

Abstract
Visceral leishmaniasis (VL) in South Asia is a serious disease affecting children and adults. Acute VL develops in only a fraction of those infected individuals, the majority being asymptomatic with the potential to transmit infection and develop disease. We followed 56 individuals characterized as being asymptomatic by sero-positivity with rk39 RDT in a hyper endemic district of Bangladesh to define the utility of Leishmania-specific antibodies and DNA in identifying infection. At baseline, 54 of the individuals were sero-positive with one or more quantitative antibody assays and antibody levels persisted at follow-up. Most sero-positive individuals (47/54) tested positive by qPCR at baseline, but only 16 tested positive at follow-up. The discrepancies among the different tests may shed light on the dynamics of asymptomatic infections of L. donovani, as well as underscore the need for standard diagnostic tools for active surveillance as well as assessing the effectiveness of prophylactic and therapeutic interventions.
Keywords: Biomarkers, vaccine, leishmania, asymptomatic infection, intracellular pathogens, PCR, ELISA
INTRODUCTION
One of the characteristics of neglected diseases is the lack of tools for successful disease management or prevention. As an example, it has only been recently that tools to detect infected individuals have begun to be applied in regions endemic for Leishmania donovani. Leishmania are vector-borne protozoan parasites spread by the bite of the infected phlebotomine sand-fly (1, 2). Two Leishmania spp. cause visceral leishmaniasis (VL; also known as kala-azar) which is the most severe form, often fatal if not treated (3). Half a million new VL cases are diagnosed every year and, with a mortality rate of 10%, VL is second only to malaria in terms of fatality (4).
In the Indian sub-continent VL is caused by L. donovani. An estimated 100,000 VL cases are reported annually in the endemic foci of northeastern India, Nepal and Bangladesh, costing 400,000 disability adjusted life years and putting 150 million people at risk for infection(5) The situation in this region, where humans are the only reservoir of the parasite, makes elimination feasible (3). In 2005 these three countries committed to a kala-azar elimination program, with the intent of decreasing incidence to 1 in 10,000 per year by 2015 (6). Goals of the program include early diagnosis and treatment, coordinated vector control and effective disease surveillance through passive and active case detection (7). A pressing concern that could interfere with this goal is the presence of large populations who harbor infection with no overt signs of disease but who can potentially develop VL and likely spread infection (8, 9). Considerably more asymptomatic individuals than those with VL disease are identified in endemic areas (10, 11).
Traditionally, both direct (detection of parasites) and indirect (detection of antibodies) tests have been used in the diagnosis of VL. The standard diagnostic method for VL has been microscopic visualization of amastigotes in splenic, lymph node or bone marrow aspirates, ethically and technically unsuitable for asymptomatic individuals(12). The kala-azar elimination program does not evaluate asymptomatic individuals because defined parameters to identify low levels of infection are lacking. A limited number of antibody detection tests including the direct agglutination test (DAT), ELISA, and immunochromatography-based rapid tests, have been standardized (12). Given the endemic nature of L. donovani infection, serological tests lack the ability to discern between uninfected, antibody-positive individuals and asymptomatic infected individuals (13, 14). Unlike the situation in Africa and Brazil, Leishmania- specific antibodies persist for extended periods of time in the Indian sub- continent, emphasizing the importance of defining biomarkers that reflect the dynamics of asymptomatic infections (14). PCR-based tests directly demonstrating the presence of parasite nucleic acids likely represent a more accurate tool for the assessment of asymptomatic individuals, with the advantage of being sensitive enough to detect very low levels of parasite DNA (15). Serologic and PCR- based techniques can complement each other to establish how these biomarkers reflect asymptomatic L. donovani infection. Given the potential availability of vaccines against L. donovani, these tests could be used to identify individuals or populations that will benefit most from vaccination.
To determine the utility of various tests, a group of asymptomatic infected individuals in a L. donovani hyper endemic region were examined over a period of 24 months with serological tests, DAT and ELISA, as well as a sensitive qPCR test to define the dynamics of these biomarkers.
METHODS
Study design, ethics and parameters
We defined an asymptomatic infected individual as a person from a VL endemic area with no past history of VL or PKDL, clinically healthy, and positive by the rK39 rapid test (Kala-azar DETECT™, Inbios, Seattle, USA) in the field using finger prick blood. We recruited 3,849 clinically healthy people in the Harirampur Union of sub-district Trishal, Mymensingh district which is hyperendemic for VL with a reported incidence of 65 per 10,000 people in 2007 (Trishal Hospital). Initial consent was obtained from the head of the household to screen household members based on past VL or PKDL history, then individual written consent was obtained before enrollment in the study. Initial screening was conducted using the kala-azar detect™ rk39 RDT and 332 were found positive, with 200 deemed fit to participate in the study based on no prior history of VL or PKDL (50% were female and 35.5% were under 15 years). Fifty six were then enrolled as study subjects based on the availability of matching serum and DNA samples when the study was initiated (baseline) and 12 months after initiation (follow-up) (Figure 1A). All serological and PCR tests were conducted on these 56 serum and DNA samples collected at baseline and 12 month follow-up (Figure 1B). The enrollees were monitored each month for clinical symptoms of VL during household visits up to 24 months after study initiation, with 3 of the 56 enrollees developing VL disease by 24 months. They were referred to the study clinic where a qualified medical officer examined them following the diagnostic criteria for VL of the National Guideline before referring to the Trishal Hospital for treatment (Figure 1B) (16).
Figure 1. Study enrollment criteria, timeline and tests performed on study subjects.
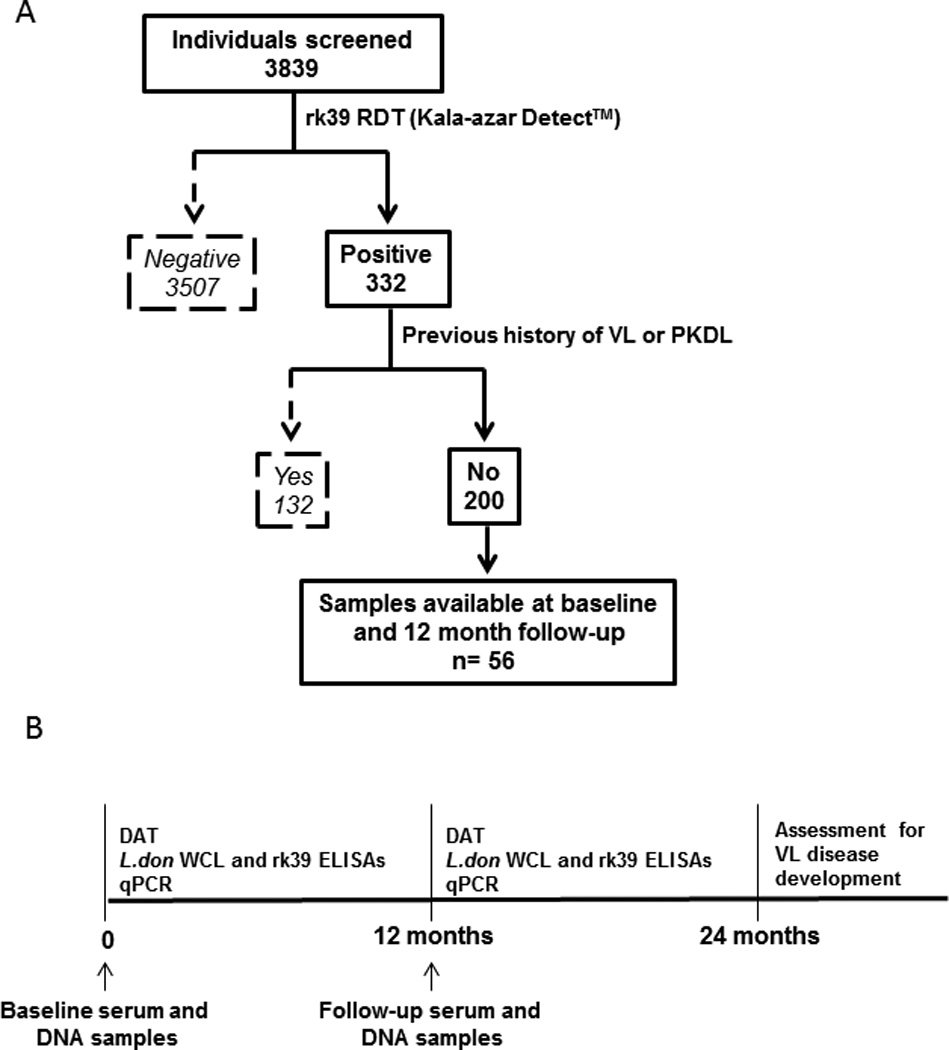
A) Out of 3839 individuals screened in the Mymensingh district of Bangladesh, 332 tested positive by the rk39 rapid test. 200 of them were eligible for the study because they had no past history of VL/ PKDL. Of these, 56 were enrolled on the basis of availability of matching serum and DNA samples at study initiation (baseline) and 12- months after initiation (follow-up). B) Timeline of sample collection and tests performed during the course of the study. Serum and DNA were extracted from the 56 study subjects at baseline and 12 month follow up. After that they were monitored for VL disease development according to the Bangladesh National Guidelines by monthly surveillance until 24 months.
Sample collection and storage
Blood specimens were collected at enrollment and then at 12 months. Blood specimens were collected by venipuncture. For DNA extraction, an aliquot of 2 ml was placed in an EDTA containing vacutainer and centrifuged at 3500rpm for 20 minutes for separation of buffy coat. Collected buffy coat was transported to the ICDDRB Parasitology Laboratory maintaining cold chain and DNA was isolated using QIAamp DNA blood mini kit (Qiagen, Hilden, Germany) as per the manufacturer’s instructions. For serum preparation, 2 mL of blood was placed in red-top vacutainers and allowed to clot at room temperature for an hour and centrifuged for 5 minutes for separation of serum. Collected serum was transported as above.
Serological tests
DAT was performed according to the manufacturer’s instructions (KIT Biomedical Research, Amsterdam, Netherlands) with minor modifications (17). Briefly, in V-bottom 96-well plates, healthy US control and test serum samples were serially diluted two-fold Ssarting at 1:400 in 0.9% sodium chloride added 0.8% β-mercaptoethanol at a final volume of 50 μL per well. Antigen re-suspended in 50 μL of 0.9% sodium chloride was then added per well. Each plate included at least two blank wells containing only sample diluent. After brief mixing, plates were covered and left undisturbed at room temperature for 18 hours before reading. Samples were run in singlet per plate and duplicated by a second researcher. Results were independently scored by three readers. Each reader recorded the titer as the last sample dilution at which agglutination was apparent. The titer for each sample was taken as the median score from the three readers, and the scores from the two plates were compiled to obtain a final titer. In the event of disagreement between duplicates, the higher of the two titers was chosen. Samples with a final titer of 1:1600 or higher were considered positive.
Serum antibodies against L. donovani whole cell lysate (WCL) and recombinant k39 were assessed by ELISA. In brief, 0.5 μg per well of WCL or 0.1 μg per well of antigens at 50 μL/well in 0.1M PBS pH 7.2 were used to coat 384-microwell plates (Corning, Tewksbury, USA) at 4°C overnight. After blocking with 1% BSA in PBS/0.05%Tween-20, serum samples at 1:100 dilutions were added to wells in triplicate and allowed to incubate at room temperature (RT) with shaking for 1.5 hours. After washing, 1:5000 diluted peroxidase conjugated donkey anti-human IgG H+L chains (Jackson ImmunoResearch, Westgrove, PA) were added for 1 hour with shaking. Plates were developed with SureBlue TMB Microwell peroxidase (Thermofisher, Lafayette, CO) for five minutes and stopped with 1N H2SO4. Plates were read immediately at 450 and 570nm (Synergy 2 plate reader, Biotek, Winooski, VT) and final optical density was recorded as the difference between the two. Mean values from triplicate readings for each sample were calculated. A cut-off value for positive results was determined as the mean OD of non- endemic control sera from healthy U.S normals added two standard deviations. Sera from 8 confirmed VL patients from Bangladesh and Sudan served as positive controls.
DNA extraction and PCR tests
Genomic DNA was extracted from the buffy coat of blood samples using the QiaAMP DNA blood mini kit (Qiagen) in the ICDDRB Parasitology laboratory. An aliquot of DNA was shipped to IDRI, Seattle. Quantitative detection of Leishmania DNA was performed on a Roche LightCycler 480 system (Roche, Indianapolis, IN) using Taqman primers and probes; 5’- GCGACGTCCGTGGAAAGAA-3’, 5‘-GGCGGGTACACATTAGCAGAA-3’ and reporter (FAM): 5’-CAACGCGTATTCCC-3’ (Applied Biosystems Inc., Foster City, CA). The custom gene assay was developed using ABI FileBuilder 3.1 (open selection) from the conserved region of Leishmania REPL repeats (L42486.1) which can be detected in L. donovani and L. infantum (18, 19). For standard curves, L. donovani promastigotes were spiked into uninfected human blood/paxgene mixtures to provide a concentration of 1000 parasites/mL in duplicate, allowed to incubate at room temperature for 4 hours, and then serially diluted with human blood/paxgene mixture to provide a range of 1000 to 10−1 parasites/mL blood. The dilution set was frozen at −20°C overnight, then DNA was extracted using the QIAamp DNA Mini Kit. No template controls and blood from healthy US donors were included in each assay. Samples were initially examined in duplicate; for those samples that did not amplify, or had very late amplification (≥ 40 cycles) the assay was repeated in triplicate. Mean Cp values of amplification were extrapolated against the standard curve to enumerate the number of parasites per mL of blood. Samples that displayed no products at >40 cycles were considered negative. A 2-tailed, paired t-test was performed to determine correlations between the circulating parasite DNA at baseline and follow-up.
RESULTS
qPCR as a measure of circulating parasite DNA
The presence of L. donovani DNA was assessed by quantitative PCR (qPCR), which is sensitive enough to detect the DNA equivalent of 0.01 L. donovani/mL of blood and can be accurately extrapolated to a standard curve to measure the presence of parasite DNA equivalents in the blood of study subjects. The number of PCR-positives fell from 47 of the 56 study subjects (84%) at baseline to 16 (28%) at 12 month follow-up (Table 1). One new PCR positive emerged at follow-up while 40 positives at intake became PCR negative. The predicted parasite burden in these samples was very low, the median being 0.1 L. donovani DNA equivalents/mL of blood. A plot of the individual trends of PCR status through the study of the 40 study subjects who became negative at follow-up and illustrates a highly significant drop in PCR positivity at follow-up is noticed with a p-value of 2.5*e−9 by a paired, 2-tailed Student’s t-test (Figure 2A). The 16 who remained positive at follow-up showed similar parasite DNA contents at baseline and follow-up with one negative gaining detectable parasite DNA at follow-up (Figure 2B). The data suggests that the presence of L.donovani DNA is transient, consistent with results obtained in a larger survey of asymptomatic children in Brazil (20).
Table 1. Summary of study outcome.
Number and percentages of study subjects who tested positive by the indicated tests at baseline and 12 month follow- up including those that became negative and new positives detected at follow- up.
| Diagnostic test | Positive at baseline (%) |
Positive at 12 month follow-up (%) |
Became negative at follow-up |
Became positive at follow-up |
|---|---|---|---|---|
| DAT | 32 (57%) | 32 (57%) | 2 | 2 |
| Ld WCL ELISA | 46 (82%) | 42 (75%) | 8 | 4 |
| rk39 ELISA | 50 (89%) | 51 (92%) | 1 | 2 |
| qPCR | 47 (84%) | 16 (28%) | 32 | 1 |
Figure 2. L.donovani DNA equivalents measured by qPCR, quantitated by extrapolating to a standard curve with known numbers of parasite.
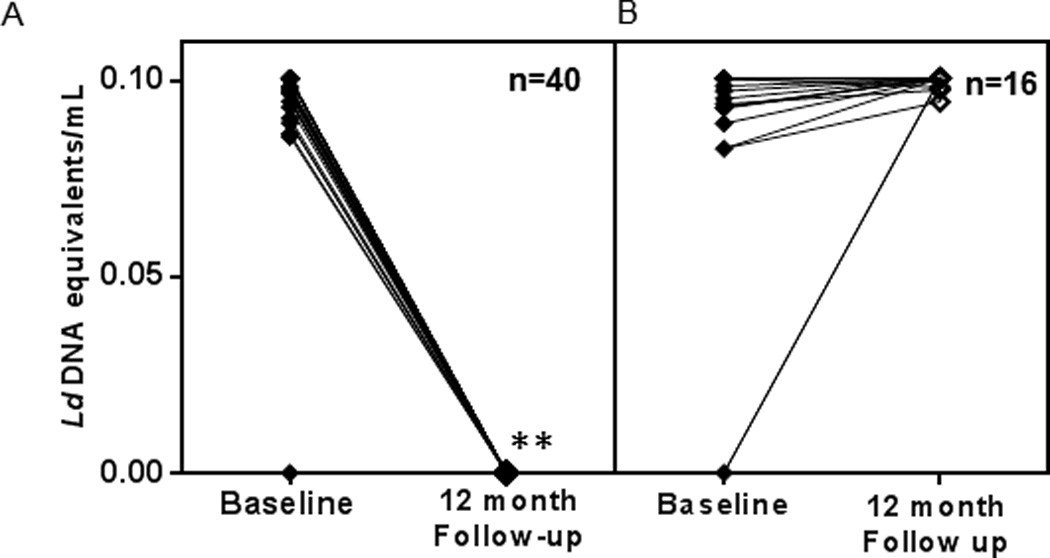
A) Progression of L.donovani DNA loads of the 40 study subjects who tested PCR negative at 12 month follow-up. B) Progression of L.donovani DNA loads of the 16 study subjects who tested PCR positive at 12 month follow-up. Each line represents an individual tested at baseline and follow-up and n represents the number of individuals. **L.donovani DNA equivalents at baseline and follow-up were significantly different as measured by a p value of 2.5*e−9 by a paired, 2- tailed t-test.
DAT provides an incomplete indication of asymptomatic infection
DAT is a standard test, developed primarily for serological confirmation of VL. When applied to sero-evaluation of asymptomatic individuals, DAT positivity was 57%, both at baseline and 12 month follow-up (Table 1). 32 study subjects were DAT positive at baseline with 30 of the 32 remaining positive at follow-up (Table 1). At follow-up, two new positives emerged. At baseline and follow-up, the median dilution at which agglutination was observed remained 3200 (Figure 3A). Correlation of DAT with PCR at baseline was about 64% while 2 PCR negative samples were DAT positive (Figure 4A). We then compared DAT and PCR profiles at follow- up to see how the dynamics of Leishmania-specific antibodies correlates to parasite DNA. DAT profiles at follow-up revealed poor correlation to PCR. 22 of the 40 PCR negatives at follow- up (55%) remained DAT positive, 2 among them being new sero-converts (Figure 4A). DAT profiles of the 16 PCR positives at follow-up reveal a 63% correlation, with 10 also remaining DAT positive (Figure 4B).
Figure 3. Distribution of sero-reactivity of asymptomatic individuals at baseline and follow-up.
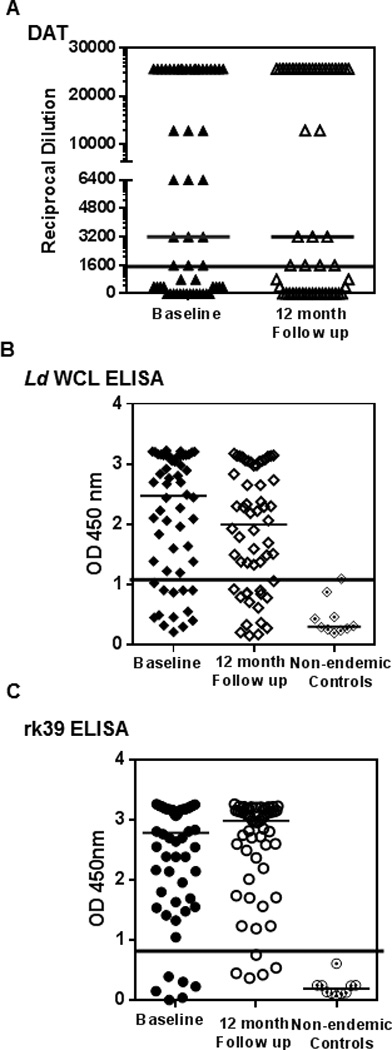
A) Comparison of the reciprocal serum dilution at which agglutination was observed by DAT of individuals at baseline (closed triangles) and follow-up (open triangles). Black line represents cut- off for positivity at 1600 B) Comparison of OD at 450 nm of individuals at baseline and follow-up by antibody detection ELISAs against L.donovani WCL (closed and open diamonds) along with non- endemic controls (dotted diamonds). Black line represents cut- off OD for positivity at 1.05 C) Comparison of OD at 450 nm of individuals at baseline and follow-up by antibody detection ELISAs against rk39 (closed circles) along with non- endemic controls (dotted circles). Black line represents cut- off OD for positivity at 0.89. Medians for each distribution are represented by black bars. No significant differences between baseline and follow-up measurements were seen for any of the serological tests.
Figure 4. Correlation between qPCR and serological tests in asymptomatic individuals.
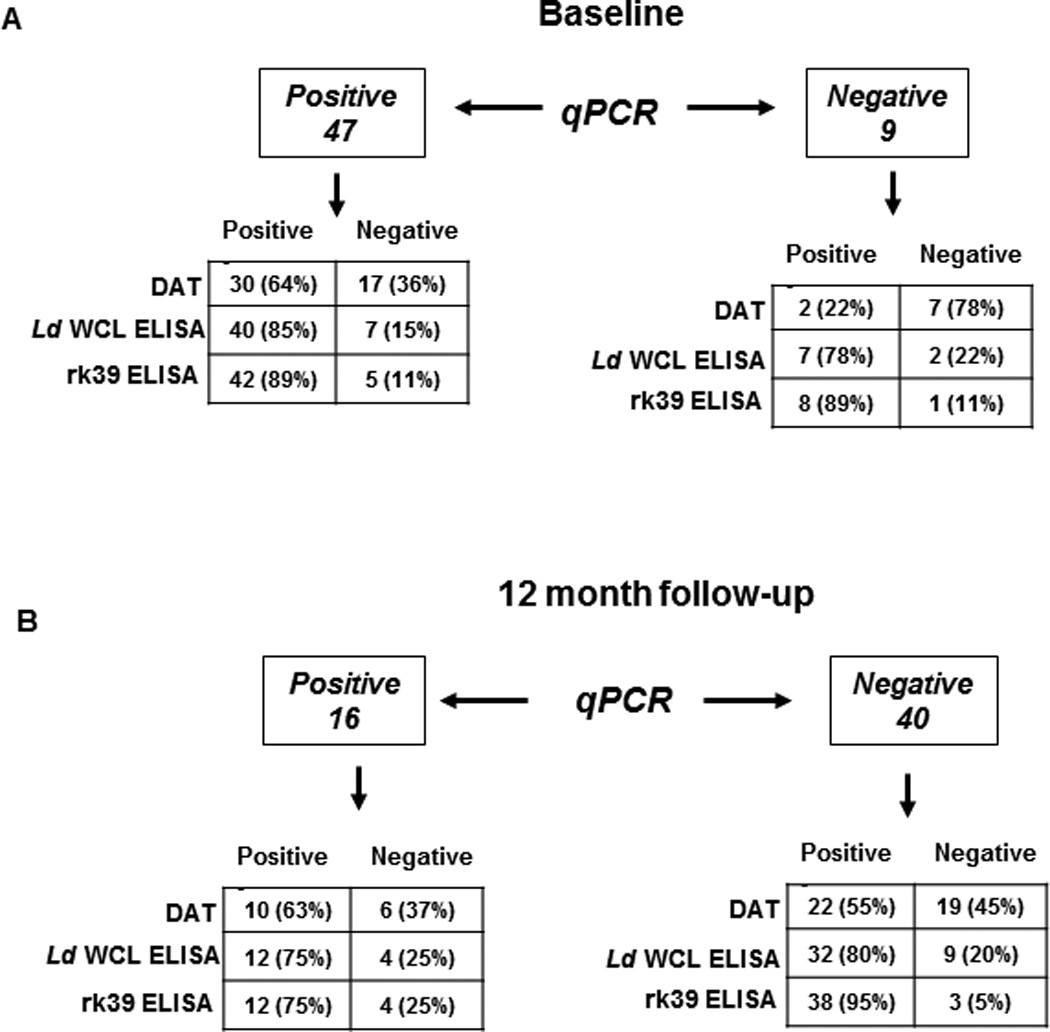
The distribution of qPCR positive and negative individuals as detected by DAT, L.donovani WCL and rk39 ELISA has been depicted. A) Correlation of qPCR positivity and negativity with DAT, L.donovani WCL and rk39 ELISA at baseline. Number of individuals positive or negative followed by percent positive or negative in parenthesis. B) Correlation of qPCR positivity and negativity with DAT L.donovani WCL and rk39 ELISA at 12 month follow-up.
ELISAs indicate persistent presence of antibodies
To provide comparability between DAT and ELISA, we used L. donovani WCL as the coating antigen to detect anti-leishmania antibodies. At baseline46 individuals were positive for antibodies against WCL (82%) of which 38 remained positive at the 12 month follow-up, with 4 new seroconverts (75%) (Table 1). Similar to the results obtained with DAT, the median OD was relatively unchanged over time, 2.5 at baseline and 2.0 at follow-up (Figure 3B). At baseline, L.donovani WCL positivity correlated well with PCR positivity, with 40 of the 47 PCR positives also testing positive for L.donovani WCL antibodies (85% correlation) but 7 of the 9 PCR negatives also tested positive for L.donovani WCL antibodies (78%) (Figure 4A). At follow-up, correlation between PCR and the L.donovani WCL ELISA was higher than with DAT, with 75% testing positive or 12 out of 16 PCR positives (Figure 4B). But 80% of PCR negatives at follow-up (32 out of 40 individuals) also remained positive by L.donovani WCL ELISA (Figure 4B).
Recombinant rk39 is a well-defined diagnostic antigen that has been used in both ELISAs and rapid detection tests (21, 22). However, tests composed of the rk39 antigen have also been used, in an off label manner for epidemiological screening, as opposed only to diagnosis. Consistent with initial inclusion of asymptomatic individuals based on rk39 RDT positivity, antibodies against rk39 were detected in sera of most study subjects at both baseline and follow-up by ELISA. At baseline, 50 (89%) tested positive by rk39 ELISA, of whom 49 remained positive at follow-up with 2 new seroconverts (92%) (Table 1). The median OD remained relatively unchanged at 2.8 and 2.9, respectively between these times (Figure 3C). Though rk39 positivity correlated well with PCR positivity at baseline, with 89% or 42 of the 47 PCR positives also being rk39 positive similar to the L.donovani WCL ELISA, 8 of the 9 PCR negatives at baseline (89%) remained rk39 positive (Figure 4, top). Among the 16 PCR positives at follow-up, 75% or 12 remained rk39 ELISA positive. But 95% of PCR negatives at follow-up (38 out of 40) still remained rk39 ELISA positive attesting to a poor correlation between rk39 ELISA and PCR, very similar to the other serological tests used in this study (Figure 4, bottom). Thus a persistent antibody response to L.donovani WCL and rk39 was seen at baseline and 12 month follow- up. The serological results of all study subjects at baseline and follow-up relative to PCR are represented in Figure 6.
Figure 6. Serological and PCR test status of all samples at baseline and 12-month follow-up, compared side-by-side.
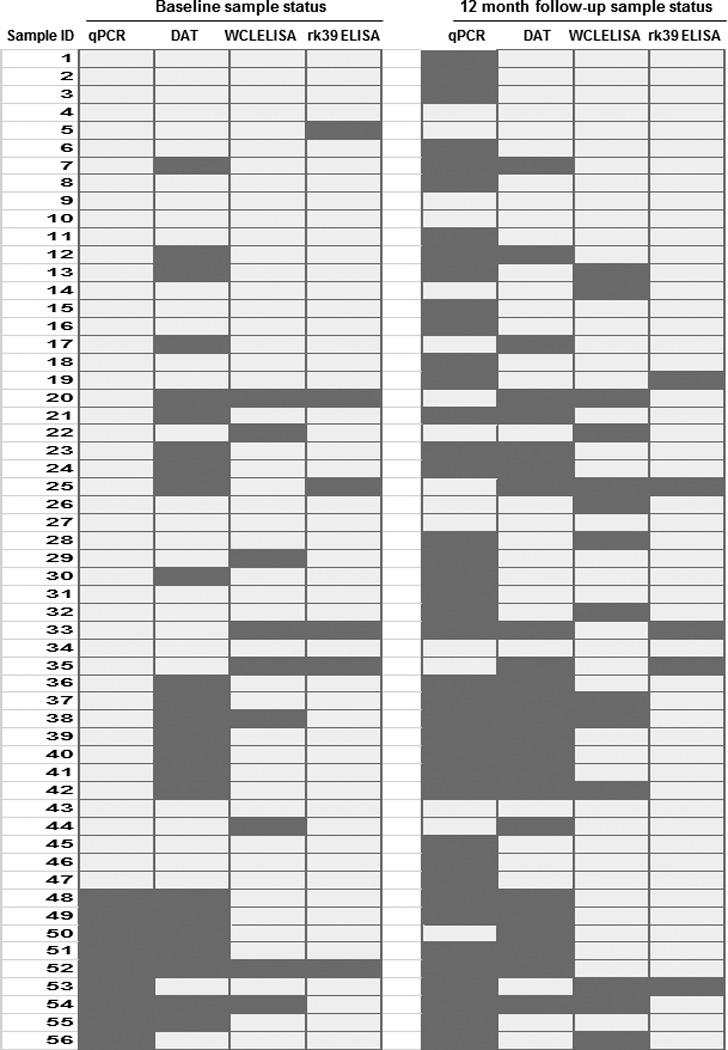
Light gray shaded- boxes indicate positive status, while dark gray boxes indicate negative status
Serological tests are complementary
We then compared the complementation among the three serological tests. L. donovani WCL ELISA detected 5 out of 6 rk39 ELISA negative individuals (83% complementarity) at baseline while DAT could detect 3/6 (50% complementarity) (Figure 5A, left). With 2 individuals being positive by both tests, 1 was detected only by DAT and 3 were detected only by L. donovani WCL ELISA (Fig 6). At follow-up, L.donovani WCL ELISA detected 3 out of 5 rk39 negative individuals (60% complementarity) compared to 2 by DAT (40% complementarity) (Figure 5A, right). Because DAT positivity was lower than ELISA positivity in this study and DAT is a widely used diagnostic test in the Indian sub-continent we asked whether ELISA could complement DAT. At baseline, 21 of the 24 DAT negatives were positive by rk39 ELISA with the trend remaining the same at follow- up (Figure 5B). 20 of the 24 DAT negatives at baseline were positive by L.donovani WCL ELISA and 18 of the 24 DAT negatives at follow-up were positive by L.donovani WCL ELISA (Figure 5B). Both DAT sero-converts at follow- up were positive by rk39 ELISA while one was positive by L. donovani WCL (Figure 6).
Figure 5. Complementation of serological tests in asymptomatic individuals.
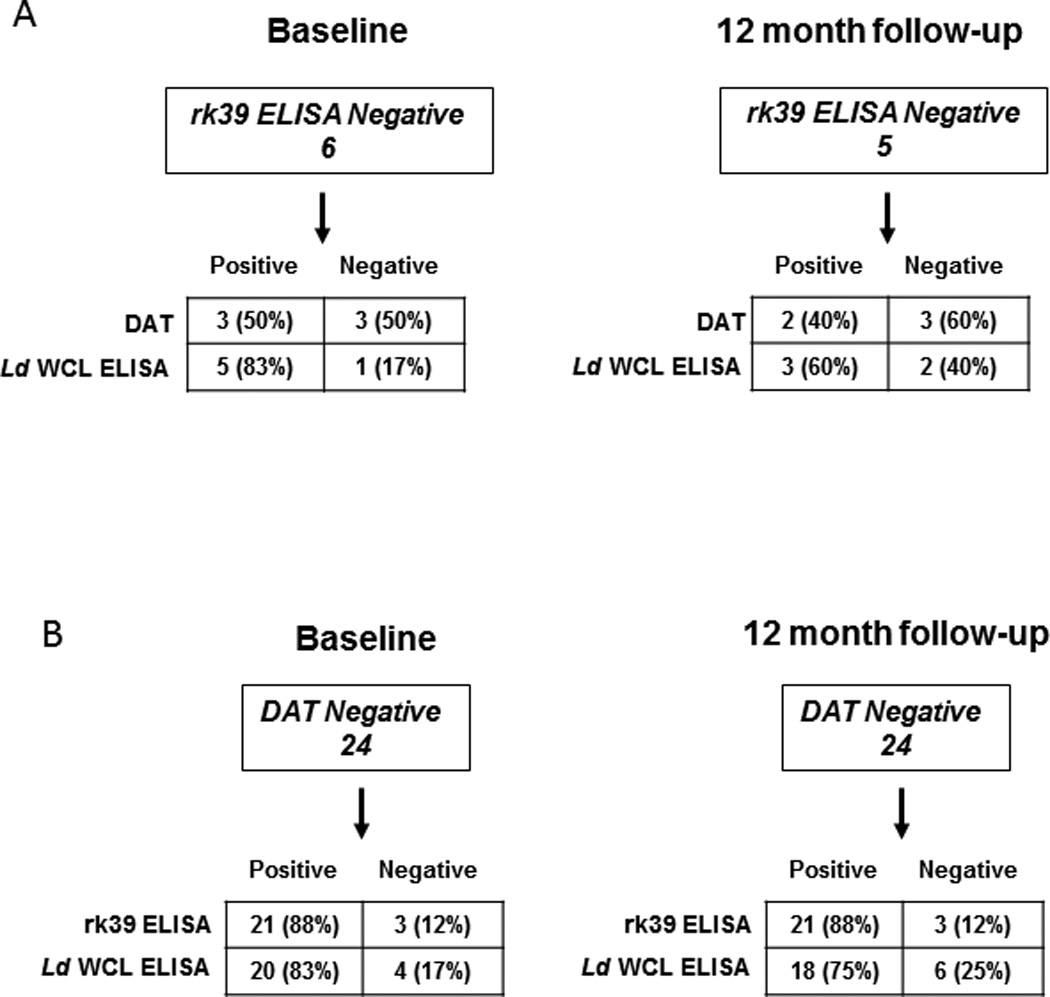
Detection ofrk39 and DAT negative individuals. A) Complementation of rk39 ELISA negativity by DAT and L.donovani WCL ELISA at baseline and 12 month follow-up. B) Complementation of DAT negativity by rk39 and L.donovani WCL ELISA at baseline and 12 month follow-up.
DISCUSSION
Disease intervention programs, particularly disease elimination programs, will benefit from proactive identification of high risk individuals. A comprehensive application of biomarker based tests for diseases in which humans are the primary or only source of infection can make such identification possible. An excellent example of a disease to which this strategy may be applied is VL. Because VL in the Indian subcontinent is anthroponotic, identifying asymptomatic infected individuals should be considered a priority within kala-azar elimination programs (23). Developing tools that can predict progression to VL disease will lead to early intervention and treatment, two goals of the elimination program (6). In previous studies to identify such tools in India, the rk39 RDT and DAT have proven to be of limited utility in predicting progression to disease, because neither did the tests correlate with each other nor did sero-conversion correlate with disease progression (11,23,27). To develop better tools to reflect the dynamics of asymptomatic infection, we wanted to determine the utility of a direct method (qPCR) and indirect methods (serology) for detecting asymptomatic L. donovani infection. We conducted a study on 56 rk39 RDT positive individuals in a VL-endemic area of Bangladesh using DAT and ELISA to detect antibodies and qPCR to detect DNA over 24 months. 54 of the 56 enrollees at baseline and 55 of 56 enrollees at follow- up tested positive by any of the serological tests used (Figure 6). This indicates that in endemic areas, antibodies to Leishmania antigens can be encountered in the absence of disease mirroring the observation that circulating antibodies are observed over extended periods after cure (13, 14). At baseline the PCR results agreed closely with serological tests, but at the 12 month follow-up the number of PCR positives, was significantly decreased while serology positives remained more or less unchanged (Table 1 and Figure 6). However, given the low parasite burden of asymptomatic infections, it is unclear whether the results reflect the actual number of individuals with active. Studies are underway to determine the robustness of PCR methods to detect low parasite burdens.
Though all 56 study subjects tested positive by the rk39 rapid test at study intake, 6 baseline samples tested negative by ELISA against rk39 (Figure 5, top). This discrepancy can be attributed to both differences in the sensitivity of the rapid test between whole blood and serum and the use of a quantitative ELISA with stringent cut-offs to identify positives, highlighting the utility of standardized tests with defined cut-offs for sensitivity when screening populations. Rapid tests may be more suitable to confirm infection in the presence of symptoms. The complementation of the few rk39 negative sera by the other serological tests underlines the utility of a broader panel of antigens provided by both lysed or freeze- dried promastigotes to detect early infection (Figures 5 and 6). Our data indicated that L. donovani WCL ELISA complemented rk39 better than DAT. A quantitative test such as ELISA may be more sensitive in detection compared to DAT in which agglutination is visually assessed.
DAT is used extensively in the Indian sub-continent for both screening and detection (11, 24, 25). Similar to other reports, our DAT results did not correlate with PCR (15, 26). A study conducted in the VL-endemic Terai region of Nepal found PCR positivity among healthy individuals with no history of past VL to be 17.6% and among treated VL patients to be 25.6%, while DAT positivity in these groups was 5.5% and 95%, respectively (26). In the same study, DAT positivity was higher and correlated well with PCR in healthy household contacts of persons with present or past VL disease, confirming previous observations that constant exposure to VL patients increases the risk of acquiring infection (10, 23, 24, 27).
Among the 56 study subjects, only 3 went on to develop VL within the 24 month study period, about 5%. Of those, 2 individuals remained consistently positive by PCR and serology, while the other was PCR positive at baseline and follow-up but consistently negative by all serological tests at baseline and positive by only rk39 ELISA at follow-up. DNA has a shorter half-life than antibodies, and its presence in blood constitutes direct evidence of ongoing infection (14, 28). Appearance of DNA could also precede the appearance of detectable antibodies, signaling recent infections. In a canine model of VL, circulating parasite DNA was detected by PCR 4 months after, while Leishmania specific antibodies were detected by ELISA 6 months after infection reflecting the different kinetics of these biomarkers (29). Because PCR positivity was a a shared feature of those that progressed to disease, sensitive and standardized DNA amplification platforms will be useful tools in active surveillance for asymptomatic infections and effectively complement standardized point- of- care serological tests. The small number of individuals who are both serology and PCR positive may represent clusters or hyper-endemic foci within endemic regions that are at the highest risk of transmitting the parasite or progressing to disease. Hence, tests that detect the presence of parasite DNA and Leishmania- specific antibodies will identify individuals likely to benefit most from a vaccine or other intervention (30). Because vaccines against VL are in development, the tests used together could help set enrollment criteria for trials as well as define end- points to assess vaccine efficacy.
A limitation of this study was the small sample size, which may be the reason for lower disease conversion rates (5%) than previously reported (11, 23). We are also aware that, because positivity in the rk39 RDT was the criterion for initial screening in this study, we may have pre-selected individuals who were already high in antibody titers against Leishmania. This study was undertaken to identify tools that can reveal how asymptomatic infection emerges, contributes to VL incidence and how to design vaccine trials (29). To clearly understand the utility of the tools we generated, they should be used in longitudinal studies on large populations within endemic regions with no pre-selection except for past VL disease. Such a study will help us better utilize these biomarkers in the detection of asymptomatic infection and design effective programs for disease control.
Acknowledgements
We would like to thank Dr. Franco Piazza for his valuable assistance in the review of the manuscript, Dr. Randall F. Howard for project management support and Dr. Ajay Bhatia and Dr. Greg C. Ireton for assisting in procuring samples from Bangladesh.
Funding:
This work was supported by grants from the Bill and Melinda Gates Foundation (631 and 39129). M.S.D, R.N.C and S.G.R were also supported by the National Institute of Allergy And Infectious Diseases of the National Institutes of Health under Award Number R01AI025038. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funding agencies were not involved in data analysis or interpretation.
Abbreviations
- VL
visceral leishmaniasis
- L. donovani / L.d.
Leishmania donovani
- DAT
direct agglutination test
- RDT
Rapid Detection Test
- ELISA
enzyme-linked immunosorbent assay
- WCL
whole cell lysate
- rk39/rk28
recombinant k39 or k28
- qPCR
Quantitative PCR
- Ln-PCR
Leishmania-specific nested PCR
REFERENCES
- 1.Herwaldt BL. Leishmaniasis. Lancet. 1999;354:1191–1199. doi: 10.1016/S0140-6736(98)10178-2. [10.1016/S0140-6736(98)10178-2]. [DOI] [PubMed] [Google Scholar]
- 2.Desjeux P. Leishmaniasis: current situation and new perspectives. Comp Immunol Microbiol Infect Dis. 2004;27:305–318. doi: 10.1016/j.cimid.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 3.van Griensven J, Diro E. Visceral leishmaniasis. Infect Dis Clin North Am. 2012 Jun;26(2):309–322. doi: 10.1016/j.idc.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 4.Desjeux P. Disease Watch Focus: Leishmaniasis. Nature Rev Microbiol. 2004;2:692–693. doi: 10.1038/nrmicro981. [DOI] [PubMed] [Google Scholar]
- 5.Sundar S, Arora R, Singh SP, Boelaert M, Varghese B. Household cost-of-illness of visceral leishmaniasis in Bihar, India. Trop Med Int Health. [Research Support, Non-U.S. Gov't] 2010 Jul;15(Suppl 2):50–54. doi: 10.1111/j.1365-3156.2010.02520.x. [DOI] [PubMed] [Google Scholar]
- 6.Dhillon GP, Sharma SN, Nair B. Kala-azar elimination programme in India. J Indian Med Assoc. 2008 Oct;106(10):664, 6–8. [PubMed] [Google Scholar]
- 7.Matlashewski G, Arana B, Kroeger A, Battacharya S, Sundar S, Das P, et al. Visceral leishmaniasis: elimination with existing interventions. Lancet Infect Dis. 2011 Apr;11(4):322–325. doi: 10.1016/S1473-3099(10)70320-0. [DOI] [PubMed] [Google Scholar]
- 8.Stauch A, Sarkar RR, Picado A, Ostyn B, Sundar S, Rijal S, et al. Visceral leishmaniasis in the Indian subcontinent: modelling epidemiology and control. PLoS Negl Trop Dis. 2011 Nov;5(11):e1405. doi: 10.1371/journal.pntd.0001405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bern C, Courtenay O, Alvar J. Of cattle, sand flies and men: a systematic review of risk factor analyses for South Asian visceral leishmaniasis and implications for elimination. PLoS neglected tropical diseases. [Review] 2010;4(2):e599. doi: 10.1371/journal.pntd.0000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bern C, Haque R, Chowdhury R, Ali M, Kurkjian KM, Vaz L, et al. The epidemiology of visceral leishmaniasis and asymptomatic leishmanial infection in a highly endemic Bangladeshi village. Am J Trop Med Hyg. 2007 May;76(5):909–914. [PubMed] [Google Scholar]
- 11.Ostyn B, Gidwani K, Khanal B, Picado A, Chappuis F, Singh SP, et al. Incidence of symptomatic and asymptomatic Leishmania donovani infections in high-endemic foci in India and Nepal: a prospective study. PLoS Negl Trop Dis. 2011 Oct;5(10):e1284. doi: 10.1371/journal.pntd.0001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srivastava P, Dayama A, Mehrotra S, Sundar S. Diagnosis of visceral leishmaniasis. Trans R Soc Trop Med Hyg. [Research Support, N.I.H., Extramural Review] 2011 Jan;105(1):1–6. doi: 10.1016/j.trstmh.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hailu A. Pre- and post-treatment antibody levels in visceral leishmaniasis. Trans R Soc Trop Med Hyg. 1990;84:673–675. doi: 10.1016/0035-9203(90)90141-z. [10.1016/0035-9203(90)90141-Z]. [DOI] [PubMed] [Google Scholar]
- 14.Gidwani K, Picado A, Ostyn B, Singh SP, Kumar R, Khanal B, et al. Persistence of Leishmania donovani antibodies in past visceral leishmaniasis cases in India. Clin Vaccine Immunol. [Research Support, Non-U.S. Gov't] 2011 Feb;18(2):346–348. doi: 10.1128/CVI.00473-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cota GF, de Sousa MR, Demarqui FN, Rabello A. The diagnostic accuracy of serologic and molecular methods for detecting visceral leishmaniasis in HIV infected patients: metaanalysis. PLoS neglected tropical diseases. [Comparative Study Evaluation Studies Meta- Analysis Research Support, Non-U.S. Gov't Review] 2012;6(5):e1665. doi: 10.1371/journal.pntd.0001665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rahman RBM, Kabir H, Naher FB, Mahboob S, Hossain M, editors. National Guideline and Training Module for Kala-azar Elimination in Bangladesh. First edition. CDC; editor. DGHS ed2008. [Google Scholar]
- 17.Meredith SE. Leish-KIT, a stable direct agglutination test based on freeze-dried antigen for serodiagnosis of visceral leishmaniasis. J Clin Microbiol. 1995;33:1742–1745. doi: 10.1128/jcm.33.7.1742-1745.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lachaud L, Marchergui-Hammami S, Chabbert E, Dereure J, Dedet JP, Bastien P. Comparison of six PCR methods using peripheral blood for detection of canine visceral leishmaniasis. Journal of Clinical Microbiology. [Comparative Study Evaluation Studies] 2002 Jan;40(1):210–215. doi: 10.1128/JCM.40.1.210-215.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schulz A, Mellenthin K, Schonian G, Fleischer B, Drosten C. Detection, differentiation, and quantitation of pathogenic leishmania organisms by a fluorescence resonance energy transfer-based real-time PCR assay. Journal of Clinical Microbiology. [Evaluation Studies Research Support, Non-U.S. Gov't] 2003 Apr;41(4):1529–1535. doi: 10.1128/JCM.41.4.1529-1535.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.dos Santos Marques LH, Gomes LI, da Rocha IC, da Silva TA, Oliveira E, Morais MH, et al. Low parasite load estimated by qPCR in a cohort of children living in urban area endemic for visceral leishmaniasis in Brazil. PLoS neglected tropical diseases. [Research Support, Non-U.S. Gov't] 2012 Dec;6(12):e1955. doi: 10.1371/journal.pntd.0001955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pattabhi S, Whittle J, Mohamath R, El-Safi S, Moulton GG, Guderian JA, et al. Design, development and evaluation of rK28-based point-of-care tests for improving rapid diagnosis of visceral leishmaniasis. PLoS neglected tropical diseases. [Comparative Study Evaluation Studies Research Support, Non-U.S. Gov't] 2010;4(9) doi: 10.1371/journal.pntd.0000822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vaish M, Bhatia A, Reed SG, Chakravarty J, Sundar S. Evaluation of rK28 antigen for serodiagnosis of visceral Leishmaniasis in India. Clin Microbiol Infect. 2012 Jan;18(1):81–85. doi: 10.1111/j.1469-0691.2011.03540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Das VN, Siddiqui NA, Verma RB, Topno RK, Singh D, Das S, et al. Asymptomatic infection of visceral leishmaniasis in hyperendemic areas of Vaishali district, Bihar, India: a challenge to kala-azar elimination programmes. Trans R Soc Trop Med Hyg. 2011 Nov;105(11):661–666. doi: 10.1016/j.trstmh.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Gidwani K, Kumar R, Rai M, Sundar S. Longitudinal Seroepidemiologic Study of Visceral Leishmaniasis in Hyperendemic Regions of Bihar, India. Am J Trop Med Hyg. 2009 Mar 1;80(3):345–346. 2009. [PubMed] [Google Scholar]
- 25.Singh SP, Picado A, Boelaert M, Gidwani K, Andersen EW, Ostyn B, et al. The epidemiology of Leishmania donovani infection in high transmission foci in India. Trop Med Int Health. [Research Support, Non-U.S. Gov't] 2010 Jul;15(Suppl 2):12–20. doi: 10.1111/j.1365-3156.2010.02519.x. [DOI] [PubMed] [Google Scholar]
- 26.Bhattarai NR, Van der Auwera G, Khanal B, De Doncker S, Rijal S, Das ML, et al. PCR and direct agglutination as Leishmania infection markers among healthy Nepalese subjects living in areas endemic for Kala-Azar. Trop Med Int Health. [Comparative Study Multicenter Study Research Support, Non-U.S. Gov't] 2009 Apr;14(4):404–411. doi: 10.1111/j.1365-3156.2009.02242.x. [DOI] [PubMed] [Google Scholar]
- 27.Topno RK, Das VN, Ranjan A, Pandey K, Singh D, Kumar N, et al. Asymptomatic infection with visceral leishmaniasis in a disease-endemic area in bihar, India. Am J Trop Med Hyg. 2010 Sep;83(3):502–506. doi: 10.4269/ajtmh.2010.09-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prina E, Roux E, Mattei D, Milon G. Leishmania DNA is rapidly degraded following parasite death: an analysis by microscopy and real-time PCR. Microbes Infect. [Research Support, Non-U.S. Gov't] 2007 Sep;9(11):1307–1315. doi: 10.1016/j.micinf.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez-Cortes A, Ojeda A, Lopez-Fuertes L, Timon M, Altet L, Solano-Gallego L, et al. A long term experimental study of canine visceral leishmaniasis. Int J Parasitol. [Research Support, Non-U.S. Gov't] 2007 May;37(6):683–693. doi: 10.1016/j.ijpara.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 30.Bern C, Hightower AW, Chowdhury R, Ali M, Amann J, Wagatsuma Y, et al. Risk factors for kala-azar in Bangladesh. Emerg Infect Dis. 2005 May;11(5):655–662. doi: 10.3201/eid1105.040718. [DOI] [PMC free article] [PubMed] [Google Scholar]