

Abstract
Environmental or occupational exposure to arsenic, a chemical element classified as metalloid, has been associated with cancer of the lung, skin, bladder, liver, etc.. Mdig (mineral dust-induced gene) is a newly identified oncogene linked to occupational lung diseases and lung cancer. It is unclear whether mdig is also involved in arsenic-induced malignant transformation of the lung cells. By using human bronchial epithelial cells and human lung cancer cell lines, we showed that arsenic was able to induce expression of mdig. We further demonstrated that this mdig induction by arsenic was partially dependent on the JNK and STAT3 signaling pathways. Disruption of the JNK or STAT3 by either chemical inhibitors or siRNAs diminished arsenic-induced accumulation of mdig mRNA and protein. Furthermore, we also showed that microRNA-21 (miR-21) and Akt were down-stream effectors of the JNK and STAT3 signaling pathways in arsenic-induced mdig expression. Transfection of the cells with anti-miR-21 or pre-treatment of the cells with Akt inhibitor blunted mdig induction by arsenic. Clinically, the levels of mdig can be applied to predict the disease progression, the first progression (FP), in non-small cell lung cancer (NSCLC) patients. Taken together, our data suggest that mdig may play important roles on the pathogenesis of arsenic-induced lung cancer and that JNK and STAT3 signaling pathways are essential in mediating arsenic-induced mdig expression.
1. Introduction
Arsenic or arsenic-containing compounds are well-established human carcinogens [1]. Epidemiological studies indicated that arsenic exposure through either ingestion of the contaminated drinking water or inhalation in some occupational settings increases the incidence rate of the non-small cell lung cancers (NSCLC), mostly the squamous cell lung cancer and adenocarcinoma [2; 3; 4]. It is believed that the inorganic form of arsenic, especially, the inorganic trivalent arsenic (As3+) is more carcinogenic and reactive with thiol-contained molecules. How As3+ causes cancer development remains to be fully elucidated. Accumulating evidence suggests that activation of several signaling pathways by As3+, such as MAPK [5], Akt [6], AP-1 [7], p53 [8], NF-κB [9], etc., is essential for As3+-induced carcinogenesis. Recently, we have revealed that As3+ is able to activate the JNK-STAT3-Akt signaling axis that leads to serine 21 phosphorylation of the EZH2, the enzymatic subunit of the polycomb repressive complex 2 (PRC2) that represses expression of the tumor suppressors and DNA repair proteins through catalyzing tri-methylation of the lysine 27 of histone H3 [10; 11].
Mineral dust-induced gene (mdig), also named myc-induced nuclear antigen 53 (mina53) and nucleolar protein 52 (NO52), was first identified from alveolar macrophages (AM) of coal miners [12; 13; 14]. In human bronchial epithelial cells, the expression of mdig can be induced by some occupational or environmental hazards, including silica particles and tobacco smoke. The human mdig gene is located on chromosome 3q11.2, which encodes a 465-amino acid protein with a calculated molecular mass of 52.7 kD. The mdig protein contains a conserved JmjC domain, the signature domain of the majority of the histone demethylases. Several reports suggest that in addition to lung cancer, mdig is also overexpression in a number of other human cancers, including breast cancer, colon cancer, esophageal squamous cell carcinoma, gastric cancer, hepatocellular carcinoma, lymphoma, renal cell carcinoma, etc.. Thus, mdig has been viewed as a potential novel human oncogene [15].
MicroRNA-21 (miR-21) is the first identified oncogenic miRNA (oncomir) that targets several important tumor suppressors, leading to aberrant activation of the oncogenic protein kinases, such as Akt, through down-regulating PTEN, PDCD4 and Spry2, the negative regulators of Akt [10; 16]. Human miR-21 is an intronic miRNA of the host gene TMEM49. Under the circumstance of malignant transformation or in cancer cells, the expression of miR-21 is regulated by STAT3 along with several other transcription factors, such as NF-κB, C/EBP-α and SRF. We had recently shown that As3+ is capable of inducing expression of miR-21 through a signaling cascade of JNK and STAT3 in bronchial epithelial cells [10]. In the present study, we further demonstrated that the JNK-STAT3-miR-21 signaling cascade is involved in the expression of mdig induced by As3+. Furthermore, we also demonstrated that increased expression of mdig is a prognostic factor for disease progression of the patients with NSCLC. These findings, thus, may provide a new insight into the mechanism of carcinogenesis resulted from environmental or occupational exposure to As3+.
2. Materials and Methods
2.1. Cell culture
The human bronchial epithelial cell line BEAS-2B and lung carcinoma cell line A549 were purchased from the American Type Culture Collection (ATCC, Manassas, VA). BEAS-2B cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 5% fetal bovine serum (Invitrogen), 1% penicillin-streptomycin and 1% L-Glutamine (Sigma). A549 cells were cultured in RPMI-1640 medium (HyClone) with 10% fetal bovine serum and 1% penicillin-streptomycin and 1% L-Glutamine at 37 C, in presence of 5% CO2.
2.2. Western blotting
Cells were lysed by sonication in 1 × RIPA buffer (Millipore). Total cellular proteins were diluted with 4 × NuPage LDS sample buffer (Invitrogen) and 50 mM DTT, then run on 10% SDS-PAGE gel and transferred onto PVDF membranes (Invitrogen). Membranes were probed with the primary antibody at a dilution of 1:1000 or 1:2000 (according to the signal intensity) overnight at 4°C. The second antibody with AP or HRP tag was applied at the dilution of 1:2000 (AP) or 1:5000 (HRP). CDP-Star Reagent (New England Biolabs, Ipswich, MA) was used for image development. The primary antibodies, including phospho-SAPK/JNK (Thr183/Tyr185), SAPK/JNK, phospho-Akt (Ser473), Akt, phospho-STAT3 (Ser727), phospho-STAT3 (Tyr705), STAT3, PTEN, PDCD4, GAPDH and β-actin, were purchased from Cell Signaling Technology, Inc. (CST, MA). The anti-mdig (mina53) primary antibody was purchased from Abcam, Inc.. Anti-spry2 was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
2.3. RT-PCR
Total RNA was prepared by lysing the cells with TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. The reverse transcription and PCR were performed using Access Quick RT-PCR System (Promega) with 1 μg of total RNA and 0.3 μM sense and anti-sense primer. The mdig primer sequences are: left primer: 5′-TCATGTCGGGCCTAAGAGAC-3′; right primer: 5′-GGCATTTGATTCTGCAAAGG-3′, which amplify a 1509 bp cDNA fragment covering the entire coding region of the mdig mRNA. The GAPDH primer sequences are: sense: 5′-CTGAACGGGAAGCTCACTGGCATGGCCT-3′; antisense: 5′-CATGAGGTCCACCACCCTGTTGCTGTAG-3′.
2.4. Real-Time PCR
Total RNA was extracted by TRIzol method and followed by reverse transcription using High-capacity cDNA Reverse Transcription Kits (Applied Biosystems). Each sample contains 0.2 μl dNTPs (100 mM, with dTTP), 1μl reverse transcriptase, 1.5 μl 10 × reverse transcription buffer, 0.2 μl RNase inhibitor (20U/ul), 1 μg total RNA, 5 × RT miR-21 primer (3 μl) and comparative volume of Nuclease-free water to the total volume of 15 μl. The miR-21 was quantified by real time PCR using TaqMan Small RNA Assays (Applied Biosystems) and 18S rRNA that was used as an endogenous control.
2.5. JNK, STAT3 or Akt Inhibitor
Cells were seeded into 6-well tissue culture plates at a density of 2 × 105/ml. When the cells reached to 80–90% confluence around 48 h of culture, cells were treated by JNK inhibitor SP600125 (Calbiochem) or STAT3 inhibitor V (InV) (Stattic, Santa Cruz) or Akt inhibitor Wortmannin (Calbiochem) to the final concentration of 20 μM for 1 or 4 h, then incubated with the indicated concentrations of arsenic chloride (As3+, Sigma-Aldrich, MO) for an additional 0.5 to 16 h. After washing once with PBS solution, cells were collected for Western blotting or PCR experiments.
2.6. siRNA transfection
Cells with a concentration of 1×105/ml were seeded into 6-well plates and allowed to grow to 70%–80% confluence. Then siRNA was forward transfected into cells using Lipofectamine RNAiMAX reagent (Invitrogen). The medium was changed by normal culture medium to get higher cell survival rate 4 to 6 h later. Twenty-four hours after transfection, As3+ was added and cultured for the indicated times. SiRNA that specifically silences JNK1 was purchased from Eurofins (Huntsville, AL); control siRNA and siRNAs specifically silencing the JNK2 and STAT3 were purchased from Santa Cruz Technology (Santa Cruz, CA).
2.7. MicroRNA inhibitor
The BEAS-2B cells were first seeded in a density of 2 × 105 /ml into 6-well plates at 37°C with 5% CO2 for 24 h, then transfected with 60 nM anti-miR-21 (Ambion) with Lipofectamine RNAiMAX reagent (Invitrogen). Opti-MEM I Reduced Serum Medium (Invitrogen) was used to dilute nucleic acid and liposome as suggested by the manufacturer. Medium was changed to normal culture medium 24 h after transfection, and then the cells were treated with or without As3+ for the indicated times. Protein levels were determined by Western blotting as described above.
2.8. Statistics
SigmaPlot statistics software was used for statistical analyses. Data were expressed as means ± standard deviation (SD) of at least three repeats. The survival analyses of the lung cancer patients were performed using the Kaplan-Meier (K-M) Plotter database [17]. K-M curves were used to compare the First Progression (FP) or Overall Survival (OS) probabilities of the NSCLC patients stratified by the levels of mdig expression. The statistical significance of the separation between the K-M curves was evaluated using a log-rank test.
3. Results
3.1. As3+ induces mdig expression in vitro
Several population-based case-control studies have shown a strong association of As3+ exposure with NSCLC and other cancers [18; 19]. Since we and others had observed an increased expression of mdig in human lung cancer [12; 15; 20], we explored the possible association of As3+ exposure with mdig expression in human lung cancer cell line, A549 cells, and human bronchial epithelial cell line, BEAS-2B cells. Based on the consideration that human cells have multiple transcripts of mdig resulted from alternative splicing of the mdig pre-mRNA, which renders difficulty in quantifying the expression levels of mdig by real-time RT-PCR that amplifies small fragments of mdig cDNA, we employed a traditional RT-PCR procedure using PCR primers that amplifies the whole open reading frame of mdig. As shown in Figs. 1A and 1B, As3+ is able to induce accumulation of mdig mRNA and protein in manners of both dose- and time-dependent in A549 cells. Both mRNA and protein of mdig could be induced by a treatment of the cells with 20 μM As3+ for 4 h, and this induction was maximized in the cells treated with 40 μM As3+ for 4 h. In time course studies, cells were treated with 40 μM As3+ over a 6 h time course for mRNA and 16 h time course for protein, respectively. Induction of mdig mRNA was detected at 2 h treatment compared with control treatment (0 h), which reached peak between 4 h and 6 h (Fig. 1A). Increased mdig protein was observed at 2 to 6 h of As3+ treatment and then returned to the control level at later time point, 16 h (Fig. 1B). A similar dose- and time-dependent induction of mdig mRNA and protein induced by As3+ could be observed in BEAS-2B cells (Figs. 1C and 1D).
Fig. 1.

As3+ induces mdig expression in A549 cells and BEAS-2B cells. A. RT-PCR analyses showed dose- and time-dependent induction of mdig mRNA by As3+. B. Dose- and time-dependent induction of mdig protein by As3+ as revealed by Westernblotting assay. C. As3+ induced mdig mRNA in BEAS-2B cells as revealed by RT-PCR. D. Westernblotting shows dose- and time-dependent induction of mdig protein by As3+ in BEAS-2B cells.
3.2. JNK and STAT3 are involved in As3+-induced mdig expression
Our previous studies had revealed that As3+ is a potent activator for the JNK and STAT3 signaling pathways [10; 21]. To delineate the potential contribution of JNK and STAT3 to the As3+-induced mdig expression, we pre-treated the cells with JNK inhibitor SP600125 (SP) and STAT3 inhibitor V (Stattic, InV), respectively. As depicted in Fig. 2, both levels of basal and As3+-induced mdig mRNA were diminished after the cells treated with JNK inhibitor or STAT3 inhibitor, suggesting that JNK and STAT3 are involved in As3+-induced mdig expression.
Fig. 2.
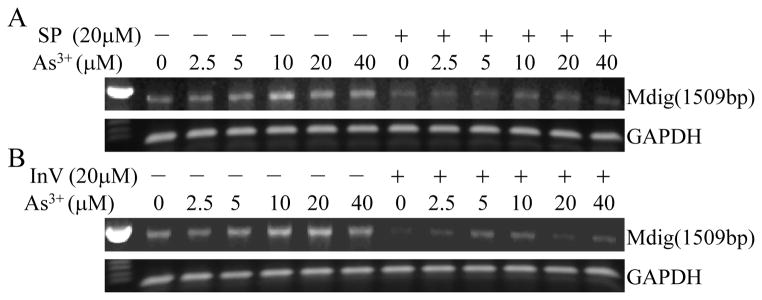
JNK and STAT3 inhibitors decrease As3+-induced mdig mRNA expression. A549 cells were cultured in the presence or absence of 20 μM JNK inhibitor SP600125 (SP, A) or STAT3 inhibitor V (InV, B) for 4 h followed by the indicated concentrations of As3+ for an additional 2 h.
3.3. JNK-STAT3-Akt signaling axis contributes to As3+-induced mdig expression
Earlier studies had confirmed that As3+ can activate the serine/threonine kinase Akt that plays a pivotal role in cell transformation and angiogenesis [22]. In our recent studies, we also demonstrated that in response to As3+ treatment, a signaling cascade from JNK, STAT3 to Akt was activated in the cells [10; 11]. In this signaling axis, JNK appears to be the upstream kinase for the serine 727 phosphorylation of STAT3 that in turn enhance Akt activation through miR-21 that downregulates endogenous Akt inhibitors, including PTEN, PDCD4 and Spry2. To determine whether this signaling axis is involved in As3+-induced mdig expression, the cells were pretreated with JNK inhibitor, SP600125, STAT3 inhibitor, InV or PI3K/Akt inhibitor, Wortmannin (Wort), followed by As3+ treatment for an addition 2 h. In agreement with our previous findings, inhibitors of JNK and STAT3 blunted As3+-induced Akt activation along with mdig expression substantially (Fig. 3A). Inhibition of Akt by Wortmannin reduced the level of mdig protein notably. However, Wortmannin showed very marginal effect on JNK and STAT3, suggesting that Akt is the downstream kinase of JNK and STAT3 in mediating As3+-induced mdig expression.
Fig. 3.

JNK, STAT3 and Akt contribute to As3+-induced mdig expression. A. BEAS-2B cells were pre-treated with 20 μM JNK inhibitor, SP600125, STAT3 inhibitor V (InV) or PI3K/Akt inhibitor, Wortmannin (Wort) for 1 h, followed by culturing the cells in the presence of 20 μM As3+ for an additional 2 h. The levels of mdig protein, activation of STAT3, JNK and Akt were determined by Westernblotting. B. Inhibition of STAT3 by STAT3 inhibitor V (InV) diminished dose-dependent activation of JNK2 and Akt, and reduced mdig expression induced by As3+ in the BEAS-2B cells.
The importance of STAT3 in As3+-induced mdig mRNA expression was revealed by treating the cells with STAT3 inhibitor V followed by different concentrations of As3+ treatment for an additional 2 h (Fig. 2B). As shown in Fig. 3B, we also noted that such a treatment reduced mdig protein in the cells treated with every dose of As3+ significantly (Fig. 3B). Intriguingly, we noted that STAT3 inhibitor V had a pronounced inhibitory effect on the activated JNK2 (pJNK2) but not pJNK1 (panels of JNKs in Fig. 3B), an unexpected phenomenon that remains to be further verified.
3.4. Silencing STAT3 or JNK by siRNA suppresses mdig induction by As3+
We used chemical inhibitors in the above experiments and indicated an involvement of STAT3 and JNK in As3+-induced mdig expression. To rule out the possible nonspecific effect of the chemical inhibitors on mRNA or protein expression, we further verified these data by siRNAs that specifically silence STAT3 and JNK, respectively. In agreement with our previous observation showing that As3+ induces serine 727 phosphorylation (pS727) but inhibits tyrosine 705 phosphorylation (pY705) of the STAT3 [10; 21], pS727 of STAT3 was detected in the As3+-treated cells along with a significant inhibition of pY705 of STAT3 (Figs. 3 and 4A). Silencing STAT3 reduced the levels of pS727 and mdig protein (Fig. 4A), suggesting pS727, but not pY705 of the STAT3, is important for As3+-induced mdig expression.
Fig. 4.
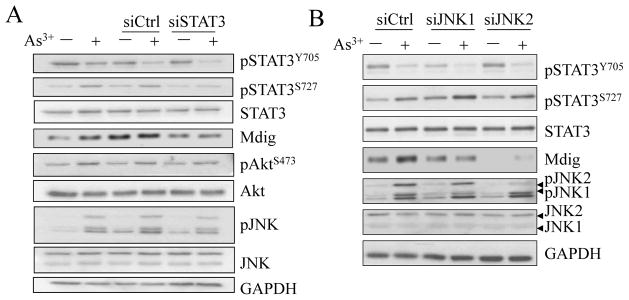
Silencing STAT3 and JNK repressed mdig expression. A. BEAS-2B cells were transfected with 50 nM control siRNA (siCtrl), or STAT3 specific siRNA (siSTAT3) and cultured for 24 h. Cells were cultured in the presence of 20 μM As3+ for an additional 2 h. The levels of STAT3 activation, mdig protein, JNK and Akt activation were determined by Westernblotting. B. Silencing JNK2, but not JNK1, reduced the levels of basal and As3+-induced mdig. Cell transfection with siRNA and treatment are the same as in A.
To additionally validate the role of JNK in As3+-induced mdig expression in the BEAS-2B cells, siRNA specifically targeting JNK1 or JNK2 was transfected into the cells. As depicted in Fig. 4B, a considerable repression of As3+-induced mdig expression was observed in the cells transfected with siRNA that silences the JNK2. This result indicates that in the case of As3+-induced mdig expression, JNK2 may be more important than the JNK1.
3.5. As3+-induced mdig requires miR-21
As an oncogenic microRNA, miR-21 has been linked to a variety of malignancies. Our earlier study had shown that As3+ is capable of inducing miR-21 [10]. In the present study, we noted that As3+ can induce miR-21 in a dose dependent manner (Fig. 5A). miR-21 was about 2-fold higher in the cells treated with 40 μM As3+ than the cells without As3+ treatment. Furthermore, this induction of miR-21 by As3+ is dependent on both JNK and STAT3, as the fact that inhibitors for JNK or STAT3 prevented both basal and As3+-induced miR-21 significantly (Fig. 5B). To determine whether miR-21 is required for As3+-induced mdig expression, the cells were transfected with 100 nM anti-miR-21 that can bind to and inhibit the activity of endogenous mature miR-21. As shown in Fig. 5C, although this anti-miR-21 itself has some activities in inducing mdig mRNA, possibly due to stress response, inhibiting miR-21 by anti-miR-21 decreased the level of mdig mRNA in the cells treated with As3+ (Fig. 5C, the last lane).
Fig. 5.

As3+-induced mdig requires miR-21. A. Dose-dependent induction of miR-21 by As3+. Relative levels (fold changes) of miR-21 were determined by quantitative real-time PCR. B. As3+ induces miR-21 through JNK and STAT3 activation. BEAS-2B cells were pretreated with inhibitors for JNK or STAT3 for 2 h and then treated with 20 μM As3+ for an additional 2 h. The levels of miR-21 were determined as in A. C. Repressing miR-21 by anti-miR-21 prevented mdig mRNA induction by As3+. BEAS-2B cells were pre-transfected with 100 nM anti-miR-21 for 24 h and then treated with As3+ for an additional 2 h. The relative levels of mdig mRNA and GAPDH mRNA were determined by regular RT-PCR.
3.6. Mdig is a new prognostic factor for human NSCLC
Given the fact that As3+ is a human carcinogen that induces the expression of mdig that is involved in cell growth regulation as we and others reported previously [12; 13], we believe that mdig may be one of the mediators of As3+ carcinogenesity. In addition, the levels of mdig expression may somehow influence the malignancy of the tumors. To explore such a possibility, a Kaplan-Meier (K-M) database [17] was used to evaluate the time delay to the first progression (FP) in NSCLC patients with higher and lower mdig expression (mdighigh and mdiglow). The K-M curves showed that mdig expression was significant in predicting earlier occurrence of FP of the adenocarcinoma (Ad) (Fig. 6A, left panel, p = 0.0024). However, mdighigh appears to be unable to predict the earlier or later FP of the squamous cell carcinoma (Sq) (Fig. 6A, right panel, p = 0.067). To determine the possible gender differences of mdig in predicting FP of the Ad patients, we applied this assay into male and female patients who had Ad, respectively. As shown in Fig. 6B, higher expression of mdig shortened the time to FP significantly in male patients who had Ad (Fig. 6B, left panel, p = 0.00027). In female Ad patients, in contrast, higher mdig expression appears to be a favorable factor in prolonging the time to FP (Fig. 6B, right panel, p = 0.00071). Thus, male patients who had Ad and higher expression of mdig were more likely to have a short time to progression compared with female patients who had Ad and higher expression of mdig.
Fig. 6.

Kaplan-Meier probability analyses of the first progression (FP) for the patients with NSCLC and higher or lower mdig expression (mdighigh or mdiglow). (A) Higher mdig expression shortens the times to FP of the patients with Adenocarcinoma (Ad, left panel) but not the patients with squamous cell carcinoma (Sq, right panel). (B) Higher mdig expression predicts earlier FP of the male patients with Ad (left panel) but later FP of the female patients with Ad (right panel).
4. Discussion
In this report, we provide evidence showing that As3+ can induce mdig in lung epithelial or lung cancer cells and further establish a mechanistic link of As3+-induced mdig expression with the signaling axis from JNK and STAT3 activation to the miR-21-mediated Akt activation (Fig. 7). Interruption of the JNK, STAT3 or Akt signaling either by relatively specific chemical inhibitors or targeting-specific siRNAs prevented mdig induction by As3+. Furthermore, by evaluating the available clinical data, we showed that increased expression of mdig predicts fast disease progression of the adenocarcinoma (Ad), esp., for the male patients.
Fig. 7.

Schematic diagram showing As3+-induced JNK-STAT3-Akt signaling on the expression of mdig that contributes to initiation and progression of the non-small cell lung cancers (NSCLC), esp., for male adenocarcinoma.
Mdig was first identified as a mineral dust-induced gene from patients with occupational lung diseases and further studies suggested its association with a number of human cancers, including lung cancer [12], colon cancer [23], esophageal cell carcinoma [24], renal cell carcinoma [25], hepatocellular carcinoma [26], breast cancer [27], and lymphoma [28]. Other studies also indicated that oncogene c-Myc plays an important role in regulating the expression of mdig and named mdig as a myc-induced nuclear antigen 53 (mina53) [13]. By immunofluorescent staining, some studies indicated that mdig is a nucleolar protein [14; 15]. Our most recent studies showed that under the conditions of ectopic overexpression or As3+-induced stress, mdig can also be localized in the entire nuclear matrix or cytoplasm [29]. Since mdig protein contains a conserved JmjC domain, a molecular signature of many histone demethylases, it has been assumed that mdig may be also involved in protein demethylation. In human bronchial epithelial cells, mdig exhibited very marginal demethylation activity toward the global histone protein. However, mdig appears to be highly capable of reducing the level of lysine 9 tri-methylation on histone H3 in the regulatory region of H19 and Oct4 gene [30]. Functionally, in addition to previous observations showing cell proliferative role of the mdig through overexpression [12; 13], we noted that overexpression of mdig reduced, but not enhanced cell migration and invasion [29]. Additional studies had also revealed that mdig/mina53 could catalyze histidyl hydroxylation of the ribosomal protein Rpl27a and Rpl8 [31]. Genetic disruption of the mdig/mina53 gene reduced allergic response of the mice [32]. In mdig heterozygous knockout mice, a significantly decrease of the silica-induced lung fibrosis was observed (Thakur, et al., unpublished observations). Clinically, increased mdig expression in the cancer tissues predicts poorer survival of the patients with lung cancer [29; 30], breast cancer [27; 30] and ovarian cancer [30]. In the present study, we also showed that mdig may be used as an additional marker for predicting disease progression of the Ad, esp, for the male patients. In patients with higher mdig expression, FP is a life-threatening condition and is worth to identify effective strategies in disease management and treatment. In this context, mdig may be considered as a valuable factor for determining NSCLC patients who are at risk for earlier disease progression.
As3+ is a well-established human carcinogen. A number of population-based case-control studies indicated strong association of As3+ exposure with small cell lung cancer and NSCLC [2; 3; 4]. An important notion is that both high and low to moderate As3+ exposures in some regions of the world have been linked to an increased risk of human lung cancers [19]. Limited number of studies suggested histopathological variations of lung cancers resulted from different exposure routes of environmental As3+. It was reported that As3+ exposure through ingestion of the contaminated drinking water or food is more likely associated with the squamous cell carcinoma [2], whereas exposure through inhalation is leaned toward the adenocarcinoma [3; 4]. It remains to be determined, however, whether the differences in cancer histology or cell type are truly resulted from exposure routes or the differences in the accumulative As3+ exposure levels. Mechanistically, As3+ can activate a number of intracellular signaling pathways, such as JNK, STAT3, Akt, and NF-κB that linked to cell growth, cell cycle transition, malignant transformation, cancer cell invasion and metastasis [10; 33; 34; 35; 36]. Importantly, As3+ is a known inducer of the oxidative stress responses in the cells due to its capability in activating NADPH oxidase and other redox regulating pathways [37]. Treatment of the cells with antioxidant prevented As3+-induced activation of JNK [38], STAT3, Akt, and the subsequent phosphorylation of the EZH2 protein (Li et al., unpublished observations). Accordingly, it is very likely that oxidative stress is involved in the mdig expression induced by As3+. It has been known that As3+ is able to induce expression of oncogenes, oncogenic kinases, DNA damages, and a number of cell-to-cell interaction signals. Expression of mdig in response to As3+ can either be an independent entity in concert with other As3+-induced oncogenic signals or a crucial component in the signaling network that mediates As3+-induced carcinogenesis. Further studies are needed to delineate how mdig itself plays essential role in the malignant transformation of the cells and carcinogenesis associated with environmental As3+ exposure. The present study demonstrated that As3+ is able to induce mdig expression through the signaling axis from JNK, STAT3 to Akt activation, which may provide an additional mechanistic explanation for human cancers resulted from environmental or occupational exposure to As3+.
Acknowledgments
This work was supported by NIH grants R01 ES017217 and R01 ES020137 to FC. JS and MY are joint-training graduate students between China Medical University and the Wayne State University, and are supported by Wayne State University start-up fund and NIH grants to FC. HZ was supported by National Natural Science Foundation of China (Grant No.: 30770956) and Bureau of Science and Technology of Shenyang (Grant No.: F12-193-9-02).
Footnotes
Conflicts of Interest
No potential conflicts of interest were disclosed.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Basu A, Mahata J, Gupta S, Giri AK. Genetic toxicology of a paradoxical human carcinogen, arsenic: a review. Mutat Res. 2001;488:171–194. doi: 10.1016/s1383-5742(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 2.Guo HR, Wang NS, Hu H, Monson RR. Cell type specificity of lung cancer associated with arsenic ingestion. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2004;13:638–643. [PubMed] [Google Scholar]
- 3.Wicks MJ, Archer VE, Auerbach O, Kuschner M. Arsenic exposure in a copper smelter as related to histological type of lung cancer. American journal of industrial medicine. 1981;2:25–31. doi: 10.1002/ajim.4700020106. [DOI] [PubMed] [Google Scholar]
- 4.Pershagen G, Bergman F, Klominek J, Damber L, Wall S. Histological types of lung cancer among smelter workers exposed to arsenic. British journal of industrial medicine. 1987;44:454–458. doi: 10.1136/oem.44.7.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bode AM, Dong Z. The paradox of arsenic: molecular mechanisms of cell transformation and chemotherapeutic effects. Crit Rev Oncol Hematol. 2002;42:5–24. doi: 10.1016/s1040-8428(01)00215-3. [DOI] [PubMed] [Google Scholar]
- 6.Wang Z, Yang J, Fisher T, Xiao H, Jiang Y, Yang C. Akt activation is responsible for enhanced migratory and invasive behavior of arsenic-transformed human bronchial epithelial cells. Environ Health Perspect. 2012;120:92–97. doi: 10.1289/ehp.1104061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong Z. The molecular mechanisms of arsenic-induced cell transformation and apoptosis. Environ Health Perspect. 2002;110(Suppl 5):757–759. doi: 10.1289/ehp.02110s5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joe Y, Jeong JH, Yang S, Kang H, Motoyama N, Pandolfi PP, Chung JH, Kim MK. ATR, PML, and CHK2 play a role in arsenic trioxide-induced apoptosis. The Journal of biological chemistry. 2006;281:28764–28771. doi: 10.1074/jbc.M604392200. [DOI] [PubMed] [Google Scholar]
- 9.Roussel RR, Barchowsky A. Arsenic inhibits NF-kappaB-mediated gene transcription by blocking IkappaB kinase activity and IkappaBalpha phosphorylation and degradation. Arch Biochem Biophys. 2000;377:204–212. doi: 10.1006/abbi.2000.1770. [DOI] [PubMed] [Google Scholar]
- 10.Chen B, Liu J, Chang Q, Beezhold K, Lu Y, Chen F. JNK and STAT3 signaling pathways converge on Akt-mediated phosphorylation of EZH2 in bronchial epithelial cells induced by arsenic. Cell Cycle. 2013;12:112–121. doi: 10.4161/cc.23030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rojanasakul Y. Linking JNK-STAT3-Akt signaling axis to EZH2 phosphorylation: a novel pathway of carcinogenesis. Cell Cycle. 2013;12:202–203. doi: 10.4161/cc.23419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Lu Y, Yuan BZ, Castranova V, Shi X, Stauffer JL, Demers LM, Chen F. The Human mineral dust-induced gene, mdig, is a cell growth regulating gene associated with lung cancer. Oncogene. 2005;24:4873–4882. doi: 10.1038/sj.onc.1208668. [DOI] [PubMed] [Google Scholar]
- 13.Tsuneoka M, Koda Y, Soejima M, Teye K, Kimura H. A novel myc target gene, mina53, that is involved in cell proliferation. The Journal of biological chemistry. 2002;277:35450–35459. doi: 10.1074/jbc.M204458200. [DOI] [PubMed] [Google Scholar]
- 14.Eilbracht J, Kneissel S, Hofmann A, Schmidt-Zachmann MS. Protein NO52--a constitutive nucleolar component sharing high sequence homologies to protein NO66. European journal of cell biology. 2005;84:279–294. doi: 10.1016/j.ejcb.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 15.Lu Y, Chang Q, Zhang Y, Beezhold K, Rojanasakul Y, Zhao H, Castranova V, Shi X, Chen F. Lung cancer-associated JmjC domain protein mdig suppresses formation of tri-methyl lysine 9 of histone H3. Cell Cycle. 2009;8:2101–2109. doi: 10.4161/cc.8.13.8927. [DOI] [PubMed] [Google Scholar]
- 16.Ling M, Li Y, Xu Y, Pang Y, Shen L, Jiang R, Zhao Y, Yang X, Zhang J, Zhou J, Wang X, Liu Q. Regulation of miRNA-21 by reactive oxygen species-activated ERK/NF-kappaB in arsenite-induced cell transformation. Free Radic Biol Med. 2012;52:1508–1518. doi: 10.1016/j.freeradbiomed.2012.02.020. [DOI] [PubMed] [Google Scholar]
- 17.Mihaly Z, Kormos M, Lanczky A, Dank M, Budczies J, Szasz MA, Gyorffy B. A meta-analysis of gene expression-based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast cancer research and treatment. 2013;140:219–232. doi: 10.1007/s10549-013-2622-y. [DOI] [PubMed] [Google Scholar]
- 18.Celik I, Gallicchio L, Boyd K, Lam TK, Matanoski G, Tao X, Shiels M, Hammond E, Chen L, Robinson KA, Caulfield LE, Herman JG, Guallar E, Alberg AJ. Arsenic in drinking water and lung cancer: a systematic review. Environ Res. 2008;108:48–55. doi: 10.1016/j.envres.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 19.Heck JE, Andrew AS, Onega T, Rigas JR, Jackson BP, Karagas MR, Duell EJ. Lung cancer in a U.S. population with low to moderate arsenic exposure. Environ Health Perspect. 2009;117:1718–1723. doi: 10.1289/ehp.0900566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Komiya K, Sueoka-Aragane N, Sato A, Hisatomi T, Sakuragi T, Mitsuoka M, Sato T, Hayashi S, Izumi H, Tsuneoka M, Sueoka E. Mina53, a novel c-Myc target gene, is frequently expressed in lung cancers and exerts oncogenic property in NIH/3T3 cells. J Cancer Res Clin Oncol. 2010;136:465–473. doi: 10.1007/s00432-009-0679-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu J, Chen B, Lu Y, Guan Y, Chen F. JNK-dependent Stat3 phosphorylation contributes to Akt activation in response to arsenic exposure. Toxicol Sci. 2012;129:363–371. doi: 10.1093/toxsci/kfs199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu LZ, Jiang Y, Carpenter RL, Jing Y, Peiper SC, Jiang BH. Role and mechanism of arsenic in regulating angiogenesis. PLoS One. 2011;6:e20858. doi: 10.1371/journal.pone.0020858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Teye K, Tsuneoka M, Arima N, Koda Y, Nakamura Y, Ueta Y, Shirouzu K, Kimura H. Increased expression of a Myc target gene Mina53 in human colon cancer. The American journal of pathology. 2004;164:205–216. doi: 10.1016/S0002-9440(10)63111-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsuneoka M, Fujita H, Arima N, Teye K, Okamura T, Inutsuka H, Koda Y, Shirouzu K, Kimura H. Mina53 as a potential prognostic factor for esophageal squamous cell carcinoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2004;10:7347–7356. doi: 10.1158/1078-0432.CCR-03-0543. [DOI] [PubMed] [Google Scholar]
- 25.Ishizaki H, Yano H, Tsuneoka M, Ogasawara S, Akiba J, Nishida N, Kojiro S, Fukahori S, Moriya F, Matsuoka K, Kojiro M. Overexpression of the myc target gene Mina53 in advanced renal cell carcinoma. Pathology international. 2007;57:672–680. doi: 10.1111/j.1440-1827.2007.02156.x. [DOI] [PubMed] [Google Scholar]
- 26.Ogasawara S, Komuta M, Nakashima O, Akiba J, Tsuneoka M, Yano H. Accelerated expression of a Myc target gene Mina53 in aggressive hepatocellular carcinoma. Hepatology research: the official journal of the Japan Society of Hepatology. 2010;40:330–336. doi: 10.1111/j.1872-034X.2009.00604.x. [DOI] [PubMed] [Google Scholar]
- 27.Thakur C, Lu Y, Sun J, Yu M, Chen B, Chen F. Increased expression of mdig predicts poorer survival of the breast cancer patients. Gene. 2013 doi: 10.1016/j.gene.2013.11.031. http://dx.doi.org/10.1016/j.gene.2013.1011.1031. [DOI] [PMC free article] [PubMed]
- 28.Teye K, Arima N, Nakamura Y, Sakamoto K, Sueoka E, Kimura H, Tsuneoka M. Expression of Myc target gene mina53 in subtypes of human lymphoma. Oncology reports. 2007;18:841–848. [PubMed] [Google Scholar]
- 29.Yu M, Sun J, Chen B, Lu Y, Zhao Z, Chen F. Paradoxical roles of mdig on cell proliferation and migration/invasion. PloS One. 2013 doi: 10.1371/journal.pone.0087998. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen B, Yu M, Chang Q, Lu Y, Thakur C, Ma D, Yi Z, Chen F. Mdig de-represses H19 large intergenic non-coding RNA (lincRNA) by down-regulating H3K9me3 and heterochromatin. Oncotarget. 2013;4 doi: 10.18632/oncotarget.1155. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ge W, Wolf A, Feng T, Ho CH, Sekirnik R, Zayer A, Granatino N, Cockman ME, Loenarz C, Loik ND, Hardy AP, Claridge TD, Hamed RB, Chowdhury R, Gong L, Robinson CV, Trudgian DC, Jiang M, Mackeen MM, McCullagh JS, Gordiyenko Y, Thalhammer A, Yamamoto A, Yang M, Liu-Yi P, Zhang Z, Schmidt-Zachmann M, Kessler BM, Ratcliffe PJ, Preston GM, Coleman ML, Schofield CJ. Oxygenase-catalyzed ribosome hydroxylation occurs in prokaryotes and humans. Nature chemical biology. 2012;8:960–962. doi: 10.1038/nchembio.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mori T, Okamoto K, Tanaka Y, Teye K, Umata T, Ohneda K, Tokuyama K, Okabe M, Tsuneoka M. Ablation of Mina53 in mice reduces allergic response in the airways. Cell Struct Funct. 2013 doi: 10.1247/csf.13006. [DOI] [PubMed] [Google Scholar]
- 33.Kennedy NJ, Davis RJ. Role of JNK in tumor development. Cell Cycle. 2003;2:199–201. [PubMed] [Google Scholar]
- 34.Lin EY. Linking inflammation to tumorigenesis in colon: Stat3, a double-edged sword? Cell Cycle. 2010;9:2485–2486. doi: 10.4161/cc.9.13.12271. [DOI] [PubMed] [Google Scholar]
- 35.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, Nicoletti F, Fagone P, Malaponte G, Mazzarino MC, Candido S, Libra M, Basecke J, Mijatovic S, Maksimovic-Ivanic D, Milella M, Tafuri A, Cocco L, Evangelisti C, Chiarini F, Martelli AM. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget. 2012;3:954–987. doi: 10.18632/oncotarget.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nogueira L, Ruiz-Ontanon P, Vazquez-Barquero A, Moris F, Fernandez-Luna JL. The NFkappaB pathway: a therapeutic target in glioblastoma. Oncotarget. 2011;2:646–653. doi: 10.18632/oncotarget.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi H, Shi X, Liu KJ. Oxidative mechanism of arsenic toxicity and carcinogenesis. Mol Cell Biochem. 2004;255:67–78. doi: 10.1023/b:mcbi.0000007262.26044.e8. [DOI] [PubMed] [Google Scholar]
- 38.Chen F, Castranova V, Li Z, Karin M, Shi X. Inhibitor of nuclear factor kappaB kinase deficiency enhances oxidative stress and prolongs c-Jun NH2-terminal kinase activation induced by arsenic. Cancer Res. 2003;63:7689–7693. [PubMed] [Google Scholar]