicate experiments. Immunoblots are representative of the results obtained by three replicate experiments. Calculated error was from three trials and reported as ±SEM. One asterisk (*) indicates p<0.05 and two asterisks (**) indicate p<0.005.
Chronic therapy with N-carbamylglutamate
Given the substantial experimental evidence that the patient’s mutation was deleterious, as well as her marked response to a short-term trial of NCG, she was prescribed daily NCG treatment.
On her clinic visit just prior to NCG administration, plasma ammonia level was 118μM, glutamine 601μM, alanine 579 μM, blood urea nitrogen 0.7 mM, and arginine 25 μM. Also, essential amino acids, including branched chain amino acids, were low, presumably as a result of protein restriction and alternate pathway therapy. At the time her dietary protein consumption was limited to 0.5g/kg/d (~30–35g/d), and she was on sodium phenylbutyrate at 18g/day.
We prescribed a continuous daily regimen of oral NCG, 2.2 g/m2/d, identical to her dose during the short-term trial. Phenylbutyrate intake was tapered off over a month, and the patient has been able to entirely liberalize her dietary protein. During the three months of therapy to date, she has been clinically well. Her most recent plasma ammonia level was 4 μM, glutamine 379 μM, alanine 374 μM, and arginine 105 μM. Blood urea nitrogen was 4.6 mM, the highest ever recorded in her.
Discussion
This is the first report of a disease-causing mutation in the regulatory region of NAGS and the second in any urea-cycle gene [20]. NAGS deficiency is the rarest of the urea cycle disorders(Tuchman, et al., 2008b). In the 2008 report from the Urea Cycle Disease Consortium Longitudinal Study, there were no enrolled patients with NAGS deficiency (Tuchman, et al., 2008b), yet there were 11 patients without a current specific diagnosis that had symptomatic hyperammonemic episodes and biochemical measures highly suggestive of a urea cycle disorder (UCD). It is likely that these patients have proximal UCDs since distal UCDs and transporter defects typically present with additional amino acid abnormalities. These undiagnosed patients may in fact have mutations in the regulatory region of NAGS or CPSI genes.
Although it is well documented that mutations in non-coding regions of a gene can result in disease (Crossley and Brownlee, 1990; Perez-Tur, et al., 1995; Sakai, et al., 1991), currently available clinical molecular diagnostics frequently does not screen for mutations in regulatory regions, and therefore such patients are likely to remain undiagnosed. In a recent report from the Human Genome Mutation Database (HGMD, www.hgmd.cf.ac.uk) (Stenson, et al., 2009), only 1.6% of all reported mutations were in regulatory regions of genes. As a preliminary survey, we searched the public version of HGMD for apparently regulatory mutations in the 8 genes of the urea cycle as well as the genes of the American College of Medical Genetics newborn screening core panel, not including congenital deafness, cystic fibrosis or the hemoglobinopathies. As of March 10, 2011, this search only identified a single (aforementioned) disease-causing regulatory mutation in the OTC promoter (Luksan, et al.), out of 3316 reported mutations in genes associated with inherited metabolic disease.
In the patient described herein, with a high clinical suspicion of NAGS deficiency, a successful diagnostic strategy involved sequencing nearly the entire NAGS gene, including the upstream regulatory regions. Continued advances in molecular genetics have allowed for more rapid and accurate sequencing of vast regions of DNA. These technologies will be applied to sequence not just exons, but entire genes, in an attempt to identify deleterious mutations in other patients with a highly-likely clinical diagnosis. However, this strategy will certainly identify numerous single base substitutions, and subsequent SNP genotyping, as well as functional studies, will be required to differentiate between common polymorphisms and disease causing mutations.
In our SNP genotyping studies, our patient’s mutation was not identified in 1086 control alleles, from individuals of varied ethnic backgrounds. Also, the patient’s parents were both heterozygous for the -3063C>A substitution, consistent with the expected autosomal recessive inheritance. These results were highly suggestive that the patient’s mutation was indeed deleterious.
Functional studies ultimately determined the deleterious effect of the HNF-1 binding-site mutation. While the role of HNF-1 in NAGS regulation is not yet entirely understood, these studies demonstrate that it plays an important role in the regulation of NAGS expression. The engineering of a consensus HNF-1 sequence resulted in increased HNF-1 binding (Figure 8). It is this enhanced affinity to HNF-1 that likely resulted in increased transcription of our reporter gene (Figure 7). Conversely, the -3063C>A mutation results in decreased binding of HNF-1 (Figure 8), and thus significantly reduces, but does not abolish transcription (Figure 7).
This patient, like others with regulatory mutations in genes coding for enzymes (Coffee and Tolan,; Luksan, et al.), presented at an age well after the neonatal period. This likely reflects the nature of promoter or enhancer mutations, which might decrease but not eliminate expression of the genes they regulate. The residual gene expression and enzymatic activity retained in these patients results in a less severe clinical presentation.
Treatment with NCG is the therapy of choice in patients with NAGS deficiency, as it corrects the metabolic defect in this condition. In affected patients, NCG has been shown to normalize blood ammonia (Elpeleg, et al., 2002; Gessler, et al.,; Guffon, et al., 2005; Pandya, et al., 1991; Tuchman, et al., 2008a) and restore ureagenesis (Tuchman, et al., 2008a). This is likely the case in our patient, who has been symptom-free while on NCG, despite discontinuing ammonia scavengers and liberalizing protein in her diet. NCG may also have a salutary effect in other conditions where hepatic NAG is reduced (Coude, et al., 1982), as well as in some patients with partial CPSI deficiency (Kuchler, et al., 1996; O’Connor, et al., 1985).
In the short-term N-carbamylglutamate trial, we used a stable isotope method to assess in vivo changes to patients’ capacity for ureagenesis in response to NCG. Stable isotopes are safe, and in this case, the analysis of isotopic enrichment in blood urea, by ion-ratio mass spectrometry, is highly sensitive, robust and reproducible (de Meer, et al., 1999; Kalhan, et al., 1986). This method reliably documented the augmentation in ureagenesis following NCG administration in previous 8 patients studied by our group, including seven patients with propionic academia and one with NAGS deficiency (Ah Mew, et al.,; Tuchman, et al., 2008a). In the previously-described patient with NAGS deficiency, there was a greater than 4-fold increase in 13C label incorporation into urea following NCG administration – a marked augmentation that was also observed in the patient described herein. In fact, in this patient, this restoration of ureagenesis provided an important diagnostic clue, and triggered an extensive mutation search in the NAGS gene, culminating in the identification of the deleterious mutation and confirmation of the regulatory function of the site affected by it. This marked increase in isotope-enriched urea following a trial of NCG is likely pathognomonic of NAGS deficiency, and may be a useful diagnostic tool in future patients where this diagnosis is suspected, but in whom no deleterious mutations are identified. Furthermore, subsequent long-term treatment with NCG appears to have corrected the metabolic defect in our patient, suggesting that the increase in ureagenesis observed during a short-term trial with NCG is predictive of the long-term response to this medication.
Acknowledgments
This work was supported by public health service grants R01HD058567, R01DK064913, P30HD026979, M01RR020359, from the National Institutes of Health. We thank Dr. Uta Lichter for her expert patient care, Dr. Robert McCarter for his expertise in statistics, Dr. Marshall Summar for providing HepG2 cells, and the Clinical Research Center staff of Children’s National Medical Center for help in performing the study.
Abbreviations
- AAP
antarctic alkaline phosphatase
- BUN
blood urea nitrogen
- CPSI
carbamyl phosphate synthetase I
- CREB
cAMP response element binding
- FBS
fetal bovine serum
- HepG2
human hepatoma cell line G2
- HGDP
Human Genome Diversity Panel
- HNF-1
Hepatic Nuclear Factor 1
- MIM
Mendelian inheritance in Man
- NAG
N-acetyl-L-glutamate
- NAGS
N-acetylglutamate synthase
- NCG
N-carbamyl-L-glutamate
- NF-Y
Nuclear Factor Y
- OTC
ornithine transcarbamylase
- PCR
polymerase chain reaction
- PICC
peripheral inserted central catheter
- Sp1
Specificity protein 1
- UCD
urea cycle disorders
- UCDC
Urea Cycle Disorders Consortium
References
- Ah Mew N, McCarter R, Daikhin Y, Nissim I, Yudkoff M, Tuchman M. N-carbamylglutamate augments ureagenesis and reduces ammonia and glutamine in propionic acidemia. Pediatrics. 126(1):e208–14. doi: 10.1542/peds.2010-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ah Mew N, Payan I, Daikhin Y, Nissim I, Tuchman M, Yudkoff M. Effects of a single dose of N-carbamylglutamate on the rate of ureagenesis. Mol Genet Metab. 2009;98(4):325–30. doi: 10.1016/j.ymgme.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann C, Brandis M, Weissenbarth-Riedel E, Burghard R, Colombo JP. N-acetylglutamate synthetase deficiency, a second patient. J Inherit Metab Dis. 1988;11(2):191–3. doi: 10.1007/BF01799871. [DOI] [PubMed] [Google Scholar]
- Bachmann C, Krahenbuhl S, Colombo JP, Schubiger G, Jaggi KH, Tonz O. N-acetylglutamate synthetase deficiency: a disorder of ammonia detoxication. N Engl J Med. 1981;304(9):543. doi: 10.1056/NEJM198102263040918. [DOI] [PubMed] [Google Scholar]
- Batshaw ML. Hyperammonemia. Curr Probl Pediatr. 1984;14(11):1–69. doi: 10.1016/0045-9380(84)90047-1. [DOI] [PubMed] [Google Scholar]
- Belanger-Quintana A, Martinez-Pardo M, Garcia MJ, Wermuth B, Torres J, Pallares E, Ugarte M. Hyperammonaemia as a cause of psychosis in an adolescent. Eur J Pediatr. 2003;162(11):773–5. doi: 10.1007/s00431-002-1126-2. [DOI] [PubMed] [Google Scholar]
- Brusilow SW, Valle DL, Batshaw M. New pathways of nitrogen excretion in inborn errors of urea synthesis. Lancet. 1979;2(8140):452–4. doi: 10.1016/s0140-6736(79)91503-4. [DOI] [PubMed] [Google Scholar]
- Caldovic L, Morizono H, Daikhin Y, Nissim I, McCarter RJ, Yudkoff M, Tuchman M. Restoration of ureagenesis in N-acetylglutamate synthase deficiency by N-carbamylglutamate. J Pediatr. 2004;145(4):552–4. doi: 10.1016/j.jpeds.2004.06.047. [DOI] [PubMed] [Google Scholar]
- Caldovic L, Morizono H, Gracia Panglao M, Gallegos R, Yu X, Shi D, Malamy MH, Allewell NM, Tuchman M. Cloning and expression of the human N-acetylglutamate synthase gene. Biochem Biophys Res Commun. 2002;299(4):581–6. doi: 10.1016/s0006-291x(02)02696-7. [DOI] [PubMed] [Google Scholar]
- Caldovic L, Morizono H, Panglao MG, Cheng SF, Packman S, Tuchman M. Null mutations in the N-acetylglutamate synthase gene associated with acute neonatal disease and hyperammonemia. Hum Genet. 2003;112(4):364–8. doi: 10.1007/s00439-003-0909-5. [DOI] [PubMed] [Google Scholar]
- Caldovic L, Morizono H, Panglao MG, Lopez GY, Shi D, Summar ML, Tuchman M. Late onset N-acetylglutamate synthase deficiency caused by hypomorphic alleles. Hum Mutat. 2005;25(3):293–8. doi: 10.1002/humu.20146. [DOI] [PubMed] [Google Scholar]
- Caldovic L, Morizono H, Tuchman M. Mutations and polymorphisms in the human N-acetylglutamate synthase (NAGS) gene. Hum Mutat. 2007;28(8):754–9. doi: 10.1002/humu.20518. [DOI] [PubMed] [Google Scholar]
- Caldovic L, Tuchman M. N-acetylglutamate and its changing role through evolution. Biochem J. 2003;372(Pt 2):279–90. doi: 10.1042/BJ20030002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann HM, de Toma C, Cazes L, Legrand MF, Morel V, Piouffre L, Bodmer J, Bodmer WF, Bonne-Tamir B, Cambon-Thomsen A, et al. A human genome diversity cell line panel. Science. 2002;296(5566):261–2. doi: 10.1126/science.296.5566.261b. [DOI] [PubMed] [Google Scholar]
- Coffee EM, Tolan DR. Mutations in the promoter region of the aldolase B gene that cause hereditary fructose intolerance. J Inherit Metab Dis. 33(6):715–25. doi: 10.1007/s10545-010-9192-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coude FX, Rabier D, Cathelineau L, Grimber G, Parvy P, Kamoun P. A mechanism for valproate-induced hyperammonemia. Adv Exp Med Biol. 1982;153:153–61. doi: 10.1007/978-1-4757-6903-6_21. [DOI] [PubMed] [Google Scholar]
- Courtois G, Baumhueter S, Crabtree GR. Purified hepatocyte nuclear factor 1 interacts with a family of hepatocyte-specific promoters. Proc Natl Acad Sci U S A. 1988;85(21):7937–41. doi: 10.1073/pnas.85.21.7937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley M, Brownlee GG. Disruption of a C/EBP binding site in the factor IX promoter is associated with haemophilia B. Nature. 1990;345(6274):444–6. doi: 10.1038/345444a0. [DOI] [PubMed] [Google Scholar]
- de Meer K, Roef MJ, Kulik W, Jakobs C. In vivo research with stable isotopes in biochemistry, nutrition and clinical medicine: an overview. Isotopes Environ Health Stud. 1999;35(1–2):19–37. doi: 10.1080/10256019908234077. [DOI] [PubMed] [Google Scholar]
- Elpeleg O, Shaag A, Ben-Shalom E, Schmid T, Bachmann C. N-acetylglutamate synthase deficiency and the treatment of hyperammonemic encephalopathy. Ann Neurol. 2002;52(6):845–9. doi: 10.1002/ana.10406. [DOI] [PubMed] [Google Scholar]
- Gessler P, Buchal P, Schwenk HU, Wermuth B. Favourable long-term outcome after immediate treatment of neonatal hyperammonemia due to N-acetylglutamate synthase deficiency. Eur J Pediatr. 169(2):197–9. doi: 10.1007/s00431-009-1006-0. [DOI] [PubMed] [Google Scholar]
- Guffon N, Schiff M, Cheillan D, Wermuth B, Haberle J, Vianey-Saban C. Neonatal hyperammonemia: the N-carbamoyl-L-glutamic acid test. J Pediatr. 2005;147(2):260–2. doi: 10.1016/j.jpeds.2005.04.059. [DOI] [PubMed] [Google Scholar]
- Haberle J, Schmidt E, Pauli S, Kreuder JG, Plecko B, Galler A, Wermuth B, Harms E, Koch HG. Mutation analysis in patients with N-acetylglutamate synthase deficiency. Hum Mutat. 2003;21(6):593–7. doi: 10.1002/humu.10216. [DOI] [PubMed] [Google Scholar]
- Hall LM, Metzenberg RL, Cohen PP. Isolation and characterization of a naturally occurring cofactor of carbamyl phosphate biosynthesis. J Biol Chem. 1958;230(2):1013–21. [PubMed] [Google Scholar]
- Heckmann M, Wermuth B, Haberle J, Koch HG, Gortner L, Kreuder JG. Misleading diagnosis of partial N-acetylglutamate synthase deficiency based on enzyme measurement corrected by mutation analysis. Acta Paediatr. 2005;94(1):121–4. doi: 10.1111/j.1651-2227.2005.tb01799.x. [DOI] [PubMed] [Google Scholar]
- Hwu WL, Chien YH, Tang NL, Law LK, Lin CY, Lee NC. Deficiency of the carnitine transporter (OCTN2) with partial N-acetylglutamate synthase (NAGS) deficiency. J Inherit Metab Dis. 2007;30(5):816. doi: 10.1007/s10545-007-0594-y. [DOI] [PubMed] [Google Scholar]
- Kalhan SC, Oliven A, King KC, Lucero C. Role of glucose in the regulation of endogenous glucose production in the human newborn. Pediatr Res. 1986;20(1):49–52. doi: 10.1203/00006450-198601000-00013. [DOI] [PubMed] [Google Scholar]
- Kuchler G, Rabier D, Poggi-Travert F, Meyer-Gast D, Bardet J, Drouin V, Cadoudal M, Saudubray JM. Therapeutic use of carbamylglutamate in the case of carbamoyl-phosphate synthetase deficiency. J Inherit Metab Dis. 1996;19(2):220–2. doi: 10.1007/BF01799434. [DOI] [PubMed] [Google Scholar]
- Luksan O, Jirsa M, Eberova J, Minks J, Treslova H, Bouckova M, Storkanova G, Vlaskova H, Hrebicek M, Dvorakova L. Disruption of OTC promoter-enhancer interaction in a patient with symptoms of ornithine carbamoyltransferase deficiency. Hum Mutat. 31(4):E1294–303. doi: 10.1002/humu.21215. [DOI] [PubMed] [Google Scholar]
- Nordenstrom A, Halldin M, Hallberg B, Alm J. A trial with N-carbamylglutamate may not detect all patients with NAGS deficiency and neonatal onset. J Inherit Metab Dis. 2007;30(3):400. doi: 10.1007/s10545-007-0454-9. [DOI] [PubMed] [Google Scholar]
- O’Connor JE, Jorda A, Grisolia S. Acute and chronic effects of carbamyl glutamate on blood urea and ammonia. Eur J Pediatr. 1985;143(3):196–7. doi: 10.1007/BF00442137. [DOI] [PubMed] [Google Scholar]
- Olsen J, Laustsen L, Troelsen J. HNF1 alpha activates the aminopeptidase N promoter in intestinal (Caco-2) cells. FEBS Lett. 1994;342(3):325–8. doi: 10.1016/0014-5793(94)80525-3. [DOI] [PubMed] [Google Scholar]
- Pandya AL, Koch R, Hommes FA, Williams JC. N-acetylglutamate synthetase deficiency: clinical and laboratory observations. J Inherit Metab Dis. 1991;14(5):685–90. doi: 10.1007/BF01799936. [DOI] [PubMed] [Google Scholar]
- Perez-Tur J, Froelich S, Prihar G, Crook R, Baker M, Duff K, Wragg M, Busfield F, Lendon C, Clark RF, et al. A mutation in Alzheimer’s disease destroying a splice acceptor site in the presenilin-1 gene. Neuroreport. 1995;7(1):297–301. [PubMed] [Google Scholar]
- Sakai T, Ohtani N, McGee TL, Robbins PD, Dryja TP. Oncogenic germ-line mutations in Sp1 and ATF sites in the human retinoblastoma gene. Nature. 1991;353(6339):83–6. doi: 10.1038/353083a0. [DOI] [PubMed] [Google Scholar]
- Schmidt E, Nuoffer JM, Haberle J, Pauli S, Guffon N, Vianey-Saban C, Wermuth B, Koch HG. Identification of novel mutations of the human N-acetylglutamate synthase gene and their functional investigation by expression studies. Biochim Biophys Acta. 2005;1740(1):54–9. doi: 10.1016/j.bbadis.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NS, Cooper DN. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1(1):13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summar M. Current strategies for the management of neonatal urea cycle disorders. J Pediatr. 2001;138(1 Suppl):S30–9. doi: 10.1067/mpd.2001.111834. [DOI] [PubMed] [Google Scholar]
- Thompson PD, Moyna N, Seip R, Price T, Clarkson P, Angelopoulos T, Gordon P, Pescatello L, Visich P, Zoeller R, et al. Functional polymorphisms associated with human muscle size and strength. Med Sci Sports Exerc. 2004;36(7):1132–9. doi: 10.1249/01.mss.0000132274.26612.23. [DOI] [PubMed] [Google Scholar]
- Toniatti C, Demartis A, Monaci P, Nicosia A, Ciliberto G. Synergistic trans-activation of the human C-reactive protein promoter by transcription factor HNF-1 binding at two distinct sites. EMBO J. 1990;9(13):4467–75. doi: 10.1002/j.1460-2075.1990.tb07897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuchman M, Caldovic L, Daikhin Y, Horyn O, Nissim I, Korson M, Burton B, Yudkoff M. N-carbamylglutamate markedly enhances ureagenesis in N-acetylglutamate deficiency and propionic acidemia as measured by isotopic incorporation and blood biomarkers. Pediatr Res. 2008a;64(2):213–7. doi: 10.1203/PDR.0b013e318179454b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuchman M, Lee B, Lichter-Konecki U, Summar ML, Yudkoff M, Cederbaum SD, Kerr DS, Diaz GA, Seashore MR, Lee HS, et al. Cross-sectional multicenter study of patients with urea cycle disorders in the United States. Mol Genet Metab. 2008b;94(4):397–402. doi: 10.1016/j.ymgme.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vockley J, Vockley CM, Lin SP, Tuchman M, Wu TC, Lin CY, Seashore MR. Normal N-acetylglutamate concentration measured in liver from a new patient with N-acetylglutamate synthetase deficiency: physiologic and biochemical implications. Biochem Med Metab Biol. 1992;47(1):38–46. doi: 10.1016/0885-4505(92)90006-k. [DOI] [PubMed] [Google Scholar]
- Wu GD, Chen L, Forslund K, Traber PG. Hepatocyte nuclear factor-1 alpha (HNF-1 alpha) and HNF-1 beta regulate transcription via two elements in an intestine-specific promoter. J Biol Chem. 1994a;269(25):17080–5. [PubMed] [Google Scholar]
- Wu KJ, Wilson DR, Shih C, Darlington GJ. The transcription factor HNF1 acts with C/EBP alpha to synergistically activate the human albumin promoter through a novel domain. J Biol Chem. 1994b;269(2):1177–82. [PubMed] [Google Scholar]