

Abstract
The Raf family of protein kinases are key signaling intermediates, acting as a central link between the membrane-bound Ras GTPases and the downstream kinases MEK and ERK. Raf kinase regulation is well-known for its complexity but only recently has it been realized that many of the mechanisms involved in Raf regulation also modulate Raf dimerization, now acknowledged to be a required step for Raf signaling in multiple cellular contexts. Recent studies have shown that Raf dimerization is necessary for normal Ras-dependent Raf kinase activation and contributes to the pathogenic function of disease-associated mutant Raf proteins with all but high intrinsic kinase activity. Raf dimerization has also been found to alter therapeutic responses and disease progression in patients treated with ATP-competitive Raf inhibitors as well as certain other kinase-targeted drugs. This demonstration of clinical significance has stimulated the recent development of biosensor assays that can monitor inhibitor-induced Raf dimerization as well as studies demonstrating the therapeutic potential of blocking Raf dimerization.
Keywords: Raf family kinases, B-Raf, C-Raf, Ras, MEK, ERK cascade
Raf Kinase Regulation: A Brief Review
The Raf family kinases are important signaling molecules, best known for their role in Ras pathway signal transduction. In mammalian cells, there are 3 Raf proteins, A-Raf, B-Raf, and C-Raf (also known as Raf-1).1 Each member of this family can bind activated Ras and function as the initiating enzyme in the 3-tiered ERK kinase cascade, comprised of the Raf, MEK, and ERK kinases. Elucidating the mechanisms that regulate Raf activity, however, has been a daunting task that has challenged investigators for years—due largely to the complexity of the process (reviewed in ref. 2). In their inactive state, all Raf proteins are found in the cytoplasm, with the N-terminal regulatory domain acting as an autoinhibitor of the C-terminal kinase domain. 14–3-3 dimers bind to phosphorylation sites present in both the N- and C-terminal regions and stabilize the autoinhibited state. Under normal signaling conditions, Ras activation recruits the Raf proteins to the plasma membrane, which induces the release of 14-3-3 from the N-terminal binding site and facilitates the dephosphorylation of inhibitory sites as well as the phosphorylation of activating sites within the kinase domain. Following catalytic activation, Raf signaling is subsequently attenuated by ERK-mediated feeback phosphorylation, which disrupts the Ras/Raf interaction and releases Raf from the plasma membrane.3,4 Strikingly, it is now known that many of the molecular mechanisms involved in Raf regulation directly impact Raf dimer formation, a key aspect of Raf activation and function. Here, we will give a historical perspective of Raf dimerization and highlight recent studies demonstrating the importance of Raf dimerization in normal and disease-associated Ras/Raf/MEK/ERK signaling. In addition, we will discuss the unexpected consequences of Raf dimerization induced by kinase-targeted drugs as well as the therapeutic potential of Raf dimerization inhibitors.
Raf Dimerization: A Historical Perspective
The first indication that dimerization might contribute to Raf regulation came in 1996, when forced homo-dimerization of C-Raf using artifical dimerization constructs and chemical agents was shown to increase C-Raf enzymatic activity.5,6 However, without evidence that the Raf proteins could dimerize under physiological conditions, the significance of these findings were unclear. It was not until 2001 that Weber and colleagues7 published the first report that activated Ras could induce complex formation between wild-type (WT) B-Raf and C-Raf proteins. Further investigation on Raf/Raf interactions appeared to stall until a paper from the laboratories of Drs Richard Marais and David Barford reported the first crystal structure of the B-Raf kinase domain and presented the characterization of various oncogenic B-Raf mutants.8 As with the Ras family GTPases, mutant Raf proteins can function as drivers of human disease states. Germline mutations in both B-Raf and C-Raf are associated with a group of related-developmental disorders known collectively as Rasopathies,9 whereas somatic mutations primarily in B-Raf are found in a variety of cancers, with ~60% of malignant melanomas containing B-Raf mutations.10 Analysis of the oncogenic B-Raf mutants revealed that some mutations, including the most prevalent V600E mutation, caused a dramatic increase in the intrinsic kinase activity of B-Raf, whereas other mutations had an intermediate activating effect, and surprisingly, a group of mutations even reduced B-Raf’s kinase activity to a level below that of the wild-type protein.8 These kinase-impaired B-Raf mutants were further shown to be dependent on endogenous C-Raf for their transforming abilities. Following this report, studies from various groups used biochemical approaches to further characterize the Raf/Raf interactions, focusing almost exclusively on B-Raf/C-Raf binding. The kinase-impaired oncogenic B-Raf proteins were found to interact constitutively with C-Raf in a Ras-independent manner.11 This interaction occurred in the cytoplasm and required that the C-terminal 14–3-3 binding site was intact. Although growth factor-induced B-Raf/C-Raf complex formation also required 14-3-3 binding to the Raf C-terminal sites, this interaction occurred at the plasma membrane and was dependent on Ras activation.4,12 In addition, ERK-mediated feedback phosphorylation on the Rafs was found to disrupt the B-Raf/C-Raf complex.3,4 Despite all the data analyzing the requirements and dynamics of the B-Raf/C-Raf interaction, however, it was still unclear whether these interactions reflected direct dimerization of the Rafs, given that binding of the dimeric 14–3-3 proteins was required. Moreover, many questions remained regarding the extent to which these interactions affected Raf kinase activity and function.
The realization that Raf proteins do directly contact one another finally came in 2009 when the B-Raf crystal structure was re-evaluated by the laboratories of Drs. Marc Therrien and Frank Sicheri. Through their work, residues conserved in all Raf proteins as well as the closely related KSR family were identified that were critical for direct side-to-side dimer formation.13 Shortly thereafter, Raf dimerization was implicated to be a critical aspect of Raf regulation through a series of studies examining the effects of treating melanoma cells with ATP-competitive Raf inhibitors.14-16 Given that the high activity V600E mutation is observed in > 95% of malignant melanomas containing B-Raf mutations,17 numerous ATP-competitive Raf inhibitors have been developed, some with high specificity toward V600E-B-Raf. These inhibitors were found to suppress ERK signaling in melanoma lines containing V600E-B-Raf; however, they paradoxically increased ERK signaling and promoted the dimerization of wild-type B-Raf and C-Raf in lines expressing Ras mutants.14-16 Moreover, even in patients possessing V600E-B-Raf mutations, Raf inhibitor treatment could promote the development of secondary cancers in cells that harbored activating Ras mutations.18 Thus, these surprising side effects of Raf inhibitor therapy demonstrated the urgent need to fully understand the role that dimerization plays in Raf activation and function.
Revisiting Raf Dimerization in Growth Factor Signaling
To address some of the outstanding questions regarding Raf dimerization, our laboratory embarked on a project to examine Raf dimerization in normal Ras-dependent signaling and mutant Raf signaling.19 In particular, we wanted to determine whether all Raf family members can dimerize under physiological conditions, whether Raf heterodimerization or homodimerization was most critical, and whether dimerization was an absolute requirement for Raf kinase activation. In studies analyzing the heterodimerization of the endogenous Raf proteins, we found that growth factor treatment primarily induced B-Raf/C-Raf heterodimerization, with only low levels of B-Raf/A-Raf binding and little to no C-Raf/A-Raf binding observed. A basal level of B-Raf homodimerization was also detected that increased approximately 2-fold following growth factor treatment. However, C-Raf homodimerization was only observed following growth factor treatment and at low levels. Protein depletion experiments further revealed that the growth factor-induced activation of C-Raf was highly dependent on the presence of B-Raf, whereas B-Raf activation was only partially dependent on C-Raf, consistent with the observation that growth factor treatment induced both heterodimerization with C-Raf as well as B-Raf homodimerization. Interestingly, A-Raf, which exhibited the lowest dimerization potential, was only weakly activated by growth factor treatment, and its depletion had no significant effect on the activation of either B-Raf or C-Raf in response to growth factor treatment.
To verify that Raf dimerization is an activating mechanism, our laboratory next turned to the structure of the Raf dimer interface and utilized a mutation in a conserved arginine residue that had been shown to disrupt Raf dimerization (R509H-B-Raf and R401H-C-Raf13). We found that the incorporation of the R > H mutation into either C-Raf or B-Raf abolished Raf heterodimerization as well as homodimerization, and blocked the growth factor-induced activation of these kinases19 (Fig. 1A). This mutation did not affect the ability of the Rafs to interact with other binding partners such as Ras, 14–3-3, Hsp90, or MEK. Moreover, the R > H mutation did not prevent the growth factor-induced phosphorylation of C-Raf on the negative charge regulatory region S338 site, which is considered to be a prerequisite for C-Raf activation (Fig. 1A). In contrast, another substitution in the dimer interface (a glutamic acid to lysine substitution, E586K-B-Raf; E478K-C-Raf13) enhanced all Raf dimerization states, coincident with increased Raf kinase activity.19 It should be noted that of the Raf dimer interface mutations, the effect of the R > H substitution was dominant over the effect of the E > K substitution, in that growth factor-induced B-Raf/C-Raf heterodimerization was disrupted for C-Raf proteins containing both the R > H and E > K mutations (Fig. 1B). Thus, the use of the dimer interface mutations confirmed the importance of Raf dimerization in normal Ras-dependent Raf kinase activation.

Figure 1. Effect of dimer interface R > H mutation and Raf inhibitor treatment on Raf dimerization. (A and B) Serum-starved HeLa cells expressing Flag-tagged WT- R > H-, E > K, R > H/E > K-C-Raf proteins were treated or not with EGF for 5 min prior to lysis. Immunoprecipitated Flag-C-Raf was probed for the binding of endogenous B-Raf (A and B) and for the phosphorylation of S338 (A). The kinase activity of purified Flag-C-Raf proteins was determined in immune complex kinase assays using recombinant MEK as a substrate (A). 32P-labeled MEK was analyzed by SDS-PAGE and autoradiography. (C) Cycling HeLa cells expressing the indicated Flag or Pyo-tagged Raf proteins were treated with PLX4720 or SB590885 for 1 h prior to lysis. Immunoprecipitated Flag-Raf proteins were probed for the binding of Pyo-Raf.
Revisiting Raf Dimerization in Human Disease States
To address the issue of Raf dimerization in disease-associated mutant Raf signaling, we incorporated the same dimer interface mutations into a panel of mutant B-Raf and C-Raf proteins possessing various intrinsic kinase activities.19 As had been observed for the WT Raf proteins, the R > H substitution disrupted Raf dimerization, whereas the E > K mutation increased dimerization for all the Raf mutants tested, regardless of their activity. In focus formation assays, Raf dimerization had little effect on the transforming function of B-Raf mutants with high kinase activity, such as V600E-B-Raf; however, the ability to dimerize profoundly affected the function of disease-associated Raf proteins with intermediate or impaired kinase activity, in that the E > K mutation caused increased transformation, whereas the R > H mutation inhibited transformation. Similar results were also reported by Roring et al.20 in studies utilizing the R > H mutation to examine MEK and ERK activation induced by mutant B-Raf proteins.
Recent studies from other laboratories have also found that Raf dimerization contributes to the function of intermediate and kinase-impaired Raf mutants. In work by Wu et al., increased heterodimerization with B-Raf was found to be the common pathogenic mechanism for Rasopathy/Noonan Syndrome (NS) C-Raf mutants.21 More specifically, Wu and coworkers found that NS C-Raf mutants with impaired (D486N) or intermediate (L613V) kinase activities displayed increased heterodimerization with B-Raf. With the use of the R > H mutation as well as Raf depletion experiments, they went on to show that the upregulation in MEK/ERK signaling induced by these NS C-Raf mutants was dependent upon the interaction with B-Raf. In a second study, L597V-B-Raf, an intermediate activity B-Raf mutant detected in both cancer and Rasopathies, was found to dimerize with C-Raf and to promote C-Raf kinase activation.22 Moreover, Raf inhibitor treatment further upregulated MEK/ERK signaling in cells expressing this mutant and the upregulation correlated with increased heterodimerization between L597V-B-Raf and C-Raf.
Interestingly, B-Raf mutants exhibiting enhanced self-homodimerization potential have also been observed in human cancer. In low-grade pediatric astrocytomas, several oncogenic KIAA1549-BRAF gene fusions have been identified that generate proteins consisting of the N-terminal region of KIAA1549 fused to the B-Raf kinase domain.23 These fusion proteins constitutively homodimerize with themselves to upregulate MEK/ERK signaling and are subject to the paradoxical activating effect of Raf inhibitor treatment. Splice variants of V600E-B-Raf that contain deletions removing the Ras binding domain also constitutively self-homodimerize.24 These splice variants were identified in melanoma patients and cell lines that had developed acquired resistance to the Raf inhibitor vemurafenib, and self-dimerization of these V600E variants was found to be required for vemurafenib resistance as the R > H mutation in their dimer interface restored drug sensitivity. Of note, several other mechanisms of Raf inhibitor resistance in melanomas expressing V600E-B-Raf also have the consequence of promoting the dimerization of wild-type Raf proteins in drug-treated cells.25
Further work from the laboratories of Drs. Therrien and Sicheri has recently provided key insight regarding how the ATP-competitive Raf inhibitors promote Raf dimerization.26 Protein kinase domains consist of N- and C-lobes, and due to their dynamic nature, there is a significant degree of flexibility between the N- and C-lobes as well as within the N-lobe itself. The side-to-side dimer interface spans both the N- and C-lobes, and by binding the ATP pocket, Lavoie and coworkers26 found that the inhibitors stabilize the closed conformation of the kinase domain, thus generating a static dimer interface surface that is conducive for dimerization. These investigators went on to engineer Raf dimerization biosensors to examine dimerization induced by a panel of ATP-competitive Raf inhibitors. They found that all of the Raf inhibitors tested strongly induced B-Raf/C-Raf heterodimerization; however, the levels of homodimerization or binding to A-Raf or the closely related KSR proteins varied with the inhibitor used. In co-immunoprecipitation assays, our laboratory has observed similar results in that 2 Raf inhibitors (PLX4720 and SB590885) strongly induced B-Raf/C-Raf heterodimerization and to a lesser extent promoted B-Raf and C-Raf homodimerization (Fig. 1C). Using the biosensor assay to screen a drug library, Lavoie et al.26 further found that certain ATP-competitive inhibitors for BCR-ABL, p38, and VEGFR also promote Raf dimerization. In addition, these inhibitors induced the paradoxical activation of ERK, indicating the clinical importance of screening kinase-targeted drugs for their potential to promote Raf dimerization.
Blocking Raf Dimerization Has Therapeutic Potential
The paradoxical activating effect of the ATP-competitive inhibitors has demonstrated the need to find alternative ways for inhibiting Raf signaling. Because our studies showed the importance of dimerization in Ras-dependent Raf activation as well as signaling from all but the high catalytic activity Raf mutants, our laboratory set out to determine whether a peptide mimetic of the Raf dimer interface could disrupt Raf dimerization. From these studies, we identified an 18 amino acid peptide, denoted as DI1, that was able to bind wild-type C-Raf and B-Raf and prevent growth factor-induced Raf dimerization and Raf-mediated MEK activation.19 In addition, the DI1 peptide was able to bind disease-associated Raf proteins as well as the closely related KSR1 protein (Fig. 2). Importantly, when administered to non-small cell lung carcinoma lines, the DI1 peptide specifically blocked Raf signaling and increased cell death in lines expressing either activated K-Ras or impaired activity B-Raf proteins, both of which require Raf dimerization for upregulated ERK signaling. Thus, this work provided the first proof-of-principle that blocking Raf dimerization is a valid therapeutic approach to inhibit Raf signaling.
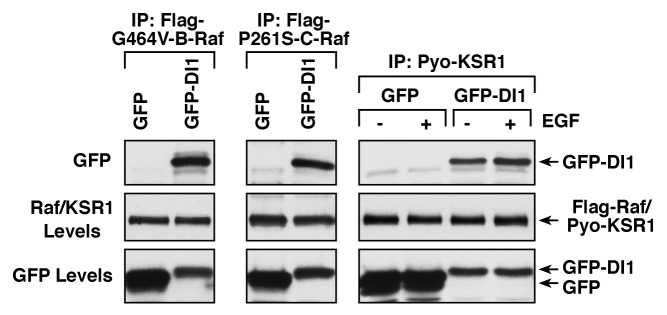
Figure 2. Binding of Dimer Interface DI1 peptide to KSR1 and disease-associated Raf mutants. Cos cells stably expressing Pyo-KSR1 or the indicated Flag-tagged B-Raf or C-Raf mutant proteins via lentivirus infection were transfected to express either GFP or GFP-DI1. Pyo-KSR1, Flag-G464V-B-Raf, or Flag-P261S-C-Raf proteins were immunoprecipitated and probed for the presence of GFP-DI1.
Conclusions
Without question Raf dimerization plays an important role in Raf biology, being required for Ras-dependent Raf activation and for the pathological function of various disease-associated Raf mutants (Fig. 3). Moreover, Raf dimerization can alter the effectiveness of certain drug therapies, and its impact must be taken into consideration as more kinase-targeted inhibitors enter the clinic for cancer therapy. Given the importance of Ras/Raf/MEK/ERK signaling in human disease, the continued development of Raf inhibitors that do not promote Raf dimerization as well as the search for drugs that actively disrupt dimerization will be the challenge of the future.

Figure 3. Raf dimerization in cell signaling. (A) In normal Ras-dependent growth factor signaling, Raf dimerization is required for Raf kinase activation and signaling to MEK (left). (B) In disease-associated states, Raf dimerization is required for upregulated MEK/ERK signaling induced by (1) disease-associated mutant Raf proteins with intermediate and impaired kinase activity, (2) mutationally-activated receptor tyrosine kinases (RTK) and RasGTPases, (3) self-homodimerizing V600E-B-Raf splice variants that mediate Raf inhibitor resistance, and (4) treatment with ATP-competitive Raf inhibitors in the presence of activated Ras. Yellow stars indicate oncogenic mutations and white star indicates a Rasopathy mutation.
Acknowledgments
This work was funded by federal funds from the National Cancer Institute.
Glossary
Abbreviations:
- ERK
extracellular signal-regulated kinase
- MEK
MAP/ERK kinase
- ATP
adenosine triphosphate
- GTP
guanosine triphosphate
- KSR
kinase suppressor of Ras
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/smallgtpases/article/26117
References
- 1.Hagemann C, Rapp UR. Isotype-specific functions of Raf kinases. Exp Cell Res. 1999;253:34–46. doi: 10.1006/excr.1999.4689. [DOI] [PubMed] [Google Scholar]
- 2.Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875–85. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- 3.Dougherty MK, Müller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, Conrads TP, Veenstra TD, Lu KP, Morrison DK. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell. 2005;17:215–24. doi: 10.1016/j.molcel.2004.11.055. [DOI] [PubMed] [Google Scholar]
- 4.Ritt DA, Monson DM, Specht SI, Morrison DK. Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Mol Cell Biol. 2010;30:806–19. doi: 10.1128/MCB.00569-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luo Z, Tzivion G, Belshaw PJ, Vavvas D, Marshall M, Avruch J. Oligomerization activates c-Raf-1 through a Ras-dependent mechanism. Nature. 1996;383:181–5. doi: 10.1038/383181a0. [DOI] [PubMed] [Google Scholar]
- 6.Farrar MA, Alberol-Ila J, Perlmutter RM. Activation of the Raf-1 kinase cascade by coumermycin-induced dimerization. Nature. 1996;383:178–81. doi: 10.1038/383178a0. [DOI] [PubMed] [Google Scholar]
- 7.Weber CK, Slupsky JR, Kalmes HA, Rapp UR. Active Ras induces heterodimerization of cRaf and BRaf. Cancer Res. 2001;61:3595–8. [PubMed] [Google Scholar]
- 8.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, et al. Cancer Genome Project Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–67. doi: 10.1016/S0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 9.Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009;19:230–6. doi: 10.1016/j.gde.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Röring M, Brummer T. Aberrant B-Raf signaling in human cancer -- 10 years from bench to bedside. Crit Rev Oncog. 2012;17:97–121. doi: 10.1615/CritRevOncog.v17.i1.70. [DOI] [PubMed] [Google Scholar]
- 11.Garnett MJ, Rana S, Paterson H, Barford D, Marais R. Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol Cell. 2005;20:963–9. doi: 10.1016/j.molcel.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 12.Rushworth LK, Hindley AD, O’Neill E, Kolch W. Regulation and role of Raf-1/B-Raf heterodimerization. Mol Cell Biol. 2006;26:2262–72. doi: 10.1128/MCB.26.6.2262-2272.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rajakulendran T, Sahmi M, Lefrançois M, Sicheri F, Therrien M. A dimerization-dependent mechanism drives RAF catalytic activation. Nature. 2009;461:542–5. doi: 10.1038/nature08314. [DOI] [PubMed] [Google Scholar]
- 14.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor SL, Vigers G, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–5. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 15.Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–21. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–30. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 18.Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, Reis-Filho JS, Kong X, Koya RC, Flaherty KT, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207–15. doi: 10.1056/NEJMoa1105358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freeman AK, Ritt DA, Morrison DK. Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol Cell. 2013;49:751–8. doi: 10.1016/j.molcel.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Röring M, Herr R, Fiala GJ, Heilmann K, Braun S, Eisenhardt AE, Halbach S, Capper D, von Deimling A, Schamel WW, et al. Distinct requirement for an intact dimer interface in wild-type, V600E and kinase-dead B-Raf signalling. EMBO J. 2012;31:2629–47. doi: 10.1038/emboj.2012.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu X, Yin J, Simpson J, Kim KH, Gu S, Hong JH, Bayliss P, Backx PH, Neel BG, Araki T. Increased BRAF heterodimerization is the common pathogenic mechanism for noonan syndrome-associated RAF1 mutants. Mol Cell Biol. 2012;32:3872–90. doi: 10.1128/MCB.00751-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andreadi C, Cheung LK, Giblett S, Patel B, Jin H, Mercer K, Kamata T, Lee P, Williams A, McMahon M, et al. The intermediate-activity (L597V)BRAF mutant acts as an epistatic modifier of oncogenic RAS by enhancing signaling through the RAF/MEK/ERK pathway. Genes Dev. 2012;26:1945–58. doi: 10.1101/gad.193458.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sievert AJ, Lang SS, Boucher KL, Madsen PJ, Slaunwhite E, Choudhari N, Kellet M, Storm PB, Resnick AC. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci U S A. 2013;110:5957–62. doi: 10.1073/pnas.1219232110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–90. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Solit DB, Rosen N. Resistance to BRAF inhibition in melanomas. N Engl J Med. 2011;364:772–4. doi: 10.1056/NEJMcibr1013704. [DOI] [PubMed] [Google Scholar]
- 26.Lavoie H, Thevakumaran N, Gavory G, Li JJ, Padeganeh A, Guiral S, Duchaine J, Mao DY, Bouvier M, Sicheri F, et al. Inhibitors that stabilize a closed RAF kinase domain conformation induce dimerization. Nat Chem Biol. 2013;9:428–36. doi: 10.1038/nchembio.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]