

Abstract
Patients with renal diseases associated with salt-losing tubulopathies categorized as Gitelman and classic form of Bartter syndrome have undergone genetic screening for possible mutation capture in two different genes: SLC12A3 and CLCNKB. Clinical symptoms of these two diseases may overlap.
Bartter syndrome and Gitelman syndrome are autosomal recessive salt-losing tubulopathies with hypokalemia, metabolic alkalosis, hyperreninemia, hyperplasia of the juxtaglomerular apparatus, hyperaldosteronism, and, in some patients, hypomagnesemia.
Here we describe four patients from an inbred family with a novel missense variant in the CLCNKB gene. All of patients are asymptomatic; yet they have the typical metabolic abnormality of salt losing tubulopathies. One of those patients had hypomagnesaemia while others not. Clinical and laboratory data of all patients was described. All 4 patients have a homozygous c.490G > T missense variant in exon 5 of the CLCNKB gene. This variant alters a glycine into a cysteine on amino acid position 164 of the resulting protein (p.Gly164Cys). The c.490G > T variant is a novel variant not previously described in other patients nor controls. Polyphen analysis predicts the variation to be possibly damaging. Analysis of SLC12A3 was normal.
Here in we are describing a novel homozygous c.490G > T missense variation was identified in exon 5 of the CLCNKB gene was identified in an Emirati patients with a mild manifestation of Bartter - Gitelman syndrome.
Keywords: Bartter syndrome, Gitelman syndrome, Mutation, Phenotype, CLCNKB gene
Introduction
Bartter syndrome (BS) and Gitelman syndrome (GS) are autosomal recessive disorders with a characteristic set of metabolic abnormalities (Amirlak and Dawson 2000; Konrad et al. 2000; Naesens et al. 2004). These include hypokalemia, metabolic alkalosis, hyperreninemia, hyperplasia of the juxtaglomerular apparatus (the source of renin in the kidney), hyperaldosteronism, and, in some patients, hypomagnesemia. (Amirlak and Dawson 2000; Naesens et al. 2004).
The main pathogenesis in BS is the defect of NaCl reabsorption in the thick ascending limb of Henle's loop (TALH). (Konrad et al. 2000) Inherited BS can be divided into five subtypes according to the different disease genes involved. Antenatal BS type 1 is due to variants in the SLC12A1 gene, whereas antenatal BS type II is due to variants in the KCNJ1 encoding the inward rectifying K + channel (ROMK). Classical Bartter syndrome (type III) is caused by variants in CLCNKB encoding the kidney-specific basolateral chloride channel for cBS. Type IV by variants in BSND encoding the β-subunit of the basolateral chloride channel for aBS (type IV), and type V by variants in the calcium-sensing receptors for autosomal dominant hypocalcemia associated with hypokalemia (Amirlak and Dawson 2000; Vargas et al. 2011) Apr as shown in Table 1. In the majority of GS patients, DNA variants are found in the SLC12A3 gene, which encodes the thiazide- sensitive NaCl co-transporter (NCC). In a small minority of GS patients, variants in the CLCNKB gene encoding the chloride channel ClC-Kb have been identified (Vargas et al. 2011) Apr. Konrad et al. (Konrad et al. 2000) and (Jeck et al. 2000) reported that variants in the CLCNKB gene not only cause classical Bartter syndrome (type III), but also phenotypes that overlap with either antenatal Bartter syndrome (Types I-II) or Gitelman syndrome. In addition, a pharmacology-based classification and pharmacotype terminology for SLTs were developed and introduced in 2008 (Seyberth 2008) Oct. This classification is based on three major types of salt-losing tubulopathy can be defined: distal convoluted tubule dysfunction (thiazide-like DCT disorders) leading to hypokalemia (currently known as Gitelman or Bartter syndrome type III), the second more-severe condition of polyuric loop dysfunction and furosemide-like loop disorders (often referred to as antenatal Bartter or hyperprostaglandin E syndrome or BS types I and II), and the most-severe condition of combined loop and distal convoluted tubule dysfunction (antenatal Bartter or hyperprostaglandin E syndrome with sensorineural deafness) or BS type IV. (Jeck et al. 2004; Seyberth and Schlingmann 2011) October.
Table 1.
Genetics and presentation of Bartter and Gitelman syndromes
| Disorder | Gene affected | Gene product | Clinical presentation |
|---|---|---|---|
| Bartter syndrome type I | SLC12A1 | NKCC2 | Antenatal Bartter syndrome (Hyperprostaglandin E syndrome) |
| Bartter syndrome type II | KCNJ1 | ROMK | Antenatal Bartter syndrome |
| Bartter syndrome type III | CLCKB | CLC-Kb | Hypochloremia., mild hypomagnesemia, FTT in infancy |
| Bartter syndrome type IVA | BSND | Barttin (B-subunit of CLC-Ka and CLC-Kb) | Antenatal Bartter syndrome (Hyperprostaglandin E syndrome) and sensorineural deafness |
| Bartter syndrome type IVB | CLCNKA and CLCNKB | CLC-Ka and CLC-Kb | Antenatal Bartter syndrome (Hyperprostaglandin E syndrome) and sensorineural deafness |
| Bartter syndrome type V• | CaSR gene | CaSR | Bartter syndrome with hypocalcemia |
| Gitelman syndrome | SLC12A3 | NCC (thiazide- sensitive NaCl co-transporter). | Hypomagnesemia, hypocalcuria, growth retardation |
There are six Bartter syndrome subtypes (I, II, III, IV, IVB, and V) corresponding to six genetic defects. NKCC2: furosemide-sensitive sodium-potassium-2 chloride cotransporter; ROMK: renal outer medullary potassium channel; CLC-Kb: chloride channel Kb; CLC-Ka: chloride channel Ka; CaSR: calcium sensing receptor; NCCT: thiazide-sensitive sodium-chloride cotransporter. Modified from Seybrerth et al. (Jeck et al. 2004) Jan.
Here in, we described a case series with mild and heterogeneous phenotype, all had novel mutation in the CLCNKB gene.
Case reports
The first patient (proband1) was a 14 years old girl was presented with prolonged hypokalemia after an attack of acute gastroenteritis. Review of all systems was negative. There was no significant perinatal history, her birth history records revealed full-term by normal spontaneous delivery and normal birth weight of 3,400 grams without antenatal polyhydramnios. There was no history of chronic drug ingestion. She was doing well in school and had normal actively level, normal hearing and vision.
On presentation her height (160 cm) and body weight (58 kg) both were around the 50th percentile. Blood pressure was 114/75 mmHg, heart rate 96 beats/min and respiratory rate 22/min. The remainder of the physical examination was unremarkable. Family history was negative for chronic diseases and sudden death.
The data of biochemical studies are shown in Tables 2 and 3. The most striking findings were hypokalemia (2.4 mmol/l), metabolic alkalosis with arterial PH of 7.49 (7.35-7.45), HCO3 33 mmol/l. (22-28(mmol/L). EKG disclosed a sinus rhythm. Abdominal sonography revealed bilateral normal size kidneys without nephrocalcinosis. Urinalysis revealed negative protein, glucose, leukocytes, and red blood cells. Urine Sodium, Potassium and chloride levels were 199, 62, and 245 mmol/L respectively (all should be less than 20 mmol/L). Average urinary volume was about 1600 ml daily. The trans-tubular potassium gradient (TTKG) was 9.6 (normally it should be less than 3 in the presence of hypokalemia). Serum magnesium (Mg) was tested twice and the value was 0.61 and 0.62 mmol/L respectively, urine Mg was high with Mg/Cr 1.27 mmol/mmol (normal < 0.9) and the fractional excretion of Mg (FeMg) was 3.8 (normal < 2). Serum calcium (Ca) levels were normal, urine Ca/Cr 0.42 mmol/mmol (normal < 0.7). Normal Prostaglandin E2 (PGE2) level (140 ng/24 hrs). Sweat chloride test was normal.
Table 2.
Clinical features in all patients
| Clinical feature | Case 1 (index) | Case 2 | Case 3 | Case 4 |
|---|---|---|---|---|
| Age in years on presentation/follow up | 14 | 8 | 11 | 8 |
| Gender | Female | Male | Female | Female |
| Weight centile on presentation/follow up | 75th/90th | <5th/10th | 15th | 50th |
| Height centile on presentation/follow up | 50th/75th | 30th | 10th | 30th |
| History of polyuria and polydipsia | Negative | Negative | Negative | Negative |
| Blood pressure | 114/75 | 100/70 | 110/68 | 105/68 |
| Nephrocalcinosis/Nephrolithiasis | Negative | Negative | Negative | Negative |
Table 3.
Laboratory features of all patients
| Laboratory findings | Case 1 | Case 2 | Case 3 | Case 4 | Normal ranges |
|---|---|---|---|---|---|
| Sodium | 130 | 138 | 138 | 137 | 135-143 (mmol/L) |
| Potassium | 2.4 | 2.3 | 2.7 | 2.3 | 3.4-4.5 (mmol/L) |
| Chloride | 89 | 96 | 96 | 89 | 98-106 (mmol/L) |
| Bicarbonate | 33 | 31 | 32 | 32 | 22-28 (mmol/L) |
| Serum creatinine | 44 | 30 | 33 | 31 | 27-53 (micromol/L) |
| Blood urea | 3.6 | 4 | 2.8 | 3.9 | 2.9-7.1 (mmol/L) |
| Magnesium | 0.61 | 0.92 | 0.71 | 0.85 | 0.74-1.03 (mmol/L) |
| Renin | 216.7 | NA | NA | NA | 30-40 ng/L (resting) |
| Aldosterone | 324 | NA | NA | NA | 115-406 ng/L |
| PGE2 | 140 | 70 | 74 | 44 | 400-620 ng/24 hours |
| Urine Ca/Cr | 0.42 | 0.55 | 0.06 | NA | <0.7 mmol/mmol |
| Urine Mg/Cr | 1.37 | 0.65 | 0.28 | NA | <0.9 mmol/mmol |
Family was counseled for the screening of other siblings for metabolic abnormalities that was found in another sibling (proband 2) with same hypokalemic metabolic alkalosis (Tables 1 and 2). Two of their cousins were having same manifestation (probands 3 and 4) as explained in the family pedigree (Figure 1). DNA analysis showed genomic DNA was extracted from the peripheral white blood cells. Sequencing of the entire coding region (exons 2-20) and all intron-exon boundaries of CLCNB gene was performed. The reference sequence and exon numbering and according to Gene bank accession number NM_000085.2, with the A of the ATG start codon on position 1. A homozygous c.490G > T missense variant was identified in exon 5 of the CLCNKB gene. This variant alters a glycine into cystine on amino acid position 164 of the resulting protein (p.Gly164Cys). The c.490G > T variant is a novel variant not previously described in other patients nor controls. Polyphen analysis predicts the variant to be "possibly damaging". All of 5 patients were homozygous for the same variant, whereas their parents were heterozygous for this missense variant. Sequencing of the entire coding region (exons 1-26) and al interons-exons boundaries of the SLC12A3 gene. The reference sequence and exon numbering are according to Genbank accession number NM_000339 with the A of the ATG start codon on position 1 was done and normal results were obtained.
Figure 1.
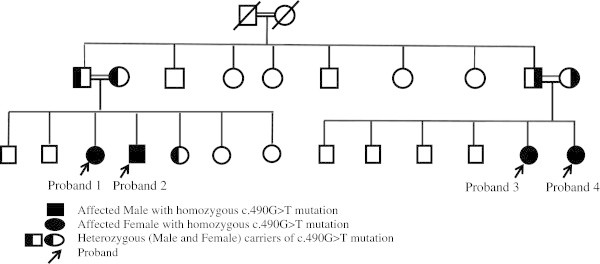
Family pedigree of the affected patient showing the affected patient proband 1 and other affected family members. Both parents are cousins and the grand parents were cousins as well. Parents of the two families were having the same mutation in a heterozygous carrier state.
Discussion
Bartter and Gitelman syndromes are genotypic and phenotypic heterogeneous diseases. Clinical manifestation and laboratory findings might be misleading in proposing diagnosis and managing appropriate treatment. (Jeck et al. 2000).
Our patients were having the typical metabolic abnormalities of BS/GS tubulopathy. Urine Na, K, and Cl were significantly high, TTKG was high in the presence of hypokalemia indicateing renal loss. Their parents and other heterozygous siblings are normal and they don’t have electrolytes abnormalities.
The main difference in the clinical presentation of Bartter and Gitelman Syndromes is explained in Table 4. (Urbanova et al. 2011) The symptoms of Bartter syndrome type III (classical Bartter) often occur in the first 2 years of life, but are usually diagnosed in school age children or even later in adolescence. Patients present with polyuria and polydipsia initially, and growth retardation later becomes evident if no early intervention is done. (Peters et al. 2002) Patients with BS are usually associated with high PGE2 production and hypercalciuria. GS usually has milder presentation with no polyuria and no failure to thrive and usually presented later in life. (Seyberth and Schlingmann 2011) October Our patients showed low PGE2 and low calcium in urine in contrary to patients with BS as they trend to have high urine PGE2 levels and hypercalciuria. (Peters et al. 2002; Brochard et al. 2009)
Table 4.
Features differentiating Bartter and Gitelman syndromes
| Features | Classic Bartter syndrome | Gitelman syndrome |
|---|---|---|
| Age at onset | Childhood (early) | Childhood or later |
| Maternal hydramnios | Rare | Absent |
| Polyuria, polydipsia | Present | Rare |
| Dehydration | Often present | Absent |
| Tetany | Rare | Present |
| Growth retardation | Present | Absent |
| Urinary calcium | Normal or high | Low |
| Nephrocalcinosis | Rare | Absent |
| Serum magnesium | Occasionally low | Low |
| Urine prostaglandins (PGE2) | High or normal | Normal |
Modified from Urbanova et al. (Peters et al. 2002).
In our patient Gitelman was initially diagnosed based on the clinical and laboratory findings and so Genetic analysis for SLC12A3 gene was done and it was normal; CLCNKB gene showed a novel mutation in the exon 5. Co-segregation of this missense variant in an inbred family with 4 affected patients suggest that this variant is pathogenic.
Clinical symptoms and biochemical markers of GS and classic form of Bartter syndrome (type III) may overlap and thus genetic analysis may specify the real cause of symptoms. (Brochard et al. 2009) Our patient had hypomagnesaemia which is due to renal loss based on the high Mg in the urine in the presence of hypomagnesaemia and the high FeMg. Mg supplement was needed for the index patient but not for the others; however other patients may develop hypomagnesaemia in the future as transition phenotypes from classical BS (cBS) to GS have also been described. (Cruz and Castro 2013 Jan).
There is a difference in both clinical and biochemical expression of CLCNKB mutations in both GS and type III BS syndromes between patients who share the same mutations suggests. (Briet et al. 2006; Uchida 2000) A modifier effect from genetic and/or environmental factors as it has been often reported in other cases of CLCNKB mutation (Dong Yan et al. 2010; Nozu et al. 2007) Sep and other human diseases such as polycystic kidney disease (Fain et al. 2005). However, the genotype–phenotype relation is variable, and mutations in the CLCNKB gene may cause overlapping phenotypes of classic/antenatal BS, cBS/GS, and GS (Cruz and Castro 2013; Fain et al. 2005; Peters et al. 2002). Several former studies tried to focus on the correlation between specific DNA mutation and phenotypic clinical outcome. In a study by Coto, many individuals carrying exactly the same mutation coming from unrelated families did not correlate in values of ionic composition in blood and urine. Their clinical symptoms also differed. (Coto et al. 2004) To date, more than 30 CLCNKB variants have been reported (Pierre Robitaille et al. 2011; Israel et al. 2003; Fukuyama et al. 2003; Fukuyama et al. 2004; Rodriguez et al. 2005; Yu et al. 2010; Xiumin et al. 2013; Gorgojo et al. 2006; Lee et al. 2012; Toshihiro et al. 2006; Enriquez et al. 2010; Konrad et al. 2000a) in patients with classical BS phenotype, atypical BS or mixed Bartter-Gitelman phenotypes as found in our patients. Our family shows that even patients and even siblings with the same DNA variants could present differences in clinical symptoms, and even mimic a different syndromes. This was the case in a study by Zelikovic, where a large Bedouin family sharing CLCNKB variant presented clinical characteristics specific for Gitelman syndrome, on the one side of the spectrum, to classic Bartter syndrome, on the other. (Zelikovic et al. 2003) Therefore, there is an indication for screening the CLCNKB gene in those patients with the Gitelman phenotype who do not have variants in the SLC12A3 gene. (Konrad and Weber 2003).
Conclusion
Our findings demonstrate intrafamilial phenotypic heterogeneity, namely the presence of Gitelman syndrome and classic Bartter syndrome phenotypes in kindred’s with CLCNKB c.490G > T mutation.
Consent
Written informed consent was obtained from the patients' parents for the publication of this report and any accompanying images.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AAS: Dr. Al Shibli conceptualized and designed the report, designed the data collection instruments, coordinated and supervised data collection, drafted the initial manuscript, and approved the final manuscript as submitted. MY: Dr. Yusuf participated in data collection, reviewed and revised the manuscript, and approved the final manuscript as submitted. IAN: Dr. AbouNajab participated in data collection, reviewed and revised the manuscript, and approved the final manuscript as submitted. PJW: Prof Willems did the genetic testing, reviewed and revised the manuscript, and approved the final manuscript as submitted.
Contributor Information
Amar Al-Shibli, Email: ashibli@tawamhospital.ae.
Madinah Yusuf, Email: myusuf@tawamhospital.ae.
Issam Abounajab, Email: iabounajab@tawamhospital.ae.
Patrick J Willems, Email: patrick.willems@genetic-diagnostic.net.
References
- Amirlak I, Dawson K. Bartter syndrome: an overview. Q J Med. 2000;93:207–215. doi: 10.1093/qjmed/93.4.207. [DOI] [PubMed] [Google Scholar]
- Briet M, Vargas-Poussou R, Lourdel S, Houillier P, Blanchard A. How Bartter's and Gitelman's syndromes, and Dent's disease have provided important insights into the function of three renal chloride channels: ClC-Ka/b and ClC-5. Nephron Physiol. 2006;103:7–13. doi: 10.1159/000090218. [DOI] [PubMed] [Google Scholar]
- Brochard K, Boyer O, Blanchard A. Phenotype- genotype correlation in antenatal and neonatal variant of Bartter syndrome. Nephrol Dial Transplant. 2009;24:1455–1464. doi: 10.1093/ndt/gfn689. [DOI] [PubMed] [Google Scholar]
- Coto E, Rodriguez Jeck N, Alvarez V, Stone R, Loris C, Rodriguez M, Fischbach M, Seyberth W, Santos F. A new mutation (intron 9 +1 G > T) in the SLC12A3 gene is linked to Gitelman syndrome in Gypsies. Kidney Int. 2004;65:25–29. doi: 10.1111/j.1523-1755.2004.00388.x. [DOI] [PubMed] [Google Scholar]
- Cruz AJ, Castro A. Gitelman or Bartter type 3 syndrome? A case of distal convoluted tubulopathy caused by CLCNKB gene mutation. BMJ Case Rep. 2013;22:2013. doi: 10.1136/bcr-2012-007929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Yan JI, Gang FQ, Xianting Z, Gengru JIANG. A novel splicing mutation in CLCNKB in a Chinese patient with Bartter syndrome type III. Chin Med J. 2010;123(21):3151–3153. [PubMed] [Google Scholar]
- Enriquez R, Adam V, Sirvent AE, García-García AB, Millán I, Amorós F. Gitelman syndrome due to p.A204T mutation in CLCNKB gene. IntUrolNephrol. 2010;42(4):1099–1102. doi: 10.1007/s11255-010-9850-4. [DOI] [PubMed] [Google Scholar]
- Fain PR, McFann KK, Taylor M, et al. Modifier genes play a significant role in the phenotypic expression of PKD1. Kidney Int. 2005;67:1256–1267. doi: 10.1111/j.1523-1755.2005.00203.x. [DOI] [PubMed] [Google Scholar]
- Fukuyama S, Okudaira S, Yamazato S, Yamazato M, Ohta T. Analysis of renal tubular electrolyte trans- porter genes in seven patients with hypokalemic metabolic alkalosis. Kidney Int. 2003;64:808–816. doi: 10.1046/j.1523-1755.2003.00163.x. [DOI] [PubMed] [Google Scholar]
- Fukuyama S, Hiramatsu M, Akagi M, Higa M, Ohta T. Novel mutations of the chloride channel Kb gene in two Japanese patients clinically diagnosed as Bartter syndrome with hypocalciuria. J Clin Endocrinol Metab. 2004;89:5847–5850. doi: 10.1210/jc.2004-0775. [DOI] [PubMed] [Google Scholar]
- Gorgojo JJ, Donnay S, Jeck N, Konrad M. A Spanish founder mutation in the chloride channel gene, CLCNKB, as a cause of atypical Bartter syndrome in adult age. Horm Res. 2006;65(2):62–68. doi: 10.1159/000090601. [DOI] [PubMed] [Google Scholar]
- Israel Z, Raymonde, Ali H, Valentina L, Ihab H, Nadine C, Farid N. A novel mutation in the chloride channel gene, CLCNKB, as a cause of Gitelman and Bartter syndromes. Kidney Int. 2003;63:24–32. doi: 10.1046/j.1523-1755.2003.00730.x. [DOI] [PubMed] [Google Scholar]
- Jeck N, Konrad M, Peters M, Weber S, Bonzel KE, Seyberth HW. Mutations in the chloride channel gene, CLCNKB, leading to a mixed Bartter-Gitelman phenotype. Pediatr Res. 2000;48:754–758. doi: 10.1203/00006450-200012000-00009. [DOI] [PubMed] [Google Scholar]
- Jeck N, Waldegger P, Doroszewicz J, Seyberth H, Waldegger S. A common sequence variation of the CLCNKB gene strongly activates ClC- Kb chloride channel activity. Kidney Int. 2004;65(1):190–197. doi: 10.1111/j.1523-1755.2004.00363.x. [DOI] [PubMed] [Google Scholar]
- Konrad M, Weber S. Recent Advances in Molecular Genetics of Hereditary Magnesium-Losing Disorders. J Am Soc Nephrol. 2003;14:249–260. doi: 10.1097/01.ASN.0000049161.60740.CE. [DOI] [PubMed] [Google Scholar]
- Konrad M, Vollmer M, Lemmink HH, et al. Mutations in the chloride channel gene CLCNKB as a cause of classic Bartter syndrome. J Am Soc Nephrol. 2000;11(8):1449–1459. doi: 10.1681/ASN.V1181449. [DOI] [PubMed] [Google Scholar]
- Lee BH, Cho HY, Lee H, et al. Genetic basis of Bartter syndrome in Korea. Nephrol Dial Transplant. 2012;27(4):1516–1521. doi: 10.1093/ndt/gfr475. [DOI] [PubMed] [Google Scholar]
- Naesens M, Steels P, Verberckmoes R, Vanrenterghem Y, Kuypers D. Bartter's and Gitelman's syndromes: from gene to clinic. Nephron Physiol. 2004;96(3):65–78. doi: 10.1159/000076752. [DOI] [PubMed] [Google Scholar]
- Nozu K, Fu XJ, Nakanishi K, Yoshikawa N, Kaito H, Kanda K, Krol RP, Miyashita R, Kamitsuji H, Kanda S, Hayashi Y, Satomura K, Shimizu N, Iijima K, Matsuo M. Molecular analysis of patients with type III Bartter syndrome: picking up large heterozygous deletions with semiquantitative PCR. Pediatr Res. 2007;62(3):364–369. doi: 10.1203/PDR.0b013e318123fb90. [DOI] [PubMed] [Google Scholar]
- Peters M, Jeck N, Reinalter S, Leonhardt A, Tonshoff B, Klaus GG, Konrad M, Seyberth HW. Clinical presentation of genetically defined patients with hypokalemic salt-losing tubulopathies. Am J Med. 2002;112(3):183–190. doi: 10.1016/S0002-9343(01)01086-5. [DOI] [PubMed] [Google Scholar]
- Pierre Robitaille AM, Ning H, York P. Bartter syndrome in two sisters with a novel mutation of the CLCNKB gene, one with deafness. ur J Pediatr. 2011;170:1209–1211. doi: 10.1007/s00431-011-1464-z. [DOI] [PubMed] [Google Scholar]
- Rodriguez J, Vallo A, Perez G. A founder variant in the CLCNKB gene causes Bartter syndrome type III in Spain. Pediatr Nephrol. 2005;20(7):891–896. doi: 10.1007/s00467-005-1867-z. [DOI] [PubMed] [Google Scholar]
- Seyberth HW. An improved terminology and classification of Bartter-like syndromes. Nat Clin Pract Nephrol. 2008;4(10):560–567. doi: 10.1038/ncpneph0912. [DOI] [PubMed] [Google Scholar]
- Seyberth HW, Schlingmann KP. Bartter- and Gitelman-like syndromes: salt-losing tubulopathies with loop or DCT defects. Pediatr Nephrol. 2011;26(10):1789–1802. doi: 10.1007/s00467-011-1871-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshihiro T, Mitsuru N, Yutaka T, Yumiko M, Shigetaka S, Masaaki Y, Mutsumi M, Masanori A, Katsuhiko T, Hiroshi M. Molecular Analysis of the CLCNKB Gene in Japanese Patients with Classic Bartter Syndrome. Endocrine Journal. 2006;53(5):647–652. doi: 10.1507/endocrj.K06-034. [DOI] [PubMed] [Google Scholar]
- Uchida S. In vivo role of CLC chloride channels in the kidney. Am J Physiol Renal Physiol. 2000;279:F802–F808. doi: 10.1152/ajprenal.2000.279.5.F802. [DOI] [PubMed] [Google Scholar]
- Urbanova M, Reiterova J, Štekrova J, Lnenicka P, Rysava R. DNA Analysis of Renal Electrolyte Transporter Genes Among Patients Suffering from Bartter and Gitelman Syndromes – Summary of Mutation Screening. Folia Biol (Praha) 2011;57:65–73. [PubMed] [Google Scholar]
- Vargas R, Dahan K, Kahila D, Venisse A, Riveira E, Debaix H, Grisart B, Bridoux F, Unwin R, Moulin B, Haymann JP, Vantyghem MC, Rigothier C, Dussol B, Godin M, Nivet H, Dubourg L, Tack I, Gimenez AP, Houillier P, Blanchard A, Devuyst O, Jeunemaitre X. Spectrum of mutations in Gitelman syndrome. J Am Soc Nephrol. 2011;22(4):693–703. doi: 10.1681/ASN.2010090907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiumin W, Zheng S, Meichun X, Junfen F, Li L. A Chinese Girl with Bartter Syndrome Type III due to a Novel Mutation and/or Single Nucleotide Polymorphisms (SNPs) in CLCNKB Gene. Iran J Pediatr. 2013;23(1):89–94. [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Xu C, Pan X, et al. Identification and functional analysis of novel mutations of the CLCNKB gene in Chinese patients with classic Bartter syndrome. Clin Genet. 2010;77(2):155–162. doi: 10.1111/j.1399-0004.2009.01288.x. [DOI] [PubMed] [Google Scholar]
- Zelikovic I, Szargel R, Hawash A, Labay V, Hatib I, Cohen N, Nakhoul F. A novel mutation in the chloride channel gene, CLCNKB, as a cause of Gitelman and Bartter syndromes. Kidney Int. 2003;63(24–32):647–652. doi: 10.1046/j.1523-1755.2003.00730.x. [DOI] [PubMed] [Google Scholar]